

SUMMARY
HLA class I (HLA-I) glycoproteins drive immune responses by presenting antigens to cognate CD8+ T cells. This process is often hijacked by tumors and pathogens for immune evasion. Because options for restoring HLA-I antigen presentation are limited, we aimed to identify druggable HLA-I pathway targets. Using iterative genome-wide screens, we uncovered that the cell surface glycosphingolipid (GSL) repertoire determines effective HLA-I antigen presentation. We show that absence of the protease SPPL3 augmented B3GNT5 enzyme activity, resulting in upregulation of surface neolacto-series GSLs. These GSLs sterically impeded antibody and receptor interactions with HLA-I and diminished CD8+ T cell activation. Furthermore, a disturbed SPPL3-B3GNT5 pathway in glioma correlated with decreased patient survival. We show that the immunomodulatory effect could be reversed through GSL synthesis inhibition using clinically approved drugs. Overall, our study identifies a GSL signature that inhibits immune recognition and represents a potential therapeutic target in cancer, infection, and autoimmunity.
In Brief
Numerous glycosphingolipids (GSLs) are expressed at the cell surface; however, their functions remain mostly unknown. Jongsma et al. identify that the protease SPPL3 destroys the GSL synthesis enzyme B3GNT5, whose GSL products negatively affect HLA-I-mediated immune responses. This pathway represents a potential therapeutic target in cancer, infection, and autoimmunity.
Graphical Abstract
INTRODUCTION
Human leukocyte antigen class I (HLA-I) glycoproteins are the primary modules recognized by CD8+ T cells determining both the induction and the effector phases of adaptive immune responses. Their primary function is to present peptide fragments from degraded proteins to the T cell receptor (TCR) of cytotoxic CD8+ T cells, leading to T cell-mediated elimination of target cells (Neefjes et al., 2011; Unanue and Cerottini, 1989). Because HLA-I molecules on tumor cells also present tumor antigen-derived peptides to cognate T cells, they play a major role in the anti-tumor activity of T cells unleashed by current immunotherapeutic strategies (Schumacher and Schreiber, 2015). As a consequence, tumors often escape from immune surveillance by downregulating one or multiple molecules critical in HLA-I antigen presentation (Chowell et al., 2018; Gettinger et al., 2017; Restifo et al., 1996; Sade-Feldman et al., 2017; Zaretsky et al., 2016). This reduction is often reversible, for example, by interferon (IFN) stimulation, ionizing radiation, or inhibition of histone deacetylases, which has led to various experimental therapies aimed at increasing tumor HLA-I surface expression (Haworth et al., 2015; Reits et al., 2006; Thor Straten and Garrido, 2016). Moreover, sensitizing tumor cells as immune targets can act synergistically with T cell activating and re-activating strategies, thereby increasing the therapeutic potential of enhancing HLA-I availability (Hähnel et al., 2008). Active suppression of HLA-I surface expression to escape T cell surveillance is also employed by an array of pathogens, such as Epstein-Barr virus, cytomegalovirus, and SARS-CoV-2 (Hansen and Bouvier, 2009; Yewdell and Hill, 2002; Zhang et al., 2020). These examples underscore the broad relevance of HLA-I-based interventions, necessitating a thorough understanding of the molecular mechanisms underpinning the HLA-I pathway.
Over the last 35 years, several key elements in HLA-I expression and antigen presentation have been identified and extensively studied. A protein complex directed by the transcriptional regulator NLRC5 drives HLA-I expression in selected tissues, whereas HLA-I translation and glycosylation are thought to be executed by general enzymes and mechanisms (Jongsma et al., 2019; Ryan and Cobb, 2012). In the endoplasmic reticulum (ER), the HLA-I heavy chain and its light chain beta-2 microglobulin (β2m) assemble and are stabilized by a unique combination of the ER chaperone proteins: tapasin (TAPBP), ERp57 (PDIA3), and calreticulin (CALR) (Rock et al., 2016). These HLA-I chaperone complexes bind the peptide transporter TAP to form the peptide-loading complex (PLC), which drives efficient ER import and loading of peptides into the HLA-I peptide binding groove (Blees et al., 2017). Subsequently, mature trimeric complexes of HLA-I heavy chain, β2m, and peptide are released from the PLC and transported to the cell surface for peptide presentation to T cells (Garstka et al., 2015; Wearsch and Cresswell, 2008). Given the multifactorial complexity of the HLA-I antigen presentation pathway, we hypothesized that additional regulatory mechanisms of this central process in adaptive immunity must exist.
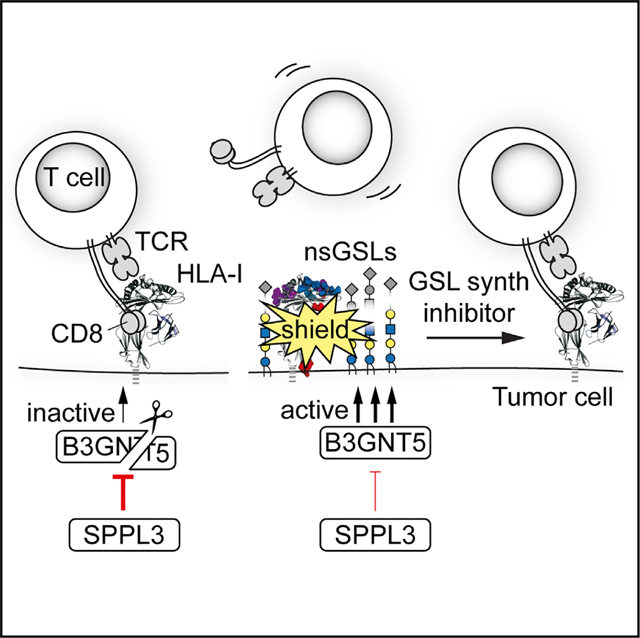
To uncover additional components involved in successful HLA-I antigen presentation, we performed a series of genome-wide haploid genetic screens. We identified the enzyme signal peptide peptidase-like 3 (SPPL3) as a positive regulator of HLA-I antigen presentation. We found that SPPL3 controlled the composition of the cell surface glycosphingolipid (GSL) repertoire by inhibiting the glycosyltransferase B3GNT5. In the absence of SPPL3, an increase in B3GNT5 activity led to a high amount of complex negatively charged neolacto-series GSLs (nsGSLs), preventing HLA-I from being accessed by several immune cell receptors and interfering with activation of CD8+ T cells. GSL synthesis in several tumors, including glioma, was skewed toward the nsGSL synthesis pathway. High activation of this pathway negatively correlated with survival of glioma patients. We show that intervention of GSL synthesis in glioma cells by U.S. Food and Drug Administration (FDA)-approved drugs led to improved anti-tumor immunity in vitro. In conclusion, nsGSL synthesis constitutes a targetable layer of immune regulation through HLA-I shielding at the cell surface.
RESULTS
A Haploid Genetic Screen Provides a High-Resolution Map of the HLA-I Antigen Presentation Pathway
To identify unknown factors regulating HLA-I antigen presentation, we performed a genome-wide insertional mutagenesis screen in haploid human fibroblast-like HAP1 cells endogenously expressing HLA-I (Brockmann et al., 2017; Carette et al., 2009). A heterogeneous pool of millions of cells, each genetically ablated for a random gene or set of genes, was generated by retroviral gene trap insertion and expanded. Mutagenized cells that were poorest or best recognized by the HLA-I-specific W6/32 antibody were sorted by flow cytometry (Figure 1A). Subsequently, we determined the relative enrichment of unique disruptive integrations per gene between the sorted populations using deep sequencing. This provided an unbiased overview of genes involved in HLA-I antigen presentation (Figure 1B). Among the highly significant positive modifiers were the known key genes directing HLA-I expression, such as the transcriptional activators and coactivators NLRC5, RFXAP, and RFX5 (Jongsma et al., 2019) and essential components for post-transcriptional assembly, glycosylation, and peptide loading: B2M, MOGS (α-glucosidase I), GANAB (α-glucosidase II), TAP1, TAP2, TAPBP (tapasin), PDIA3 (ERp57), and CALR (Figure 1B) (Wearsch and Cresswell, 2008). Thus, this single genetic map identified components of the HLA-I pathway previously discovered through decades of research. In addition, HLA-I regulators identified in human B cell lymphoma CRISPR-Cas9 screens by Dersh et al., such as SUSD6, SND1, ANKRD33B, and EZH2, were confirmed as highly significant hits in our haploid genetic screen (Dersh et al., 2020). The most prominent hit from our screen in the W6/32lo-sorted population was the gene encoding SPPL3, a protease that had not been previously described in the context of antigen presentation (Figure 1B). SPPL3 is an ER- and Golgi-localized transmembrane protein of the family of intramembrane-cleaving aspartyl proteases (Fluhrer and Haass, 2007).
Figure 1. A Haploid Genetic Screen Reveals SPPL3 as a Regulator of Antibody Accessibility to Membrane-Proximal HLA-I Regions.

(A) Schematic overview of a genome-wide haploid genetic screen using HLA-I-specific W6/32 antibody.
(B) Fish-tail plot showing per gene the mutation index (ratio of integrations mapped in the specified populations) against the amount of mapped integrations. A two-sided false discovery rate (FDR) (Benjamini-Hochberg) corrected Fisher’s exact test was applied. The symbol legend is indicated.
(C) (Left) Titration curves of four HLA-I-specific antibodies on mixed barcoded HAP1 cells (see Figure S1E). The individual antibody binding epitopes are shown on the HLA-I structure. (Right) Flow cytometry histograms of nonsaturating antibody stain as indicated by the arrow (close to EC50 values). MFI, mean fluorescence intensity.
(D) Quantification of (C) using the ratio of WT and SPPL3−/− EC50 values. Mean ± SD is plotted (n = 5–8 independent experiments).
(E) SPPL3-susceptibility of different epitopes is plotted on the HLA-I/β2m crystal structure (individual epitopes in Figures 1C and S2A).
See also Figures S1 and S2.
SPPL3 Enables Antibody Binding to Membrane-Proximal Regions of HLA-I
To validate that HLA-I cell surface expression was altered by SPPL3, we created SPPL3−/− HAP1 cells using CRISPR-Cas9 (Table S1) and performed flow cytometry using the W6/32 antibody. HLA-I surface expression in the absence of SPPL3 turned out identical to those of wild-type (WT) cells (Figure S1A). Likewise, the total HLA-I content of SPPL3−/− and WT cells was similar, as determined by immunoblot analysis (Figure S1B). Furthermore, SPPL3 deficiency did not alter HLA-I turnover or stability of the peptide-HLA-I interaction (Figures S1C and S1D). Because the anti-HLA antibody W6/32 was used under nonsaturating staining conditions in the genome-wide screen, these seemingly contradictory outcomes may have resulted from reduced accessibility of the W6/32 epitope in the absence of SPPL3.
To test this hypothesis, we titrated the W6/32 antibody for binding to WT, SPPL3−/−, and control HLA-A−/−B−/−C−/− (HLA-I-deficient) and TAPBP−/− cells (Table S1), which were individually color barcoded and combined in a single well for optimal comparison of staining intensity (Figure S1E). In contrast to saturating W6/32 concentrations, lower W6/32 concentrations resulted in decreased binding to SPPL3−/− cells compared with WT cells, indicating that accessibility of the HLA-I epitope recognized by W6/32 was indeed hindered (Figures 1C and 1D). A similar result was apparent using monovalent W6/32 Fab fragments, validating this conclusion (Figure S2A). To further define SPPL3-dependent antibody accessibility to HLA-I, we performed additional titrations using 13 HLA-I-specific antibodies recognizing distinct HLA-I epitopes and one targeting a β2m epitope (Figures 1C and S2A; Table S2). The binding of three antibodies (clones W6/32, TP25.99, and ROU9A6) was markedly affected by the absence of SPPL3 (Figures 1C, 1D, and S2A). By superimposing critical amino acid positions for binding of the individual antibodies onto an HLA-I structure, we defined the SPPL3-susceptible region as relatively proximal to the cellular membrane, an area that is largely conserved among HLA-I alleles (Figure 1E). Binding of several other antibodies (e.g., B1.23.2) was not affected by SPPL3, supporting that HLA-I surface expression is not targeted by SPPL3 and providing unique intra-molecular controls for further experiments. To determine whether SPPL3 differentially affected HLA-A, HLA-B, or HLA-C alleles, we reconstituted HLA-A−/−B−/−C−/− cells on a WT or SPPL3−/− background (Table S1) with the single original HLA-I alleles and analyzed their accessibility for antibodies. Each allele showed a difference in HLA-I accessibility between WT and SPPL3-deficient cells comparable to that detected by W6/32 (Figure S2B). In addition, other cell lines exhibited a similar decrease in W6/32 accessibility to HLA-I after small interfering RNA (siRNA) silencing or CRISPR-Cas9 genetic ablation of SPPL3, indicating that regulation of HLA-I accessibility is not solely restricted to HAP1 cells or their HLA-I haplotype (Figures S2C–S2E).
SPPL3 Expression Promotes Receptor Binding to HLA-I and CD8+ T Cell Activation
SPPL3 modulates antibody reactivity toward specific regions of HLA-I molecules, which raises the question of whether the HLA-Ifunction is affected. The CD8 coreceptor, which on most T cells is essential for sensitizing responsiveness by supporting TCR docking to its cognate peptide-HLA-I complex, ligates closely to the region of HLA-I that is affected by SPPL3 (as depicted in Figures 1E and 2A) (Gao et al., 1997; Purbhoo et al., 2004; Roszkowski et al., 2003). We evaluated whether SPPL3 enhances HLA-I antigen presentation to CD8+ T cells by stimulating multiple HLA-A*02:01-restricted T cell clones specific for different tumor-expressed antigens endogenously expressed by WT and SPPL3−/− cells (Amir et al., 2011; van Bergen et al., 2007, 2010). All clones were more reactive to SPPL3-expressing WT cells, as determined by their IFN-γ or granulocyte-macrophage colony-stimulating factor (GM-CSF) production (Figure 2B), indicating that SPPL3 increases functional access to HLA-I. The effect of SPPL3 on T cell function was confirmed in 51Cr release assays showing reduced killing of SPPL3−/− cells compared with WT cells (Figures 2C and S3A).
Figure 2. SPPL3 Expression Promotes LIR-1 Binding to HLA-I and Enhances CD8+T Cell Activation.

(A) CD8 and LIR-1 interaction sites mapped on the HLA-I/β2m crystal structure.
(B) IFN-γ production by T cells recognizing the specified endogenously derived antigens after coculture with the indicated HAP1 cells (n = 3).
(C) Normalized 51Cr release (specific lysis) of the indicated HAP1 cells targeted by specified T cells at different effector-target (E:T) ratios (n = 3) (see also Figure S3A).
(D) (Left) Representative titration curves of LIR-1 Fc fusion protein on indicated HAP1 cells (n = 2). (Right) Histogram of LIR-1 Fc binding at the indicated concentration (arrow).
