

Abstract
Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer comprised of cells that lack expression of targetable biomarkers. Nucleic acid aptamers are a group of molecular ligands that can specifically bind to their targets with high affinity. The ssDNA aptamer PDGC21-T recognizes poorly differentiated cancer cells and tumor tissues through an unidentified cell surface target(s). Because TNBC tumor cells are poorly differentiated, the aptamer PDGC21-T is a promising therapeutic candidate to target TNBC tumor cells. In vitro study revealed that synthetic aptamer probes selectively targeted TNBC cell lines. To assess aptamer immunotherapeutic targeting capability, we generated aptamer-engineered NK cells (ApEn-NK) using aptamer probes as a targeting ligand and NK cells as a therapeutic agent. Cell clustering formation assays revealed that ApEn-NK bound both suspended and adherent TNBC cells with high affinity. In a functional study, ApEn-NK treatment triggered apoptosis and death of cultured TNBC cells. Finally, systemic administration of ApEn-NK in mice harboring TNBC xenografts resulted in significant inhibition of lung metastasis relative to parental NK cell treatments. Unlike chemotherapy, ApEn-NK treatment did not affect body weight in treated mice. We demonstrate a novel approach for targeted TNBC immunotherapy.
Keywords: Aptamer, natural killer cell, triple-negative breast cancer (TNBC), targeted therapy
Introduction
Breast cancer is the second leading cause of death in women and the most commonly diagnosed cancer worldwide. With an estimated 2.3 million new cases in 2020, breast cancer accounted for 11.7% of all new cancer cases[1]. Triple-negative breast cancer (TNBC) cells lack expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)[2]. TNBC accounts for 10–20% of all breast cancer cases and is considered the most aggressive breast cancer subtype, with a mortality rate of approximately 40% within the first five years after diagnosis[3, 4]. As TNBC cells lack the expression of known druggable cell-surface targets, conventional chemotherapy is the only established therapeutic option for TNBC patients to improve disease outcomes[5]. Despite encouraging results of emerging therapies such as immune check-point inhibition, there continues to be a reliance on systemic chemotherapy as the clinical state of the art. Due to the limited efficacy and high rates of side effects associated with the current standard of care, there is a need for new treatment options for TNBC[6].
Nucleic acid aptamers are short single-stranded (ss) oligonucleotides with high affinity and specificity for a broad range of target molecules[7–9]. Aptamers are artificially selected ligands that can be easily synthesized and modified[10]. Their advantages include rapid and inexpensive synthesis, low immunogenicity, and efficient delivery to a variety of cells[11, 12]. Aptamers are often referred to as “chemical antibodies” but compared to conventional antibodies, they are artificially synthesized and screened using an in vitro selection method called systemic evolution of ligands by exponential enrichment (SELEX)[13, 14]. Thus, aptamers can be easily modified and may be useful for targeted diagnostic and therapeutic applications in cancer treatment[14]. Importantly, by altering target cell selection and off-target cell elimination, aptamers may identify subtle differences among different cell types and thereby identify unique biomarkers on cells of interest[15–17]. A recent study used SELEX to select an ssDNA aptamer (PDGC21-T) that targeted poorly differentiated gastric cancer tissue, though the identity of the molecule it targets on tumor cells remains unknown[18]. Aptamer PDGC21-T also recognized poorly differentiated colorectal cancer cells[18]. Notably, TNBC cells are poorly differentiated without targetable biomarker expression[6, 19, 20].
In this study, we assessed the capacity of aptamer PDGC21-T to target TNBC tumor cells and then generated aptamer-engineered NK cells (ApEn-NK) for targeted TNBC immunotherapy. In vitro study revealed that ApEn-NK selectively bound TNBC cells and induced apoptosis/death of targeted cells. Further, systemic administration of ApEn-NK in mice harboring TNBC xenografts resulted in significant inhibition of lung metastasis.
