

Abstract
The aim of this review article is to describe the specific features of Stargardt disease and ABCA4 retinopathies (ABCA4R) using multimodal imaging and functional testing and to highlight their relevance to potential therapeutic interventions. Standardised measures of tissue loss, tissue function and rate of change over time using formal structured deep phenotyping in Stargardt disease and ABCA4R are key in diagnosis, and prognosis as well as when selecting cohorts for therapeutic intervention. In addition, a meticulous documentation of natural history will be invaluable in the future to compare treated with untreated retinas. Despite the familiarity with the term Stargardt disease, this eponymous classification alone is unhelpful when evaluating ABCA4R, as the ABCA4 gene is associated with a number of phenotypes, and a range of severity. Multimodal imaging, psychophysical and electrophysiologic measurements are necessary in diagnosing and characterising these differing retinopathies. A wide range of retinal dystrophy phenotypes are seen in association with ABCA4 mutations. In this article, these will be referred to as ABCA4R. These different phenotypes and the existence of phenocopies present a significant challenge to the clinician. Careful phenotypic characterisation coupled with the genotype enables the clinician to provide an accurate diagnosis, associated inheritance pattern and information regarding prognosis and management. This is particularly relevant now for recruiting to therapeutic trials, and in the future when therapies become available. The importance of accurate genotype-phenotype correlation studies cannot be overemphasised. This approach together with segregation studies can be vital in the identification of causal mutations when variants in more than one gene are being considered as possible. In this article, we give an overview of the current imaging, psychophysical and electrophysiological investigations, as well as current therapeutic research trials for retinopathies associated with the ABCA4 gene.
Keywords: ABCA4, autosomal recessive, fundus flavimaculatus, inherited retinal degeneration, inherited retinal dystrophies, phenotyping, STGD1, therapy
Introduction
Background
Stargardt disease (STGD1) was first described in 1909 by Karl Stargardt, who reported a macular dystrophy surrounded by yellow-white pisciform flecks in seven patients from two families. 1 In 1965, Franceschetti used the term fundus flavimaculatus to describe the widespread presence of flecks (see Figure 1).2,3 Initially, it was not clear whether STGD1 and fundus flavimaculatus were part of the same disease spectrum or different diseases 3 but genetic linkage in the early 1990s localised STGD1 and fundus flavimaculatus to the same locus on the short arm of chromosome 1. Thus, it was confirmed that they were both part of the same disease spectrum.4,5 In 1997, Allikmets et al. 6 identified mutations in the ABCA4 gene in association with STGD1. Initially, the ABCA4 transcript was only detected in rod photoreceptors,6–8 but later studies detected its presence in cones 9 and more recently within the retinal pigment epithelium (RPE). 10 The ABCA4 gene encodes a protein located in the outer segment disc membranes of rod and cone photoreceptors. ABCA4 has been shown to translocate N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine from the lumen to the cytoplasmic leaflet of the photoreceptor cell membrane. 11 This action removes waste retinoid compounds from photoreceptors. Mutations in ABCA4 lead to a decrease in transport activity. 11
Figure 1.

Colour fundus photograph (a), autofluorescence (b) and optical coherence tomography (OCT) imaging (c) of the same patient showing the typical features of STGD1. (a, b) Flecks are indicated by the green arrows, atrophic macula by the red arrows and the spared peripapillary region by the blue arrow. (c) The extent of the atrophy is shown between the red arrows on the OCT image and the blue arrow demonstrates the spared retinal layers in the peripapillary region.
STGD1, Stargardt disease.
Epidemiology
STGD1 (OMIM# 248200), also called Stargardt macular dystrophy, juvenile macular degeneration or fundus flavimaculatus, is the most common childhood onset inherited retinal disease (IRD). 12 Spiteri Cornish et al. 13 carried out a British Ophthalmological Surveillance Unit (BOSU) study regarding the incidence of STGD1 in the United Kingdom and found an annual incidence between 0.110 and 0.127 per 100,000 individuals per year. In the United States, Blacharski estimated that the incidence of STGD1 was between 1/8000 and 1/10,000. 12 This figure is commonly quoted in the literature,14–17 but it should be noted that this was derived from an assumption that STGD1 was more common than retinoblastoma but less common than retinitis pigmentosa (RP), which respectively have an incidence of 1/15,000 and 1/5000. 12 Recently, Hanany et al. 18 calculated that the worldwide general prevalence of STGD1 and ABCA4R is 1/6578 and that approximately 1.4 million individuals have an ABCA4R based on parsing data from six worldwide populations.
Natural history
The age of onset in STGD1 is variable and patients with early onset disease typically have more severe symptoms and faster disease progression.16,19–21 A later age of onset (>45 years) is associated with milder symptoms,22,23 better visual acuity (VA),19,20 foveal sparing disease22–25 and slower disease progression.
Inheritance in STGD1
STGD1/ABCA4R are inherited in an autosomal recessive pattern. The phenotypes are variable but with some key distinguishing features. To date, more than 2000 ABCA4 variants have been reported in the literature (www.lovd.nl/ABCA4).
The estimated carrier frequency for ABCA4 disease-causing variants has been reported to range between 4.5% and 10%; these studies included individuals of mainly European descent.26–29 Moreover, Hanany et al. 18 recently proposed that European subpopulations had the highest carrier frequency of 2.5% based on genotype data from six worldwide populations.
Presentation and natural history
Patients typically present due to a reduction in their central vision, because of either blurring of vision typically characterised by increasing difficulty in carrying out tasks requiring detailed vision4,30 or the presence of a central scotoma.31,32 Other symptoms include delayed dark adaptation,33–35 photosensitivity, photopsia and abnormal colour vision.32,36 The typical phenotype in STGD1 is characterised by three features: flecks, macular atrophy and sparing of the peripapillary region.37–39
ABCA4R
ABCA4R include a range of different clinical appearances, or phenotypes, namely STGD1, bull’s eye maculopathy (BEM)40,41 and chorioretinal atrophy. 42 These clinical phenotypes can be further characterised regarding which cells of the retina are affected. Thus, an ABCA4R may be also called a macular dystrophy, or a cone-rod dystrophy depending on which cells in the retina are affected combined with the clinical appearance. In the former, only the macula is affected, and in the latter, there is cone and rod dysfunction as defined by electrophysiological testing.
Specific features of ABCA4R
Flecks
Flecks are usually present at some point in the disease course. They typically develop centrifugally from within the macular region. In some patients, they progress beyond the vascular arcades. Flecks may appear later in patients with a BEM phenotype, (Figure 2) but may be an early feature in patients presenting with chorioretinal atrophy.
Figure 2.

Fundus autofluorescence imaging showing a mild ring of ↑ AF signal shown by the yellow arrow (a) 4-year follow-up image shows ↓ foveal AF (increased atrophy) surrounded by flecks developed since the first image (see red arrow) and a ring of ↑ AF signal indicated by the yellow arrow (b).
AF, autofluorescence.
Macular atrophy
The macular region is typically affected in STGD1 and the atrophic changes are usually progressive. The severity of the macular atrophy can be variable. Some only have a small region of atrophy within the central macular region, while those with more severe disease can develop atrophic regions beyond the arcades or widespread scalloped pattern of atrophy.43,44 The atrophy initially occurs in the RPE and the ellipsoid zone (EZ) layer, but this can progress to involve the choriocapillaris (CC) 43 and the choroidal layers 45 with eventual exposure of the sclera in some cases. 42
Peripapillary sparing
Sparing of the peripapillary retina is considered a diagnostic feature in STGD1.37–39 In ABCA4R, the peripapillary region typically has a normal autofluorescence (AF) signal 39 with a preserved EZ on optical coherence tomography (OCT). 46 However, peripapillary involvement in ABCA4R has been reported.46,47 Burke et al. 46 found that peripapillary involvement was typically seen in patients with cone and rod dysfunction on electrodiagnostic testing (EDT), signifying more severe disease and Hwang et al. 48 proposed that certain combinations of ABCA4 variants might lead to peripapillary involvement. Figure 3 shows loss of peripapillary sparing.
Figure 3.
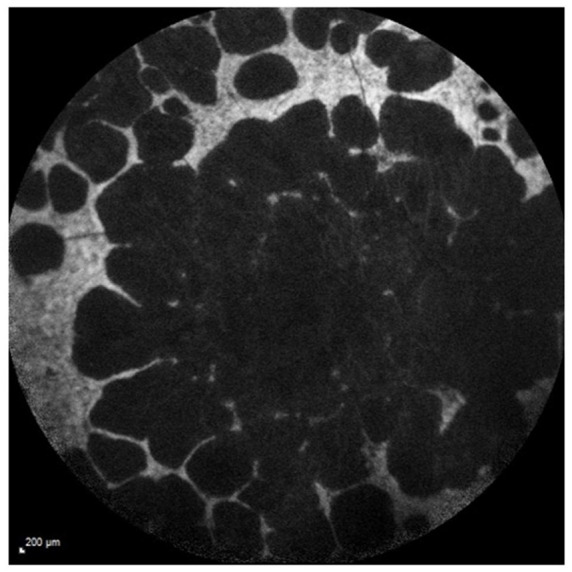
Fundus autofluorescence image showing widespread scalloped pattern atrophy of the RPE and disease involving the peripapillary region.
RPE, retinal pigment epithelium.
BEM
BEM may be seen in STGD1 as shown in Figure 4. This particular phenotype has also been reported in other macular dystrophies and in hydroxychloroquine (HCQ) toxic retinopathy.40,41 Flecks may appear later in the disease course in patients with ABCA4 mutations who present with a BEM (Figure 4). Some patients with STGD1 presenting with a BEM have been reported to be asymptomatic or to have milder disease. 41
Figure 4.

FAF image showing bull’s eye maculopathy (a) baseline imaging showing ↓ foveal AF signal (yellow arrow) surrounded by a ring of ↑ AF (orange arrow). (b) Follow-up imaging showing area of atrophy ↓ AF signal (yellow arrow) surrounded by new flecks (red arrow).
AF, autofluorescence; FAF, fundus autofluorescence.
Foveal sparing disease
Foveal sparing in STGD1 has been reported to occur in between 21% and 59% of patients (Figure 5).23,49–54 It is usually associated with a later age of disease onset (in adulthood and in some cases >45 years of age)49,50 and a milder phenotype. 49 In patients with foveal sparing disease, the fovea usually becomes involved later in the disease course.
Figure 5.
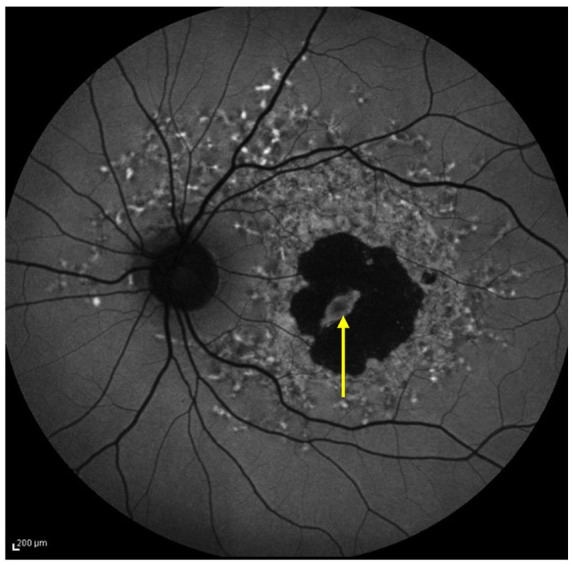
FAF image showing a spared fovea, highlighted by yellow arrow.
FAF, fundus autofluorescence.
Chorioretinal degeneration
The most severe ABCA4 phenotype is characterised by chorioretinal degeneration and CC atrophy16,42,55 (Figure 6). In severe cases, the sclera may be visible. Widespread nummular and sparse bone spicule pigmentary deposits may also be observed. 42 Severe early onset disease may be associated with vascular attenuation, peripapillary disease involvement and pigment deposition, sometimes leading to a diagnosis of RP. 56 However, in ABCA4R, there is usually retinal sparing beyond the mid periphery with a clear edge at the nummular areas of atrophy, both features differing from typical RP. 57 Tanaka et al. proposed the term ‘rapid-onset chorioretinopathy’ to describe a distinct ABCA4 phenotype, caused by deleterious bilallelic null mutations. This group is usually characterised by abnormal cone and/or rod function on electrophysiology testing, symptomatic onset before 10 years of age and significant choroidal thinning within the first 20 years of life.29,56,58,59
Figure 6.

Retinal imaging showing a severe chorioretinal atrophy phenotype in a Stargardt patient. (a) The colour fundus image shows widespread macular atrophy extensive mid perihpeary involvement with pigmentary deposition. (b) The autofluoresence image shows the extent of the RPE atrophy with scalloped edges with some sparing of the peripapillary region and a heterogenous background AF signal in nonatrophic areas.
AF, autofluorescence; RPE, retinal pigment epithelium.
Subretinal fibrosis
Subretinal fibrosis can occur following minor ocular trauma in patients with STGD1.60–65 Initially, an orange pigmented region (thought to be lipofuscin) is observed, which gives way to subretinal fibrosis and RPE hypertrophy. 64 Grandinetti et al. 65 suggest that ocular trauma causes a release of excess lipofuscin which disrupts the RPE and leads to subretinal fibrosis and RPE hypertrophy. Similarly, Gass et al. 63 suggest that stored lipofuscin leads to engorgement and hypertrophy of the RPE and that trauma causes RPE disruption. Del Buey et al. 62 have linked subretinal fibrosis to the release of growth factors from the RPE in response to injury. However, subretinal fibrosis has also been reported in patients without a history of ocular trauma.60,64 Rossi et al. 60 found that patients without a history of ocular trauma played competitive sports and inferred that microtrauma could potentially lead to subretinal fibrosis. Patients with STGD1 should be given lifestyle advice regarding use of eye protection when playing competitive sports to avoid even minor ocular trauma which can potentially lead to RPE damage.60,65
Other rarer phenotypic features
Rare phenotypic features previously described in STGD1 include small parafoveal white dots, 49 hypopigmentary macular changes, 66 bone spicules 67 and peripheral pigmentary deposition. 68 Very rarely, choroidal neovascularisation (CNV) can occur in association with STGD1. A CNV, identified in one eye in a patient with two ABCA4 variants, was reported by Van-Westeneng et al. 23 However, in the majority of the cases, only one variant or no ABCA4 variants have been identified so they may not be true ABCA4-related phenotypes.69–71
Genotype-phenotype correlation in ABCA4R
It was initially thought that disease severity was inversely proportional to the amount of functional ABCA4 protein. For example, cone-rod dystrophy, 72 RP,59,73 CC dystrophy 43 and rapid-onset chorioretinopathy 57 have all been associated with severe variants, such as nonsense mutations, while missense variants that produce some functioning protein are noted to be associated with a milder phenotype.27,74,75 However, the relationship is more complex than this. Certain variants, such as deep intronic variants originally thought to not affect splicing of the protein, have now been identified to have a significant effect on protein function and have been linked to severe disease.76–78 Genotype-phenotype correlation is beyond the scope of this review and is investigated in further detail by our group in a genotype review. Genetic modifiers and gene-to-gene interactions are expected to have a significant role in phenotype variation. 79
Phenocopies
A number of IRDs share similar features to STGD1 which makes distinguishing them difficult. This phenomenon is known as phenocopies. They differ in their genotype and often some aspect of their phenotype. Phenocopies relevant to ABCA4R include STGD 3 and 4. STGD3 is associated with variants in elongation of very long chain fatty acids protein 4 (ELOVL4) and is inherited in an autosomal dominant manner.80,81 STGD4 is associated with variants in Prominin 1 (PROM1),82,83 as well as HCQ retinopathy, but this can be excluded by a history of HCQ ingestion and negative ABCA4 testing. Natural history studies to investigate these phenocopies will be important in identifying features that may distinguish them from ABCA4R. Currently, the natural history of PROM1-related retinopathy is being investigated in the ProgStar-4 study. 84
Peripherin 2 (PRPH2) is associated with both pattern and pseudofundus-flavimaculatus type phenotypes, and in this context is the most commonly encountered ABCA4 phenocopy. PRPH2 is inherited as autosomal dominant and shows reduced penetrance with variable expressivity. Other phenocopies are seen associated with variants in CDHR1, CERKL, CRX and RPE65. 82 Differing features aid in distinguishing these conditions: the inheritance pattern, and certain specific features, for example, lack of AF signal in RPE65, 85 bone spicule pigmentation extending to the far periphery (not usually seen in ABCA4R) among others. The presence of flecks in a family with an autosomal dominant inheritance makes an ABCA4R unlikely, unless there is consanguinity with pseudo-autosomal dominant inheritance.