(E) Flow cytometry staining of HLA-C*05:01-transduced SPPL3−/−HLA-A−/−B−/−C−/− or depicted control HAP1 cells with nonsaturating concentrations of KIR2DL1 and KIR2DL2 Fc fusion proteins. Gray, unstained control.
(F) Normalized quantification (median fluorescence intensity [median FI]) of HLA-I binding by the indicated fusion proteins (including data from D, E, and Figures 3G and 4E) (n = 3–6).
(G) Predicted protein structure of SPPL3 with its catalytic residues magnified.
(H) Histogram (cells from the RFP+ gate for transduced samples) and quantification (MFI normalized to the RFP− gate) of nonsaturating W6/32 stain on indicated HAP1 cells transduced with RFP-empty vector (EV), RFP-SPPL3, or catalytically inactive RFP-SPPL3 D271A (n = 5). See Figure S3C for B1.23.2 stain.
(I) IFN-γ secretion in supernatant after coculture of the indicated T cells with RFP+ fluorescence-activated cell sorting (FACS)-sorted HAP1 SPPL3−/− cells transduced as in (H) or with unsorted WT or HLA-I-deficient (gray) cells (n = 2–3). Mean ± SD of n independent experiments is plotted in (D), (F), and (H). A representative of n experiments is shown in (B), (C), and (I). ns, not significant; * p<0.05; ** p<0.01; *** p<0.001. See also Figure S3.
Disturbed receptor accessibility to HLA-I may affect not only T cells but also other immune cells that express HLA-I-interacting proteins, including the inhibitory leukocyte immunoglobulin (Ig)-like receptor (LIR) and killer cell Ig-like receptor (KIR) families that suppress unwanted immune cell activation (Saverino et al., 2000; Valiante et al., 1997). The defined SPPL3-susceptible region on the HLA-I protein highly overlaps with the binding site of LIR-1, which is expressed by monocytes, B cells, and small subsets of natural killer (NK) cells and T cells (Borges et al., 1997; Colonna et al., 1997; Cosman et al., 1997) (Figure 2A). Even more pronounced than for SPPL3-affected antibodies, binding of a recombinant LIR-1 Fc fusion protein (Gonen-Grosset al., 2010) to HLA-I was strongly decreased on SPPL3−/− cells compared with WT cells, suggesting that various immune cell functions can be affected by SPPL3 (Figures 2D and 2F). Similarly, we investigated the NK cell receptors KIR2DL1 and KIR2DL2 by evaluating the binding of their recombinant Fc fusion proteins to overexpressed HLA-C*05:01 on HLA-A−/−B−/−C−/− cells on a WT or SPPL3−/− background (Figure S3B) (Anfossi et al., 2006; Moesta et al., 2008). KIR2DL1 binding was not influenced by SPPL3 depletion, whereas KIR2DL2 binding was significantly reduced in the absence of SPPL3 (Figures 2E and 2F). Thus, the effect of SPPL3 on KIR and LIR binding to HLA-I is variable, indicating diverse, not further defined, effects on signaling toward different immune cell subsets.
Accessibility to HLA-I Depends on SPPL3-Mediated Proteolytic Cleavage
SPPL3 contains two aspartate residues embedded in conserved YD and GxGD motifs located in transmembrane helices 6 and 7, respectively, forming the catalytic unit for proteolysis (Voss et al., 2013) (Figure 2G). To investigate whether SPPL3 catalytic activity was critical in controlling protein accessibility to HLA-I and function, we expressed WT SPPL3 or a catalytically inactive SPPL3 mutant (D271A) in SPPL3−/− cells (Voss et al., 2012). Flow cytometry analysis showed that only SPPL3 D271A failed to restore the accessibility of HLA-I for W6/32, indicating that SPPL3 proteolytic activity is required for antibody accessibility to HLA-I (Figures 2H and S3C). This lack of rescue was confirmed on a functional level, because expression of active SPPL3, but not inactive SPPL3, in SPPL3−/− cells partially restored their capacity to activate T cells (Figure 2I).
SPPL3 has previously been reported to affect protein N-glycosylation by proteolytic inactivation of glycosyltransferases in the ER and Golgi (Kuhn et al., 2015; Voss et al., 2014). Variations in the N-linked glycan of HLA-I, located at position N86 close to the SPPL3-susceptible region (Figure S3D), can affect its availability for proteins (Barbosa et al., 1987; Neefjes et al., 1990). However, liquid chromatography-mass spectrometry (LC-MS) (reversephase [RP] nano-LC-ESI-MS(/MS)) of HLA-I N-linked glycans revealed no differences between WT and SPPL3−/− cells (Figure S3E), indicating that the HLA-I N-glycan structure is not regulated by SPPL3 activity. To exclude a contribution of the HLA-I N-glycan to SPPL3-modulated HLA-I accessibility for several receptors and antibodies, we inhibited complex N-glycan formation on SPPL3−/− cells using the α-mannosidase I and II inhibitors kifunensine and swainsonine. The decrease of complex N-glycans failed to alter the accessibility of the W6/32 epitope on SPPL3−/− cells (Figures S3F and S3G). This result was confirmed in cells genetically engineered to lack complex (HLA-I) N-glycosylation through ablation of the gene encoding GANAB (Table S1), which resulted in lower overall HLA-I surface expression as visualized by decreased W6/32 and B1.23.2 signals (Figure S3H). Comparison of these antibody stainings between WT and SPPL3−/− cells showed that W6/32 accessibility to HLA-I was still impaired in the absence of SPPL3 (Figure S3H). As we ruled out a role for protein glycosylation, our finding that SPPL3 activity affects HLA-I at the cell surface suggests the involvement of at least one unknown SPPL3 target.
SPPL3-Controlled GSLs Modulate Receptor Accessibility to HLA-I
To elucidate how SPPL3 controls protein accessibility to HLA-I, we followed two genome-wide screening strategies to specifically identify targets that are either positively or negatively regulated by SPPL3. An SPPL3-activated target affecting HLA-I would likely be a hit in the original W6/32 screen, just like SPPL3 (Figure 1B). However, the identification of such a target was complicated by the long list of significant hits. To distinguish SPPL3-activated targets from other candidates, we complemented the original screen with a new genome-wide haploid screen using a different HLA-I-specific antibody that was only mildly affected by the absence of SPPL3 (antibody BB7.2) (Figures 3A and S2A). This additional screen yielded another high-resolution snapshot of HLA-I antigen presentation (Figure S4A). A comparison of the two screens showed that SPPL3 was the only factor selectively affecting W6/32 binding, implying that no other gene was as strongly required for W6/32 accessibility to HLA-I (Figure 3B).
Figure 3. SPPL3-Controlled GSLs Modulate Receptor Accessibility to HLA-I.

(A–C) Schematic outlines of screening strategies to discover potential HLA-I regulators activated (A) or inactivated (C) by SPPL3. (B) Rocket plot of haploid genetic screens comparing the number of unique disruptive integrations per gene in BB7.2lo- and W6/32lo-sorted HAP1 populations. Two-sided FDR corrected Fisher’s exact test was applied. The symbol legend is indicated (see also Figure S4A).
(D) Fish-tail plot of the haploid genetic screen in SPPL3−/− HAP1 cells, showing per gene the mutation index (ratio of integrations mapped in indicated populations) against the amount of integrations. Statistics and color legend as in (B) (see also Figure S4B).
(E) Schematic overview of the core enzymes involved in the synthesis of GSL subtypes. The putative SPPL3-targeted branch is shown in orange. PM, plasmamembrane.
(F and G) Histograms of W6/32, B1.23.2 (F), and LIR-1 Fc (G) surface staining of the indicated HAP1 cells. Quantification (MFI, F; or median fluorescence intensity [median FI], G) is shown as mean ± SD (n = 3 independent experiments). Gray, unstained controls; S−/−, SPPL3−/−.
ns, not significant; ** p<0.01; *** p<0.001. See also Figure S4.
We then searched for potential genes negatively regulated by SPPL3 to affect HLA-I. To this end, we performed a genome-wide haploid screen in SPPL3−/− HAP1 cells. In these cells, which potentially lacked SPPL3-mediated suppression of the sought target, the gene trap mutagenesis of such a target or its associated pathway should improve W6/32 accessibility to HLA-I (Figure 3C). The hits from this screen converged to the GSL synthesis pathway (Figure 3D). The enzymes UGCG, B4GALT5, and B3GNT5 catalyze the synthesis of GSLs in the Golgi membrane by consecutive linkage of sugar residues derived from uridine diphosphate (UDP)-glucose, UDP-galactose, and UDP-N-acetylglucosamine (GlcNAc) donors on ceramide molecules (Figure 3E) (Allende and Proia, 2014). The latter two carbohydrate donors are transported from the cytoplasm into the Golgi by SLC35A2 and SLC35A3, respectively, which were also identified in the screen (Figure 3D) (Caffaro and Hirschberg, 2006). Other hits from the screen included proteins andcomplexes associated with Golgi homeostasis, such as the members of the component of oligomeric Golgi (COG) and Golgi-associated retrograde protein (GARP) complexes that facilitate GSL synthesis and trafficking (Fröhlich et al., 2015; Kingsley et al., 1986) (Figure 3D). None of these hits related to GSL metabolism emerged in the original screen with W6/32 on SPPL3-containing WT cells, strongly suggesting that in WT cells, the GSL synthesis or biosynthetic transport pathway is suppressed by SPPL3 (Figure S4B). Collectively, these observations revealed the existence of a pathway comprising GSL-mediated regulation of HLA-I access and function controlled by SPPL3.
To validate that SPPL3 reduces protein accessibility to HLA-I through manipulation of GSL synthesis, we generated GSL-deficient SPPL3−/−cells by additional genetic ablation of the first enzyme of the GSL synthesis pathway, UGCG (Table S1). In these SPPL3−/− UGCG−/− cells, we observed full rescue of the W6/32 HLA-I epitope accessibility without affecting SPPL3-independent B1.23.2 staining (Figure 3F), pointing toward an essential role for GSLs in shielding specific HLA-I epitopes. Importantly, GSL-deficient SPPL3−/− cells regained the capacity to engage the HLA-I binding receptor LIR-1 (Figure 3G), underscoring the physiological relevance of GSL-mediated epitope shielding of HLA-I.
B3GNT5 Tunes the Capacity of HLA-I to Interact with Its Natural Receptors
The synthesis of GSLs is probably best illustrated as a chain of sugar moiety transfers catalyzed by different Golgi enzymes (Figure 3E). UGCG initiates the GSL synthesis pathway by transferring a glucose to a ceramide on the cytosolic leaflet of the Golgi membrane (Allende and Proia, 2014). After this glucosylceramide is flipped into the Golgi lumen, a galactose moiety is added by B4GALT5 or B4GALT6 to generate lactosylceramide (LacCer). This neutral GSL then serves as a substrate for various glycosyltransferases responsible for the generation of different GSL series: A4GALT (globo-series), A3GALT2 (isoglobo-series), B3GNT5 (lacto-series and nsGSLs), B4GALNT1 (gangliosides, o series), and ST3GAL5 (gangliosides, a, b, and c series) (Figure 3E) (Allende and Proia, 2014; Zhang et al., 2019). Our screening data suggested that lacto-series GSL or nsGSL production, through B3GNT5 activity, could diminish protein accessibility to HLA-I (Figures 3D and S4A). To confirm this specificity, we generated polyclonal cell lines on the SPPL3−/− background, each CRISPR-Cas9 targeted for one of the five branching enzymes, and then analyzed W6/32 binding by flow cytometry. W6/32 accessibility to HLA-I in SPPL3−/− cells was restored only by ablation of B3GNT5 or control UGCG, confirming B3GNT5 as the sole branching enzyme involved in HLA-I epitope shielding (Figures 4A, 4B, and S4C). This effect was selective for SPPL3-deficient cells, because HLA-I accessibility for W6/32 was unaffected on WT cells with corresponding genetic ablations (Figures 4C and S4D). Single-cell-derived B3GNT5−/−and SPPL3−/− B3GNT5−/− cell lines were generated to corroborate a pivotal role for B3GNT5 in HLA-I epitope shielding (Table S1). As expected, additional genetic B3GNT5 depletion in SPPL3−/− cells restored not only W6/32 binding to its epitope but also accessibility of other SPPL3-susceptible epitopes recognized by TP25.99 and ROU9A6 (Figure 4D). Accessibility to SPPL3-independent epitopes and total HLA-I surface expression were not affected by additional B3GNT5 depletion (Figure 4D). Moreover, the lack of B3GNT5 expression in SPPL3−/− cells restored both binding of LIR-1 to HLA-I and their potential to activate T cells (Figures 4E and 4F). Altogether, our results suggest that active SPPL3 disrupts the B3GNT5 protein, which tunes the capacity of HLA-I to interact with its receptors.
Figure 4. B3GNT5 Function Determines HLA-I Accessibility for Its Natural Receptors.

(A) W6/32 surface staining of guide RNA (gRNA)-transduced (GFP+; gRNAs in Table S4) or control (GFP−) SPPL3−/− HAP1 cells.
(B and C) Quantification showing normalized MFIs of the depicted antibody binding to SPPL3−/− (B) or WT (C) cells combined data from two experiments with 2–4 gRNAs per gene (examples in A) (Figures S4C and S4D).
(D and E) Histograms and quantifications (MFI, D, or median fluorescence intensity, E, median FI) of surface staining of the indicated HAP1 cells using antibodies recognizing SPPL3-susceptible (W6/32, TP25.99, and ROU9A6) or SPPL3-independent (B1.23.2, WK4E3, and SN230G6) HLA-I epitopes (D; n = 4–7) or using LIR-1 Fc fusion protein (E; n = 2). S−/−, SPPL3−/−.
(F) IFN-γ or GM-CSF secretion by depicted T cells after coculture with the indicated HAP1 cells. Representative of n = 3.
Gray histograms are unstained controls. Mean ± SD of n independent experiments is plotted in (B)–(E). ns, not significant; * p<0.05; ** p<0.01; *** p<0.001. See also Figure S4.
SPPL3 Controls the Generation of nsGSLs by Proteolytically Inactivating B3GNT5
To detect a direct interaction between SPPL3 and its putative target B3GNT5, we performed coimmunoprecipitation of overexpressed epitope-tagged proteins. We coisolated B3GNT5 predominantly with the catalytically inactive SPPL3 D271A mutant, suggesting a transient interaction between SPPL3 and its substrate (Figure 5A). Cleavage of B3GNT5 by the intramembrane protease SPPL3 was confirmed in total lysate by a small reduction in the molecular weight of B3GNT5, reflecting proteolytic removal of the 1.5- to 4-kDa cytosolic tail, and by the presence of luminal B3GNT5 fragments in the supernatant (Figures 5A and 5B). Two other branching enzymes of the GSL synthesis pathway, B4GALNT1 and ST3GAL5, were poorly coisolated with SPPL3 (Figure 5A). Furthermore, cleavage products were not detected in the supernatant, indicating that B3GNT5 is a specific substrate of SPPL3 (Figure 5A).
Figure 5. SPPL3 Controls the Generation of nsGSLs by Targeting B3GNT5.