Materials and methods
Cell lines and cell culture
Twelve breast cancer cell lines (T-47D, MCF-7, MDA-MB-468, MDA-MB-436, HCC70, HCC1937, MDA-MB-157, SUM159PT, BT-549, Hs 578T, MDA-MB-231, and HCC38), one cervical cancer cell line (HeLa), one prostate cancer cell line (VCaP), and a mouse embryonic fibroblast cell line (NIH/3T3) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) medium (Corning Inc., Corning, NY, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Corning Inc., Corning, NY, USA) and 100 units/mL penicillin-streptomycin (Corning Inc., Corning, NY, USA). Other cell lines included U-937 (Histiocytic lymphoma), Jurkat (acute T cell leukemia), L-428, HDLM-2, and KM-H2 (Hodgkin lymphoma), CA46 (Burkitt lymphoma), Karpas 299 (T-cell lymphoma), Maver-1, Jeko-1, and Mino (mantle cell lymphoma), SUDHL-1 (diffuse histiocytic lymphoma), AsPC-1 (pancreatic cancer), LNCaP (prostate cancer), and NCI-H1666 (lung cancer). Cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Corning Inc., Corning, NY, USA) containing 10% (v/v) heat-inactivated FBS (Corning Inc., Corning, NY, USA) and 100 units/mL penicillin-streptomycin (Corning Inc., Corning, NY, USA). NK-92 cells (natural killer cell) were purchased from ATCC and maintained in Alpha Minimum Essential Medium (α-MEM) (Gibco, Waltham, MA, USA) supplemented with 12.5% FBS (Corning Inc., Corning, NY, USA), 12.5% horse serum (ATCC, Manassas, VA, USA), 0.2 mM inositol (Sigma-Aldrich, St. Louis, MO, USA), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich), 0.02 mM folic acid (Sigma-Aldrich), 200 units/mL recombinant interleukin-2 (IL-2) (Peprotech, Rocky Hill, NJ, USA), and 100 units/mL penicillin-streptomycin (Corning Inc., Corning, NY, USA). MDA-MB-231-luciferase cells with stable luciferase expression were generated by transducing the luciferase expression lentiviral packaging particles LVP009 (Amsbio LLC, Cambridge, MA, USA) into the parental MDA-MB-231 cells. The luciferase-expressing stable cell line was established after blasticidin selection following the protocols provided by the company. Cells under exponential growth conditions were used for experiments. All cells were kept in a humidified incubator at 37°C and 5% CO2.
Cell-binding assay and flow cytometry
ssDNA aptamer PDGC21-T was synthesized based on the reported sequence: ACACCAAAATCGTCCGTTTCGTTTTAGTCCGTCTCTTTAGGGTGT[18]. For tracking purposes, aptamers were labeled with fluorochrome Cy3 at the 5′ end (IDT, Integrated DNA Technologies, Coralville, IA, USA). The cell-binding assay was performed as reported previously[21]. Briefly, Cy3-labeled aptamers were diluted in binding buffer and heated at 95°C for 5 min and cooled on ice for 10 min before mixing with the tested cell lines. The aptamer-cell mixture was then incubated in the dark at RT for 30 min. The mixture was then washed twice with washing buffer and subjected to flow cytometry analysis with a BD LSR II cytometer (BD Biosciences, San Jose, CA, USA). Another set of harvested cells were treated with random ssDNA sequences of the same length and the tested aptamer probes were used as controls. Data were analyzed using FlowJo v10.0.7 software (FlowJo, Ashland, OR, USA). The mean fluorescence intensity (MFI) was calculated using data collected from the flow cytometry assay.
PDGC21-T ApEn-NK cell production
To achieve optimal aptamer-anchor efficiency for NK-92 cell engineering, the Cy3-labeled (5’ end) PDGC21-T aptamer was conjugated with double C18 hydrocarbon chains at the 3’ end, as reported previously[21]. The Cy3-PDGC21-T-2xC18 aptamer-anchor conjugates were synthesized and purified by IDT (Integrated DNA Technologies, Coralville, IA, USA). Conjugates were stored in nuclease-free water at −20°C until use. To produce the PDGC21-T aptamer-engineered NK (ApEn-NK) cells, human NK-92 cells (5 × 105) were harvested in DPBS (Corning Inc., Corning, NY, USA) and gently mixed with the Cy3-PDGC21-T-2xC18 conjugates (final concentration: 1 μM). The mixture was then transferred to a 37°C water bath for 30 min (the reaction tube was flicked every 5–10 min) to complete the anchoring reaction. Various conditions, including the reaction time, reaction temperature, and aptamer-anchor concentrations were tested before the optimal condition was set for the PDGC21-T ApEn-NK production process. After the reaction, the ApEn-NK cells were resuspended in DPBS for future use.