Imaging relevant to STGD1
Multimodal imaging is instrumental in diagnosing ABCA4R, and typically includes colour fundus photographs, fundus autofluorescence (FAF) imaging and spectral domain OCT (SD-OCT). The role of OCT angiography (OCTA) is yet to be determined. Since the introduction of FAF, fundus fluorescein angiography (FA) is very rarely used now in the diagnosis of IRD. Indocyanine green angiography (ICGA) has not been found to be particularly helpful in characterising ABCA4 phenotypes. High-resolution retinal imaging with adaptive optics scanning laser ophthalmoscopy (AOSLO) has been recently introduced and is at present primarily a research tool. However, it can provide a highly sensitive assessment of photoreceptor structure and has been used to identify areas of increased photoreceptor spacing, which suggests photoreceptor loss, in retinal areas appearing normal on OCT and FAF. 86
Colour fundus photography
The first fundus camera was made commercially available following its development by Carl Zeiss in 1926. Fundus cameras traditionally use a white flash, provide images with realistic colour and capture a 30° or 45° field of view. 87 Widefield retinal cameras became commercially available in 1997 and used a contact lens with fibreoptic illumination to capture up to 130° of the retina. 88 The most recent advance in fundus photography includes the development of ultra-widefield (UWF) imaging systems. These systems can image the far peripheral retina as they have a field of view of up to 200°. Widefield imaging systems typically use a confocal scanning laser ophthalmoscope with combinations of green, red and blue laser light. 89 The benefits of these imaging systems are that they do not rely on pupillary dilation or a contact lens to take the image. 89 Some disadvantages of different types available include inaccuracy in retinal colour reproduction, image clarity, apparent vignetting and difficulty in obtaining accurate measurements due to a lack of image standardisation with respect to a specific axis of the eye. 89 A comprehensive review of the types of cameras available is beyond the scope of this review, but Chen’s comparison of four UWF viewing systems is very informative. 90
Colour fundus photography provides documentation of the type of retinal changes observed and extent of the disease. The Fishman classification is most frequently used and is based on the presence of the flecks and macular atrophy 91 (Table 1 and Figure 7).
Table 1.
The Fishman classification for colour fundus photographs. 91
| Class 1 | Macular atrophy with flecks isolated to the foveal and parafoveal regions with a ‘beaten metal appearance’ |
| Class 2 | Macular flecks present throughout the posterior pole (within or beyond the arcades or nasal to the optic disc) |
| Class 3 | Resorption of flecks in association with widespread atrophy |
| Class 4 | Further resorption of flecks and extensive atrophy of the choriocapillaris and retinal pigment epithelium (RPE) |
Figure 7.

Colour photographs illustrating the Fishman classification. (a) Fishman class 1 with foveal atrophy and flecks within the parafoveal area. (b) Fishman class 2 showing central macular atrophy and widespread flecks that extend beyond the vascular arcades. (c) Fishman class 3 showing macular atrophy with widespread flecks that extend beyond the vascular arcades and resorption of the central flecks. (d) Fishman class 4 showing extensive macular atrophy, resorption of the flecks and pigmentary deposition. 91
FAF imaging
The two main methods used to capture FAF images are confocal scanning laser ophthalmoscopy (cSLO) and fundus photography using a standard fundus camera with modification of the excitation and barrier filters. 92 cSLO is the more frequently used method and acquires images using a low power laser beam, which is swept across the retina in a raster pattern. 93 By contrast, the fundus camera takes one photograph and summates the AF signal simultaneously emitted from the retina and RPE. 92 However, a phenomenon of pseudo-AF caused by scattered light from outside the retina can be achieved when using a fundus camera. 94
Light is used to excite fluorophores and different wavelengths of light can be used depending on the object of interest. FAF with short wavelength light [short-wavelength fundus autofluorescence (SW-FAF); 488-nm excitation, 500-nm cut-off filter] is the most commonly used, and is thought to be derived primarily from lipofuscin, a by-product of the visual cycle. FAF with near-infrared light [near infrared-wavelength fundus autofluorescence (NIR-FAF); 787-nm excitation, 830-nm cut-off filter] appears to arise mainly from the melanin in the RPE and choroid. 95
UWF imaging systems provide FAF imaging of up to 200° using green light rather than the blue light used in SW-FAF images. 96 UWF images are affected by distortion (horizontal image stretching) and regional variations in the contrast level (limited contrast superiorly and inferiorly). 97 Care needs to be taken when comparing UWF-FAF images with SW-FAF images due to these differences and the different wavelengths of light used for FAF. For example, green light is better at imaging the fovea compared with blue light 98 because green light is less absorbed by the macular pigment 99
On SW-AF images, regions with increased AF signal have higher levels of lipofuscin, while areas with reduced AF signal signify atrophy or degeneration of the RPE. 100 FAF imaging is increasingly being used in the diagnosis of IRD as it is noninvasive and has almost completely replaced angiography for this indication.101,102 FAF imaging is a valuable tool to assess and characterise IRDs. The specific features of AF aid in diagnosis and help to guide genetic testing. FAF imaging is especially valuable in assessing STGD1 patients because it can detect retinal changes manifested by an increase in AF signal in the retina before the onset of symptoms, and before changes on fundoscopy and colour fundus imaging become visible. 103 Detecting the presence of flecks before they become apparent on fundoscopy or colour imaging 104 enables patients to receive an early diagnosis. AF can also be used as a sensitive tool to record change over time.105,106
Flecks typically have an increased AF signal on SW-FAF. Later in the disease process, they can undergo reabsorption, which is then associated with a decreased AF signal. FAF highlights disease features that are not always apparent on colour imaging or fundoscopy, such as the true extent and distribution of flecks and areas of RPE degeneration and atrophy. One of the classic features of STGD1 is peripapillary sparing; FAF imaging is the best imaging modality to demonstrate this (Figure 8(a)).37–39 Care needs to be taken when interpreting FAF images, for example, decreased AF signal can arise due to masking of the retina or RPE as seen with a retinal bleed, fibrosis or increased pigmentation. An increased AF signal may be associated with intraretinal and subretinal fluid or drusen.96,100
Figure 8.

FAF images in patients with ABCA4R. (a) Atrophic macula surrounded by a region of heterogenous background AF with flecks with relatively normal AF signal in peripapillary area. (b) Extensive macular atrophy involving the peripapillary region with flecks in the peripheral retina. Despite the severe phenotype, there is a remnant of preserved peripapillary tissue nasally. The spared peripapillary area is marked by the yellow arrow.
ABCA4R, ABCA4 retinopathies; AF, autofluorescence; FAF, fundus autofluorescence.
Both SW-FAF and NIR-FAF (Figure 9) are useful in monitoring for disease progression in ABCA4R. This is because the AF signal is emitted from different parts of the retina. On SW-FAF imaging, flecks usually have an increased AF signal and areas of RPE degeneration or atrophy have reduced or absent AF signal, respectively.105–111 Using NIR-FAF imaging, flecks are associated with a decreased AF signal and are occasionally surrounded by a halo of reduced AF signal. On NIR-FAF imaging compared with SW-FAF imaging (Table 2), flecks appear larger, 112 can be more visible in the periphery and appear earlier.53,99,112–114 Sparrow et al. 114 detected an earlier reduction in AF signal from resorbing central flecks using NIR-FAF when compared with SW-FAF. The area of RPE atrophy has also been observed to be larger on NIR-FAF compared with SW-FAF.99,115
Figure 9.

(a) Short wavelength-AF image showing flecks associated with a ↑ AF signal pertaining to the flecks (see red arrows), which extend beyond the vascular arcades and a central region of macular atrophy (absent AF signal) with a small foveal remnant. The foveal remnant is indicated by the yellow arrow, atrophic regions are highlighted by blue arrows, a fleck with a ↑ AF signal is shown by red arrows (b) Near infrared AF image of the same patient showing flecks with predominantly ↓ AF (see orange arrow) that are more eccentric. The areas with ↓ and abnormal AF signal are also larger compared with the SW-AF image. The spared fovea can be seen in both images. The spared fovea is shown by the yellow arrow, atrophic regions by blue arrows and fleck with ↑ signal (red arrow). (c and d) Comparison between the areas of abnormal AF on (c) SW-AF imaging and (d) NIR-AF in the same eye of a different patient. The area of abnormal AF is larger and more eccentric on the NIR-AF image compared with the SW-AF image.
AF, autofluorescence; SW-AF, short-wavelength autofluorescence; NIR-AF, near infrared-wavelength autofluorescence.
Table 2.
Summary of retinal appearance comparing SW-AF and NIR-AF in patients with STGD1.
| Feature | SW-AF | NIR-AF |
|---|---|---|
| Flecks | Predominantly raised AF signal | Predominantly reduced AF signal |
| Central flecks emit lower AF signal compared with more eccentric flecks | Central flecks emit lower AF signal compared with more eccentric flecks | |
| Smaller and less eccentric | Larger and more eccentric than on SW-AF | |
| Appear after flecks on NIR-AF | Precede appearance on SW-AF | |
| Atrophy | Clearly delineates atrophic region | Atrophic region is larger than that seen on SW-AF |
| Foveal sparing | Macular pigment causes masking of the spared fovea | Better at identifying foveal sparing disease |
NIR-AF, near infrared-wavelength autofluorescence; SW-AF, short-wavelength autofluorescence.
A demarcation line in the inferonasal area that separates a region of reduced nasal AF and increased temporal AF can be seen on both NIR-FAF and SW-FAF imaging in patients with STGD1,99,116 and this demarcation line is respected by both the flecks and areas of mottled AF. 117 Duncker et al. 118 proposed that this demarcation line represents the closed optic fissure. Comparison of AF imaging in RP patients similarly showed this demarcation line in both modalities, while in normal healthy individuals, the demarcation line was only identifiable on SW-AF imaging which led the Duncker et al. 117 to suggest that this sign could be used as a marker for disease. UWF-FAF imaging has enabled visualisation of peripheral retinal changes in STGD1 not visible on standard imaging.116,119 NIR-FAF and UWF-FAF imaging are better than SW-FAF at identifying foveal sparing disease and it is thought to be the case that the 787-nm wavelength of light is absorbed less by the macular pigment thus causing less masking.53,99
There are a number of AF-based STGD1 phenotype classifications,99,120,121 the most widely used is from Fujinami et al. who describe the AF appearance on SW-AF images of the posterior pole. Their classification comprises three different groups according to specific AF features (Table 3 and Figure 10).107,122 This classification is helpful in categorising the severity of STGD1 on its clinical appearance and can be mapped to retinal function. 107 A phenotypic classification for UWF-FAF images has been described by Klufas et al., 123 which classifies disease severity into three groups based on specific retinal features (Table 4). Kuehlewein et al. have also used the terms definitely decreased AF (DDAF) and questionably decreased AF (QDAF) to describe the AF level compared with the darkness level of the optic nerve head. DDAF describes an area with greater than or equal to 90% of the darkness of the optic nerve head and QDAF to describe areas with a darkness level between 50% and 90% of the optic nerve head darkness. These areas can be semi-automatically measured using RegionFinder module of Heidelberg Eye Explorer software 124 and is now an established way of grading the AF appearance in STGD1 and is being used in the ProgStar study. 17 This grading has been used to determine suitable patients for Emixustat treatment in the Safety and Efficacy of Emixustat in STGD1 (SeaSTAR) trial. 125
Table 3.
| Type 1 | Decreased foveal AF signal with/without flecks and surrounded by a background of homogenous AF |
| Type 2 | Decreased macular AF with flecks that within the posterior pole that extend to the vascular arcades and surrounded by a background of heterogenous AF signal |
| Type 3 | Presence of multiple areas of decreased AF signal within the posterior pole with/without flecks and associated with a background of heterogenous AF signal |
AF, autofluorescence.
Figure 10.

Images from our cohort using the Fujinami AF classification. (a) Type 1 showing ↓ foveal AF signal with a small number of flecks and a homogeneous background AF. (b) Type 2 showing ↓ macular AF signal surrounded by a heterogenous background AF and widespread flecks extending beyond the vascular arcades. (c) Type 3 showing multiple areas of ↓ AF surrounded by a heterogenous background AF and widespread flecks extending beyond the vascular arcades. 122
AF, autofluorescence.
Table 4.
The autofluorescence types in UWF-AF described by Klufas et al. 123
| Type 1 | Lesions isolated to the macular region |
| Type 2 | Atrophic macula with peripheral flecks |
| Type 3 | Atrophic macula associated with peripheral atrophy |
UWF-AF, ultra-widefield autofluorescence.
Several studies have looked at the progression of atrophy in macular disease. This is a useful way of assessing the rate of disease progression and severity. This is likely to form part of the routine assessment of response to treatment and is an ideal modality to capture this information. An area of macular atrophy can be measured over time to monitor the progression of disease.24,105,109,126–129 Measuring the growth of the atrophic lesions on FAF imaging is the primary outcome measure of the largest natural history study in STGD1 to date, the ProgStar study. 17 Faster progression has been observed with the following features: early onset disease,105,126 larger area of RPE atrophy at baseline imaging,105,109 presence of a heterogenous background AF signal,105,109 presence of flecks beyond the vascular arcades,105,109,121 a worse VA 116 and larger scotomas. 130 In the majority of cases, longer disease duration is associated with larger areas of RPE atrophy.105,131 However, the main finding in the ProgStar study was that the progression rate corresponded to the area of decreased AF and that the differences described earlier were not observed when analysis was corrected for the baseline size of the lesion.105,109 The area of RPE atrophy is usually symmetrical126–128,132 meaning that one eye can be used as a control in therapeutic trials. Lambertus et al. 132 found that there was better correlation in the area of atrophy in patients with early onset disease and suggested that patients with early onset disease would be more suitable for assessing treatment efficacy in therapeutic trials.
Cideciyan et al. 133 were the first to describe the use of reduced illuminance SW-FAF and NIR-FAF imaging in STGD1 patients due to concerns that frequent SW-FAF imaging could theoretically lead to an increased rate of lipofuscin accumulation and/or increase its toxicity. They observed that reduced illuminance FAF imaging provided similar information on the disease profile compared with the conventional FAF imaging. 133 Strauss et al. 134 observed that reduced illuminance FAF was similar to conventional FAF imaging when assessing the area of DDAF but that there was a difference between the two modalities when assessing areas with poorly demarcated QDAF.
Both SW-FAF and NIR-FAF imaging modalities have a role in assessing patients in the clinic setting. Both imaging modalities will be highly relevant to therapeutic trials as SW-AF enables clear delineation of retinal changes and NIR-AF can be used to identify earlier disease. Predicting future disease progression could help identify diseased areas earlier in patients recruited for therapeutic trials.
An early phenotype that the authors have noticed in some STGD1 patients is the presence of increased foveal AF signal in patients with very mild symptoms (see Figure 11). This could either represent an early phenotype in STGD1 that will progress to the more typical features associated with the disease, or a mild phenotype detected in patients now because of sophisticated imaging. How it progresses and its rate of progression needs to be evaluated.
Figure 11.
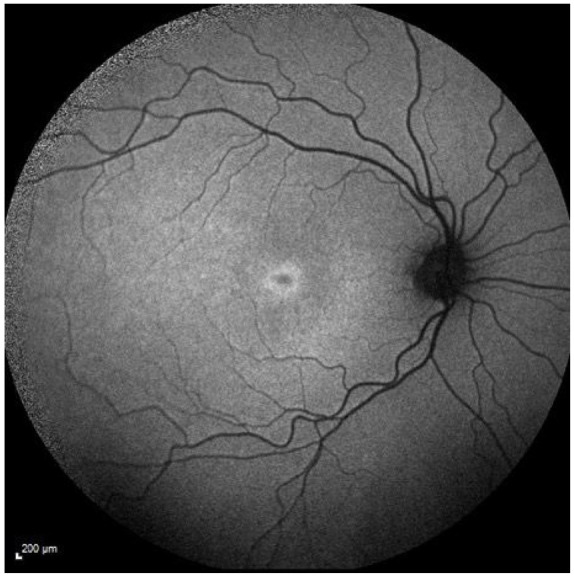
Fundus autofluorescence image showing discrete focal parafoveal area of ↑ AF signal.
AF, autofluorescence.