(A) Coimmunoprecipitation of the indicated FLAG-tagged proteins with RFP-EV (RFP), RFP-SPPL3 (R-SPPL3), or RFP-SPPL3 D271A (R-D271A). Sup, supernatant; TL, total lysate. Representative of n = 2.
(B) Schematic model of B3GNT5 proteolysis by SPPL3.
(C and D) TLC of the indicated HAP1 cell lysates incubated with UDP-GlcNAc and boron-dipyrromethene (BODIPY)-LacCer substrate to detect B3GNT5 activity (n = 3). BODIPY-Lc3Cer quantification (see Figure S5A for LC-MS validation) (C) and an example chromatogram (D) are shown.
(E) Base peak chromatograms of porous graphitized carbon (PGC) LC-MS on total GSL glycans isolated from indicated HAP1 cells (n = 3). Proposed glycan structures and their relative abundance are listed in (F), Table S3 and Figure S5B.
(F) Quantified relative abundance of the three major GSL types of the indicated cells.
(G) Histograms and normalized MFI of cholera toxin B (CTB) and C3D-1 binding to indicated HAP1 cells (n = 3). Gray, unstained control; S−/−, SPPL3−/− (see Figure S5C).
Mean ± SD of n independent experiments is plotted in (C), (F), and (G). ns, not significant; * p<0.05; ** p<0.01; *** p<0.001. See also Figure S5.
To investigate whether SPPL3 affects B3GNT5 activity, we performed a B3GNT5 enzymatic assay. Lysates of indicated WT and genetically ablated cells were incubated with a BODIPY-conjugated analog of the B3GNT5 substrate LacCer and the donor sugar UDP-N-GlcNAc, followed by thin-layer chromatography (TLC) of extracted GSLs. The B3GNT5 product lactotriaosylceramide (BODIPY-Lc3Cer), as confirmed by LC-MS, was generated in increased amounts in SPPL3−/− compared with WT cell lysates (Figures 5C, 5D, and S5A). Inaddition, no Lc3Cer was synthesized in lysates of B3GNT5−/− cells, demonstrating that B3GNT5 is the sole producer of Lc3Cer in HAP1 cells. Because SPPL3 inhibits B3GNT5 activity, we next addressed the extent to which SPPL3 defines the cellular GSL profile. Glycan portions of the GSL repertoire of WT, SPPL3−/−, and SPPL3−/− B3GNT5−/− cells were isolated and analyzed by LC-MS. We found an extensive shift in the relative GSL abundance toward B3GNT5-produced nsGSLs, from 44% in WT cells to 82% in SPPL3−/− cells (Figures 5E and 5F; Table S3). The increasewasmostevident forcomplex nsGSLs containing six or more sugar residues, as determined by relative quantification of individual GSLs, suggesting that epitope shielding of HLA-I is mediated by complex nsGSLs (Figure S5B; Table S3). To validate the shift in GSL repertoire in living cells, we conducted flow cytometry-based experiments using cholera toxin subunit B, which binds the ganglioside GM1, and an antibody against the nsGSL SSEA-1 epitope. GSL-deficient UGCG−/− cells were negative for all probes, demonstrating probe specificity toward GSLs on our cells (Figure S5C). Compared with WT cells, SPPL3−/− cells expressed increased amounts of SSEA-1 nsGSLs, which were generated by B3GNT5, and decreased amounts of GM1 gangliosides (Figures 5G and S5C). Consistent with our relative quantification of individual glycans detected by LC-MS, these data demonstrate that SPPL3 dictates the composition of the GSL repertoire by inhibiting the nsGSL biosynthesis activity of B3GNT5.
Sialic Acid Residues on nsGSL Acid Residues for HLA-I Shielding
GSLs are major constituents of membrane microdomains (Sezgin et al., 2017). A change in GSL composition may then disturb membrane protein localization, mobility, and function. We therefore investigated the mobility of HLA-I in SPPL3−/− cells by single-particle tracking. The mobile fraction and diffusion constant of BB7.2 Fab labeled HLA-I molecules were equal between SPPL3−/− and WT cells, indicating that HLA-I membrane dynamics were not detectably affected by potential alterations in membrane microdomain organization or by SPPL3 (Figure S6). This renders unlikely a scenario in which HLA-I associates with another protein in the absence of SPPL3, because this would reduce the HLA-I diffusion rate. Instead, our data suggested that decreased receptor accessibility to HLA-I was a direct consequence of interactions with nsGSLs. Such GSL-protein interactions can occur between gangliosides and hormone receptors through a charge-based linkage of GSL-derived sialic acid with positively charged amino acids (D’Angelo et al., 2013). Further analyses of the GSL signature of SPPL3−/− compared with WT cells revealed that the nsGSL glycan chains more frequently contain α−2,3- and α−2,6-linked sialic acid residues, as well as noncharged fucoses (Figures 5D and 6A; Table S3). To test the requirement of these nsGSL-localized sugar residues, we inhibited all sialyltransferase and fucosyltransferase activity and found that dose-dependent inhibition of sialylation, but not fucosylation, restored W6/32 accessibility to HLA-I in SPPL3−/− cells (Figures 6B–6E). The requirement for sialic acids was substantiated by the genetic ablation of CMP-sialic acid synthetase (CMAS), which also recovered W6/32 accessibility to HLA-I in SPPL3−/− cells (Figures 6B and 6F). Finally, the enzymatic removal of sialic acid residues at the cell surface by neuraminidase treatment diminished HLA-I shielding (Figures 6B and 6G). Thus, the B3GNT5-generated GSLs shield HLA-I through its sialic acids, likely via a direct charge-based interaction.
Figure 6. Sialic Acid Residues on nsGSLs Are Required for HLA-I Shielding.

(A) Quantified relative abundance of sialylated and fucosylated nsGSLs in WT or SPPL3−/− HAP1 cells. Data from Figure 5E were reused.
(B) Schematic view of targetable steps in nsGSL sialylation and fucosylation. NA, neuraminidase.
(C) Antibody staining of the indicated HAP1 cells cultured with a serial dilution of sialyltransferase (SiaT) or fucosyltransferase (FucT) inhibitors.
(D and E) Histograms (D) and normalized MFI (E) of the depicted antibody binding to HAP1 cells precultured with 100 μM inhibitors as in (C) (n = 6).
(F) Histograms and quantification of the depicted antibody staining of the indicated HAP1 cells (n = 2).
(G) Histograms and quantification of W6/32 and B1.23.2 binding to NA-treated HAP1 cells (n = 4).
Gray histograms are unstained controls. S−/−, SPPL3−/−. Mean ± SD of n independent experiments is plotted in (A) and (E)–(G). ns, not significant; * p<0.05; ** p<0.01; *** p<0.001.
Pharmacological Inhibition of GSL Synthesis in Glioma Enhances Anti-tumor Immune Activation In vitro
After determining that nsGSL-rich target cells suppress T cell activity, we examined the effect of increased nsGSL expression or downmodulated SPPL3 activity in tumors. Because of the complexity of identifying (large) nsGSLs, there is a limited amount of data available on their tissue expression, including tumors (Merrill and Sullards, 2017; Zhang et al., 2019). Nonetheless, elevated amounts of nsGSLs or their synthesis enzyme B3GNT5 have been observed on several tumor types, including glioma, acute myeloid leukemia (AML), and adenocarcinomas (Furukawa et al., 2015; Hakomori, 1984; Wang et al., 2012; Wikstrand et al., 1991). In addition, The Cancer Genome Atlas (TCGA) analyses demonstrated that high B3GNT5 expression in low-grade glioma correlates with decreased overall patient survival (Figure 7A). In line with our findings, the reverse held true for the B3GNT5-suppressing SPPL3 (Figure 7B). Moreover, analyses of the combined effect of B3GNT5 and SPPL3 expression showed only lower survival rates for patients with high B3GNT5 and low SPPL3 expression (Figures 7C and S7A), probably reflecting that nsGSL expression is elevated in tumors from this group. Because this indicated that gliomas may limit immune detection by exploiting the SPPL3-B3GNT5 axis, we tested the role of SPPL3 and nsGSLs in the glioblastoma cell line U-251. Overexpression of SPPL3 increased HLA-I accessibility to W6/32 without altering HLA-I expression (Figure 7D). Next, genetic depletion of GSLs (including nsGSLs) from U-251 cells resulted in a specific increase in W6/32 binding to HLA-I (Figures 7E and S7B). Moreover, in the absence of GSLs, U-251 cells were better activators of T cells (Figure 7F).
Figure 7. Pharmacological Inhibition of GSL Synthesis in Glioma Enhances Anti-tumor Immune Responses.

(A–C) TCGA-derived Kaplan-Meier curves showing the survival of patients with tumors expressing low or high B3GNT5 (A), low or high SPPL3 (B), or any of the four combinations thereof (C) (see Figure S7A).
(D) Histograms of W6/32 and B1.23.2 binding to U-251 glioblastoma cells overexpressing GFP-SPPL3 or RFP-EV, combined in a single well. Quantification (MFIGFP+ cells/MFI RFP+ cells) includes a GFP-EV control (n = 5).
(E) Histograms and normalized MFI of the depicted antibody staining of WT and UGCG−/− U-251 cells (n = 4–5) (see Figure S7B).
(F) IFN-γ secretion by the indicated T cells against WT or UGCG−/− U-251 cells (n = 3).
(G) Normalized MFI of the depicted antibody staining of the indicated HAP1 cells precultured with specified UGCG inhibitors (n = 2–7) (see Figures S7C–S7F). (H and I) IFN-γ or GM-CSF secretion by the indicated T cells cocultured with WT or SPPL3−/− HAP1 cells that were precultured with or without the specified UGCG inhibitor (n = 3) (see Figure S7G for more T cell clones).
(J) Histogram and normalized MFI of W6/32 binding to WT U-251 cells precultured with the indicated UGCG inhibitors (n = 3) (see Figure S7H).s
(K) IFN-γ secretion by the indicated T cells after coculture with the depicted inhibitor-pretreated U-251 cells (n = 3).
Gray histograms are unstained controls. Mean ± SD of n independent experiments is plotted in (D), (E), (G), and (J). A representative of n experiments is shown in (F), (H), (I), and (K). ns, not significant; * p<0.05; ** p<0.01; *** p<0.001. See also Figure S7.
To downregulate nsGSL expression in patients, the clinically approved GSL synthesis inhibitors miglustat and eliglustat may be used (Stirnemann et al., 2017). These drugs are being used as substrate reduction therapy in Gaucher disease. We first explored whether these small-molecule drugs affect accessibility to HLA-I epitopes that are shielded in SPPL3−/− cells. The miglustat mimics MZ21 and MZ31, with fewer off-target effects, were also included (Ghisaidoobe et al., 2014). All GSL synthesis inhibitors fully restored W6/32 accessibility to HLA-I, despite a small proportion of GSLs remaining detectable on the cell surface (Figures 7G and S7C–S7F). Moreover, these inhibitors increased the capacity of SPPL3−/− cells to activate T cells (Figures 7H, 7I, and S7G). This was also observed for U-251 cells, for which HLA-I shielding was alleviated and their capacity to activate T cells was increased (Figures 7J, 7K, and S7H). Altogether, these data demonstrate that these inhibitors can boost immune responses against tumor cells that display excess nsGSLs.
DISCUSSION
The process of HLA-I antigen presentation has been a topic of long-standing interest, giving rise to a detailed understanding of this complex pathway. We here describe an additional element for the equation of successful antigen presentation, namely, the SPPL3-B3GNT5 pathway responsible for the production of a subset of GSLs. GSLs are present on every cell, yet their functional roles in the cell membrane remain largelyunknown. By iteratively conducting sensitive genome-wide screens, we uncovered a role for a subset of GSLs in immunity controlled by the aspartyl protease SPPL3. These nsGSLs shield HLA-I molecules, limiting their interactions with several immune cell receptors and decreasing CD8+ T cell responses. We identified SPPL3 as a switch controlling the expression of nsGSLs through proteolytic inhibition of the nsGSL-synthesizing enzyme B3GNT5. Altogether, our study reveals a layer of immune regulation that acts late in the HLA-I antigen presentation pathway by shielding critical HLA-I epitopes at the cell surface.
Understanding nsGSL function at a molecular level and in physiological and pathophysiological settings is challenging given that their isolation, their analytical dissection, and in particular their experimental manipulation have been extraordinarily demanding to date (Merrill and Sullards, 2017; Zhang et al., 2019). Hence, no validated methods are available to study nsGSL-protein interactions, restricting options to directly probe the nsGSL-HLA-I interaction. Our current data indicate that the interaction between nsGSLs and HLA-I molecules must be transient, because a high antibody dose can overcome limited accessibility to HLA-I. In addition, we show that this interplay is independent of carbohydrate-carbohydrate interactions (D’Angelo et al., 2013) between nsGSLs and HLA-I N-glycans. However, the profound shielding of large HLA-I patches by nsGSLs can be explained by the ability of nsGSLs, in contrast to other GSL subtypes, to carry huge glycan chains of up to 60 sugar residues (Miller-Podraza et al., 1993, 1997). These long carbohydrate trees may reach HLA-I domains involved in the interaction with receptors such as LIR-1. The nsGSLs may sterically compete with proteins for accessibility to HLA-I or restrict protein accessibility by altering HLA-I orientation toward the cellular membrane (Mitra et al., 2004). In addition, our data do not exclude a direct interaction between the GSL ceramide and the HLA-I transmembrane domain (Contreras et al., 2012), which could contribute to the positioning the nsGSL glycan chain close to HLA-I. Finally, we show that sialic acid residues on GSLs are essential for HLA-I shielding. The negatively charged sialylated nsGSLs may establish ionic interactions with HLA-I, which has abundant positively charged patches at its molecular surface (Li et al., 2012). Similar GSL-protein interactions have been found between sialic acids on short GSL glycans and exposed positively charged amino acid residues close to the plasma membrane (D’Angelo et al., 2013). This shows that sialylated nsGSLs may shield cell surface receptors other than HLA-I and possibly affect their cognate interactions. This assumption is supported by SPPL3 being identified in genome-wide CRISPR-Cas9 screens for surface detection of butyrophilin (BTN) molecules by a functional Vγ9Vδ2+ γδ TCR or CD47 and CD59 by antibodies (Davis et al., 2015; Logtenberg et al., 2019; Rigau et al., 2020). In these cases, the underlying molecular mechanism of the SPPL3 effect was not resolved, yet these cases suggest that the SPPL3-B3GNT5 pathway constitutes a mechanism to finetune communication between cells, including a functional tumor cell-T cell interaction, as we report here.