ApEn-NK cell characterization
To confirm the cell surface anchoring feature of the Cy3-PDGC21-T-2xC18 conjugates on ApEn-NK cells, a confocal microscope (FV3000; Olympus America Inc., Center Valley, PA, USA) was used to image ApEn-NK cells in either bright field, Cy3, or Hoechst channels to evaluate cell morphology, the presence and location of Cy3-conjugated DNA aptamer, and the presence and location of the cell nucleus, respectively. In an additional validation step, ApEn-NK cells were treated with 100 units/mL DNase I (Sigma-Aldrich, St. Louis, MO, USA) in DPBS at 37 °C for 10 min. ApEn-NK cells were then washed with DPBS twice before confocal imaging. Hoechst 33258 dye (1 mg/mL, Sigma-Aldrich, St. Louis, MO, USA) was used to stain cell nuclei. Finally, fluorescent signals derived from surface-anchored aptamer conjugates on ApEn-NK cells with/without DNase treatment were analyzed by flow cytometry. The longevity of the fluorescent cell surface signals was also analyzed by flow cytometry. For storage, ApEn-NK products were added into fresh culture medium containing 10% Fetal Bovine Serum (FBS) and kept at 37°C. To monitor stability, stored ApEn-NK was washed twice with DPBS and signals of cell surface-anchored aptamers were measured kinetically by flow cytometry up to 72 hr. The mean intensity of the fluorescent signals at each time point were quantified and analyzed.
Cell proliferation assay
The cell counting KIT-8 assay (CCK-8, WST-8[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2 H-tetrazolium, monosodium salt]) (APExBIO, Houston, TX, USA) was used to assess NK-92 cell proliferation under various conditions. Briefly, parental NK-92 cells (1 × 104 cells per well) were either engineered by the Cy3-PDGC21-T-2xC18 conjugates (under identical conditions to produce ApEn-NK cells), treated with equal doses of PDGC21-T aptamer, or not treated (controls). After treatment, cells were seeded in 96-well plates with complete NK culture medium and grown in the cell incubator for 24, 48, or 72 h. At each time point, 10 μL of CCK-8 solution was added to 100 μL cell suspension (1 × 104 cells per well) in each well and the mixture was incubated at 37°C for 2 h. The absorbance in each well was measured by a microplate reader at 450 nm (Molecular Devices, San Jose, CA, USA). Each experiment was performed in triplicate. Background readings from the media were subtracted from the well readings to standardize the results.
NK-cancer cell-binding assay
Cancer cells, including HCC1937, MDA-MB-231, or MCF-7 cells were seeded in 96-well plates (5 × 103 cells per well) for 24 h before use. PDGC21-T ApEn-NK cells were produced as earlier described. For tracking purposes, NK cells, including ApEn-NK cells and parental NK-92 cells, were pre-stained with 100 nM Calcein Red-Orange AM dye (Life Technologies, Carlsbad, CA, USA) before being mixed with cancer cells at defined NK to tumor cell ratios (ranging from 1:3 to 3:1). After incubating at 37°C for 1 h, cell mixtures were centrifuged at 1500 rpm for 5 min before removing unbound NK cells from wells. Cells were imaged in both bright field and fluorescent channels using the Incucyte S3 Live-Cell Imaging and Analysis System (Essen BioScience Inc., Ann Arbor, MI, USA). Cell numbers of the remaining NK cells and the targeted cancer cells to which they were bound, were automatically quantified by the analysis system. Cell binding capacity was calculated by formula: ApEn-NK that bound to tumor cells / total tumor cells in the same cultures × 100%. In control experiments, non-specific binding of parental NK-92 cells to tumor cells was calculated as baseline for normalization. Finally, increased tumor cell binding (folds) of ApEn-NK was determined by comparing to baseline of parental NK-92 cells in each group.
NK-cancer cell cluster quantification
This assay was performed as previously described[21]. Briefly, cancer cells (HCC1937, MDA-MB-231, or MCF-7) were pre-stained with 100 nM Calcein-AM dye (Life Technologies, Carlsbad, CA, USA) and NK cells were tracked using signals derived from either cell surface aptamer conjugates (ApEn-NK) or the Calcein Red-Orange AM dye (parental NK-92). For the cell-binding assay, equal amounts (1 × 105) of effector and target cells were mixed in 500 μL DMEM complete media. This mixture was incubated at RT for 30 min on a rotator (Thermo Fisher Scientific, Waltham, MA, USA) to mimic circulation and minimize non-specific cell clotting. Fluorescent imaging of cell clusters was performed using the microscope (Olympus America Inc., Center Valley, PA, USA). Cell clusters were quantified by mixing equal amounts of target cells (3 × 104) and variable numbers of either ApEn-NK or parental NK-92 cells to achieve different NK to tumor cell ratios (ranging from 1:3 to 3:1). Different groups of cell mixtures were then incubated in a 12-well plate with constant gentle shaking for 30 min before the formed NK-tumor cell clusters were counted under a fluorescent microscope. NK-tumor cell clusters were defined as a group of cells containing more than four cells and including both the effector (red) and target (green) cells.