Quantitative AF imaging
Quantitative AF (qAF) was described by Delori et al. 135 in 2011 and introduced a standardised approach to assessing lipofuscin levels in the retina. qAF requires bleaching of the photoreceptors (20 s for rods and 30 s for cones) to reduce the absorption of photopigment, followed by the acquisition of SW-FAF images from which the AF signal intensity is calculated. 135 In normal patients, qAF levels increase with age, 136 are highest in the superotemporal quadrant 136 and are lowest in the inferonasal quadrant.137,138 Greenberg et al. 136 found that White patients had a higher qAF compared with Black and Asian patients, and that females have a significantly higher qAF level compared with males. The distribution of qAF level in different regions of the retina has been shown to be similar between STGD1 patients and normal patients meaning that these changes seen in ABCA4R can be compared with normal controls.137,138
In ABCA4R, the qAF levels are typically raised even when the fundus appears normal. 137 qAF imaging may be useful in diagnosing ABCA4R when there is uncertainty regarding the diagnosis. It could be useful in patient with BEM or cone dystrophy where an ABCA4R is suspected but only one ABCA4 variant has been identified. 40 This is because a higher qAF signal may be seen in early disease ABCA4R compared with other genetic causes of these phenotypes. Sparrow’s group investigated the levels of qAF and found ABCA4 patients had higher qAF values than those seen in PRPH2 patients. 139 However, Gliem et al. 140 noted that elevated qAF is present in different IRDs in particular those caused by variants in ABCA4, PRPH2, PROM1, CRX and CDHR1, but that it depended on the stage of disease. It is clear that qAF levels are highest in the initial stages of disease40,137,141 and decrease in later stages of disease as atrophic changes predominate. 142 Therefore, in severe ABCA4R, qAF levels do not increase with age as seen in healthy controls.40,137,141 However Müller et al. 142 observed that patients carrying two null variants had substantially elevated qAF levels and that milder ABCA4 variants were linked to a mild elevation in qAF. Patients carrying the p.(Gly1961Glu) and p.(Asn1868IIle) variants can have qAF levels that fall within the upper end of the normal range; therefore, this imaging modality is not completely reliable in confirming ABCA4R.44,142
However, qAF could be a useful biomarker for assessing lipofuscin levels following therapeutic interventions to aid assessment of efficacy of therapies. It is important to note though that qAF imaging using the 488-nm wavelength bleaches the photoreceptors and may thus be potentially retinotoxic. 133 NIR-AF may be a potential safer alternative. Another limitation is variability in test–retest results which was found to be ±9.4% in normal eyes under standardised conditions. 136
Fluorescence lifetime imaging ophthalmoscopy
Fluorescence lifetime imaging ophthalmoscopy (FLIO) enables measurement of the lifetime of fluorophores by measuring the average time taken between absorption of light that excites the fluorophores, causing them to emit light at a longer wavelength, and the time for the fluorophores to return to their normal energy level. A 473-nm laser is used to excite fluorophores and two emission spectra can be detected, the short wavelength between 498 and 560 nm and the long wavelength between 560 and 720 nm. 143 A shorter lifetime AF is shown as red AF signal and longer lifetime AF is shown as a green AF signal. 143 In healthy eyes, the lifetime AF is shortest within the foveal region, higher in the peripheral retina and increases with age. 144
In STGD1, flecks initially emit red AF signal and gain lifetime AF with time. Older flecks within the central retina typically have a longer lifetime AF, while peripheral flecks have a shorter lifetime AF. Resorbing flecks have a central longer lifetime AF surrounded by shorter lifetime AF. 145 The atrophic regions emit both short and long lifetime AF signals. Dysli et al. 145 suggested that the long lifetime AF arose from underlying structures such as the choroid and the short lifetime AF was emitted from macular pigment. Again, the usefulness of this modality may be limited by the wavelength and its potential effects on the retina.
Angiography
FA is a method of imaging the retinal and choroidal circulation by injecting fluorescein dye into the venous circulation. Fluorescein absorbs blue light (465–490 nm) and emits green light (520–530 nm). 146 The high molecular weight of the dye means that it is too large to escape from the retinal circulation and is unable to diffuse through the RPE, 147 thus enabling clear imaging of the vasculature.
ICGA is primarily used to image the choroidal circulation. 146 Indocyanine green dye is injected into the venous circulation. The dye is excited using an infrared excitation light of 800 nm and emits light with an 835-nm wavelength. This wavelength can penetrate the RPE and ocular pigments and enables good visualisation of the choroidal circulation because 98% of indocyanine green is bound to protein meaning that there is minimal diffusion through the CC. 148
Fundus FA in STGD1 and ABCA4R
Historically, the appearance of a dark choroid (Figure 12) on FFA was considered a diagnostic feature of STGD1. It occurs due to the raised lipofuscin levels which block the fluorescence from the choroid.47,149,150 However, the dark choroid sign is not always present; it is has been reported to be absent in 35% of paediatric patients 104 and 33.3–80%52,23 of adult patients. It is also less frequently seen in patients with the p.(Gly1961Glu) variant. 151
Figure 12.
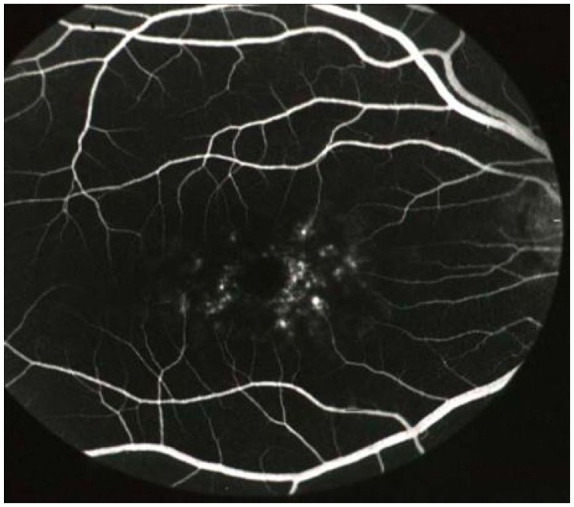
Fluorescein angiogram illustrating the dark (silent) choroid (absence of any choroidal vessel filling in the background). Image courtesy of Professor Alan Bird, Moorfields.
On FFA, flecks may be seen as hypofluorescent regions blocking the transmission of the fluorescein signal, 152 or hyperfluorescent 4 or appear as window defects. 52 The flecks can be seen on FFA before their appearance on fundoscopy and colour imaging, which is why FFA is used to diagnose STGD1/ABCA4R in children with symptoms but no obvious fundus features. 153 Regions of RPE atrophy are hyperfluorescent 39 with clearly demarcated borders. 39 FFA has since been replaced by noninvasive FAF imaging.
ICGA in STGD1
ICGA is now very rarely, if ever, used to assess patients with STGD1. In the fundus flavimaculatus phenotype, Schwoerer et al. 39 observed areas of decreased signal that progressed and resulted in reticular patterns with small areas of normal choroid surrounded by a clearly demarcated network of curvilinear lesions with low signal. The reticular pattern of decreased fluorescence was observed to have a polygonal shape similar to watershed zones between terminal choroidal arterioles supplying the CC. Flecks were present in all eyes but did not always correspond to yellow flecks seen on fundus examination. Regions of atrophic RPE appear as dark regions154,155 on ICGA.
OCT
OCT measures the back-reflected light from the retina to produce two-dimensional cross-sectional images (B-scan) that can be stacked to produce a three-dimensional (3D) volume. Representations of tissue structure within OCT images are based on interference and the interference fringes detected by a Michaelson interferometer. 156 The back-reflected light is combined with a reference beam: if the distance travelled by light in both paths is the same, this leads to positive interference; if the distance differs by half of the wavelength, this leads to destructive interference. As the interference pattern is sensitive to small changes in the distance travelled by light, it provides superior axial (depth) resolution. This enables high-resolution imaging of the retinal layers.
OCT imaging technology has developed enormously over the last 25 years. Time domain OCT has largely been superseded by frequency domain OCT. Frequency domain OCT includes SD-OCT and swept source OCT (SS-OCT). Currently in the clinical setting, SD-OCT is the most frequently used. It provides an axial resolution of 5–7 microns.157,158 The choroid can also be imaged with SD-OCT using enhanced depth imaging (EDI) protocols. 159 SS-OCT uses a longer wavelength of light that enables imaging of the choroidoscleral regions and optic nerve head, and has better imaging ability through cataracts and ocular opacities. 160
OCTA enables visualisation of the blood flow within the retinal and choroidal circulation by analysing successive B-scans, which capture different light reflection and scattering due to blood flow. Segmentation of the 3D volumes derived from OCTA imaging allows the creation of an en-face image that enables assessment of blood flow within specific layers. 161
Although a number of observations have been made regarding the appearance of specific disease features of STGD1 on OCT, there is currently no widely accepted classification for STGD1/ABCAR using OCT imaging.
Retinal layers
OCT enables the structural assessment of individual retinal layers (Figure 13). In ABCA4R, thinning mostly occurs within the outer retinal layers.162,163 Disruption or loss of the EZ or RPE can be observed and flecks are observed as hyperreflective deposits.14,164
Figure 13.

OCT images in patients with STGD1. (a) A region of macular atrophy where the outer retinal thickness is ↓ and the EZ is absent. The yellow arrows show where the EZ is absent. (b) Preservation of the EZ layer within the peripapillary region. The outer retina is lost in the region of macular atrophy. The accompanying infrared scanning laser ophthalmoscopy image also shows a hyperreflective region of macular atrophy and normal reflectance within the spared peripapillary region. (c) The spared fovea with is shown between the two yellow arrows and the spared EZ layer is shown by the two red arrows.
EZ, ellipsoid zone; OCT, optical coherence tomography; STGD1, Stargardt disease.
The hypothesis of RPE apoptosis and consequent photoreceptor degeneration is widely thought to be the mechanism of vision loss in STGD1. 165 However, disruption and loss of the EZ on OCT can occur in areas with normal AF signal on SW-FAF images which suggests that photoreceptor degeneration may precede RPE degeneration in STGD1.16,110,111,129,166 It is also possible as proposed by Ritter et al. 167 that photoreceptor and RPE degeneration occur simultaneously in STGD1; they noticed regions of irregular AF corresponding to areas with abnormal RPE that had skip lesions and were associated with an absence or disruption of photoreceptor cells. Progressive loss in the width of the EZ is also seen in RP168,169,170,171 and its optical intensity has also been suggested as an early biomarker for disease in RP. 172 Similarly, Romano et al. 173 found that in Best disease, an increased baseline reflectivity was also associated with a lower VA and faster progression. In choroideremia, it appears that the RPE loss precedes photoreceptor degeneration and is more extensive.174,175 Careful evaluation at various disease stages using high-resolution imaging is helping to resolve these questions.
Preservation of the EZ has also been shown to correlate with the VA in STGD1110,120 and other IRDs, thus highlighting the importance of the EZ towards predicting the visual function in patients and potentially means its health could be used to monitor efficacy of therapeutic treatments.
Flecks correspond to hyperreflective deposits on OCT imaging.112,114,152,176 In an early study using OCT, Querques et al. described two types of flecks. Type 1 flecks comprise dome shaped deposits in the inner part of the RPE and type 2 flecks are small linear deposits in the outer nuclear layer (ONL) not involving the RPE. 152 More recently, Voigt et al. 176 devised an OCT classification for flecks comprising five subtypes (Table 5) and suggested that the differences between the flecks could reflect different disease stages of disease. Flecks with the highest AF signal were also observed to penetrate the ONL on OCT. 176 The EZ and external limiting membrane (ELM) are less reflective in areas adjacent to the flecks 114 and the ONL is thinner in areas with longer flecks.112,114 Resorbing flecks are hyporeflective and progressively interrupt rather than displace the interdigitation zone (IZ), EZ, ELM 114 and ONL 112 which led Sparrow et al. 114 to suggest that flecks expanded from the outer to the inner retinal layers.
Table 5.
The Voigt classification for flecks on OCT imaging. 176
| Voigt class | Appearance on OCT | Proportion of patients with fleck type (%) |
|---|---|---|
| Class A | Presence of hyperreflective deposit at the level of the OS, RPE interdigitations and RPE/BM complex | 100 |
| Class B | The hyperreflective deposits occurring in class A progressing through the IS and OS layer up until the ELM | 100 |
| Class C | Breakthrough of the hyperreflective deposits into the ONL | 81 |
| Class D | Presence of hyperreflective deposits only within the ONL | 55 |
| Class E | Presence of pigment epithelial detachments that resemble drusen | 29 |
BM, Bruch membrane; IS, inner segments; ELM, external limiting membrane; OCT, optical coherence tomography; ONL, outer nuclear layer; OS, outer segments; RPE, retinal pigment epithelium.
Huang et al. 177 found that progressive thinning of the ONL was associated with thickening of the inner nuclear layer but could not assess the effect of this retinal remodelling on the visual function due to the absence of photoreceptors. Inner retinal thickening in association with thinning of the ONL has similarly been reported in other IRDs which include X-linked RP,178,179 RP caused by variants in RHO 180 and CERKL, 181 Usher syndrome 1B (USH1B) with variants in MYO7A 182 and Leber congenital amaurosis with variants in AIPL1. 183 This highlights that measuring the retinal thickness in IRD is complicated and the focus should be on measuring individual layers rather than the overall retinal thickness due to retinal modelling linked to the disease process. Careful segmentation and evaluation are important when doing this. Indeed, volumetric measurements of the macular layers using OCT-based imaging could potentially be used to monitor STGD1 and assess the efficacy of therapeutic interventions as Strauss et al. 184 identified a significant decrease in total macular volume (TMV) over time. However, measuring TMV is limited by its need for significant manual correction for the software algorithms 184 and changes to the preferred retinal locus (PRL) which means that a different location could be imaged; thus, the follow-up function of the software cannot be used in these patients. 185 Moreover, Kong et al. 186 reported difficulties in measuring the individual retinal layers in STGD1 eyes due to factors such as the heterogenous and sporadic EZ disruption and outer retinal disorganisation. Velaga et al. 187 proposed that using an adaptive method to select a subset of B-scan images in STGD1 can help facilitate measuring the area and volume using manual segmentation.
Thinning of the peripapillary retinal nerve fibre layer has also been reported in ABCA4R.188,189
Thickening of the ELM (Figure 14) is thought to be an early sign of STGD1 in paediatric patients.16,103,104,190–193 It is most pronounced in younger patients, 192 occurs in asymptomatic patients103,104 and precedes changes on both fundus examination16,104 and FAF imaging. 16 Interestingly, the only adult to have this sign was a male in his 40s with a BEM phenotype on FAF imaging but this sign was not present in his older brother carrying the same variants who was also in his 40s. 86 Burke et al. 190 suggested that the thickened ELM could represent a Muller cell response to structural changes occurring within the photoreceptors. However, Pang et al. proposed that it could signify ‘migration and retraction of the inner segment ellipsoid back to the ELM during degeneration of photoreceptors’ and, that the thickened hyperreflective appearance could be arising from ‘densely packed mitochondria within the inner segment ellipsoid’. They suggest that this sign does not last as it precedes photoreceptor loss but disappears after progressive outer retinal degeneration. 191 At the time of writing this review, thickened ELM has only been reported in patients with ELOVL4 by Palejwala et al. 194 and in patients with USH1B due to MYO7A variant. 182
Figure 14.

The OCT images in (a), (b) and (c) show areas of thickened ELM particularly within the perifoveal region, examples indicated by the yellow arrow, associated with a normal EZ in (c) and disruptions/vitelliform lesions in (a) and (b), corresponding to the flecks on the IR image, arising from the RPE and either disrupting or elevating the EZ and ELM indicated by the red arrows.
OCT, optical coherence tomography; ELM, external limiting membrane; EZ, ellipsoid zone; IR, infrared.
The optical gap appearance (Figure 15) is also thought to represent an early sign in ABCA4R 111 and has also been linked to the BEM phenotype and milder ABCA4 variants (such as p.(Gly1961Glu). 195 This feature was first described in achromatopsia196,197 and Leng et al. 198 reported it in patients with cone dysfunction and reduced multifocal electroretinogram (mfERG) amplitudes. The optical gap is also reported in cone dystrophies,199,200 occult macular dystrophy,201–203 tamoxifen-induced retinopathy,204,205 ‘poppers maculopathy’,206,207 laser pointer retinopathy208,209 and solar retinopathy. 210 This suggests that it should not be considered a specific feature. Of note, Oh et al. found that the optical gap width increased the most per year in STGD1 patients but the gap height decreased in STGD1 patients. By contrast, the height increased in patients with occult macular dystrophy and achromatopsia. 200 The optical gap is characterised by a disruption of the EZ, thinning of its proximal ONL followed by cavitation in areas with preserved RPE. Noupuu et al. described the progression of the optical gap over three stages that are summarised in Table 6.
Figure 15.
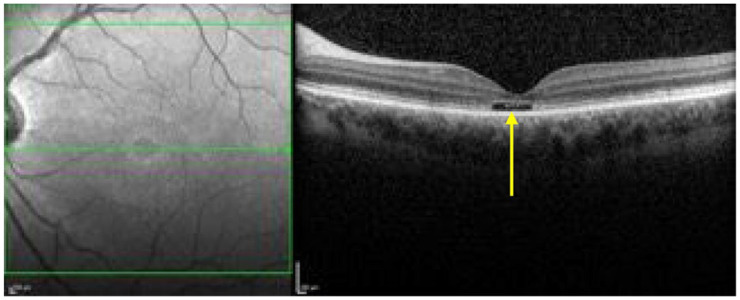
The OCT shows an optical gap (see yellow arrow) in the foveal region containing photoreceptor debris.
OCT, optical coherence tomography.
Table 6.
Summary of the three stages of the optical gap appearance described by Noupuu et al. 195
| Stage 1 | Mild disruption of the EZ in the foveal region |
| Stage 2 | Appearance of the optical gap due to photoreceptor loss following loss of the EZ |
| Stage 3 | Collapse of the inner retinal layers into the optical gap and ultimately resulting in RPE disease and loss |
EZ, ellipsoid zone; RPE, retinal pigment epithelium.
En-face OCT imaging
En-face OCT imaging illustrates the different retinal layers as OCT slabs. The atrophic RPE appears as hyperreflective areas211,212 and the EZ loss appears as abnormal hyporeflective areas.211,212 The area of RPE atrophy tends to be smaller than the area of EZ loss in STGD1.211–214 Indeed, Alabduljalil et al. 213 found that the area of EZ loss was on average 1.6 times larger than the area of RPE atrophy and Sodi et al. found that the area of EZ loss exceeded the RPE atrophy in 96% of STGD1 eyes they assessed 211 and this has similarly been observed in patients with atrophic age-related macular degeneration (AMD). 215
En-face OCT can also be used to measure the area of preserved EZ in RP, thus allowing for monitoring of the disease over time. 216
The choroid
SD-OCT can be used to image the choroid and its related structures. This can be improved by using an EDI OCT protocol. This enables accurate measurements of choroidal thickness. However, blood flow through the choroidal layers is best imaged using OCTA.