Various malignant cells exhibit alterations in their GSL surface repertoire, to which several specific functions have been attributed. Some GSLs can serve as signaling molecules to control cellular processes such as apoptosis and proliferation, whereas other GSL species can confer anti-cancer drug resistance by inhibiting proteins that facilitate their membrane transport (Liu et al., 2013; Ogretmen and Hannun, 2004). We here propose that changes in the tumor GSL repertoire, in particular increments of sialic-acid-containing nsGSLs, limit HLA-I signaling to T cells as a means to evade immune surveillance. In support of this hypothesis, our in vitro data show that GSLs diminish the capacity of CD8+ T cells to respond to glioma, a tumor type with high expression of nsGSLs (Furukawa et al., 2015). Furthermore, HLA-I-related NK cell activation against tumors lacking SPPL3 may be restricted according to recent genome-wide CRISPR-Cas9 screens (Pech et al., 2019). Thus, nsGSL upregulation by tumors such as glioma might allow T cell escape while marginalizing NK cell recognition. In addition to in vitro experimentation, analyses involving glioma patients revealed worst overall survival when the SPPL3 and B3GNT5 expression signature of the tumor suggests high nsGSL synthesis. Such correlation with patient outcome may have been influenced by covariates, which potentially include nsGSL-mediated shielding of other immune or nonimmune receptors or membrane turnover (Catalaa et al., 2006; Righi et al., 2009). Other tumor types, including AML, colorectal carcinoma, adenocarcinoma, and ductal carcinoma in situ (DCIS), also overexpress B3GNT5 and its product nsGSLs (Hakomori, 1984; Potapenko et al., 2015; Wang et al., 2012; Wikstrand et al., 1991), suggesting that nsGSL overexpression is a general strategy for tumor survival. Furthermore, pathogens such as cytomegalovirus, respiratory syncytial virus, and HIV alter the GSL composition of the host cell, potentially inducing immune evasion through HLA-I shielding (Fantini et al., 2000; Moore et al., 2008; Radsak and Wiegandt, 1984). Except for low-resolution data concerning cytomegalovirus-induced nsGSL expression upon infection (Andrews et al., 1989; Radsak and Wiegandt, 1984), little is known about which viral infections influence complex nsGSL expression.
In this study, we have presented GSLs as highly relevant molecules affecting the efficiency of immune responses. nsGSLs and their molecular switch SPPL3 represent an unexplored avenue for therapeutic intervention in cancer, infection, and autoimmune diseases. Two small-molecule drugs inhibiting GSL synthesis are registered: miglustat (Zavesca) and eliglustat (Cerdelga). These structurally different UGCG inhibitors (Platt et al., 1994; Shayman, 2010) have been approved for the treatment of patients with lysosomal storage disorders, such astype I Gaucher disease and Niemann-Pick disease type C (Lachmann, 2003; Wraith and Imrie, 2009). Therefore, therapeutic application can be extended efficiently to include immune enhancement against tumors or pathogen-infected cells. GSL synthesis inhibition may even be combined successfully with existing immunotherapies, such as PD-1 blockade, because of the potential synergy between enhanced tumor cell immunogenicity and simultaneous T cell activation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robbert Spaapen, Ph.D (r.spaapen@sanquin.nl).
Materials Availability
Plasmids and genetically ablated cell lines generated in this study will be available from the lead contact with a completed Material Transfer Agreement (MTA).
Data and Code availability
The published article includes all data generated or analyzed during this study, except for the processed screen results, which are accessible in an interactive database (https://phenosaurus.nki.nl/). The accession number for the raw sequence data of the screens reported in this paper is NCBI Sequence Read Archive: PRJNA665349. The bioSample accession number for these data in NCBI Sequence Read Archive: SAMN16252402.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell lines and T cell clones
HAP1 (HLA-A*02:01, HLA-B*40:01 and HLA-Cw*03:04, near-haploid, derived from the male chronic myeloid leukemia KBM7 cell line with a fibroblast-like phenotype), MelJuSo (HLA-A*01:01, B*08:01 and C*07:01, female melanoma authenticated at Eurofins), SW620 (HLA-A*24:02, A*02:01, B*07, B*15 and C*07:04, male colon, derived from metastatic site: lymph node) (kindly provided by Dr. T. de Gruijl (Amsterdam UMC, the Netherlands)) and U-251 (HLA-A*02:01, male glioblastoma) (kindly provided by Dr. H. Versteeg (LUMC)) cell lines were cultured in IMDM (GIBCO and Lonza) supplemented with 10% FCS and antibiotics (PenStrep; Invitrogen) at 37°C and 5% CO2. HEK293T (ATCC Cat# CRL-3216, RRID: CVCL_0063) and its derivative Phoenix ampho (female embryonic kidney) cells were cultured in similarly supplemented DMEM (GIBCO). The HLA-A*02:01-restricted CD8+ T cell clones reactive against antigens derived from the tumor-expressed proteins USP11, FDPS, VPS13B, ADIR, and SSR1 that are all endogenously expressed by HAP1 cells were previously described (Amir et al., 2011; van Bergen et al., 2007, 2010) and expanded using a feeder cell-cytokine mixture in medium with human serum (Sanquin) as described (Oostvogels et al., 2014).
METHOD DETAILS
Haploid genetic screening
Genome-wide haploid genetic screening was performed in either early passage WT or CRISPR/Cas9 generated SPPL3−/− HAP1 cells using directly conjugated W6/32 or BB7.2 antibodies. Retroviral mutagenesis was performed on ∼100 × 106 cells using GT-GFP or GT-BFP plasmids as previously described (Brockmann et al., 2017). 2 × 109 expanded mutagenized cells were fixed, stained and sorted into two separate populations based on the fluorescent intensity of the respective HLA-I antibody staining (Brockmann et al., 2017). Gene trap integration sites were amplified using a LAM-PCR with a biotinylated primer on genomic DNA isolated from sorted cells. Biotinylated products were captured on streptavidin-coated beads followed by a single-stranded DNA linker ligation and a subsequent amplification step using two primers to generate fragments that include a genomic region flanking the insertion site in addition to adaptors for deep sequencing. Deep sequencing reads were aligned to the human genome (hg19) and intersected with protein coding genes to obtain the numbers of unique disruptive integrations mapped per gene in both populations either lowly or highly recognized by the respective HLA-I antibody. Enrichment of mutations in genes was assessed using a Fisher’s exact test corrected for false discovery (Benjamini-Hochberg). The approach is described in detail in Brockmann et al. (2017).
Plasmids
pMXs-puro vector (Cell Biolabs) was equipped with a novel multiple cloning site with or without N- or C-terminal tags (RFP, GFP and FLAG). SPPL3 and its inactive mutant SPPL3 D271A (kind gift from Dr. R. Flührer) (Voss et al., 2012) were recloned into pMXs-puro-RFP or -GFP using XhoI/BamHI restriction sites. B3GNT5 and B4GALNT1 were PCR amplified from IMAGE:202800754 and IMAGE:202800771 and cloned into pMXs-puro-FLAG-C or -N by EcoRI/BamHI restriction sites. ST3GAL5 was amplified from IMAGE:202759803 and cloned into pMXs-puro-FLAG-C using EcoRI/BclI digestion into an EcoRI/BamHI digested plasmid. The pLZRS-based retroviral vectors containing HLA-A*02:01/B*40:01/C*03:03-IRES-ΔNGFR were described before (Griffioen et al., 2012; Van Bergen et al., 2010). Generation of retroviral supernatants and transduction of cells were performed as described (Spaapen et al., 2008). HLA-C*05:01 (IMGT/HLA database) with a mutated signal peptide from HLA-A*02:01 (M4V) was purchased as a gBlock gene fragment (Integrated DNA Technologies, Inc.) and used as a template for amplification. The PCR product was cloned into the puc2CL6IN lentiviral vector using NheI and BamHI. gRNAs (Table S4) were cloned into the pX330 expression vector or the lentiviral vectors lentiCRISPR_v2 or pLCRISPR.efs.GFP (Addgene) as described (Heckl et al., 2014; Sanjana et al., 2014).
Genome editing
SPPL3−/− and TAPBP−/− HAP1 cells were created by in frame integration of a blasticidin-resistance gene after cotransfection of pX330 with TIA-2Ablast (using Extremegene HP, Sigma) as described for other targets (Blomen et al., 2015; de Waard et al., 2020). Clones growing after blasticidin selection (10μg/mL, Life Technologies) were sequence verified for specific genome editing by Sanger sequencing (primers in Table S5). Lentiviral constructs containing gRNAs targeting SPPL3, UGCG, CMAS and the five core GSL-enzymes were cotransfected into HEK293T with the packaging enzymes psPAX2, pVSVg, pAdVAntage using polyethylenimine (PEI; Polyscience) for virus production. Filtered viral supernatants were used for transduction by spinoculation in the presence of 8μg/mL protamine sulfate. Cells were selected using puromycin (0.25μg/mL; GIBCO), blasticidin (10μg/mL; GIBCO), geneticin (550μg/mL; Formedium) or gated on based on the coexpression of GFP or RFP. Polyclonal gene-deficient populations without selection (core GSL-enzymes for HAP1 cells) or after puromycin selection (SPPL3 and CMAS for HAP1 cells and UGCG for U-251 cells) were used for flow cytometry or T cell assays, or genetically ablated clones were made by limiting dilution and sequence verified. HLA-A−/−HLA-B−/−HLA-C−/− cells were generated by pX330 transfection followed by single cell sort by flow cytometry using W6/32 and sequence verification of clones (de Waard et al., 2020).
Sanger sequencing
Constructs were sequenced using BigDye Terminator Kit (Applied Biosystems). Genomic DNA isolated from cell lines using DirectPCR (Cell) lysis reagent (Viagen) supplemented with Proteinase K (Sigma) was amplified using primers mentioned in Table S5 and directly sequenced using BigDye Terminator Kit (Applied Biosystems). Sequences were analyzed using Snapgene (GSL Biotech). Sequence decomposition to assess the size of deletions or insertions of genome-edited diploid cells was performed using Tide (Brinkman et al., 2014).
siRNA transfections
Gene silencing was performed in a 96F well plate using 5 μL siRNA (500nM stock) mixed with 0.1 μL DharmaFECT1 #1 (Dharmacon) in 4.9 μL IMDM. The mixture was incubated for 20min on a shaker at RT and mixed with 4700 cells/200ul. After three days cells were analyzed using flow cytometry. siGENOME human SPPL3 (D-006042, Dharmacon) and β2m (M-004366, Dharmacon) siRNAs were used to silence SPPL3 and β2m respectively. Non-targeting siRNA (siCTRL, D-001206–13-20, Dharmacon) was used as a negative control.
Inhibitors and enzymes
UGCG inhibitors MZ31 (used concentration: 2μM), MZ21 (2μM), miglustat (100μM) were produced as previously described (Ghisaidoobe et al., 2014) and eliglustat (200nM) was obtained from Bio-Connect. Swainsonine (20μg/mL) and kifunensine (20μM) were obtained from Sigma. N-glycosylation inhibitor activity was confirmed by incubating W6/32 immunoprecipitated HLA-I molecules with Endoglycosidase H (Sigma) in a 20μl reaction mixture (50μM Sodiumcitrate (pH5.5), 0.2% SDS, 2μl Endoglycosidase H and protease inhibitors) for 18h at 37°C followed by HLA-I detection on immunoblot. Sialyltransferase inhibitor (3Fax-peracetyl Neu5Ac, 100μM) was obtained from Sigma and fucosyltransferase inhibitor (2-Deoxy-2-fluoro-L-fucose, 100μM) was obtained from Carbosynth. Cells were cultured for 2 or 3 days at 37°C with inhibitors and analyzed by flow cytometry. Cells were incubated with neuraminidase (N2876, Sigma, 225mU/mL) for 1h at 37°C.
Fab fragment production and labeling
W6/32 Fab fragments were prepared by 1h pepsin (1 μg/μL, pH3.5) treatment at 37°C in a pH3.5 buffer containing citric acid (0.07M) and sodium citrate (0.03M), followed by reduction using DTT (2.5mM, Sigma-Aldrich). BB7.2 Fab fragments were prepared using a papain:antibody ratio of 1:100 (16 μg/mL papain) for 20h at 37°C. All Fab fragments were purified by gel filtration (Superdex 200, 10/300 GL, GE Healthcare Life Sciences). Monomeric Fab fragments were either conjugated to Alexa Fluor® 555 NHS Ester (AF555; ThermoFisher Scientific) or Alexa Fluor® 647 NHS Ester (AF647; ThermoFisher Scientific) according to the manufacturer’s labeling protocol. To remove unconjugated fluorophores, the labeled Fab fragments were further purified by gel filtration (Superdex 75, 10/300 GL, GE Healthcare Life Sciences). Fractions containing monomeric fluorophore-conjugated Fab fragments were concentrated to a protein concentration of 0.2–0.5μg/μL using Amicon Ultra-4 centrifugal filters (10 kDa cut off, Merck Millipore) and then stored in 50% glycerol at −20°C. The protein to dye ratios were determined by spectrophotometry at 280nm and the corresponding absorption maximum of the dyes at 555nm and 650nm. The protein to dye ratio of the randomly conjugated Fab fragments were 1.0 (BB7.2 Fab-AF555), 0.94 (W6/32 Fab-AF647) and 0.4 (W6/32 Fab-AF555).
Flow cytometry using antibodies
Trypsinized cells were incubated with specific antibodies diluted in PBS for 30min at 4°C and washed up to five times in PBS (with or without 0.5% BSA) before incubation with secondary antibodies if required (30min at 4°C). All stains for which no titrations are shown were performed using non-saturating antibody concentrations. Stained cells were fixed in PBS containing 1% formaldehyde (Merck). DAPI (1 μM, Sigma-Aldrich) was used to exclude dead cells from analyses. Cells were fluorescently barcoded using the fluorescent dyes CFSE (125nM, Invitrogen), Alexa Fluor 350 NHS Ester (40 μM, Thermo Fisher Scientific) or Violet proliferation dye 450 (2.5 μM, BD Horizon) diluted in PBS. Cells were incubated with a fluorescent dye for 15min (vortexed every 5min) and washed three times in ice-cold complete IMDM (see above). Barcoded cells were mixed prior to plating in 96V-bottom wells for antibody staining. Stained samples (barcoded or not) were analyzed or sorted on BD flow cytometers (Canto II, Fortessa, LSR II or ARIA II). Flow cytometry data was analyzed using FlowJo (Tree Star, Inc).
Flow cytometry using other proteins
Fusion proteins LIR-1 Fc (kindly provided by Ofer Mandelboim), KIR2DL1-Fc and KIR2DL2-Fc (R&D Biosystems) were reconstituted in PBS (100μg/mL) and stored at −80°C. Cholera toxin B subunit (CTB)-FITC (Sigma) was reconstituted in sterile water (500μg/mL) and used at 10 μg/mL. Trypsinized cells were washed in PBS with 3% FCS or 0.5% BSA and stained in 40μL (fusion) protein dilution for 30min to 2h on ice. For fusion proteins, cells were washed twice and incubated on ice in 40μl secondary antibody APC AffiniPure F(ab’)2 Fragment Goat Anti-Human IgG, Fcγ fragment specific (Jackson ImmunoResearch) (KIR2DL1, KIR2DL2) or mouse anti-human IgG (MH161–1, Sanquin) in-house conjugated to DL650 (Thermo Fisher Scientific) for 30–45min. After two washes cells were resuspended in PBS/3%FCS containing DAPI and analyzed on BD flow cytometers.