The choroidal thickness is reduced in STGD145,121,217–219 and this is most pronounced within the subfoveal region121,217 and the areas temporal and superior to the fovea. 121 Adhi et al. 217 also found that 64% of their STGD1 patients had an irregularly shaped choroidoscleral interface characterised by a concave-convex-concave appearance rather than the bowl-shaped contour in healthy controls. Arrigo et al. 220 used OCTA to describe the progression of choroidal disease based on four patterns which are summarised in Table 7. Choroidal thinning has also been reported in RP221,222 and cone dystrophy. 223 Sabbaghi et al. recently reported that the choroidal thinning pattern was different when comparing patients with STGD1, RP, Usher syndrome and cone dystrophy. They found that patients with RP and Usher syndrome had a significantly thinner choroid than patients with STGD1 and cone dystrophy which led the authors to suggest that IRDs might have different alterations to blood flow 224 or it may be that there is secondary reduction to flow in more severe diseases. The appearance of choroidal caverns is also previously described as a rare finding in AMD patients with geographic atrophy and was suggested by Querques et al. 225 to represent nonperfused vessels and the remnant stromal pillars.
Table 7.
The choroidal patterns described by Arrigo et al. 220 .
| Arrigo et al. choroidal pattern 220 | Appearance on OCTA | Proportion of patients (%) 220 |
|---|---|---|
| Pattern 1 | Normal choroid | 15 |
| Pattern 2 | Reduced Sattler or Haller layer | 29 |
| Pattern 3 | Reduction in both Sattler and Haller layers | 26 |
| Pattern 4 | Reduction in both Sattler and Haller layers in association with choroidal caverns | 30 |
OCTA, optical coherence tomography angiography.
The CC is observed to be atrophic within areas of RPE atrophy.226–228 The area of atrophic RPE seen on FAF images is larger than the area of CC atrophy seen on OCT which suggests that RPE loss precedes CC involvement. 108 There is some controversy as to whether RPE and neuronal loss occurs before CC loss45,108,155,226 or whether the choroid is affected before RPE loss. 213 Of note, Xue et al. 175 have proposed that the choroid is thinned in choroideremia in the early stages of the disease and that loss of the RPE precedes and results in the degeneration of the photoreceptors.
The large choroidal vessels (LCV) tend to be preserved121,217 and Ratra et al. 121 found that they were thickened in 40% of STGD1 eyes and suggested that this could be compensatory dilation to preserve the blood supply following the degeneration of the CC. By contrast, the small choroidal vessels tend to be significantly thinner in all STGD1 eyes. 121 Reich et al. 226 found that the foveal avascular zone is enlarged in both the superficial capillary plexus and deep capillary plexus, but the area on the superior capillary plexus is larger than the deep capillary plexus, in contrast to healthy controls. However, Battaglia et al. 227 found that only the foveal avascular zone on the superior capillary plexus was significantly larger compared with controls, while the larger foveal avascular zone on the deep capillary plexus was no different from controls.
AO
High-resolution retinal imaging with AO enables in vivo visualisation of individual cells in the living eye.229,230 AO retinal imaging systems are based on technology initially used for astronomical imaging and typically consist of a wavefront sensor and a wavefront corrector (commonly a deformable mirror) coupled together by a control system. The naturally occurring optical aberrations of the eye are measured by the wavefront sensor and corrected by the wavefront corrector, enabling AO retinal imaging systems to improve transverse optical resolutions to approximately 2 microns. 231 AO imaging enables visualisation of cone photoreceptors. Rods are smaller, so not easily visualised, but recent improvements have enabled better imaging of rod photoreceptors229,232 and the RPE. 230
The addition of AO to a scanning laser ophthalmoscope (AOSLO) has enabled the capture of high-resolution retinal images with increased contrast. The standard confocal imaging geometry of the AOSLO allows for optical sectioning. The standard, confocal, imaging geometry of the AOSLO provides high contrast en-face images of the highly reflective elements in the photoreceptor outer segments. 233 More recently, novel imaging geometries have been developed (split-detector AOSLO), which allow simultaneous imaging of other retinal structures, such as photoreceptor inner segments by using nonconfocal back-scattered light. 234 Nonconfocal imaging can also be used to assess the RPE. 235 AO imaging has been shown to detect retinal changes before FAF and OCT. 103 This is important to understand early disease stages and for the development of cell-based biomarkers of retinal disease for use as sensitive outcomes in therapeutic trials.
Investigations of early STGD1 with in vivo AOSLO imaging by Song et al. 86 have demonstrated evidence of centrifugal disease progression from the fovea. They observed areas of increased rod and cone photoreceptor spacing in areas of normal conventional clinical imaging, suggesting that photoreceptor loss precedes clinically detectable RPE disease. 86 Longitudinal studies of AOSLO imaging in patients with STGD1 have shown that the earliest cone photoreceptor spacing abnormalities occur in areas of homogeneous SW-FAF. This indicates an absence of RPE disease and these early features can be seen in the context of normal vision and normal outer retinal structures on SD-OCT. 236 These early abnormalities may support the hypothesis that photoreceptor degeneration is a primary event and does not occur secondarily to RPE cell death. These changes are followed by a heterogeneous increase in SW-FAF signal with cone loss, and then by a reduction of SW-FAF signal with cone and RPE cell death. 236 In vivo AF AOSLO imaging in patients with STGD1 has revealed patterns of AF that appear to colocalise with photoreceptors. 237 These observations support histologic reports of photoreceptor bisretinoid accumulation 238 and may be useful as an early biomarker in patients with STGD1. New methods to facilitate the interpretation of complex multimodal imaging datasets from patients with STGD1 with comparison of structural and functional information have also been described. 239
Correlation of multimodal imaging findings and retinal function with electrophysiology and psychophysical evaluation
EDT
EDT is a key tool in diagnosis, prognosis and for counselling in inherited retinal dystrophies and particularly so for ABCA4R as there may be early photoreceptor involvement in asymptomatic patients. It is also the case that repeat evaluation may be necessary. A full assessment of retinal function in ABCA4R is essential as changes in the electrophysiological responses can precede symptoms and changes on both colour and FAF imaging. 103
Electrophysiology testing in a clinical environment when investigating IRD typically includes International Society for Clinical Electrophysiology of Vision (ISCEV) standard pattern electroretinogram (PERG), mfERG, full field electroretinogram (ffERG) (scotopic, photopic as well as a 30-Hz flicker) and electro-oculogram (EOG).240,241 The PERG is used to obtain information regarding the macular and retinal ganglion cell function, represented by the P50 and N95 responses, respectively. The mfERG similarly assesses the central retinal function. The ffERG investigates the general function of the rod and cone photoreceptor cells and can be performed under scotopic conditions to test rod photoreceptors and under photopic conditions for cone photoreceptors. On ffERG, the negative a wave signifies the photoreceptor response, the next positive b wave signifies the bipolar cell and horizontal cell response and the positive c wave signifies the RPE cell response. The EOG assesses the function of the RPE by measuring the difference between the electrically negative retina and the electrically positive cornea.240–243
Different components of retinal function are tested using specific methods; the mfERG for central retinal function, ffERG for general retinal function of the rod and cone photoreceptor cells which can be performed under scotopic conditions to investigate rod photoreceptors and photopic conditions to assess cone photoreceptors
A detailed overview of electrophysiology is not within the scope of this review, but Robson et al. 244 cover this in their publication entitled ‘an ISCEV guide to visual electrodiagnostic procedures’.
In STGD1, the PERG is typically abnormal or absent.37,245 Regarding the more general assessment of the retinal function, Lois et al.’s classification is widely used for STGD1/ABCA4R. This classifies the general retinal function in STGD1 patients into three groups (Table 8); group 1 have an abnormal PERG but a normal ffERG, group 2 have isolated cone dysfunction and group 3 have cone and rod dysfunction. 246 Patients who are more likely to progress more quickly or have more severe disease are in the more severe ERG group.15,126,247 Patients with early onset disease are more likely to be in group 3; late onset disease is more likely to be allocated to group 1. 247 However, a small number of cases can progress in severity, necessitating reallocation to a different group. 15
Table 8.
Groups 1–3 mild to severe based on electrophysiology findings, described by Lois et al. 246
| Lois group | Electrophysiology findings |
|---|---|
| Group 1 | Abnormal PERG with normal ffERG |
| Group 2 | Cone dysfunction |
| Group 3 | Cone and rod dysfunction |
ffERG, full field electroretinogram; PERG, pattern electroretinogram.
Those in Lois group 3 and particularly those with more severe abnormalities of their ffERG are noted to have larger areas of RPE atrophy, 46 a faster RPE atrophy progression rate, 126 peripapillary involvement 46 and are usually in Fishman class 3 and 4167,248 (Table 2). Müller et al. 142 found that patients with a group 1 and 2 ERG had raised qAF levels in all age groups, while the qAF levels were only raised in young patients with group 3 ERG.
Correlation to OCT images shows that loss of the EZ only within the macular region is associated with a group 1 ERG, while widespread EZ loss is seen in group 2 and 3 ERGs.130,214 Moreover, macular thinning and loss of the EZ is associated with abnormalities in the PERG 245 and assessment of a larger field testing of the PERG is thought to be useful for monitoring disease progression despite its lower diagnostic sensitivity and specificity compared with the standard field.
The optical gap phenotype is associated with a normal ffERG but the mfERG shows photoreceptor dysfunction beyond the regions with retinal changes seen on both OCT and FAF111,195 meaning that the functional changes precede structural changes 195 and that abnormalities on mfERG are an early marker of disease. 111 The ERG is completely extinguished in patients with chorioretinal degeneration. 42 It can be seen from this overview that electrophysiology is extremely important in determining the severity of disease, and key in providing useful prognostic advice.
Psychophysical testing (VA and visual field assessment)
Psychophysical evaluation can be very informative when assessing an individual’s visual function and in assessing the functional impact of disease. Electrophysiology can indicate the severity of retinal disease but is not always a good guide to a patient’s visual performance. Psychophysics including VA and visual field testing enables evaluation of visual performance. Visual acuities are usually carried out by assessing a patient’s ability to see a chart at a prerequisite distance wearing their appropriate refractive correction. It is also possible to carry out this type of VA testing using digital optotypes that test VA using a screen where single letters appear and are framed by a crowding bar. 249 There is a wide range of testing equipment, and it is of key importance to ensure testing conditions are kept as uniform as possible for follow-up measurements. LogMAR vision charts are now preferred over Snellen charts as the testing is more reproducible and avoids the issue of ‘crowding’ and enables equal weighting per letter.250,251 Visual field testing can include central and peripheral visual fields. 252 There are a number of standard protocols for this type of testing. In addition, more refined specific types of testing such as fine matrix mapping, microperimetry, mesopic and scotopic perimetry testing may be undertaken on a research basis. 253 It is very important to assess the visual fields for any peripheral or central loss, or scotomata in key regions. This enables us to infer visual function together with the VA. 253 More specific functional visual testing in different environments with obstacles to navigate around in varying lighting conditions have been tailored to test different types of visual loss in different lighting conditions. These are being developed as part of research trials, particularly for severe visual impairment, as standard testing may not deliver any useful information.
VA
VA gives an objective measure of visual function. However, a VA measurement early in disease may not be a good indicator of the potential severity of disease, and speed of progression. In addition, it does not always give an accurate ‘picture’ of how well an individual is functioning in their daily life as this test is generally performed in optimal conditions.
A reduced VA and presence of central scotomas are typically seen in STGD1.31,254 The VA is lower in patients with a more severe Fishman class (Table 1),255,256 and more severe AF type. 24 A better best corrected visual acuity (BCVA) is associated with preservation of the EZ layer,110,120 while a lower BCVA is associated with a wider transverse loss of the photoreceptor layer, reduced foveal thickness110,164 and reduced macular volume. 256 BCVA is relatively preserved in patients with foveal sparing disease22,23,49,51,120,162,176 who can sometimes be asymptomatic. 49 A poorer VA on presentation is generally associated with a more severe disease course. However, the ProgStar study showed that the VA does not correlate with the area of decreased AF signal. 24 Also as the VA may not significantly change over a 2-year period, 257 it is thus not sensitive enough to be used as a primary outcome measure in clinical trials. 20
Visual fields
Testing of both central and peripheral visual fields is important when assessing patients with ABCA4R. STGD1 typically have a progressive central scotoma.31,153 Loss of mid and peripheral visual fields is usually associated with a more severe Fishman class (Table 1), more severe disease with peripheral involvement seen on UWF-AF, and Lois group 3 (Table 8).31,119,254 Schroeder et al. 130 classified the visual field severity into three groups with VF group 1 patients having a central scotoma within 10°, VF group 2 patients having central scotomas of 10°–35° and VF group 3 patients only having ‘temporal residues’ and correlation to colour fundus photographs in these groups typically showed that group 1 patients had isolated pigmentary changes within the macula, group 2 patients had extensive atrophy within the posterior pole that was associated with pigmentary changes in approximately half the patients and group 3 patients had large regions of atrophy that in some patients were associated with ‘bone corpuscle pigmentations’, vascular attenuation and optic nerve head pallor. 130
Microperimetry
More recently, fine mapping of the central retina using microperimetry has been found to be very useful in assessing central visual function. Microperimetry tests retinal sensitivity, which is very useful for assessing patients with macular disease and also allows for assessment of the fixation location and stability258,259 which can be reviewed over time to investigate disease progression.260,261 STGD1 patients with disease confined to the macula (Lois group 1) show varying degrees of reduced sensitivity depending on the stage of disease, but the retinal sensitivity in the peripheral retina is normal.262,263 Microperimetry testing in both scotopic and photopic conditions can be helpful in determining rod and cone involvement, respectively. Microperimetry under different lighting conditions and with different colour stimuli are useful potential outcome measures for therapeutic trials.264,265
Superimposing the macular sensitivities onto the FAF images can be correlated with areas of abnormal or atrophic retina. Retinal sensitivity is undetectable in areas with an absent AF signal, 66 and areas with decreased AF signal have reduced retinal sensitivity compared with regions with homogeneous AF. 53 The sensitivity is also reduced in areas with flecks 266 with the most pronounced difference in retinal sensitivity being between the hyperfluorescent flecks and their adjacent area.53,266 The mean sensitivity is higher in the more eccentric flecks on NIR-AF. 53 It is preserved in the spared fovea 52 and higher in nasal region in patients with extramacular disease, and higher in spared peripapillary area. 166 Changes in retinal sensitivity compared with healthy controls were also found to have a higher correlation with the thickness of the OS compared with the ONL and outer plexiform layer (OPL) thickness. 166 Reduction of the mean sensitivity in patients with a normal RPE and EZ layer led Salvatore et al. 35 to propose that microperimetry can help identify which regions to treat as functional changes affecting the photoreceptors detected by microperimetry might precede structural changes. The ProgStar study found that the mean yearly change in mean sensitivity was −0.68 dB/year. 263 The mean sensitivity at the border of a dense scotoma, termed edge mean sensitivity, is lower than the mean sensitivity (yearly progression rate of mean sensitivity was 1.5 dB/year compared with 2.9 dB/year for the edge mean sensitivity), which led Schönbach et al. 265 to propose that the edge mean sensitivity could potentially be used as a sensitive outcome measure over shorter periods of time in clinical trials. The ProgStar study also recently used software algorithms to create a 3D model of the volume of hill of vision from the microperimetry results which can identify changes in visual function and potentially be used as end points in clinical trials. 267 Josan et al. have similarly found that microperimetry measurements of the volumetric hill of vision were useful in STGD1, RPGR-related RP and choroideremia as it avoided averaging floor effects observed in standard microperimetry. They observed that volume measures might be able to detect sensitivity changes that would be missed by the standard mean sensitivity microperimetry approach. 268
The role of fixation stability in monitoring disease progression remains unknown as the recent results from the ProgStar study have suggested that the fixation stability might not be a sensitive outcome measure for therapeutic trials but can give useful information on visual function. 261
Discussion
Retinal imaging and functional testing are key to diagnosing and characterising severity of STGD1 and ABCA4R. ABCA4R comprise quite distinct phenotypes, but with variation in presentation and progression. Multimodal imaging and functional testing can detect and highlight progressive changes, which can be used to describe the natural history of the disease. Information derived from imaging and functional testing is invaluable in counselling patients regarding diagnosis and prognosis. Some of which are already employed as outcome measures in therapeutic trials. FAF is particularly important for diagnosing STGD1 as it is better at detecting the characteristic flecks and macular atrophy than colour photography or fundoscopy, and in some cases can even detect changes in asymptomatic patients who show no retinal changes on colour photographs or fundoscopy. AF imaging can thus highlight early onset severe disease and together with vision function testing and genotyping can enable informed counselling regarding prognosis. In addition, it will help define cohorts of patients for intervention. AF is an important tool, which can be used with automated software to track progression in patients by measuring atrophy progression. NIR-AF imaging can detect flecks and a larger area of RPE atrophy not detected by SW-AF imaging; thus, it is a useful tool to predict earlier changes and future progression in patients. In addition, it confers the benefit of less light toxicity during examination. qAF imaging could prove very useful in aiding diagnosis in suspected ABCA4R, particularly in those in whom only a single ABCA4 variant has been identified, supporting further genetic testing to identify a second variant. However, the short wavelength of light used in qAF is potentially toxic to the photoreceptors, which limits this modality’s use. In addition, abnormalities of qAF are observed in other types of IRD. OCT imaging provides complementary information, allowing a detailed view of the retinal layers. OCT is also informative regarding the behaviour of disease; a preserved EZ layer is typically associated with a better VA and retinal sensitivity. This observation is useful as a marker for prognosis, or to identify regions for gene therapy treatment, and to inform response to intervention. Early signs of disease such as a thickened ELM can be used to diagnose asymptomatic patients, for example, early onset in children. The changes seen in retinal imaging also correlate to EDT which can be used to predict the progression in patients. Patients with rod and cone-rod involvement typically have more severe progressive RPE atrophy and changes to the EZ layer. More recently, AO has demonstrated that in STGD1, there is an increase in cone spacing, a decrease in cone density and enlargement of rod photoreceptors compared with healthy control eyes. These findings correlate with the changes seen on OCT and AF imaging.236,237 Meticulous characterisation of the phenotype with multimodal imaging and functional testing in STGD1 will be important towards assessing the efficacy of ongoing therapeutic approaches and help towards identifying suitable patients for inclusion in therapeutic trials which include methods such as modification of visual cycle modulators, gene therapy, complement system inhibition and cell replacement-based therapy.