T cell assays
Target cells were cocultured with T cells in a 1:1 ratio for 18h as previously described (Spaapen et al., 2008). Cytokine content of cell free supernatants was determined using standard sandwich ELISA according to the manufacturer’s instructions (IFN-γ and GM-CSF; Sanquin and Biolegend). Some target cells were precultured for 2 days in the presence or absence of UGCG inhibitors. For cytotoxicity experiments, target cells were loaded with 100 μCi 51Chromium (Perkin-Elmer) for 90min at 37°C and then washed twice with PBS. 4000 target cells were then cocultured with T cells at indicated effector target (E:T) ratios for 4–6h in round-bottom plates after which 30 μL of the supernatant was harvested and analyzed for radioactivity using a gamma counter (Wallac). Medium and 0.1% Triton X-100 served as spontaneous and maximal release controls. The percentage of specific lysis was calculated as [(experimental cpm – spontaneous cpm) / (maximal cpm – spontaneous cpm)] × 100%.
Immunoprecipitation
The medium of transiently transfected HEK293T cells was filtered through a puradisc 30 syringe filter (0.2μM, FP 30/0.2) to remove cell material. Before sample buffer was added. HEK293T or CRISPR/Cas9 edited HAP1 cells (confluent 6cm dish) were lysed for 20min in lysis buffer containing 0.8% NP-40, 10% glycerol, 150μM NaCl, 50μM Tris-HCl pH8.0, 1mM EDTA, 5mM MgCl2 and protease inhibitors (Roche Diagnostics, EDTA free). Lysate was centrifuged for 20min at 12.000rpm and the supernatant was incubated with RFP-Trap beads (Chromotek) or antibody coated Protein-γ Sepharose beads for 2h. Beads were washed four times in lysis buffer before addition of Laemmli Sample Buffer (containing 5% β-mercaptoethanol) followed by 5min incubation at 95°C. Coimmunoprecipitated proteins were separated by SDS-PAGE for western blotting and detected by antibody staining.
SDS-PAGE and western blotting
Samples were separated by SDS-PAGE (10% or 12% acrylamide gel) and transferred to a PVDF membrane (Immobilon-P, 0.45 μm, Millipore) at 300mA for 3h. The membranes were blocked in PBS/5% Milk (Skim milk powder, Oxoid) and incubated with a primary antibody for 1h diluted in PBS/0.1% Tween/5% Milk, washed thrice for 10min in PBS/0.1% Tween and incubated with the secondary antibody for 45min diluted in PBS/0.1% Tween/5% Milk and washed thrice again in PBS/0.1% Tween. The filter was incubated with ECL reagent (SuperSignal West Dura Extended Duration Substrate, Thermo Fisher Scientific) and the signal was detected using the Chemidoc XRS+ imager (Bio-Rad) or Amersham Imager 600.
BFA assay
To determine HLA-I turnover, HAP1 cells were seeded in 96w plates (80% confluent) and cultured in the presence of Brefeldin A (0.5μg/mL BFA, Sigma Aldrich) at 37°C for indicated times. BFA containing medium was removed and the cells were washed in cold PBS, trysinized and harvested in PBS/0.5% BSA. Cells were kept on ice and stained for flow cytometry using non-saturating amounts of W6/32 and B1.23.2 antibodies (see flow cytometry section).
Crystal structures
Structural prediction software Phyre2 was used to create a model of SPPL3 using the primary consensus sequence CCDS9208.1 (Kelley et al., 2015). Models of HLA-A*02:01 were made using the crystal structure 3MRG courtesy of the RCSB PDB (Reiser et al., 2014; Winn et al., 2011). The structure 3QZW was used in conjunction with the CCP4 program ARIAIMOL (Berman et al., 2000) to determine the hydrogen bonding contacts between human HLA-A*24:02 and human CD8 alpha-alpha dimer (Shi et al., 2011). A similar method was used to assess the contacts between LIR-1 and HLA-A*02:01 using the structure 1P7Q (Berman et al., 2000; Willcox et al., 2003). All figures have been produced using the PyMOL molecular graphics software (Version 2.0 Schrödinger, LLC).
B3GNT5 activity assay
5 × 106 HAP1 cells were harvested by trypsinization and cells were lysed in lysis buffer (2% Triton X-100, 50μM sodium cacodylate pH7.4, 10μM MnCl2, and protease inhibitors (Roche Diagnostics, EDTA free)) by incubating on ice for 30min. Nuclei were precipitated by centrifugation at 15,000xg. Equal volumes of lysis buffer containing 2μM BODIPY-C5-Lactosylceramide complexed to BSA (Thermo Fisher Scientific) and/or 1mM UDP-N-acetyl-D-glucosamine (Santa Cruz) were added to post-nuclear supernatants containing equal amounts of protein in 50μL lysis buffer. Samples were incubated at 37°C for 4h and subjected to lipid extraction using Bligh-Dyer method (Bligh and Dyer, 1959). 100μL 2% NaCl, 250μL chloroform, and 500μL methanol were added to the reactions and samples were vortexed. Phase separation was induced by addition of 250μl 0.45% NaCl and 250μL chloroform and lower phases were collected. Upper phases were re-extracted twice more and collected lower phases were dried under a nitrogen flow. Dried lipids were resuspended in chloroform:methanol (2:1 v/v) solution and spotted on TLC plates. TLC plates were developed in chloroform:methanol:water (60:25:4 v/v/v) and imaged using Typhoon FLA9500 (GE Healthcare) scanner equipped with a 473nm laser and BPB1 filter (530DF30). Identity of BODIPY-Lc3Cer was confirmed by MS/MS. Structures were assigned based on MS/MS fragmentation pattern in negative mode following nomenclature from Domon and Costello (1988).
GSL extraction and purification by RP-SPE
GSLs were extracted from 1 × 107 HAP1 WT, SPPL3−/− and SPPL3−/−B3GNT5−/− cell lines in triplicate in glass vials equipped with a Teflon-lined screw cap. Cells were washed three times in 1mL of water followed by centrifugation at 2000xg for 30min. The supernatant was removed and replaced by 300 μL of 2-propanol. The samples were vortexed for 5min and incubated for 15min at 75°C. A volume of 350 μL of MTBE (Sigma-Aldrich) was added to the samples followed by 15min sonication. 200 μL of water was added to the cell pellets and incubated for 4h with shaking at room temperature. The upper phase containing GSLs was collected after centrifugation at 2700xg for 20min. Then, 400 μL of MTBE was added, followed by sonication and centrifugation. The upper phase was collected and pooled to the previous sample. The process of adding MTBE, sonication, centrifugation and removing upper phase was repeated another two times. The combined upper phases were dried under vacuum in an Eppendorf Concentrator 5301 (Eppendorf) at 30°C. Before purification of the GSLs using RP-solid phase extraction (SPE), the samples were dissolved in 200 μL methanol and vortexed for 10min, followed by addition of 400 μL water. TC18-RP-catridges were prewashed with 2mL of chloroform/methanol (2:1, v/v), 2mL of methanol followed by equilibration with 2mL methanol/water (1:2, v/v). The extracted GSLs were loaded to the cartridge for 3 times and washed with 3mL methanol/water (1:2, v/v). The GSLs were eluted from the column with 2mL methanol and 2mL chloroform/methanol (2:1, v/v). The samples containing the eluate were evaporated under nitrogen for 1h and dried under vacuum in an Eppendorf Concentrator at 30°C. The collection and dry of GSLs eluate were performed in glass tube.
GSL glycan release by EGCase I and purification
To release the glycans from the GSLs, a mixture of 2 μL Endoglycoceramidase I (EGCase I recombinant clone derived from Rhodococcus triatomea and expressed in Escherichia coli (12mU, NEB)), 4 μL 10x EGCase I buffer (500mM HEPES, 1M NaCl, 20mM DTT and 0.1% Brij 35, pH5.2, NEB) and 34 μL water (pH5.2) was added to each sample and incubated for 16h at 37°C. The released glycans were collected and applied to a TC18-RP-cartridges (Waters) which was preconditioned with 2mL of methanol and 2mL of water. The sample vials were washed with 200μL of water and residual glycans were loaded to the cartridge. Then, 500 μL of water was added to the cartridge to wash the glycans from the column. The flow-through and wash fractions were pooled and dried in an Eppendorf Concentrator at 30°C.
Reduction, desalting and carbon SPE cleanup of GSL glycans
The reduction was carried out as described previously with slight modifications (Jensen et al., 2012). In brief, GSL glycans were reduced to alditols in 20 μL of sodium borohydride (500mM, Sigma-Aldrich) in potassium hydroxide (50μM, Sigma-Aldrich) for 2h at 50°C. Subsequently, 2 μL of 100% glacial acetic acid was added to neutralize the solution and quench the reaction. The desalting of GSL glycans was performed on cation exchange columns (Sigma-Aldrich) which consist of 60 μL of AG50W-X8 resin beads deposited onto reversed phase μC18 ZipTips (Perfect Pure, Millipore) as previously described (Jensen et al., 2012). Glycan alditols were eluted with 50 μL of water twice. The combined flow-through and eluate were pooled and dried under vacuum in an Eppendorf Concentrator at 30°C. The carbon SPE clean-up was performed and the purified glycan alditols were resuspended in 10 μL water for porous graphitized carbon (PGC) LC-ESI-MS/MS analysis.
Analysis of GSL glycans using PGC LC-ESI-MS/MS
Porous graphitized carbon (PGC) LC-ESI-MS/MS analysis of GSL glycan alditols was performed on a Dionex Ultimate 3000 nano-LC system equipped with a Hypercarb PGC trap column (5 μm Hypercarb Kappa, 32 μm × 30mm, Thermo Fisher Scientific) and a Hypercarb PGC nano-column (3 μm Hypercarb Kappa, 75 μm × 100μM, Thermo Fisher Scientific) coupled to an amaZon speed ion trap mass spectrometer (Bruker Daltonics). Mobile phase A consisted of 10mM ammonium bicarbonate (Sigma-Aldrich). Mobile phase B was 60% (v/v) acetonitrile (Biosolve) / 10mM ammonium bicarbonate (Sigma-Aldrich). To analyze glycans, 2 μL injections were performed and separation was achieved with a gradient of B (1%–71% at 0.7%/min) followed by a 10min wash step using 95% of B at a flow of rate of 0.6 μL/min. MS scans from m/z 340 to 1700 were recorded in enhanced mode using negative ion mode. MS/MS spectra were recorded selecting the top 3 highest intensity peaks. Glycan structures were assigned based on glycan composition obtained from accurate mass, relative PGC elution position, MS/MS fragmentation pattern in negative-ion mode and general glycobiological knowledge (Karlsson et al., 2004), with help of Glycoworkbench (Ceroni et al., 2008) and Glycomod (Cooper et al., 2001) software tools. Extracted ion chromatograms were used to integrate area under the curve (AUC) for each individual glycan isomer using Compass Data Analysis software v.5.0. The most abundant peaks in the glycan profile were manually picked and integrated. Relative quantitation of individual glycans was performed on the total area of all included glycans within one sample normalizing it to 100%.
HLA-I glycan analysis, in-gel tryptic digestion
The glycopeptide generation and analysis were performed as described previously with slight modifications (Plomp et al., 2014). BB7.2 immunoprecipitated samples were loaded on SDS-PAGE. Bands containing HLA-I were excised and cut into pieces. The gel pieces were washed with 20μM ammonium bicarbonate, dehydrated with acetonitrile (ACN) and reduced in-gel for 30min at 55°C with 100 μL 10mM DTT in 20μM ammonium bicarbonate solution. Thereafter they were dehydrated in ACN followed by cysteine alkylation for 20min with 100 μL of a 55mM iodoacetamide (Sigma-Aldrich) in 20μM ammonium bicarbonate solution in the dark for 45min. This was repeated twice, and the gel pieces were subsequently dried in a centrifugal vacuum concentrator at 30°C for 10min. Enzymatic digestion of trypsin was performed by adding 50 μL of 20μM ammonium bicarbonate containing 0.6 μg of trypsin (sequencing grade modified trypsin, Promega) to the dried gel particles. The samples were kept on ice for 1h and were subsequently incubated overnight at 37°C. The solution surrounding the gel pieces was collected and stored at −20°C. 20 μL of 20mM ammonium bicarbonate was added to the remaining gel pieces, and incubated at 37°C for another hour. The solution was again collected and added to the first fraction prior to freezing.
Glycopeptide analysis by reverse-phase (RP) nanoLC-ESI-MS(/MS)
Glycopeptides were analyzed by RP nanoLC-ESI-MS(/MS) on an Ultimate 3000 RSLCnano system (Dionex / Thermo Fisher Scientific) coupled to an HCTultra-ESIion trap-MS (Bruker Daltonics). 5 μL sample was injected and concentrated on a trap column (Acclaim PepMap100 C18 column, 100 μm 3 2cm, C18 particle size 5 mm, pore size 100Å, Dionex / Thermo Fisher Scientific) before separation on an Acclaim PepMap RSLC nanocolumn (75 μm × 15cm, C18 particle size 2 μm, pore size 100Å, Dionex / Thermo Fisher Scientific). A flow rate of 700nL/min was applied. Solvent A consisted of 0.1% formic acid in water; solvent B, 0.1% formic acid in 95% ACN and 5% water. A linear gradient was applied with the following conditions: t = 0min, 3% solvent B; t = 5min, 3% solvent B; t = 20min, 27% solvent B; t = 21min, 70% solvent B; t = 23min, 70% solvent B; t = 24min, 3% solvent B; t = 43min, 3% solvent B. Samples were ionized in positive ion mode with an online nanospray source (4500V) using fused-silica capillaries and a Distal Coated SilicaTip Emitter (New Objective) with an internal diameter of 20 μm (10 μm at the tip) and a length of 5cm. Solvent evaporation was performed at 220°C with a nitrogen flow of 3L/min. For the detection of glycopeptides, the MS ion detection window was set at m/z 500–1800, and the MS/MS detection window at m/z 140–2200, with automated selection of the three highest peaks in the spectrum for MS/MS analysis. The LC-MS/MS results were analyzed using DataAnalysis 4.0 software (Bruker Daltonics) and screened manually for the masses of common oxonium fragment ions (m/z 366.1, [1 hexose + 1 GlcNAc + H]+; m/z 657.2, [1 hexose + 1 GlcNAc + 1 N-acetylneuraminic acid + H]+; m/z 528.2, [2 hexoses + 1 GlcNAc + H]+), which are characteristic for fragmentation spectra of glycopeptides. Glycopeptide MS/MS spectra were further analyzed manually to derive the oligosaccharide structure and the mass of the peptide moiety.