There are at least 10 visual cycle modulator compounds currently undergoing evaluation: deuterated vitamin A,269–271 retinal binding protein antagonists (fenretinide and A1120),272,273 isotretinoin,274,275 amine-based drugs, 276 soraprazan, 277 ticagrelor, 278 VX-809, 279 saffron, 280 docosahexaenoic acid (DHA) supplementation 281 and omega 3 fatty acid supplementation. 282
It is expected that treatments that lower the lipofuscin levels will be best monitored using FAF and qAF imaging. OCT imaging is likely to be useful as well as it can provide information on the preservation of the retinal architecture with markers such as the preservation of the EZ layer and thickness of the retinal layers. Deuterated vitamin A is predicted to slow the production and accumulation of the toxic A2E269,270 and response to this treatment could be monitored by measuring qAF. 271 Fenretinide and A1120 limit the entry of vitamin A into retinal cells by competitively binding to the retinal binding protein,272,283 thus limiting the amount of all-trans-retinol reaching the RPE. 273 Both have been shown to reduce A2E/lipofuscin levels in Abca4 KO mice, and mice treated with fenretinide had a reduction in AF levels.272,283 Isotretinoin has also been demonstrated to reduce A2E levels in Abca4 KO mice on electron microscopy 275 and also protect photoreceptors from light-induced damage. 274 Amine-based drugs can also be used to prevent the formation of A2E, and the retinal architecture was found to be more preserved in treated Abca4 KO mice compared with untreated mice. 276 Soraprazan is a treatment for gastroeosophageal reflux disease, 284 which has been observed to reduce the lipofuscin levels in treated monkeys 277 and mice. 285
Ticagrelor is a P2Y12 receptor inhibitor that has been found to decrease the lysosomal pH in Abca4 KO mice. OCT imaging, FAF imaging and electrophysiology will be useful in monitoring the response to ticagrelor as treated Abca4 KO mice had a thicker ONL on both OCT imaging and histological assessment, 286 had lower AF signal 278 and an improvement in a- and b-wave responses on electrophysiology testing. 286
Saffron treatment has not been shown to significantly improve the focal ERG or the VA. 280 DHA supplementation has not been found to improve retinal function in STGD1 281 and omega 3 fatty acid supplementation is currently under evaluation 282 (NCT03297515). VX-809 treatment used for cystic fibrosis has been shown to increase levels of ABCA4 expression in HEK293 cells. 279
Gene therapy approach whereby a vector is used to introduce a functioning gene to the diseased cells is a promising approach. RPE65 retinal degeneration was the first licenced gene therapy treatment for an IRD. This is delivered using an adeno-associated virus (AAV) vector. Other gene therapy or gene targeted approaches currently under investigation include a dual vector approach,287,288 using nanoparticles as a vector, 289 complement modulation290–292 and antisense oligonucleotides.293,294
Gene therapy trials to treat AMD to increase expression of complement receptor 1-like protein y (CRRY) 290 and the C5 complement inhibition 291 by Zimura 292 are underway with the aim of modifying complement activation. OCT imaging will play an important role in assessing these patients; the Zimura trial is assessing the mean rate of change of the EZ over an 18-month period.292,295 FAF is important to image change over time. Mice treated with the vector containing CRRY were noted to have a reduction in bisretinoid levels, in addition to a slower degeneration of photoreceptor cells. 290 Antisense oligonucleotides are short sequences of RNA that can modulate splicing defects 296 and have been shown to correct splicing defects caused by deep intronic variants in the ABCA4 gene.297,298
For these gene therapy approaches, OCT imaging can provide information on the extent of preservation of the retinal layers. The extent of disruption of the EZ and ELM layer has been shown to correlate with the BCVA, retinal sensitivity on microperimetry and responses on ERG and mfERG111,120,164 which suggests that assessment of these layers will be important in assessing retinal function in gene therapy trials. Recently, the ONL thickness was also shown to have a higher correlation with the BCVA when compared with the area of photoreceptor loss, and area of RPE atrophy measured on en-face OCT. This suggests that measuring the ONL may also be important in assessing clinical trial results. 299 The Zimura trial will specifically assess the rate of degeneration of the EZ. At the time of writing this review, the results of this trial were not yet available.292,295 Measurement of the retinal layers is performed using segmentation software. Automatic segmentation software is available, but errors may occur when measurements are affected by outer retinal disruptions, 300 and the presence of poorly defined hyperreflective lesions such as flecks. 187 Currently manual segmentation is required to correct these errors.186,301 Improvements in segmentation software will be vital in enabling objective assessment of the retinal layers in gene therapy trials. 187 FAF and qAF imaging are ideal techniques to assess changes to lipofuscin accumulation. AO imaging will be important in assessing the structural changes to photoreceptors and counting cones. Microperimetry will enable tracking of retinal sensitivity and temporal changes in sensitivity, as well as an accurate assessment of the central visual field.
Stem cell-based therapies in ABCA4R are aimed at replacing diseased RPE cells with RPE derived either from human embryonic stem cells (hESCs) or from induced pluripotent stem cells (iPSCs). The value of hESCs intervention for patients with ABCA4R and AMD is being evaluated in a trial.302–304 Colour photographs are useful in assessing treatment outcomes as hyperpigmentation in the treated region is thought to indicate survival of transplanted cells.302,305 These hyperpigmented regions correspond to hyperreflective areas between the photoreceptor cells and Bruch’s membrane. The role of FAF is yet to be determined for this therapeutic approach, as AF was shown to be absent up to 12 months following treatment in the trial by Mehat et al. 305 Oner et al. 306 transplanted adipose tissue derived from mesenchymal stem cells into the suprachoroidal space in STGD1 and AMD patients with some improvements in their VA, visual field and mfERG.
The number of potential interventions for ABCA4R is burgeoning with a range of different approaches. The role of imaging and functional testing will also evolve in response to this, with a drive to employ more sophisticated methods of assessing early signs of disease. The use of AOSLO, and microperimetry under different lighting conditions with different stimuli, and digital methods of assessing reading speed and acuity are examples of recent innovations. The advent of automated reading of imaging on a large scale with the introduction of artificial intelligence (AI) to this field will enable accurate evaluation of large datasets. These innovations will be key in revolutionising our ability to assess multiple images in large cohorts over time enabling an objective assessment of response to disease.
Conclusion
A comprehensive understanding of multimodal imaging (Table 9) and functional testing with access to these investigations remains key in diagnosis, prognosis and genetic counselling, as well as in assisting the complex tasks of distinguishing phenocopies, evaluating the role of genetic modifiers, identifying cohorts for therapies and assessing response to interventions.
Table 9.
Summary of the typical findings identified on multimodal imaging in Stargardt disease.
| Feature | Colour fundus imaging | Fluorescence angiography | ICGA | SW-AF imaging | NIR-AF imaging | Infrared image | Optical coherence tomography | Optical coherence tomography angiography | Adaptive optics |
|---|---|---|---|---|---|---|---|---|---|
| Flecks | Yellow-white flecks | Usually hypofluorescent flecks but can sometimes be hyperfluorescent | Hypofluorescent and do not always correspond to flecks seen in colour photographs | Typically hyperfluorescent and resorbed flecks can be hypofluorescent | Typically hypofluorescent | Typically, hyperreflective, while resorbing flecks are hyporeflective | hyper-reflective deposits | Hyperreflective deposits | Highly reflective structures that are not arranged in a contigious mosaic |
| Atrophy | ‘Beaten bronze’ appearance. The atrophic area that can sometimes be associated with visible choroid and pigmentary deposition | Hyperfluorescent | Dark region surrounded by a ring of increased signal | Decreased AF signal | Decreased AF signal | Hyperreflective region that is usually surrounded by a darker less reflective ring | Disruption/ loss of the RPE layer (difficult to discern) and loss of the EZ | Disruption/loss of the RPE layer (difficult to discern) and loss of the EZ | Brightly oversaturated |
| Other features identified specifically by modality | UWF imaging can show peripheral pigmented lesions in some patients | Dark choroid sign | Reticular pattern of decreased fluorescence | Flecks and atrophy usually more advanced than those seen in SW-AF | Thickening of the ELM in early disease | Absence of choriocapillaris in macular areas of EZ loss | Increased cone spacing Enlarged photoreceptors Increased cone: rod ratio |
AF, autofluorescence; ELM, external limiting membrane; EZ, ellipsoid zone; ICGA, indocyanine green angiography; NIR-AF, near infrared-wavelength autofluorescence; RPE, retinal pigment epithelium; SW-AF, short-wavelength autofluorescence; UWF, ultra-widefield.
Acknowledgments
The authors would like to thank the Eye Research Group Oxford (ERGO) for their support of this study, in particular Alexina Fantato, Clare Arnison-Newgass, Sophie Marlowe and Lidia Milla, as well as the patients who agreed to participate in this study.
Footnotes
Author contributions: Conceptualization, SH and SMD; investigation, SA-K, SB, SH and SMD; writing–original draft preparation, SA-K, SB, MS, SH and SMD; writing–review and editing, SA-K, SB, CRF, MS, JY, SH and SMD; supervision, SH and SMD. All authors have read and agreed to the published version of the manuscript.
Conflict of interest statement: The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The Thames Valley and South Midlands Clinical Research Network provided funding support for the clinical aspects of this study. Saoud Al-Khuzaei is supported by a scholarship from the Qatar National Research Fund (GSRA6-1-0329-19010). Jing Yu is supported by funding from the UK inherited Retinal Disease Consortium Project (UKIRDC) (RetinaUK) (HMR03950) and Winstanley Family Donation for Inherited Macular Diseases (HMD00280).
Ethics and declarations: This study was conducted in accordance with the Declaration of Helsinki with ethics approval obtained from the local research ethics committee (reference 08/H0302/96).
Consent for publication: Informed written consent was obtained from all patients
ORCID iDs: Mital Shah  https://orcid.org/0000-0001-7496-6121
https://orcid.org/0000-0001-7496-6121
Susan M. Downes
https://orcid.org/0000-0001-7373-2665
Contributor Information
Saoud Al-Khuzaei, Oxford Eye Hospital, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, UK; Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK.
Mital Shah, Oxford Eye Hospital, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, UK.
Charlotte R. Foster, Pathlab, Tauranga, New Zealand
Stephanie Halford, Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK.
Susan M. Downes, Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences, University of Oxford, Level 6 John Radcliffe Hospital, Headley Way, Oxford OX3 9DU, UK; Oxford Eye Hospital, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, UK.
References
- 1. Stargardt K. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Albrecht Von Graefes Archiv Für Ophthalmologie 1909; 71: 534–550. [Google Scholar]
- 2. Franceschetti A, Francois J. Fundus flavimaculatus. Arch Ophtalmol Rev Gen Ophtalmol 1965; 25: 505–530. [PubMed] [Google Scholar]
- 3. Franceschetti A. A special form of tapetoretinal degeneration: fundus flavimaculatus. Trans Am Acad Ophthalmol Otolaryngol 1965; 69: 1048–1053. [PubMed] [Google Scholar]
- 4. Noble KG, Carr RE. Stargardt’s disease and fundus flavimaculatus. Arch Ophthalmol 1979; 97: 1281–1285. [DOI] [PubMed] [Google Scholar]
- 5. Hadden OB, Gass JD. Fundus flavimaculatus and Stargardt’s disease. Am J Ophthalmol 1976; 82: 527–539. [DOI] [PubMed] [Google Scholar]
- 6. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat Genet 1997; 15: 236–246. [DOI] [PubMed] [Google Scholar]
- 7. Illing M, Molday LL, Molday RS. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem 1997; 272: 10303–10310. [DOI] [PubMed] [Google Scholar]
- 8. Sun H, Nathans J. Stargardt’s ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet 1997; 17: 15–16. [DOI] [PubMed] [Google Scholar]
- 9. Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet 2000; 25: 257–258. [DOI] [PubMed] [Google Scholar]
- 10. Lenis TL, Hu J, Ng SY, et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci USA 2018; 115: E11120–E11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun 2012; 3: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blacharski P. Retinal dystrophies and degenerations. New York: Raven Press, 1988, pp. 135–159. [Google Scholar]
- 13. Spiteri Cornish K, Ho J, Downes S, et al. The epidemiology of Stargardt disease in the United Kingdom. Ophthalmol Retina 2017; 1: 508–513. [DOI] [PubMed] [Google Scholar]
- 14. Burke TR, Tsang SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet 2011; 32: 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujinami K, Lois N, Davidson AE, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol 2013; 155: 1075–1088. [DOI] [PubMed] [Google Scholar]
- 16. Lambertus S, van Huet RA, Bax NM, et al. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology 2015; 122: 335–344. [DOI] [PubMed] [Google Scholar]
- 17. Strauss RW, Ho A, Munoz B, et al. The natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies: design and baseline characteristics: progstar report no. 1. Ophthalmology 2016; 123: 817–828. [DOI] [PubMed] [Google Scholar]
- 18. Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci USA 2020; 117: 2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kong X, Strauss RW, Michaelides M, et al. Visual acuity loss and associated risk factors in the retrospective progression of Stargardt disease study (ProgStar report no. 2). Ophthalmology 2016; 123: 1887–1897. [DOI] [PubMed] [Google Scholar]
- 20. Kong X, Strauss RW, Cideciyan AV, et al. Visual acuity change over 12 months in the prospective progression of atrophy secondary to Stargardt disease (ProgStar) study: ProgStar report number 6. Ophthalmology 2017; 124: 1640–1651. [DOI] [PubMed] [Google Scholar]
- 21. Miraldi Utz V, Coussa RG, Marino MJ, et al. Predictors of visual acuity and genotype-phenotype correlates in a cohort of patients with Stargardt disease. Br J Ophthalmol 2014; 98: 513–518. [DOI] [PubMed] [Google Scholar]
- 22. Fujinami K, Sergouniotis PI, Davidson AE, et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am J Ophthalmol 2013; 156: 487–501. [DOI] [PubMed] [Google Scholar]
- 23. Van-Westeneng Haaften SC, Boon CJ, Cremers FP, et al. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012; 119: 1199–1210. [DOI] [PubMed] [Google Scholar]
- 24. Kong X, West SK, Strauss RW, et al. Progression of visual acuity and fundus autofluorescence in recent-onset Stargardt disease: ProgStar study report #4. Ophthalmol Retina 2017; 1: 514–523. [DOI] [PubMed] [Google Scholar]
- 25. Testa F, Melillo P, Di Iorio V, et al. Macular function and morphologic features in juvenile stargardt disease: longitudinal study. Ophthalmology 2014; 121: 2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yatsenko AN, Shroyer NF, Lewis RA, et al. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet 2001; 108: 346–355. [DOI] [PubMed] [Google Scholar]
- 27. Maugeri A, van Driel MA, van de Pol DJ, et al. The 2588G–>C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 1999; 64: 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaakson K, Zernant J, Külm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003; 22: 395–403. [DOI] [PubMed] [Google Scholar]
- 29. Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 2000; 67: 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology 2003; 110: 1151–1158. [DOI] [PubMed] [Google Scholar]
- 31. Fishman GA, Stone EM, Grover S, et al. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol 1999; 117: 504–510. [DOI] [PubMed] [Google Scholar]
- 32. Klevering BJ, Blankenagel A, Maugeri A, et al. Phenotypic spectrum of autosomal recessive cone–rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci 2002; 43: 1980–1985. [PubMed] [Google Scholar]
- 33. Scholl HP, Besch D, Vonthein R, et al. Alterations of slow and fast rod ERG signals in patients with molecularly confirmed Stargardt disease type 1. Invest Ophthalmol Vis Sci 2002; 43: 1248–1256. [PubMed] [Google Scholar]
- 34. Kang Derwent JJ, Derlacki DJ, Hetling JR, et al. Dark adaptation of rod photoreceptors in normal subjects, and in patients with Stargardt disease and an ABCA4 mutation. Invest Ophthalmol Vis Sci 2004; 45: 2447–2456. [DOI] [PubMed] [Google Scholar]
- 35. Salvatore S, Fishman GA, McAnany JJ, et al. Association of dark-adapted visual function with retinal structural changes in patients with Stargardt disease. Retina 2014; 34: 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vandenbroucke T, Buyl R, De Zaeytijd J, et al. Colour vision in Stargardt disease. Ophthalmic Res 2015; 54: 181–194. [DOI] [PubMed] [Google Scholar]
- 37. Lois N, Halfyard AS, Bird AC, et al. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol 2004; 138: 55–63. [DOI] [PubMed] [Google Scholar]
- 38. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci 2005; 46: 4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwoerer J, Secretan M, Zografos L, et al. Indocyanine green angiography in Fundus flavimaculatus. Ophthalmologica 2000; 214: 240–245. [DOI] [PubMed] [Google Scholar]
- 40. Duncker T, Tsang SH, Lee W, et al. Quantitative fundus autofluorescence distinguishes ABCA4-associated and Non–ABCA4-associated Bull’s-eye maculopathy. Ophthalmology 2015; 122: 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Noupuu K, Lee W, Zernant J, et al. Recessive Stargardt disease phenocopying hydroxychloroquine retinopathy. Graefes Arch Clin Exp Ophthalmol 2016; 254: 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee W, Zernant J, Nagasaki T, et al. Deep scleral exposure: a degenerative outcome of end-stage stargardt disease. Am J Ophthalmol 2018; 195: 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bertelsen M, Zernant J, Larsen M, et al. Generalized choriocapillaris dystrophy, a distinct phenotype in the spectrum of ABCA4-associated retinopathies. Invest Ophthalmol Vis Sci 2014; 55: 2766–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee W, Schuerch K, Zernant J, et al. Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur J Hum Genet 2017; 25: 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muller PL, Fimmers R, Gliem M, et al. Choroidal alterations in ABCA4-related retinopathy. Retina 2017; 37: 359–367. [DOI] [PubMed] [Google Scholar]
- 46. Burke TR, Allikmets R, Smith RT, et al. Loss of peripapillary sparing in non-group I Stargardt disease. Exp Eye Res 2010; 91: 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jayasundera T, Rhoades W, Branham K, et al. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. Am J Ophthalmol 2010; 149: 656–660. [DOI] [PubMed] [Google Scholar]
- 48. Hwang JC, Zernant J, Allikmets R, et al. Peripapillary atrophy in Stargardt disease. Retina 2009; 29: 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Huet RA, Bax NM, Westeneng-Van Haaften SC, et al. Foveal sparing in Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 7467–7478. [DOI] [PubMed] [Google Scholar]
- 50. Muller PL, Pfau M, Mauschitz MM, et al. Comparison of green versus blue fundus autofluorescence in ABCA4-related retinopathy. Transl Vis Sci Technol 2018; 7: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Battaglia Parodi M, Sacconi R, Romano F, et al. Hyperreflective foci in Stargardt disease: 1-year follow-up. Graefes Arch Clin Exp Ophthalmol 2019; 257: 41–48. [DOI] [PubMed] [Google Scholar]
- 52. Nakao T, Tsujikawa M, Sawa M, et al. Foveal sparing in patients with Japanese Stargardt’s disease and good visual acuity. JPN J Ophthalmol 2012; 56: 584–588. [DOI] [PubMed] [Google Scholar]
- 53. Muller PL, Birtel J, Herrmann P, et al. Functional relevance and structural correlates of near infrared and short wavelength fundus autofluorescence imaging in ABCA4-related retinopathy. Transl Vis Sci Technol 2019; 8: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arrigo A, Romano F, Aragona E, et al. OCTA-based identification of different vascular patterns in Stargardt disease. Transl Vis Sci Technol 2019; 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klevering BJ, Deutman AF, Maugeri A, et al. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol 2005; 243: 90–100. [DOI] [PubMed] [Google Scholar]
- 56. Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet 1998; 7: 355–362. [DOI] [PubMed] [Google Scholar]
- 57. Tanaka K, Lee W, Zernant J, et al. The rapid-onset chorioretinopathy phenotype of ABCA4 disease. Ophthalmology 2018; 125: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Briggs CE, Rucinski D, Rosenfeld PJ, et al. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci 2001; 42: 2229–2236. [PubMed] [Google Scholar]
- 59. Martínez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998; 18: 11–12. [DOI] [PubMed] [Google Scholar]
- 60. Rossi S, Testa F, Attanasio M, et al. Subretinal fibrosis in Stargardt’s disease with fundus flavimaculatus and ABCA4 gene mutation. Case Rep Ophthalmol 2012; 3: 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ober RR, Limstrom SA, Simon RM. Traumatic retinopathy in Stargardt’s disease. Retina 1997; 17: 251–254. [DOI] [PubMed] [Google Scholar]
- 62. Del Buey MA, Huerva V, Minguez E, et al. Posttraumatic reaction in a case of fundus flavimaculatus with atrophic macular degeneration. Ann Ophthalmol 1993; 25: 219–221. [PubMed] [Google Scholar]
- 63. Gass JD, Hummer J. Focal retinal pigment epithelial dysplasia associated with fundus flavimaculatus. Retina 1999; 19: 297–301. [DOI] [PubMed] [Google Scholar]
- 64. De Laey JJ, Verougstraete C. Hyperlipofuscinosis and subretinal fibrosis in Stargardt’s disease. Retina 1995; 15: 399–406. [DOI] [PubMed] [Google Scholar]
- 65. Grandinetti AA, Portella E, Arana J, et al. Subretinal fibrosis in Stargardt’s disease: case report. Arq Bras Oftalmol 2011; 74: 449–451. [DOI] [PubMed] [Google Scholar]
- 66. Anastasakis A, Fishman GA, Lindeman M, et al. Infrared scanning laser ophthalmoscope imaging of the macula and its correlation with functional loss and structural changes in patients with Stargardt disease. Retina 2011; 31: 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oh KT, Weleber RG, Oh DM, et al. Clinical phenotype as a prognostic factor in Stargardt disease. Retina 2004; 24: 254–262. [DOI] [PubMed] [Google Scholar]
- 68. Zhao PY, Abalem MF, Nadelman D, et al. Peripheral pigmented retinal lesions in Stargardt disease. Am J Ophthalmol 2018; 188: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Battaglia Parodi M, Munk MR, Iacono P, et al. Ranibizumab for subfoveal choroidal neovascularisation associated with Stargardt disease. Br J Ophthalmol 2015; 99: 1268–1270. [DOI] [PubMed] [Google Scholar]
- 70. Roy R, Kumar A, Ghosh S, et al. All that glitters are not flecks: inflammatory choroidal neovascularization in fundus flavimaculatus. Ocul Immunol Inflamm 2015; 23: 188–190. [DOI] [PubMed] [Google Scholar]
- 71. Koh V, Naing T, Chee C. Fundus flavimaculatus and choroidal neovascularization in a young patient with normal electroretinography: case report. Can J Ophthalmol 2012; 47: e3–e5. [DOI] [PubMed] [Google Scholar]
- 72. Simonelli F, Testa F, Zernant J, et al. Association of a homozygous nonsense mutation in the ABCA4 (ABCR) gene with cone-rod dystrophy phenotype in an Italian family. Ophthalmic Res 2004; 36: 82–88. [DOI] [PubMed] [Google Scholar]
- 73. Fukui T, Yamamoto S, Nakano K, et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 2002; 43: 2819–2824. [PubMed] [Google Scholar]
- 74. van Driel MA, Maugeri A, Klevering BJ, et al. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet 1998; 19: 117–122. [DOI] [PubMed] [Google Scholar]
- 75. Shroyer NF, Lewis RA, Allikmets R, et al. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res 1999; 39: 2537–2544. [DOI] [PubMed] [Google Scholar]
- 76. Zernant J, Schubert C, Im KM, et al. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci 2011; 52: 8479–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sangermano R, Bax NM, Bauwens M, et al. Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461-10T→C mutation in Stargardt disease. Ophthalmology 2016; 123: 1375–1385. [DOI] [PubMed] [Google Scholar]
- 78. Aukrust I, Jansson RW, Bredrup C, et al. The intronic ABCA4 c.5461-10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmologica 2017; 95: 240–246. [DOI] [PubMed] [Google Scholar]
- 79. Al-Khuzaei S, Broadgate S, Foster CR, et al. An overview of the genetics of ABCA4 retinopathies, an evolving story. Genes (Basel) 2021; 12: 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stone EM, Nichols BE, Kimura AE, et al. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol 1994; 112: 765–772. [DOI] [PubMed] [Google Scholar]
- 81. Agbaga M-P, Brush RS, Mandal MNA, et al. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci USA 2008; 105: 12843–12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wolock CJ, Stong N, Ma CJ, et al. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet Med 2019; 21: 2336–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kniazeva M, Chiang MF, Morgan B, et al. A new locus for autosomal dominant Stargardt-like disease maps to chromosome 4. Am J Hum Genet 1999; 64: 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Strauss RW, Muñoz B, Ahmed MI, et al. The progression of the Stargardt disease type 4 (ProgStar-4) study: design and baseline characteristics (ProgStar-4 report no. 1). Ophthalmic Res 2018; 60: 185–194. [DOI] [PubMed] [Google Scholar]
- 85. Lorenz B, Wabbels B, Wegscheider E, et al. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 2004; 111: 1585–1594. [DOI] [PubMed] [Google Scholar]
- 86. Song H, Rossi EA, Latchney L, et al. Cone and rod loss in Stargardt disease revealed by adaptive optics scanning light ophthalmoscopy. JAMA Ophthalmol 2015; 133: 1198–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Panwar N, Huang P, Lee J, et al. Fundus photography in the 21st century–a review of recent technological advances and their implications for worldwide healthcare. Telemed J E Health 2016; 22: 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shoughy S, Arevalo J, Kozak I. Update on wide- and ultra-widefield retinal imaging. Indian J Ophthalmol 2015; 63: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Witmer MT, Kiss S. Wide-field imaging of the retina. Surv Ophthalmol 2013; 58: 143–154. [DOI] [PubMed] [Google Scholar]
- 90. Chen J. Comparison of the performance of four Fundus Cameras in clinical practice. Invest Ophthalmol Vis Sci 2019; 60: 6121. [Google Scholar]
- 91. Fishman GA. Fundus flavimaculatus: a clinical classification. Arch Ophthalmol 1976; 94: 2061–2067. [DOI] [PubMed] [Google Scholar]
- 92. Spaide RF. Fundus autofluorescence and age-related macular degeneration. Ophthalmology 2003; 110: 392–399. [DOI] [PubMed] [Google Scholar]
- 93. Webb RH, Hughes GW, Delori FC. Confocal scanning laser ophthalmoscope. Appl Opt 1987; 26: 1492–1499. [DOI] [PubMed] [Google Scholar]
- 94. Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. Int J Retina Vitreous 2016; 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Keilhauer CN, Delori FC. Near-infrared autofluorescence imaging of the fundus: visualization of ocular melanin. Invest Ophthalmol Vis Sci 2006; 47: 3556–3564. [DOI] [PubMed] [Google Scholar]
- 96. Oishi A, Miyata M, Numa S, et al. Wide-field fundus autofluorescence imaging in patients with hereditary retinal degeneration: a literature review. Int J Retina Vitreous 2019; 5: 23–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Oishi A, Hidaka J, Yoshimura N. Quantification of the image obtained with a wide-field scanning ophthalmoscope. Invest Ophthalmol Vis Sci 2014; 55: 2424–2431. [DOI] [PubMed] [Google Scholar]
- 98. Wolf-Schnurrbusch UE, Wittwer VV, Ghanem R, et al. Blue-light versus green-light autofluorescence: lesion size of areas of geographic atrophy. Invest Ophthalmol Vis Sci 2011; 52: 9497–9502. [DOI] [PubMed] [Google Scholar]
- 99. Chen L, Lee W, de Carvalho JRL, et al. Multi-platform imaging in ABCA4-associated disease. Sci Rep 2019; 9: 6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schmitz-Valckenberg S, Holz FG, Bird AC, et al. Fundus autofluorescence imaging: review and perspectives. Retina 2008; 28: 385–409. [DOI] [PubMed] [Google Scholar]
- 101. Frampton GK, Kalita N, Payne L, et al. Fundus autofluorescence imaging: systematic review of test accuracy for the diagnosis and monitoring of retinal conditions. Eye 2017; 31: 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Delori FC, Dorey CK, Staurenghi G, et al. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci 1995; 36: 718–729. [PubMed] [Google Scholar]
- 103. Khan KN, Kasilian M, Mahroo OAR, et al. Early patterns of macular degeneration in ABCA4-associated retinopathy. Ophthalmology 2018; 125: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bax NM, Lambertus S, Cremers FPM, et al. The absence of fundus abnormalities in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2019; 257: 1147–1157. [DOI] [PubMed] [Google Scholar]
- 105. Strauss RW, Muñoz B, Ho A, et al. Progression of Stargardt disease as determined by fundus autofluorescence in the retrospective progression of Stargardt disease study (ProgStar report no. 9). JAMA Ophthalmol 2017; 135: 1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Strauss RW, Muñoz B, Ho A, et al. Incidence of atrophic lesions in Stargardt Disease in the progression of atrophy secondary to Stargardt disease (ProgStar) study: report no. 5. JAMA Ophthalmol 2017; 135: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fujinami K, Lois N, Mukherjee R, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci 2013; 54: 8181–8190. [DOI] [PubMed] [Google Scholar]
- 108. Guduru A, Lupidi M, Gupta A, et al. Comparative analysis of autofluorescence and OCT angiography in Stargardt disease. Br J Ophthalmol 2018; 102: 1204–1207. [DOI] [PubMed] [Google Scholar]
- 109. Strauss RW, Kong X, Ho A, et al. Progression of Stargardt disease as determined by fundus autofluorescence over a 12-month period: ProgStar report no. 11. JAMA Ophthalmology 2019; 137: 1134–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ergun E, Hermann B, Wirtitsch M, et al. Assessment of central visual function in Stargardt’s disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest Ophthalmol Vis Sci 2005; 46: 310–316. [DOI] [PubMed] [Google Scholar]
- 111. Gomes NL, Greenstein VC, Carlson JN, et al. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci 2009; 50: 3953–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Paavo M, Lee W, Allikmets R, et al. Photoreceptor cells as a source of fundus autofluorescence in recessive Stargardt disease. J Neurosci Res 2019; 97: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cukras CA, Wong WT, Caruso R, et al. Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Archives of Ophthalmology 2012; 130: 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sparrow JR, Marsiglia M, Allikmets R, et al. Flecks in recessive Stargardt disease: short-wavelength autofluorescence, near-infrared autofluorescence, and optical coherence tomography. Invest Ophthalmol Vis Sci 2015; 56: 5029–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Duncker T, Marsiglia M, Lee W, et al. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 8134–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kumar V. Insights into autofluorescence patterns in Stargardt macular dystrophy using ultra-wide-field imaging. Graefes Arch Clin Exp Ophthalmol 2017; 255: 1917–1922. [DOI] [PubMed] [Google Scholar]
- 117. Duncker T, Lee W, Tsang SH, et al. Distinct characteristics of inferonasal fundus autofluorescence patterns in Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 2013; 54: 6820–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Duncker T, Greenberg JP, Sparrow JR, et al. Visualization of the optic fissure in short-wavelength autofluorescence images of the fundus. Invest Ophthalmol Vis Sci 2012; 53: 6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Abalem MF, Otte B, Andrews C, et al. Peripheral visual fields in ABCA4 Stargardt disease and correlation with disease extent on ultra-widefield fundus autofluorescence. Am J Ophthalmol 2017; 184: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Testa F, Rossi S, Sodi A, et al. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: implications for gene therapy. Invest Ophthalmol Vis Sci 2012; 53: 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ratra D, Jaishankar D, Sachidanandam R, et al. Swept-source optical coherence tomography study of choroidal morphology in Stargardt disease. Oman J Ophthalmol 2018; 11: 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. McBain VA, Townend J, Lois N. Progression of retinal pigment epithelial atrophy in Stargardt disease. Am J Ophthalmol 2012; 154: 146–154. [DOI] [PubMed] [Google Scholar]
- 123. Klufas MA, Tsui I, Sadda SR, et al. Ultrawidefield autofluoresence in ABCA4 Stargardt disease. Retina 2018; 38: 403–415. [DOI] [PubMed] [Google Scholar]
- 124. Kuehlewein L, Hariri AH, Ho A, et al. Comparison of manual and semiautomated fundus autofluorescence analysis of macular atrophy in Stargardt disease phenotype. Retina 2016; 36: 1216–1221. [DOI] [PubMed] [Google Scholar]
- 125. Safety and efficacy of emixustat in Stargardt disease (SeaSTAR), https://clinicaltrials.gov/ct2/show/NCT03772665
- 126. Georgiou M, Kane T, Tanna P, et al. Prospective Cohort study of childhood-onset Stargardt disease: fundus autofluorescence imaging, progression, comparison with adult-onset disease, and disease symmetry. Am J Ophthalmol 2020; 211: 159–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chen B, Tosha C, Gorin MB, et al. Analysis of Autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res 2010; 91: 143–152. [DOI] [PubMed] [Google Scholar]
- 128. Lambertus S, Lindner M, Bax NM, et al. Progression of late-onset Stargardt disease. Invest Ophthalmol Vis Sci 2016; 57: 5186–5191. [DOI] [PubMed] [Google Scholar]
- 129. Cai CX, Light JG, Handa JT. Quantifying the rate of ellipsoid zone loss in Stargardt disease. Am J Ophthalmol 2018; 186: 1–9. [DOI] [PubMed] [Google Scholar]
- 130. Schroeder M, Kjellstrom U. Full-field ERG as a predictor of the natural course of ABCA4-associated retinal degenerations. Mol Vis 2018; 24: 1–16. [PMC free article] [PubMed] [Google Scholar]
- 131. Valkenburg D, Runhart EH, Bax NM, et al. Highly variable disease courses in siblings with Stargardt disease. Ophthalmology 2019; 126: 1712–1721. [DOI] [PubMed] [Google Scholar]