Preparation of fibronectin-coated glass slides for single particle tracking experiments
Glass slides (24mm × 50mm #1.5 borosilicate, VWR) were immersed in a 1:1 mixture of concentrated sulfuric acid (Sigma) and 30% hydrogen peroxide (Sigma) for at least 30min, rinsed with deionized water, air-dried and glued with picodent twinsil extrahart (Picodent) to the bottom of 8-well LabTek chambers (Nunc). Slides were coated with 20μg/mL fibronectin (Sigma-Aldrich) in PBS for 1–2h at 37°C and rinsed with 1 X PBS.
Single particle tracking of HLA-I molecules on HAP1 WT and SPPL3−/− cells
0.2 × 106 HAP1 WT and SPPL3−/− cells were stained with a single molecule dilution of the BB7.2-Fab-AF555 on ice for 30min and washed twice in imaging buffer (HBSS, GIBCO, 1% FCS, 2μM MgCl2 and CaCl2). Cells were kept on ice or seeded onto fibronectin-coated glass slides for imaging at room temperature (23–27°C) and in TIRF mode. AF555 was excited with a 532 nm laser (Obis) with a power density of 0.8kW/cm2 and the emission channel was cleaned up with a TRITC filter (605/52) installed within the fast emission filter wheel (Sutter Instrument Company). We recorded single HLA-I (BB7.2 Fab-AF555) trajectories over 500 frames with an illumination time of 16ms and a total time lag of 20.6ms between two adjacent images (100 X 100 pixel ROI size). Microscopy images were processed and analyzed with the open-source image processing package Fiji (Schindelin et al., 2012). XY localization, intensity and positional accuracy of single fluorescence emitters was calculated with the Fiji plugin ThunderSTORM (Ovesný et al., 2014). After determining the localization of every fluorophore in the image stack we combined these localizations to trajectories based on a published approach (Gao and Kilfoil, 2009) with custom-made algorithms written in MATLAB (MathWorks). We calculated the mean square displacement (MSD) describing the average of the square displacements between two points of the trajectory according to MSD(tlag) = < (r.(t + tlag)-r(t))2 >. The first three MSD values as a function of time lag (tlag) were used to calculate the diffusion coefficient (D) for each trajectory according by fitting MSD = 4·D·tlag+·σxy2, with σxy representing the localization precision (Wieser et al., 2007). Multiple fractions (i.e., a fast and a slow moving fraction of molecules) were discriminated by analyzing the step-size distributions of square displacements for several time lags (Schütz et al., 1997). By assuming free Brownian motion of one mobile fraction, the cumulative probability for finding a square displacement smaller than r2 is given by P = 1 − exp((− r2 /4Dtlag)); two different fractions α and (1-α) with diffusion coefficients D1 (e.g., fast) and D2 (e.g., slow/immobile) can be distinguished by fitting the bi-exponential function P = 1 − α·exp((− r2 /4D1tlag)) − (1 − α)·exp((− r2 /4D2tlag)). We calculated the fraction of mobile and slow/immobile molecules, the diffusion rate of mobile molecules and the diffusion rate of slow/immobile molecules of all HLA-I trajectories present on a single cell (Brameshuber et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
All error bars correspond to the standard deviation of the mean. For all figures, the number of independent experiments (n) are described in the legend. For quantifications of flow cytometry data we plotted data from median fluorescent intensities (fusion proteins, no normal distribution) or mean fluorescent intensities (antibody and CTB stainings) as clarified in the legends. Data from genome-wide screens were analyzed using two-sided Fisher’s exact test followed by FDR (Benjamini-Hochberg) correction of the p value. Other statistical evaluations were done by a Student’s t test (analysis of two data groups), one-way ANOVA (three groups or more), two-way ANOVA (two variables), Mann-Whitney U test (non-parametric analyses) or Log-rank (survival analyses) with Prism-Graphpad software (http://www.graphpad.com). TCGA survival and expression data were retrieved from OncoLnc.org (Anaya, 2016). We subdivided patients with the 50% lowest SPPL3 or B3GNT5 expressing tumors versus the 50% highest expressing tumors. *p < 0.05, **p < 0.01, ***p < 0.001 and ns (= not significant). EC50 values of titrations were calculated using non-linear four parameter fit modeling with Prism software.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-HLA-ABC-PerCP-eFluor710 (W6/32) | Thermo Fisher Scientific | Cat# 46-9983-42; RRID:AB_10804486 |
| Mouse monoclonal anti-HLA-BC- APC (B1.23.2) | Thermo Fisher Scientific | Cat# 17-5935-42; RRID:AB_11151509 |
| Mouse monoclonal anti-HLA-A2- APC (BB7.2) | Thermo Fisher Scientific | Cat# 17-9876-42; RRID:AB_11149299 |
| Mouse monoclonal anti-CD15-FITC (C3D-1) | Millipore | Cat# FCMAB182F; RRID:AB_11214339 |
| Goat anti-mouse IgG (H+L) - Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A28181; RRID:AB_2536165 |
| Mouse APC anti-human IgM (MHM-88) | Biolegend | Cat# 314510; RRID:AB_493011 |
| Mouse monoclonal anti-human IgG (MH161-1) | Sanquin home made | Hybridoma; RRID:AB_1286031 |
| Rabbit polyclonal anti-RFP | Netherlands Cancer Institute home made | Rocha et al., 2009 |
| Mouse monoclonal anti-HLA-ABC (W6/32) | Dr. J. Neefjes (NKI, the Netherlands) | Hybridoma; RRID:AB_964524 |
| Mouse monoclonal anti-HLA-BC (HC10) | Dr. J. Neefjes (NKI, the Netherlands) | Hybridoma; RRID:AB_2728622 |
| Mouse monoclonal anti-HLA-A (HCA2) | Dr. J. Neefjes (NKI, the Netherlands) | Hybridoma |
| Mouse monoclonal anti-HLA-A2 (BB7.2) | Dr. M. Heemskerk(LUMC, the Netherlands) | Hybridoma; RRID:AB_627935 |
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Mouse monoclonal anti-b-actin (AC-15) | Sigma-Aldrich | Cat# A5441; RRID:AB_476744 |
| Mouse monoclonal anti-NGFR(CD271)-PE/Cy7 (ME20.4) | Biolegend | Cat# 345109; RRID:AB_11204073 |
| Goat anti-rabbit IgG (H+L)-HRP | Thermo Fisher Scientific | Cat# G-21234; RRID:AB_2536530 |
| Mouse monoclonal anti-FLAG M2-HRP antibody | Sigma-Aldrich | Cat# A8592; RRID:AB_439702 |
| APC AffiniPure F(ab’)2 Fragment Goat Anti-Human IgG, Fcg fragment specific | Jackson ImmunoResearch | Cat# 109-136-098; RRID:AB_2337693 |
| HLA-I/B2M specific antibodies, see Table S3 | N/A | |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| LIR-1 Fc fusion protein (human IgG1) | Dr. O. Mandelboim (Hebrew University Hadassah Medical School, Israel) | Gonen-Gross et al., 2G1G |
| Recombinant human KIR2DL1/CD158a Fc chimera protein | R&D systems | Cat# 1844-KR |
| Recombinant human KIR2DL2/CD158b1 Fc chimera protein | R&D systems | Cat# 3G15-KR |
| Alexa Fluor® 555 NHS Ester | Thermo Fisher Scientific | Cat# A2GGG9 |
| Alexa Fluor® 647 NHS Ester | Thermo Fisher Scientific | Cat# A37573 |
| CFSE | Invitrogen | Cat# C1157 |
| Alexa Fluor 350 NHS Ester | Thermo Fisher Scientific | Cat# A1G168 |
| Violet proliferation dye 450 | BD Horizon | Cat# 562158 |
| DyLight 650 NHS ester | Thermo Fisher Scientific | Cat# 62265 |
| Cholera Toxin B Subunit from Vibrio cholerae-FITC | Sigma | Cat# C1655; CAS: 131G96-89-4 |
| Animal-Free Recombinant Human IFN-g | Peprotech | Cat# AF-3GG-G2 |
| Sialyltransferase inhibitor (3Fax-peracetyl Neu5Ac) | Sigma | Cat# 566224; CAS: 1174G5-58-G |
| Fucosyltransferase inhibitor (2-Deoxy-2- fluoro-L-fucose) | Carbosynth | MD06089; CAS: 70763-62-1 |
| Neuraminidase from Closteridium perfringens | Sigma-Aldrich | N2876; CAS: 9001-67-6 |
| Endoglycoceramidase I | New England Biolabs | Cat# P0773S |
| Miglustat | Dr. H. Overkleeft (Leiden University, the Netherlands) | Ghisaidoobe et al., 2014 |
| Eliglustat | Bio-Connect | Cat# HY-14885A |
| MZ21 | Dr. H. Overkleeft (Leiden University, the Netherlands) | Ghisaidoobe et al., 2014 |
| MZ31 | Dr. H. Overkleeft (Leiden University, the Netherlands) | Ghisaidoobe et al., 2014 |
| Kifunensine (mannosidase inhibitor) | Santa Cruz | Cat# sc-201364; CAS: 109944-15-2 |
| Swainsonine | Sigma-Aldrich | Cat# S8195; CAS: 72741-87-8 |
| Brefeldin A | Sigma-Aldrich | Cat#B7651; CAS: 20350-15-6 |
| BODIPY FL C5-Lactosylceramide complexed to BSA | Thermo Fisher Scientific | Cat# B34402 |
| UDP-N-acetyl-D-glucosamine disodium salt | Santa Cruz | Cat# sc-286851 |
| Endoglycoceramidase I (derived from Rhodococcus triatomea and expressed in Escherichia coli) | New England Biolabs | Cat# P0773S |
| Endoglycosidase H from Streptomyces plicatus | Sigma-Aldrich | Cat# A0810; CAS: 37278-88-9 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| IFN-γ ELISA Kit | Sanquin | Cat# M9333 |
| GMCSF-ELISA Kit | Biolegend | Cat# 432002 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw sequence data | NCBI Sequence Read Archive | PRJNA665349 (SAMN16252402) |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human: HAP1 | Dr. T. Brummelkamp (NKI, the Netherlands) | |
| Human: MelJuSo | Dr. J. Neefjes (LUMC, the Netherlands) | Authenticated: 19 ZE 000486 (2019) |
| Human: U-251 | Laboratory of Dr. H. Versteeg (LUMC, the Netherlands) | N/A |
| Human: SW620 | Dr. T. de Gruijl (Amsterdam UMC, the Netherlands) | N/A |
| Human: HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Human: Phoenix Ampho | Dr. J. Neefjes (LUMC, the Netherlands) | N/A |
| Murine: BB7.2 cell line (hybridoma) | Dr. M. Heemskerk(LUMC, the Netherlands) | N/A |
| Murine: W6/32 cell line (hybridoma) | Dr. J. Neefjes (LUMC, the Netherlands) | N/A |
| Human: CD8+ T cell clone reactive to USP11 peptide | Dr. M. Heemskerk(LUMC, the Netherlands) | Amir et al., 2011 |
| Human: CD8+ T cell clone reactive to FDPS peptide | Dr. M. Heemskerk(LUMC, the Netherlands) | Amir et al., 2011 |
| Human: CD8+ T cell clone reactive to VPS13B peptide | Dr. M. Heemskerk(LUMC, the Netherlands) | Amir et al., 2011 |
| Human: CD8+ T cell clone reactive to SSR1 peptide | Dr. M. Griffioen (LUMC, the Netherlands) | Van Bergen et al., 2010 |
| Human: CD8+ T cell clone reactive to ADIR peptide | Dr. M. Griffioen (LUMC, the Netherlands) | van Bergen et al., 2007 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siGENOME Human SPPL3 siRNA | Dharmacon | D-006042-02,03,04 |
| siGENOME Human B2M siRNA (SMARTpool) | Dharmacon | M-004366-00 |
| siGENOME Non-Targeting siRNA Pool#1 | Dharmacon | D-001206-13 |
| gRNA sequences used for CRISPR/Cas9 mediated genome editing, see Table S4 | This paper | N/A |
| PCR and sequencing primers for KO validation, see Table S5. | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pX330 | Addgene | Plasmid #42230 |
| TIA-2A-blast | Dr. T. Brummelkamp (NKI, the Netherlands) | Blomen et al., 2015 |
| LentiCRISPR_v2 | Addgene | Plasmid #52961 |
| pL-CRISPR.EFS.GFP | Addgene | Plasmid #57818 |
| pMXs-puro retroviral expression vector | Cell Biolabs | Cat# RTV-012 |
| pMXs-puro-GFP | This paper | N/A |
| pMXs-puro-GFP-SPPL3 | This paper | Voss et al., 2012 |
| pMXs-puro-GFP-SPPL3DA | This paper | Voss et al., 2012 |
| pMXs-puro-FLAG-N | This paper | N/A |
| pMXs-puro-FLAG-C | This paper | N/A |
| pMXs-puro-FLAG-B3GNT5 | This paper | IMAGE:202800754 |
| pMXs-puro-B3GNT5-FLAG | This paper | IMAGE:202800754 |
| pMXs-puro-RFP | This paper | N/A |
| pMXs-puro-RFP-SPPL3 | This paper | Voss et al., 2012 |
| pMXs-puro-RFP-SPPL3DA | This paper | Voss et al., 2012 |
| pMXs-puro-FLAG-B4GALNT1 | This paper | IMAGE:202800771 |
| pMXs-puro-FLAG-ST3GAL5 | This paper | IMAGE:202759803 |
| puc2CL6IN-HLA-C*05:01 (with mutated signal peptide from HLA-A*02:01 [M4V]) | Dr. A. Halenius (University Medical Center Freiburg) | N/A |
| pLZRS-HLA-A*02:01-IRES-ΔNGFR | Dr. M. Griffioen (LUMC, the Netherlands) | (Griffioen et al., 2012; Van Bergen et al., 2010) |
| pLZRS-HLA-B*40:01-IRES-ΔNGFR | Dr. M. Griffioen (LUMC, the Netherlands) | (Griffioen et al., 2012; Van Bergen et al., 2010) |
| pLZRS-HLA-C*03:03-IRES-ΔNGFR | Dr. M. Griffioen (LUMC, the Netherlands) | (Griffioen et al., 2012; Van Bergen et al., 2010) |
|
| ||
| Software and Algorithms | ||
|
| ||
| Phyre2 | http://www.sbg.bio.ic.ac.uk/ | Kelley et al., 2015 |
| PyMOL v2.0 | http://pymol.org | Schrödinger, LCC |
| Compass Data Analysis v4.0 and v5.0 software | http://www.bruker.com | Bruker Daltonics |
| FlowJo Single Cell Analysis v10 software | http://www.flowjo.com | FlowJo, LLC |
| Fiji - plugin ThunderSTORM | Ovesny et al., 2014 | |
| Glycoworkbench | Ceroni et al., 2008 | |
| Glycomod | Cooper et al., 2001 | |
| TCGA survival and expression data | http://oncoLnc.org | Anaya, 2016 |
| Graphpad Prism | http://www.graphpad.com | N/A |
|
| ||
| Other | ||
|
| ||
| RFP-Trap_A antibody | ChromoTek | Cat# rta-20, RRID:AB_2631362 |
| Acclaim PepMap100 C18 column, 100 μm × 2cm, C18 particle size 5 μm, pore size 100υ. | Dionex/Thermo Fisher Scientific | Cat# 164199 |
| Acclaim PepMap RSLC nanocolumn, 75 μm × 15cm, C18 particle size 2 μm, pore size 100Å | Dionex/Thermo Fisher Scientific | Cat# TF164S34 |
| 51Cr, sodium chromate in normal saline (pH 8-10) | Perkin-Elmer | Cat# NEZG3G |
| Hypercarb KAPPA Column, 30 × 0.32 mm, particle size 5 μm, pore size 250Å. | Thermo Scientific | Cat# G31IG4SG |
| Hypercarb KAPPA Column, 100 mm × 75 μm, particle size 3 μm, pore size 250Å. | Thermo Scientific | Cat# G31IG4SG |
Highlights.
Iterative KO screens reveal a new pathway controlling HLA-I antigen presentation
SPPL3 suppresses B3GNT5 activity affecting the cell surface GSL repertoire
B3GNT5-generated GSLs limit the capacity of HLA-I to interact with natural ligands
Inhibition of GSL synthesis in glioma enhances anti-tumor immune activation
ACKNOWLEDGMENTS
We thank Dr. R. Fluhrer (Ludwig Maximilians University, Germany) for providing SPPL3 plasmids; Dr. M. Griffioen (LUMC, the Netherlands) for providing T cell clones; Dr. O. Mandelboim (Hebrew University Hadassah Medical School, Israel) for providing LIR-1 Fc fusion protein; Dr. T. Rispens (Sanquin, the Netherlands) for expert advice for Fab generation and protein purification; E. Mul, S. Tol, and M. Hoogenboezem (Sanquin) for assistance with flow cytometry; Dr. D. Amsen (Sanquin) and Dr. I. Berlin (LUMC) for editorial help; Dr. Y. Rombouts (Université de Toulouse, France) for contributing to MTBE extraction; and C.A.M. Koeleman and A.L. Hipgrave Ederveen (LUMC) for technical support with LC-MS. This work was supported by the Dutch Research Council (NWO-VENI 016.131.047, NWO-VIDI 91719369, and ZonMw-ETH 435004024 to R.M.S. and NWO-Vici 016.Vici.170.033 to T.R.B.), KWF (Alpe d’HuZes Bas Mulder Award 2015-7982 to R.M.S. and NKI2015-7609 to T.R.B.) the Landsteiner Foundation for Blood Transfusion Research (LSBR fellowship 1842F to R.M.S.), EMBO (ASTF 10-2016 to M.L.M.J.), the Cancer Genomics Center (to T.R.B.), Ammodo (KNAW Award 2015 for Biomedical Sciences to T.R.B.), the Boehringer Ingelheim Fonds (predoctoral fellowship to R.Platzer), the Vienna Science and Technolgy Fund (WWTF LS14-031 to J.B.H.), DOD (W81XWMH-16-1-0500 to S.F.), and NIH (R01DE028172, R03CA216114, RO3CA223886, and RO3CA231766 to S.F.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2020.11.003.
DECLARATION OF INTERESTS
T.R.B. is a cofounder and SAB member of Haplogen GmbH and a cofounder and director of Scenic Biotech BV.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Achdout et al. (2008); de Groot et al. (2016); Desai et al. (2000); Doench et al. (2016); Duquesnoy et al. (2012); Duquesnoy et al. (2013); Hiby et al. (2010); Hogan and Brown (1992); Hsu et al. (2013); Ladasky et al. (1999); Marrari et al. (2010); Mulder et al. (2010); Taketani et al. (1983); Trymbulak and Zeff (1997).
REFERENCES
- Achdout H,Manaster I,andMandelboim O(2008).Influenza virus infection augments NK cell inhibition through reorganization of major histocompatibility complex class I proteins.J. Virol 82,8030–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende ML,andProia RL(2014).Simplifying complexity: genetically resculpting glycosphingolipid synthesis pathways in mice to reveal function.Glycoconj. J 31,613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir AL,van der Steen DM,Hagedoorn RS,Kester MG,van Bergen CA,Drijfhout JW,de Ru AH,Falkenburg JH,van Veelen PA,andHeemskerk MH(2011). Allo-HLA-reactive T cells inducing graft-versus-host disease are single peptide specific. Blood 118,6733–6742. [DOI] [PubMed] [Google Scholar]
- Anaya J(2016).OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci 2,e67. [Google Scholar]
- Andrews PW,Gönczöl E,Fenderson BA,Holmes EH,O’Malley G,Hakomori S,andPlotkin S(1989).Human cytomegalovirus induces stage-specific embryonic antigen 1 in differentiating human teratocarcinoma cells and fibroblasts. J. Exp. Med 169,1347–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfossi N,André P,Guia S,Falk CS,Roetynck S,Stewart CA,Breso V,Frassati C,Reviron D,Middleton D,et al. (2006).Human NK cell education by inhibitory receptors for MHC class I. Immunity 25,331–342. [DOI] [PubMed] [Google Scholar]
- Barbosa JA,Santos-Aguado J,Mentzer SJ,Strominger JL,Burakoff SJ,andBiro PA(1987).Site-directed mutagenesis of class I HLA genes. Role of glycosylation in surface expression and functional recognition. J. Exp. Med 166,1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM,Westbrook J,Feng Z,Gilliland G,Bhat TN,Weissig H,Shindyalov IN,andBourne PE(2000).The Protein Data Bank. Nucleic Acids Res. 28,235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blees A,Januliene D,Hofmann T,Koller N,Schmidt C,Trowitzsch S,Moeller A,andTampé R(2017).Structure of the human MHC-I peptide-loading complex. Nature 551,525–528. [DOI] [PubMed] [Google Scholar]
- Bligh EG,andDyer WJ(1959).A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37,911–917. [DOI] [PubMed] [Google Scholar]
- Blomen VA,Májek P,Jae LT,Bigenzahn JW,Nieuwenhuis J,Staring J,Sacco R,van Diemen FR,Olk N,Stukalov A,et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350,1092–1096. [DOI] [PubMed] [Google Scholar]
- Borges L,Hsu ML,Fanger N,Kubin M,andCosman D(1997).A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules.J. Immunol 159,5192–5196. [PubMed] [Google Scholar]
- Brameshuber M,Kellner F,Rossboth BK,Ta H,Alge K,Sevcsik E,Göhring J,Axmann M,Baumgart F,Gascoigne NRJ,et al. (2018).Monomeric TCRs drive T cell antigen recognition.Nat. Immunol 19,487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK,Chen T,Amendola M,andvan Steensel B(2014).Easy quantitative assessment of genome editing by sequence trace decomposition.Nucleic Acids Res.42,e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann M,Blomen VA,Nieuwenhuis J,Stickel E,Raaben M,Bleijerveld OB,Altelaar AFM,Jae LT,andBrummelkamp TR(2017).Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling.Nature 546,307–311. [DOI] [PubMed] [Google Scholar]
- Caffaro CE,andHirschberg CB(2006).Nucleotide sugar transporters of the Golgi apparatus: from basic science to diseases.Acc. Chem. Res 39,805–812. [DOI] [PubMed] [Google Scholar]
- Carette JE,Guimaraes CP,Varadarajan M,Park AS,Wuethrich I,Godarova A,Kotecki M,Cochran BH,Spooner E,Ploegh HL,andBrummelkamp TR(2009).Haploid genetic screens in human cells identify host factors used by pathogens.Science 326,1231–1235. [DOI] [PubMed] [Google Scholar]
- Catalaa I,Henry R,Dillon WP,Graves EE,McKnight TR,Lu Y,Vigneron DB,andNelson SJ(2006).Perfusion, diffusion and spectroscopy values in newly diagnosed cerebral gliomas.NMR Biomed.19,463–475. [DOI] [PubMed] [Google Scholar]
- Ceroni A,Maass K,Geyer H,Geyer R,Dell A,andHaslam SM(2008).Glyco-Workbench: a tool for the computer-assisted annotation of mass spectra of glycans.J. Proteome Res 7,1650–1659. [DOI] [PubMed] [Google Scholar]
- Chowell D,Morris LGT,Grigg CM,Weber JK,Samstein RM,Makarov V,Kuo F,Kendall SM,Requena D,Riaz N,et al. (2018).Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Navarro F, Bellón T, Llano M, García P, Samaridis J, Angman L, Cella M, and López-Botet M (1997). A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med 186, 1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras FX, Ernst AM, Haberkant P, Bjorkholm P, Lindahl E, Gonen B, Tischer C, Elofsson A, von Heijne G, Thiele C, et al. (2012). Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 481, 525–529. [DOI] [PubMed] [Google Scholar]
- Cooper CA, Gasteiger E, and Packer NH (2001). GlycoMod—a software tool for determining glycosylation compositions from mass spectrometric data. Proteomics 1, 340–349. [DOI] [PubMed] [Google Scholar]
- Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, and Hsu ML (1997). A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7, 273–282. [DOI] [PubMed] [Google Scholar]
- D’Angelo G,Capasso S,Sticco L,andRusso D(2013).Glycosphingolipids: synthesis and functions. FEBS J. 280, 6338–6353. [DOI] [PubMed] [Google Scholar]
- Davis EM, Kim J, Menasche BL, Sheppard J, Liu X, Tan AC, and Shen J (2015). Comparative Haploid Genetic Screens Reveal Divergent Pathways in the Biogenesis and Trafficking of GlycophosphatidylinositolAnchored Proteins. Cell Rep. 11, 1727–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot NG, Heijmans CM, van der Wiel MK, Blokhuis JH, Mulder A, Guethlein LA, Doxiadis GG, Claas FH, Parham P, and Bontrop RE (2016). Complex MHC Class I Gene Transcription Profiles and Their Functional Impact in Orangutans. J. Immunol 196, 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard AA, Verkerk T, Jongsma MLM, Hoefakker K, Sethumadhavan S, Gerke C, Bliss S, Janssen GMC, de Ru AH, Claas FHJ, et al. (2020). PAKC: A novel Panel of HLA class I Antigen presentation machinery Knockout Cells from the same genetic origin. Eur. J. Immunol 10.1002/eji.202048599. [DOI] [PMC free article] [PubMed]
- Dersh D, Phelan JD, Gumina ME, Wang B, Arbuckle JH, Holly J, Kishton RJ, Markowitz TE, Seedhorn MO, Fridlyand N, et al. (2020). Genome-wide Screens Identify Lineage- and TumorSpecific Genes Modulating MHC-I- and MHC-IIRestricted Immunosurveillance of Human Lymphomas. Immunity. 10.1016/j.immuni.2020.11.002. [DOI] [PMC free article] [PubMed]
- Desai SA, Wang X, Noronha EJ, Zhou Q, Rebmann V, Grosse-Wilde H, Moy FJ, Powers R, and Ferrone S (2000). Structural relatedness of distinct determinants recognized by monoclonal antibody TP25.99 on beta 2-microglobulin-associated and beta 2-microglobulin-free HLA class I heavy chains. J. Immunol 165, 3275–3283. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, and Costello CE (1988). Structure elucidation of glycosphingolipids and gangliosides using high-performance tandem mass spectrometry.Biochemistry 27, 1534–1543. [DOI] [PubMed] [Google Scholar]
- Duquesnoy RJ, Marrari M, Jelenik L, Zeevi A, Claas FH, and Mulder A (2013). Structural aspects of HLA class I epitopes reacting with human monoclonal antibodies in Ig-binding, C1q-binding and lymphocytotoxicity assays.Hum. Immunol 74, 1271–1279. [DOI] [PubMed] [Google Scholar]
- Duquesnoy RJ, Marrari M, Mulder A, Claas FH, Mostecki J, and Balazs I (2012). Structural aspects of human leukocyte antigen class I epitopes detected by human monoclonal antibodies. Hum. Immunol 73, 267–277. [DOI] [PubMed] [Google Scholar]
- Fantini J, Hammache D, Pieroni G, and Yahi N (2000). Role of glycosphingolipid microdomains in CD4-dependent HIV-1 fusion. Glycoconj. J 17, 199–204. [DOI] [PubMed] [Google Scholar]
- Fluhrer R, and Haass C (2007). Signal peptide peptidases and gamma-secretase: cousins of the same protease family? Neurodegener. Dis 4, 112–116. [DOI] [PubMed] [Google Scholar]
- Fröhlich F, Petit C, Kory N, Christiano R, Hannibal-Bach HK, Graham M, Liu X, Ejsing CS, Farese RV, and Walther TC (2015). The GARP complex is required for cellular sphingolipid homeostasis. eLife 4, e08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa J, Tsuda M, Okada K, Kimura T, Piao J, Tanaka S, and Shinohara Y (2015). Comprehensive Glycomics of a Multistep Human Brain Tumor Model Reveals Specific Glycosylation Patterns Related to Malignancy. PLoS ONE 10, e0128300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, Stuart DI, Bell JI, Jones EY, and Jakobsen BK (1997). Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature 387, 630–634. [DOI] [PubMed] [Google Scholar]
- Gao Y, and Kilfoil ML (2009). Accurate detection and complete tracking of large populations of features in three dimensions. Opt. Express 17, 4685–4704. [DOI] [PubMed] [Google Scholar]
- Garstka MA, Fish A, Celie PH, Joosten RP, Janssen GM, Berlin I, Hoppes R, Stadnik M, Janssen L, Ovaa H, et al. (2015). The first step of peptide selection in antigen presentation by MHC class I molecules. Proc. Natl. Acad. Sci. USA 112, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. (2017). Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 7, 1420–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisaidoobe AT, van den Berg RJ, Butt SS, Strijland A, DonkerKoopman WE, Scheij S, van den Nieuwendijk AM, Koomen GJ, van Loevezijn A, Leemhuis M, et al. (2014). Identification and development of biphenyl substituted iminosugars as improved dual glucosylceramide synthase/neutral glucosylceramidase inhibitors. J. Med. Chem 57, 9096–9104. [DOI] [PubMed] [Google Scholar]
- Gonen-Gross T, Goldman-Wohl D, Huppertz B, Lankry D, Greenfield C, Natanson-Yaron S, Hamani Y, Gilad R, Yagel S, and Mandelboim O (2010). Inhibitory NK receptor recognition of HLA-G: regulation by contact residues and by cell specific expression at the fetal-maternal interface. PLoS ONE 5, e8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen M, Honders MW, van der Meijden ED, van Luxemburg-Heijs SA, Lurvink EG, Kester MG, van Bergen CA, and Falkenburg JH (2012). Identification of 4 novel HLA-B*40:01 restricted minor histocompatibility antigens and their potential as targets for graft-versus-leukemia reactivity.Haematologica 97, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hähnel PS, Thaler S, Antunes E, Huber C, Theobald M, and Schuler M (2008). Targeting AKT signaling sensitizes cancer to cellular immunotherapy.Cancer Res. 68, 3899–3906. [DOI] [PubMed] [Google Scholar]
- Hakomori S (1984). Tumor-associated carbohydrate antigens. Annu. Rev. Immunol 2, 103–126. [DOI] [PubMed] [Google Scholar]
- Hansen TH, and Bouvier M (2009). MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol 9, 503–513. [DOI] [PubMed] [Google Scholar]
- Haworth KB, Leddon JL, Chen CY, Horwitz EM, Mackall CL, and Cripe TP (2015). Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr. Blood Cancer 62, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol 32, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiby SE, Apps R, Sharkey AM, Farrell LE, Gardner L, Mulder A, Claas FH, Walker JJ, Redman CW, Morgan L, et al. (2010). Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J. Clin. Invest 120, 4102–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan KT, and Brown SL (1992). Localization and characterization of serologic epitopes on HLA-A2. Hum. Immunol 33, 185–192. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, Karlsson NG, Kolarich D, and Packer NH (2012). Structural analysis of N- and O-glycans released from glycoproteins. Nat. Protoc 7, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Jongsma MLM, Guarda G, and Spaapen RM (2019). The regulatory network behind MHC class I expression. Mol. Immunol 113, 16–21. [DOI] [PubMed] [Google Scholar]