- 132. Lambertus S, Bax NM, Groenewoud JM, et al. Asymmetric inter-eye progression in Stargardt disease. Invest Ophthalmol Vis Sci 2016; 57: 6824–6830. [DOI] [PubMed] [Google Scholar]
- 133. Cideciyan AV, Swider M, Aleman TS, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis 2007; 24: 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Strauss RW, Muñoz B, Jha A, et al. Comparison of short-wavelength reduced-illuminance and conventional autofluorescence imaging in Stargardt macular dystrophy. Am J Ophthalmol 2016; 168: 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Delori F, Greenberg JP, Woods RL, et al. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci 2011; 52: 9379–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Greenberg JP, Duncker T, Woods RL, et al. Quantitative fundus autofluorescence in healthy eyes. Invest Ophthalmol Vis Sci 2013; 54: 5684–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Burke TR, Duncker T, Woods RL, et al. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Müller PL, Gliem M, Mangold E, et al. Monoallelic ABCA4 mutations appear insufficient to cause retinopathy: a quantitative autofluorescence study. Invest Ophthalmol Vis Sci 2015; 56: 8179–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Duncker T, Tsang SH, Woods RL, et al. Quantitative fundus autofluorescence and optical coherence tomography in PRPH2/RDS- and ABCA4-associated disease exhibiting phenotypic overlap. Invest Ophthalmol Vis Sci 2015; 56: 3159–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gliem M, Müller PL, Birtel J, et al. Quantitative fundus autofluorescence and genetic associations in macular, cone, and cone–rod dystrophies. Ophthalmol Retina 2020; 4: 737–749. [DOI] [PubMed] [Google Scholar]
- 141. Duncker T, Stein GE, Lee W, et al. Quantitative fundus autofluorescence and optical coherence tomography in ABCA4 carriers. Invest Ophthalmol Vis Sci 2015; 56: 7274–7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Müller PL, Gliem M, McGuinnes M, et al. Quantitative fundus autofluorescence in ABCA4-related retinopathy – functional relevance and genotype-phenotype correlation. Am J Ophthalmol 2020; 222: 340–350. [DOI] [PubMed] [Google Scholar]
- 143. Dysli C, Wolf S, Berezin MY, et al. Fluorescence lifetime imaging ophthalmoscopy. Prog Retin Eye Res 2017; 60: 120–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dysli C, Quellec G, Abegg M, et al. Quantitative analysis of fluorescence lifetime measurements of the macula using the fluorescence lifetime imaging ophthalmoscope in healthy subjects. Invest Ophthalmol Vis Sci 2014; 55: 2106–2113. [DOI] [PubMed] [Google Scholar]
- 145. Dysli C, Müller PL, Birtel J, et al. Spectrally resolved fundus autofluorescence in ABCA4-related retinopathy. Invest Ophthalmol Vis Sci 2019; 60: 274–281. [DOI] [PubMed] [Google Scholar]
- 146. Yannuzzi LA, Sorenson JA, Guyer DR, et al. Indocyanine green videoangiography: current status. Eur J Ophthalmol 1994; 4: 69–81. [DOI] [PubMed] [Google Scholar]
- 147. Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5: 1–23. [PubMed] [Google Scholar]
- 148. Flower RW. Choroidal angiography today and tomorrow. Retina 1992; 12: 189–190. [DOI] [PubMed] [Google Scholar]
- 149. Fish G, Grey R, Sehmi KS, et al. The dark choroid in posterior retinal dystrophies. Br J Ophthalmol 1981; 65: 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Fishman GA, Farber M, Patel BS, et al. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology 1987; 94: 809–814. [DOI] [PubMed] [Google Scholar]
- 151. Genead MA, Fishman GA, Stone EM, et al. The natural history of Stargardt disease with specific sequence mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci 2009; 50: 5867–5871. [DOI] [PubMed] [Google Scholar]
- 152. Querques G, Leveziel N, Benhamou N, et al. Analysis of retinal flecks in fundus flavimaculatus using optical coherence tomography. Br J Ophthalmol 2006; 90: 1157–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Armstrong JD, Meyer D, Xu S, et al. Long-term follow-up of Stargardt’s disease and fundus flavimaculatus. Ophthalmology 1998; 105: 448–457; discussion 457. [DOI] [PubMed] [Google Scholar]
- 154. Giani A, Pellegrini M, Carini E, et al. The dark atrophy with indocyanine green angiography in Stargardt disease. Invest Ophthalmol Vis Sci 2012; 53: 3999–4004. [DOI] [PubMed] [Google Scholar]
- 155. Pellegrini M, Acquistapace A, Oldani M, et al. Dark atrophy: an optical coherence tomography angiography study. Ophthalmology 2016; 123: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 156. de Boer JF. Spectral/Fourier domain optical coherence tomography. In: Drexler W, Fujimoto JG. (eds) Optical coherence tomography: Technology and applications. Berlin; Heidelberg: Springer, 2008, pp. 147–175. [Google Scholar]
- 157. Adhi M, Duker JS. Optical coherence tomography–Current and future applications. Curr Opin Ophthalmol 2013; 24: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Grulkowski I, Liu JJ, Potsaid B, et al. Retinal, anterior segment and full eye imaging using ultrahigh speed swept source OCT with vertical-cavity surface emitting lasers. Biomed Opt Express 2012; 3: 2733–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Spaide RF, Koizumi H, Pozzoni MC. Enhanced depth imaging spectral-domain optical coherence tomography. Am J Ophthalmol 2008; 146: 496–500. [DOI] [PubMed] [Google Scholar]
- 160. Potsaid B, Baumann B, Huang D, et al. Ultrahigh speed 1050nm swept source/Fourier domain OCT retinal and anterior segment imaging at 100,000 to 400,000 axial scans per second. Opt Express 2010; 18: 20029–20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Spaide RF, Fujimoto JG, Waheed NK, et al. Optical coherence tomography angiography. Prog Retin Eye Res 2018; 64: 1–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Abed E, Placidi G, Calandriello L, et al. Correlation of macular focal electroretinogram with ellipsoid zone extension in Stargardt disease. J Ophthalmol 2017; 2017: 3643495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Fujinami K, Sergouniotis PI, Davidson AE, et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 2013; 120: 2324–2331. [DOI] [PubMed] [Google Scholar]
- 164. Berisha F, Feke GT, Aliyeva S, et al. Evaluation of macular abnormalities in Stargardt’s disease using optical coherence tomography and scanning laser ophthalmoscope microperimetry. Graefes Arch Clin Exp Ophthalmol 2008; 247: 303–309. [DOI] [PubMed] [Google Scholar]
- 165. Koenekoop RK. The gene for Stargardt disease, ABCA4, is a major retinal gene: a mini-review. Ophthalmic Genet 2003; 24: 75–80. [DOI] [PubMed] [Google Scholar]
- 166. Burke TR, Rhee DW, Smith RT, et al. Quantification of peripapillary sparing and macular involvement in Stargardt disease (STGD1). Invest Ophthalmol Vis Sci 2011; 52: 8006–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ritter M, Zotter S, Schmidt WM, et al. Characterization of Stargardt disease using polarization-sensitive optical coherence tomography and fundus autofluorescence imaging. Invest Ophthalmol Vis Sci 2013; 54: 6416–6425. [DOI] [PubMed] [Google Scholar]
- 168. Birch DG, Locke KG, Wen Y, et al. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol 2013; 131: 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Tee JJL, Carroll J, Webster AR, et al. Quantitative analysis of retinal structure using spectral-domain optical coherence tomography in RPGR-associated retinopathy. Am J Ophthalmol 2017; 178: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cai CX, Locke KG, Ramachandran R, et al. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and x-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 2014; 55: 7417–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Iftikhar M, Usmani B, Sanyal A, et al. Progression of retinitis pigmentosa on multimodal imaging: the PREP-1 study. Clin Exp Ophthalmol 2019; 47: 605–613. [DOI] [PubMed] [Google Scholar]
- 172. Gong Y, Chen LJ, Pang CP, et al. Ellipsoid zone optical intensity reduction as an early biomarker for retinitis pigmentosa. Acta Ophthalmol 2021; 99: e215–e221. [DOI] [PubMed] [Google Scholar]
- 173. Romano F, Arrigo A, Leone PP, et al. Altered ellipsoid zone reflectivity and deep capillary plexus rarefaction correlate with progression in Best disease. Br J Ophthalmol 2020; 104: 461–465. [DOI] [PubMed] [Google Scholar]
- 174. Jain N, Jia Y, Gao SS, et al. Optical coherence tomography angiography in choroideremia: correlating choriocapillaris loss with overlying degeneration. JAMA Ophthalmol 2016; 134: 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Xue K, Oldani M, Jolly JK, et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Invest Ophthalmol Vis Sci 2016; 57: 3674–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Voigt M, Querques G, Atmani K, et al. Analysis of retinal flecks in fundus flavimaculatus using high-definition spectral-domain optical coherence tomography. Am J Ophthalmol 2010; 150: 330–337. [DOI] [PubMed] [Google Scholar]
- 177. Huang WC, Cideciyan AV, Roman AJ, et al. Inner and outer retinal changes in retinal degenerations associated with ABCA4 mutations. Invest Ophthalmol Vis Sci 2014; 55: 1810–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Aleman TS, Cideciyan AV, Sumaroka A, et al. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Invest Ophthalmol Vis Sci 2007; 48: 4759–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Jolly JK, Menghini M, Johal PA, et al. Inner retinal thickening affects microperimetry thresholds in the presence of photoreceptor thinning in patients with RPGR retinitis pigmentosa. Br J Ophthalmol. Epub ahead of print 30 October 2020. DOI: 10.1136/bjophthalmol-2020-317692. [DOI] [PubMed] [Google Scholar]
- 180. Aleman TS, Cideciyan AV, Sumaroka A, et al. Retinal laminar architecture in human retinitis pigmentosa caused by Rhodopsin gene mutations. Invest Ophthalmol Vis Sci 2008; 49: 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Aleman TS, Soumittra N, Cideciyan AV, et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci 2009; 50: 5944–5954. [DOI] [PubMed] [Google Scholar]
- 182. Jacobson SG, Aleman TS, Sumaroka A, et al. Disease boundaries in the retina of patients with Usher syndrome caused by MYO7A gene mutations. Invest Ophthalmol Vis Sci 2009; 50: 1886–1894. [DOI] [PubMed] [Google Scholar]
- 183. Jacobson SG, Cideciyan AV, Aleman TS, et al. Human retinal disease from AIPL1 gene mutations: foveal cone loss with minimal macular photoreceptors and rod function remaining. Invest Ophthalmol Vis Sci 2011; 52: 70–79. [DOI] [PubMed] [Google Scholar]
- 184. Strauss RW, Muñoz B, Wolfson Y, et al. Assessment of estimated retinal atrophy progression in Stargardt macular dystrophy using spectral-domain optical coherence tomography. Br J Ophthalmol 2016; 100: 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ervin AM, Strauss RW, Ahmed MI, et al. A Workshop on measuring the progression of atrophy secondary to Stargardt disease in the progstar studies: findings and lessons learned. Transl Vis Sci Technol 2019; 8: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kong X, Ho A, Munoz B, et al. Reproducibility of measurements of retinal structural parameters using optical coherence tomography in Stargardt disease. Transl Vis Sci Technol 2019; 8: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Velaga SB, Nittala MG, Jenkins D, et al. Impact of segmentation density on spectral domain optical coherence tomography assessment in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2019; 257: 549–556. [DOI] [PubMed] [Google Scholar]
- 188. Pasadhika S, Fishman GA, Allikmets R, et al. Peripapillary retinal nerve fiber layer thinning in patients with autosomal recessive cone-rod dystrophy. Am J Ophthalmol 2009; 148: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Genead MA, Fishman GA, Anastasakis A. Spectral-domain OCT peripapillary retinal nerve fibre layer thickness measurements in patients with Stargardt disease. Br J Ophthalmol 2011; 95: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Burke TR, Yzer S, Zernant J, et al. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic Genet 2013; 34: 75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Pang CE, Suqin Y, Sherman J, et al. New insights into Stargardt disease with multimodal imaging. Ophthalmic Surg Lasers Imaging Retina 2015; 46: 257–261. [DOI] [PubMed] [Google Scholar]
- 192. Lee W, Noupuu K, Oll M, et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 6139–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Fujinami K, Singh R, Carroll J, et al. Fine central macular dots associated with childhood-onset Stargardt Disease. Acta Ophthalmol 2014; 92: e157–e159. [DOI] [PubMed] [Google Scholar]
- 194. Palejwala NV, Gale MJ, Clark RF, et al. Insights into autosomal dominant Stargardt-like macular dystrophy through multimodality diagnostic imaging. Retina 2016; 36: 119–130. [DOI] [PubMed] [Google Scholar]
- 195. Noupuu K, Lee W, Zernant J, et al. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest Ophthalmol Vis Sci 2014; 55: 7217–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Barthelmes D, Sutter FK, Kurz-Levin MM, et al. Quantitative analysis of OCT characteristics in patients with achromatopsia and blue-cone monochromatism. Invest Ophthalmol Vis Sci 2006; 47: 1161–1166. [DOI] [PubMed] [Google Scholar]
- 197. Greenberg JP, Sherman J, Zweifel SA, et al. Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA Ophthalmology 2014; 132: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Leng T, Marmor MF, Kellner U, et al. Foveal cavitation as an optical coherence tomography finding in central cone dysfunction. Retina 2012; 32: 1411–1419. [DOI] [PubMed] [Google Scholar]
- 199. Mukherjee R, Robson AG, Holder GE, et al. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene. Eye 2014; 28: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Oh JK, Ryu J, Lima de Carvalho JR, Jr, et al. Optical gap biomarker in cone-dominant retinal dystrophy. Am J Ophthalmol 2020; 218: 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Park SJ, Woo SJ, Park KH, et al. Morphologic photoreceptor abnormality in occult macular dystrophy on spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci 2010; 51: 3673–3679. [DOI] [PubMed] [Google Scholar]
- 202. Saffra N, Seidman CJ, Rakhamimov A, et al. ERG and OCT findings of a patient with a clinical diagnosis of occult macular dystrophy in a patient of Ashkenazi Jewish descent associated with a novel mutation in the gene encoding RP1L1. BMJ Case Rep 2017; 2017: bcr2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Ahn SJ, Ahn J, Park KH, et al. Multimodal imaging of occult macular dystrophy. JAMA Ophthalmol 2013; 131: 880–890. [DOI] [PubMed] [Google Scholar]
- 204. Doshi RR, Fortun JA, Kim BT, et al. Pseudocystic foveal cavitation in tamoxifen retinopathy. Am J Ophthalmol 2014; 157: 1291–1298. [DOI] [PubMed] [Google Scholar]
- 205. Lee S, Kim H-A, Yoon YH. OCT angiography findings of tamoxifen retinopathy: similarity with macular telangiectasia type 2. Ophthalmol Retina 2019; 3: 681–689. [DOI] [PubMed] [Google Scholar]
- 206. Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of ‘poppers maculopathy’. Eye 2012; 26: 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Luis J, Virdi M, Nabili S. Poppers retinopathy. BMJ Case Rep 2016; 2016: bcr2016214442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Dirani A, Chelala E, Fadlallah A, et al. Bilateral macular injury from a green laser pointer. Clin Ophthalmol 2013; 7: 2127–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Zhang L, Zheng A, Nie H, et al. Laser-induced photic injury phenocopies macular dystrophy. Ophthalmic Genet 2016; 37: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Jain A, Desai RU, Charalel RA, et al. Solar retinopathy: comparison of optical coherence tomography (OCT) and fluorescein angiography (FA). Retina 2009; 29: 1340–1345. [DOI] [PubMed] [Google Scholar]
- 211. Sodi A, Mucciolo DP, Cipollini F, et al. En face OCT in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2016; 254: 1669–1679. [DOI] [PubMed] [Google Scholar]
- 212. Greenstein VC, Nunez J, Lee W, et al. A comparison of en face optical coherence tomography and fundus autofluorescence in Stargardt disease. Invest Ophthalmol Vis Sci 2017; 58: 5227–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Alabduljalil T, Patel RC, Alqahtani AA, et al. Correlation of outer retinal degeneration and choriocapillaris loss in Stargardt disease using en face optical coherence tomography and optical coherence tomography angiography. Am J Ophthalmol 2019; 202: 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Melillo P, Testa F, Rossi S, et al. En face spectral-domain optical coherence tomography for the monitoring of lesion area progression in stargardt disease. Invest Ophthalmol Vis Sci 2016; 57: OCT247–OCT252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Nunes RP, Gregori G, Yehoshua Z, et al. Predicting the progression of geographic atrophy in age-related macular degeneration with SD-OCT en face imaging of the outer retina. Ophthalmic Surg Lasers Imaging Retina 2013; 44: 344–359. [DOI] [PubMed] [Google Scholar]
- 216. Hariri AH, Zhang HY, Ho A, et al. Quantification of ellipsoid zone changes in retinitis pigmentosa using en face spectral domain-optical coherence tomography. JAMA Ophthalmol 2016; 134: 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Adhi M, Read SP, Ferrara D, et al. Morphology and vascular layers of the choroid in Stargardt disease analyzed using spectral-domain optical coherence tomography. Am J Ophthalmol 2015; 160: 1276–1284.e1271. [DOI] [PubMed] [Google Scholar]