- Karlsson NG, Wilson NL, Wirth HJ, Dawes P, Joshi H, and Packer NH (2004). Negative ion graphitised carbon nano-liquid chromatography/mass spectrometry increases sensitivity for glycoprotein oligosaccharide analysis. Rapid Commun. Mass Spectrom 18, 2282–2292. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley DM, Kozarsky KF, Segal M, and Krieger M (1986). Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol 102, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn PH, Voss M, Haug-Kröper M, Schröder B, Schepers U, Bräse S, Haass C, Lichtenthaler SF, and Fluhrer R (2015). Secretome analysis identifies novel signal Peptide peptidase-like 3 (Sppl3) substrates and reveals a role of Sppl3 in multiple Golgi glycosylation pathways. Mol. Cell. Proteomics 14, 1584–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann RH (2003). Miglustat. Oxford GlycoSciences/Actelion. Curr. Opin. Investig. Drugs 4, 472–479. [PubMed] [Google Scholar]
- Ladasky JJ, Shum BP, Canavez F, Seuánez HN, and Parham P (1999). Residue 3 of beta2-microglobulin affects binding of class I MHC molecules by the W6/32 antibody. Immunogenetics 49, 312–320. [DOI] [PubMed] [Google Scholar]
- Li L, Muzahim Y, and Bouvier M (2012). Crystal structure of adenovirus E319K bound to HLA-A2 reveals mechanism for immunomodulation. Nat. Struct. Mol. Biol 19, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Hill RA, and Li YT (2013). Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res 117, 59–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logtenberg MEW, Jansen JHM, Raaben M, Toebes M, Franke K, Brandsma AM, Matlung HL, Fauster A, Gomez-Eerland R, Bakker NAM, et al. (2019). Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPa axis and a target for cancer immunotherapy. Nat. Med 25, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrari M, Mostecki J, Mulder A, Claas F, Balazs I, and Duquesnoy RJ (2010). Human monoclonal antibody reactivity with human leukocyte antigen class I epitopes defined by pairs of mismatched eplets and self-eplets.Transplantation 90, 1468–1472. [DOI] [PubMed] [Google Scholar]
- Merrill AH Jr., and Sullards MC (2017). Opinion article on lipidomics: Inherent challenges of lipidomic analysis of sphingolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 774–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Podraza H, Andersson C, and Karlsson KA (1993). New method for the isolation of polyglycosylceramides from human erythrocyte membranes.Biochim. Biophys. Acta 1168, 330–339. [DOI] [PubMed] [Google Scholar]
- Miller-Podraza H, Stenhagen G, Larsson T, Andersson C, and Karlsson KA (1997). Screening for the presence of polyglycosylceramides in various tissues: partial characterization of blood group-active complex glycosphingolipids of rabbit and dog small intestines. Glycoconj. J 14, 231–239. [DOI] [PubMed] [Google Scholar]
- Mitra AK, Célia H, Ren G, Luz JG, Wilson IA, and Teyton L (2004). Supine orientation of a murine MHC class I molecule on the membrane bilayer. Curr. Biol 14, 718–724. [DOI] [PubMed] [Google Scholar]
- Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, and Parham P (2008). Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J. Immunol 180, 3969–3979. [DOI] [PubMed] [Google Scholar]
- Moore ML, Chi MH, Goleniewska K, Durbin JE, and Peebles RS Jr. (2008). Differential regulation of GM1 and asialo-GM1 expression by T cells and natural killer (NK) cells in respiratory syncytial virus infection. Viral Immunol. 21, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder A, Kardol MJ, Arn JS, Eijsink C, Franke ME, Schreuder GM, Haasnoot GW, Doxiadis II, Sachs DH, Smith DM, and Claas FH (2010). Human monoclonal HLA antibodies reveal interspecies crossreactive swine MHC class I epitopes relevant for xenotransplantation. Mol. Immunol 47, 809–815. [DOI] [PubMed] [Google Scholar]
- Neefjes J, Jongsma ML, Paul P, and Bakke O (2011). Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol 11, 823–836. [DOI] [PubMed] [Google Scholar]
- Neefjes JJ,De Bruijn ML,Boog CJ,Nieland JD,Boes J,Melief CJ,andPloegh HL(1990).N-linked glycan modification on antigen-presenting cells restores an allospecific cytotoxic T cell response.J. Exp. Med 171,583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B,andHannun YA(2004).Biologically active sphingolipids in cancer pathogenesis and treatment.Nat. Rev. Cancer 4,604–616. [DOI] [PubMed] [Google Scholar]
- Oostvogels R,Lokhorst HM,Minnema MC,van Elk M,van den Oudenalder K,Spierings E,Mutis T,andSpaapen RM(2014).Identification of minor histocompatibility antigens based on the 1000 Genomes Project.Haematologica 99,1854–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesný M,Křížek P,Borkovec J,Svindrych Z,andHagen GM(2014).ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging.Bioinformatics 30,2389–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech MF,Fong LE,Villalta JE,Chan LJ,Kharbanda S,O’Brien JJ,McAllister FE,Firestone AJ,Jan CH,andSettleman J(2019).Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance.eLife 8,e47362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM,Neises GR,Dwek RA,andButters TD(1994).N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis.J. Biol. Chem 269,8362–8365. [PubMed] [Google Scholar]
- Plomp R,Hensbergen PJ,Rombouts Y,Zauner G,Dragan I,Koeleman CA,Deelder AM,andWuhrer M(2014).Site-specific N-glycosylation analysis of human immunoglobulin e.J. Proteome Res 13,536–546. [DOI] [PubMed] [Google Scholar]
- Potapenko IO,Lűders T,Russnes HG,Helland Å,Sørlie T,Kristensen VN,Nord S,Lingjærde OC,Børresen-Dale AL,andHaakensen VD (2015).Glycan-related gene expression signatures in breast cancer subtypes; relation to survival.Mol. Oncol 9,861–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purbhoo MA,Irvine DJ,Huppa JB,andDavis MM(2004).T cell killing does not require the formation of a stable mature immunological synapse.Nat. Immunol 5,524–530. [DOI] [PubMed] [Google Scholar]
- Radsak K,andWiegandt H(1984).Glycosphingolipid synthesis in human fibroblasts infected by cytomegalovirus.Virology 138,300–309. [DOI] [PubMed] [Google Scholar]
- Reiser JB,Legoux F,Gras S,Trudel E,Chouquet A,Léger A,Le Gorrec M,Machillot P,Bonneville M,Saulquin X,andHousset D (2014).Analysis of relationships between peptide/MHC structural features and naive T cell frequency in humans.J. Immunol 193,5816–5826. [DOI] [PubMed] [Google Scholar]
- Reits EA,Hodge JW,Herberts CA,Groothuis TA,Chakraborty M,Wansley EK,Camphausen K,Luiten RM,de Ru AH,Neijssen J,et al. (2006).Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy.J. Exp. Med 203,1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP,Marincola FM,Kawakami Y,Taubenberger J,Yannelli JR,andRosenberg SA(1996).Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy.J. Natl. Cancer Inst 88,100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigau M,Ostrouska S,Fulford TS,Johnson DN,Woods K,Ruan Z,McWilliam HEG,Hudson C,Tutuka C,Wheatley AK,et al. (2020).Butyrophilin 2A1 is essential for phosphoantigen reactivity by gd T cells.Science 367,eaay5516. [DOI] [PubMed] [Google Scholar]
- Righi V,Roda JM,Paz J,Mucci A,Tugnoli V,Rodriguez-Tarduchy G,Barrios L,Schenetti L,Cerdan S,andGarcía-Martín ML(2009).1H HRMAS and genomic analysis of human tumor biopsies discriminate between high and low grade astrocytomas.NMR Biomed.22,629–637. [DOI] [PubMed] [Google Scholar]
- Rocha N,Kujil C,van der Kant R,Janssen L,Houben D,Janssen H,Zwart W,andNeefjes J(2009).Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning.J Cell Biol 185,1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL,Reits E,andNeefjes J(2016).Present Yourself! By MHC Class I and MHC Class II Molecules.Trends Immunol.37,724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszkowski JJ,Yu DC,Rubinstein MP,McKee MD,Cole DJ,andNishimura MI(2003).CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell.J. Immunol 170,2582–2589. [DOI] [PubMed] [Google Scholar]
- Ryan SO,andCobb BA(2012).Roles for major histocompatibility complex glycosylation in immune function.Semin. Immunopathol 34,425–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saverino D, Fabbi M, Ghiotto F, Merlo A, Bruno S, Zarcone D, Tenca C, Tiso M, Santoro G, Anastasi G, et al. (2000). The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J. Immunol 165, 3742–3755. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. [DOI] [PubMed] [Google Scholar]
- Schütz GJ, Schindler H, and Schmidt T (1997). Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J 73, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Levental I, Mayor S, and Eggeling C (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayman JA (2010). ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease. Drugs Future 35, 613–620. [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Qi J, Iwamoto A, and Gao GF (2011). Plasticity of human CD8aa binding to peptide-HLA-A*2402. Mol. Immunol 48, 2198–2202. [DOI] [PubMed] [Google Scholar]
- Spaapen RM, Lokhorst HM, van den Oudenalder K, Otterud BE, Dolstra H, Leppert MF, Minnema MC, Bloem AC, and Mutis T (2008). Toward targeting B cell cancers with CD4+ CTLs: identification of a CD19-encoded minor histocompatibility antigen using a novel genome-wide analysis. J. Exp. Med 205, 2863–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, et al. (2017). A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci 18, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketani S, Krangel MS, Pious D, and Strominger JL (1983). Structural analysis of HLA-A2 antigen from immunoselected mutant 8.6.1: further definition of an HLA-A2-specific serological determinant. J. Immunol 131, 2935–2938. [PubMed] [Google Scholar]
- Thor Straten P, and Garrido F (2016). Targetless T cells in cancer immunotherapy. J. Immunother. Cancer 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trymbulak WP Jr., and Zeff RA (1997). Mutants of human beta2-microglobulin map an immunodominant epitope within the three-stranded beta-pleated sheet. Transplantation 64, 640–645. [DOI] [PubMed] [Google Scholar]
- Unanue ER, and Cerottini JC (1989). Antigen presentation. FASEB J. 3, 2496–2502. [DOI] [PubMed] [Google Scholar]
- Valiante NM, Uhrberg M, Shilling HG, Lienert-Weidenbach K, Arnett KL, D’Andrea A, Phillips JH, Lanier LL, and Parham P (1997). Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity 7, 739–751. [DOI] [PubMed] [Google Scholar]
- van Bergen CA, Kester MG, Jedema I, Heemskerk MH, van Luxemburg-Heijs SA, Kloosterboer FM, Marijt WA, de Ru AH, Schaafsma MR, Willemze R, et al. (2007). Multiple myeloma-reactive T cells recognize an activation-induced minor histocompatibility antigen encoded by the ATP-dependent interferon-responsive (ADIR) gene. Blood 109, 4089–4096. [DOI] [PubMed] [Google Scholar]
- Van Bergen CA, Rutten CE, Van Der Meijden ED, Van Luxemburg-Heijs SA, Lurvink EG, Houwing-Duistermaat JJ, Kester MG, Mulder A, Willemze R, Falkenburg JH, and Griffioen M (2010). High-throughput characterization of 10 new minor histocompatibility antigens by whole genome association scanning. Cancer Res. 70, 9073–9083. [DOI] [PubMed] [Google Scholar]
- Voss M, Fukumori A, Kuhn PH, Ku€nzel U, Klier B, Grammer G, HaugKroper M, Kremmer E, Lichtenthaler SF, Steiner H, et al. (2012). Foamy virus envelope protein is a substrate for signal peptide peptidase-like 3 (SPPL3). J. Biol. Chem 287, 43401–43409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M, Ku€nzel U, Higel F, Kuhn PH, Colombo A, Fukumori A, HaugKroper M, Klier B, Grammer G, Seidl A, et al. (2014). Shedding of glycanmodifying enzymes by signal peptide peptidase-like 3 (SPPL3) regulates cellular N-glycosylation. EMBO J. 33, 2890–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M, Schroder B, and Fluhrer R (2013). Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases.Biochim. Biophys. Acta 1828, 2828–2839. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J, Wang Y, Liu Z, Liu H, Wu D, et al. (2012). High expression of lactotriaosylceramide, a differentiation-associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology 22, 930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearsch PA, and Cresswell P (2008). The quality control of MHC class I peptide loading. Curr. Opin. Cell Biol 20, 624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser S, Moertelmaier M, Fuertbauer E, Stockinger H, and Schutz GJ€. (2007). (Un)confined diffusion of CD59 in the plasma membrane determined by high-resolution single molecule microscopy. Biophys. J 92, 3719–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrand CJ, He XM, Fuller GN, Bigner SH, Fredman P, Svennerholm L, and Bigner DD (1991). Occurrence of lacto series gangliosides 3′-isoLM1 and 3′,6′-isoLD1 in human gliomas in vitro and in vivo. J. Neuropathol. Exp. Neurol 50, 756–769. [DOI] [PubMed] [Google Scholar]
- Willcox BE, Thomas LM, and Bjorkman PJ (2003). Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat. Immunol 4, 913–919. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE, and Imrie J (2009). New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. Ther. Clin. Risk Manag 5, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, and Hill AB (2002). Viral interference with antigen presentation. Nat. Immunol 3, 1019–1025. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, HuLieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, de Waard AA, Wuhrer M, and Spaapen RM (2019). The Role of Glycosphingolipids in Immune Cell Functions. Front. Immunol 10, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Chen Y, Luo B, Yuan Y, Huang F, Yang T, Yu F, Liu J, Liu B, et al. (2020). The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. bioRxiv. 10.1101/2020.05.24.111823. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study, except for the processed screen results, which are accessible in an interactive database (https://phenosaurus.nki.nl/). The accession number for the raw sequence data of the screens reported in this paper is NCBI Sequence Read Archive: PRJNA665349. The bioSample accession number for these data in NCBI Sequence Read Archive: SAMN16252402.