- 218. Yeoh J, Rahman W, Chen F, et al. Choroidal imaging in inherited retinal disease using the technique of enhanced depth imaging optical coherence tomography. Graefes Arch Clin Exp Ophthalmol 2010; 248: 1719–1728. [DOI] [PubMed] [Google Scholar]
- 219. Vural E, Acar U, Sevinc MK, et al. Choroidal thickness in patients with Stargardt disease. Retina 2018; 38: 614–619. [DOI] [PubMed] [Google Scholar]
- 220. Arrigo A, Grazioli A, Romano F, et al. Choroidal patterns in Stargardt disease: correlations with visual acuity and disease progression. J Clin Med 2019; 8: 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Ayton LN, Guymer RH, Luu CD. Choroidal thickness profiles in retinitis pigmentosa. Clin Exp Ophthalmol 2013; 41: 396–403. [DOI] [PubMed] [Google Scholar]
- 222. Sodi A, Lenzetti C, Murro V, et al. EDI-OCT evaluation of choroidal thickness in retinitis pigmentosa. Eur J Ophthalmol 2018; 28: 52–57. [DOI] [PubMed] [Google Scholar]
- 223. Ayyildiz O, Ozge G, Kucukevcilioglu M, et al. Is there a relationship between outer retinal destruction and choroidal changes in cone dystrophy. Arq Bras Oftalmol 2016; 79: 315–318. [DOI] [PubMed] [Google Scholar]
- 224. Sabbaghi H, Ahmadieh H, Jalili J, et al. Choroidal thickness in different types of inherited retinal dystrophies. J Ophthalmic Vis Res 2020; 15: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Querques G, Costanzo E, Miere A, et al. Choroidal caverns: a novel optical coherence tomography finding in geographic atrophy. Invest Ophthalmol Vis Sci 2016; 57: 2578–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Reich M, Glatz A, Cakir B, et al. Characterisation of vascular changes in different stages of Stargardt disease using double swept-source optical coherence tomography angiography. BMJ Open Ophthalmol 2019; 4: e000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Battaglia Parodi M, Cicinelli MV, Rabiolo A, et al. Vascular abnormalities in patients with Stargardt disease assessed with optical coherence tomography angiography. Br J Ophthalmol 2017; 101: 780–785. [DOI] [PubMed] [Google Scholar]
- 228. Müller PL, Pfau M, Möller PT, et al. Choroidal flow signal in late-onset Stargardt disease and age-related macular degeneration: an OCT-angiography study. Invest Ophthalmol Vis Sci 2018; 59: AMD122–AMD131. [DOI] [PubMed] [Google Scholar]
- 229. Dubra A, Sulai Y, Norris JL, et al. Noninvasive imaging of the human rod photoreceptor mosaic using a confocal adaptive optics scanning ophthalmoscope. Biomed Opt Express 2011; 2: 1864–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Rossi EA, Chung M, Dubra A, et al. Imaging retinal mosaics in the living eye. Eye 2011; 25: 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Gill JS, Moosajee M, Dubis AM. Cellular imaging of inherited retinal diseases using adaptive optics. Eye 2019; 33: 1683–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Cooper RF, Dubis AM, Pavaskar A, et al. Spatial and temporal variation of rod photoreceptor reflectance in the human retina. Biomed Opt Express 2011; 2: 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Georgiou M, Kalitzeos A, Patterson EJ, et al. Adaptive optics imaging of inherited retinal diseases. Br J Ophthalmol 2018; 102: 1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Scoles D, Sulai YN, Langlo CS, et al. In vivo imaging of human cone photoreceptor inner segments. Invest Ophthalmol Vis Sci 2014; 55: 4244–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Scoles D, Sulai YN, Dubra A. In vivo dark-field imaging of the retinal pigment epithelium cell mosaic. Biomed Opt Express 2013; 4: 1710–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Chen Y, Ratnam K, Sundquist SM, et al. Cone photoreceptor abnormalities correlate with vision loss in patients with Stargardt disease. Invest Ophthalmol Vis Sci 2011; 52: 3281–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Song H, Rossi EA, Yang Q, et al. High-resolution adaptive optics in vivo autofluorescence imaging in Stargardt disease. JAMA Ophthalmology 2019; 137: 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Birnbach CD, Järveläinen M, Possin DE, et al. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology 1994; 101: 1211–1219. [DOI] [PubMed] [Google Scholar]
- 239. Razeen MM, Cooper RF, Langlo CS, et al. Correlating photoreceptor mosaic structure to clinical findings in Stargardt disease. Transl Vis Sci Technol 2016; 5: 6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015; 130: 1–12. [DOI] [PubMed] [Google Scholar]
- 241. Marmor MF, Brigell MG, McCulloch DL, et al. ISCEV standard for clinical electro-oculography (2010 update). Doc Ophthalmol 2011; 122: 1–7. [DOI] [PubMed] [Google Scholar]
- 242. Hood DC, Bach M, Brigell M, et al. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc Ophthalmol 2012; 124: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Bach M, Brigell MG, Hawlina M, et al. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc Ophthalmol 2013; 126: 1–7. [DOI] [PubMed] [Google Scholar]
- 244. Robson AG, Nilsson J, Li S, et al. ISCEV guide to visual electrodiagnostic procedures. Doc Ophthalmol 2018; 136: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Lenassi E, Jarc-Vidmar M, Glavac D, et al. Pattern electroretinography of larger stimulus field size and spectral-domain optical coherence tomography in patients with Stargardt disease. Br J Ophthalmol 2009; 93: 1600–1605. [DOI] [PubMed] [Google Scholar]
- 246. Lois N, Holder GE, Bunce C, et al. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Archives of Ophthalmology 2001; 119: 359–369. [DOI] [PubMed] [Google Scholar]
- 247. Simonelli F, Testa F, Zernant J, et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res 2005; 37: 159–167. [DOI] [PubMed] [Google Scholar]
- 248. Chun R, Fishman GA, Collison FT, et al. The value of retinal imaging with infrared scanning laser ophthalmoscopy in patients with Stargardt disease. Retina 2014; 34: 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Beck RW, Moke PS, Turpin AH, et al. A computerized method of visual acuity testing: adaptation of the early treatment of diabetic retinopathy study testing protocol. Am J Ophthalmol 2003; 135: 194–205. [DOI] [PubMed] [Google Scholar]
- 250. Bailey IL, Lovie-Kitchin JE. Visual acuity testing. From the laboratory to the clinic. Vis Res 2013; 90: 2–9. [DOI] [PubMed] [Google Scholar]
- 251. Tsou BC, Bressler NM. Visual acuity reporting in clinical research publications. JAMA Ophthalmol 2017; 135: 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Talib M, Dagnelie G, Boon CJF. Recording and analysis of Goldmann kinetic visual fields. Methods Mol Biol 2018; 1715: 327–338. [DOI] [PubMed] [Google Scholar]
- 253. Bagdonaite-Bejarano L, Hansen RM, Fulton AB. Microperimetry in three inherited retinal disorders. Semin Ophthalmol 2019; 34: 334–339. [DOI] [PubMed] [Google Scholar]
- 254. Aaberg TM. Stargardt’s disease and fundus flavimaculatus: evaluation of morphologic progression and intrafamilial co-existence. Trans Am Ophthalmol Soc 1986; 84: 453–487. [PMC free article] [PubMed] [Google Scholar]
- 255. Mastropasqua R, Toto L, Borrelli E, et al. Optical coherence tomography angiography findings in Stargardt disease. PLoS ONE 2017; 12: e0170343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Hargitai J, Zernant J, Somfai GM, et al. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci 2005; 46: 4402–4408. [DOI] [PubMed] [Google Scholar]
- 257. Kong X, Fujinami K, Strauss RW, et al. Visual acuity change over 24 months and its association with foveal phenotype and genotype in individuals with Stargardt disease: ProgStar study report no. 10. JAMA Ophthalmol 2018; 136: 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Schönbach EM, Ibrahim MA, Strauss RW, et al. Fixation location and stability using the MP-1 microperimeter in Stargardt disease: ProgStar report no. 3. Ophthalmol Retina 2017; 1: 68–76. [DOI] [PubMed] [Google Scholar]
- 259. Schönbach EM, Wolfson Y, Strauss RW, et al. Macular sensitivity measured with microperimetry in Stargardt disease in the progression of atrophy secondary to Stargardt disease (ProgStar) Study: report no. 7. JAMA Ophthalmol 2017; 135: 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Schönbach EM, Strauss RW, Kong X, et al. Longitudinal changes of fixation location and stability within 12 months in Stargardt disease: ProgStar report no. 12. Am J Ophthalmol 2018; 193: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Schönbach EM, Strauss RW, Cattaneo M, et al. Longitudinal changes of fixation stability and location within 24 months in Stargardt disease: ProgStar report no. 16: short title: longitudinal changes of fixation in Stargardt disease. Am J Ophthalmol. 20 July 2021. DOI: 10.1016/j.ajo.2021.07.013. [DOI] [PubMed] [Google Scholar]
- 262. Schonbach EM, Ibrahim MA, Kong X, et al. Metrics and acquisition modes for fixation stability as a visual function biomarker. Invest Ophthalmol Vis Sci 2017; 58: Bio268–Bio276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Schönbach EM, Strauss RW, Muñoz B, et al. Longitudinal microperimetric changes of macular sensitivity in Stargardt disease after 12 months: ProgStar report no. 13. JAMA Ophthalmol 2020; 138: 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Strauss RW, Kong X, Bittencourt MG, et al. Scotopic microperimetric assessment of rod function in Stargardt disease (SMART) study: design and baseline characteristics (report no. 1). Ophthalmic Res 2019; 61: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Schönbach EM, Strauss RW, Ibrahim MA, et al. Faster sensitivity loss around dense scotomas than for overall macular sensitivity in Stargardt disease: ProgStar report no. 14. Am J Ophthalmol 2020; 216: 219–225. [DOI] [PubMed] [Google Scholar]
- 266. Verdina T, Tsang SH, Greenstein VC, et al. Functional analysis of retinal flecks in Stargardt disease. J Clin Exp Ophthalmol 2012; 3: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Schönbach EM, Janeschitz-Kriegl L, Strauss RW, et al. The progression of Stargardt disease using volumetric hill of vision analyses over 24 months: ProgStar report no.15. Am J Ophthalmol 2021; 230: 123–133. [DOI] [PubMed] [Google Scholar]
- 268. Josan AS, Buckley TMW, Wood LJ, et al. Microperimetry hill of vision and volumetric measures of retinal sensitivity. Transl Vis Sci Technol 2021; 10: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Kaufman Y, Ma L, Washington I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem 2011; 286: 7958–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Ma L, Kaufman Y, Zhang J, et al. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem 2011; 286: 7966–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Charbel Issa P, Barnard AR, Herrmann P, et al. Rescue of the Stargardt phenotype in ABCA-4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci USA 2015; 112: 8415–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Berni R, Formelli F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett 1992; 308: 43–45. [DOI] [PubMed] [Google Scholar]
- 273. Radu RA, Han Y, Bui TV, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci 2005; 46: 4393–4401. [DOI] [PubMed] [Google Scholar]
- 274. Sieving PA, Chaudhry P, Kondo M, et al. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci USA 2001; 98: 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci USA 2003; 100: 4742–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Maeda A, Golczak M, Chen Y, et al. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat Chem Biol 2011; 8: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Julien S, Schraermeyer U. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol Aging 2012; 33: 2390–2397. [DOI] [PubMed] [Google Scholar]
- 278. Lu W, Gómez NM, Lim JC, et al. The P2Y(12) receptor antagonist ticagrelor reduces lysosomal pH and autofluorescence in retinal pigmented epithelial cells from the ABCA4(-/-) mouse model of retinal degeneration. Front Pharmacol 2018; 9: 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Liu Q, Sabirzhanova I, Bergbower EAS, et al. The CFTR Corrector, VX-809 (Lumacaftor), rescues ABCA4 trafficking mutants: a potential treatment for Stargardt disease. Cell Physiol Biochem 2019; 53: 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Piccardi M, Fadda A, Martelli F, et al. Antioxidant saffron and central retinal function in ABCA4-related Stargardt macular dystrophy. Nutrients 2019; 11: 2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. MacDonald IM, Sieving PA. Investigation of the effect of dietary docosahexaenoic acid (DHA) supplementation on macular function in subjects with autosomal recessive Stargardt macular dystrophy. Ophthalmic Genet 2018; 39: 477–486. [DOI] [PubMed] [Google Scholar]
- 282. NCT03297515, https://clinicaltrials.gov/ct2/show/NCT03297515
- 283. Dobri N, Qin Q, Kong J, et al. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci 2013; 54: 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Simon WA, Herrmann M, Klein T, et al. Soraprazan: setting new standards in inhibition of gastric acid secretion. J Pharmacol Exp Ther 2007; 321: 866–874. [DOI] [PubMed] [Google Scholar]
- 285. Julien-Schraermeyer S, Illing B, Tschulakow A, et al. Penetration, distribution, and elimination of remofuscin/soraprazan in Stargardt mouse eyes following a single intravitreal injection using pharmacokinetics and transmission electron microscopic autoradiography: implication for the local treatment of Stargardt’s disease and dry age-related macular degeneration. Pharmacol Res Perspect 2020; 8: e00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Lu W, Campagno KE, Tso HY, et al. Oral delivery of the P2Y12 receptor antagonist ticagrelor prevents loss of photoreceptors in an ABCA4-/- mouse model of retinal degeneration. Invest Ophthalmol Vis Sci 2019; 60: 3046–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Dyka FM, Molday LL, Chiodo VA, et al. Dual ABCA4-AAV Vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive Stargardt disease. Hum Gene Ther 2019; 30: 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. McClements ME, Barnard AR, Singh MS, et al. An AAV dual vector strategy ameliorates the Stargardt phenotype in adult Abca4(-/-) mice. Hum Gene Ther 2019; 30: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet 2005; 6: 299–310. [DOI] [PubMed] [Google Scholar]
- 290. Lenis TL, Sarfare S, Jiang Z, et al. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc Natl Acad Sci USA 2017; 114: 3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Cao S, Wang JCC, Gao J, et al. CFH Y402H polymorphism and the complement activation product C5a: effects on NF-κB activation and inflammasome gene regulation. Br J Ophthalmol 2016; 100: 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Csaky KG, Bok D, Radu RA, et al. Complement C5 inhibition as a potential treatment for autosomal recessive Stargardt disease (STGD1): design of a clinical trial assessing a novel treatment and primary outcome measure. Invest Ophthalmol Vis Sci 2018; 59: 1569. [Google Scholar]
- 293. Duijkers L, van den Born LI, Neidhardt J, et al. Antisense oligonucleotide-based splicing correction in individuals with leber congenital amaurosis due to compound heterozygosity for the c.2991+1655A>G mutation in CEP290. Int J Mol Sci 2018; 19: 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. NCT01367444, https://clinicaltrials.gov/ct2/show/NCT01367444
- 295. NCT03364153, https://clinicaltrials.gov/ct2/show/NCT03364153
- 296. Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res 2018; 46: 1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Albert S, Garanto A, Sangermano R, et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am J Hum Genet 2018; 102: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Garanto A, Duijkers L, Tomkiewicz TZ, et al. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in Stargardt disease. Genes 2019; 10: 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Mucciolo DP, Lippera M, Giorgio D, et al. Outer nuclear layer relevance in visual function correlated to quantitative enface OCT parameters in Stargardt disease. Eur J Ophthalmol. Epub ahead of print 28 January 2021. DOI: 10.1177/1120672121990579. [DOI] [PubMed] [Google Scholar]
- 300. Sadda SR, Wu Z, Walsh AC, et al. Errors in retinal thickness measurements obtained by optical coherence tomography. Ophthalmology 2006; 113: 285–293. [DOI] [PubMed] [Google Scholar]
- 301. Keane PA, Liakopoulos S, Jivrajka RV, et al. Evaluation of optical coherence tomography retinal thickness parameters for use in clinical trials for neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci 2009; 50: 3378–3385. [DOI] [PubMed] [Google Scholar]
- 302. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015; 385: 509–516. [DOI] [PubMed] [Google Scholar]
- 303. Song WK, Park K-M, Kim H-J, et al. Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Reports 2015; 4: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Sung Y, Lee MJ, Choi J, et al. Long-term safety and tolerability of subretinal transplantation of embryonic stem cell-derived retinal pigment epithelium in Asian Stargardt disease patients. Br J Ophthalmol 2020; 105: 829–837. [DOI] [PubMed] [Google Scholar]
- 305. Mehat MS, Sundaram V, Ripamonti C, et al. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells in macular degeneration. Ophthalmology 2018; 125: 1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Oner A, Gonen ZB, Sevim DG, et al. Suprachoroidal adipose tissue-derived mesenchymal stem cell implantation in patients with dry-type age-related macular degeneration and Stargardt’s macular dystrophy: 6-month follow-up results of a phase 2 study. Cell Reprogram 2018; 20: 329–336. [DOI] [PubMed] [Google Scholar]