NK-cancer cell killing assay
To assess the cytotoxicity of ApEn-NK cells in cancer cells, three breast cancer cell lines (HCC1937, MDA-MB-231, or MCF-7) were seeded in a 12-well plate at a density of 6 × 104 cells per well and incubated at 37°C overnight. Both the PDGC21-T ApEn-NK cells and the parental NK-92 cells were pre-stained with calcein red-orange AM dye (Life Technologies, Carlsbad, CA, USA). Variable numbers of either ApEn-NK cells or parental NK-92 cells were added to wells with target cancer cells to achieve different NK to tumor cell ratios (ranging from 1:3 to 3:1). After co-incubation for 1 h, unbound NK cells were removed with gently washing from the wells and the remaining cell mixtures were incubated in fresh culture medium at 37°C for an additional 24 h. Cells were then collected and stained with FITC Annexin V (BD Biosciences, San Jose, CA, USA) to highlight apoptotic cells and dye eFluor 450 (eBioscience Inc., San Diego, CA, USA) to mark dead cells for 30 min before flow cytometry analysis. Tumor cells were then gated using flow cytometry by separating them from ApEn-NK with fluorescent signal derived from surface-engineered aptamers. Subsequently, apoptotic and dead tumor cell subpopulations were gated and quantified using flow cytometry based on the cellular signals of FITC Annexin V and eFluor 450, respectively. Apoptotic and death rates of tumor cells were calculated as a percentage of total tumor cells (%).
To rule out cellular effect of PDGC21-T aptamer, the same set of cultured tumor cells were treated with aptamer or random sequences alone at a final concentration of 100 nM for 24 h. Resultant changes in apoptosis and death rates (%) of the treated cells were measured as described above.
Lung metastasis mouse model for breast cancer
The lung metastasis mouse model for breast cancer has been previously described[22–24]. For bioluminescent tracking, human TNBC MDA-MB-231 cells with stable luciferase expression were generated as earlier described. Five to six-week-old female NSG (NOD/SCID/IL2rγ−/−) mice were used in compliance with the Animal Care and Use Committee of Houston Methodist Research Institute guidelines. To induce lung metastasis, 3 × 105 luciferase-transduced human MDA-MB-231 cells (100 μl) were injected into the lateral tail vein of NSG mice. Within an hour of cell inoculation, cellular bioluminescent signals were visualized using the International Veterinary Information Service (IVIS) imaging system (PerkinElmer, MA, USA) to confirm successful settlement of the MDA-MB-231 luciferase-expressing cells in the lungs. Three days after cell inoculation, cellular bioluminescent signals were imaged using the IVIS imaging system (PerkinElmer, Waltham, MA, USA). Bioluminescent signals in the lung tissues of each mouse were quantified to facilitate mouse grouping. With a cutting threshold of 1 × 106 total flux (p/s) per mouse, 24 tumor-bearing mice were randomly divided into three groups (8 mice per group): control, NK-92-treated, and ApEn-NK-treated.
NK cell therapy in vivo
ApEn-NK cells were generated as earlier described. Following the anchoring reaction, ApEn-NK cells were washed once and resuspended in DPBS. Both the ApEn-NK and parental NK-92 cells were supplemented with IL-2 (1000 units per 100 μL cell suspension) before the in vivo treatment. The third week after cell inoculation, tumor-bearing mice were treated once a week for eight weeks. Mice were left untreated for two weeks before they reached the study endpoint. Each group of mice was intravenously administered DPBS as vehicle control (100 μL), NK-92 cells (NK-92-treated group, 5×105 parental NK-92 cells in 100 μL DPBS), or ApEn-NK cells (ApEn-NK-treated group, 5×105 ApEn-NK cells in 100 μL DPBS), respectively. The tumor signals of total flux values were acquired using bioluminescent imaging. Bioluminescent signals for each mouse were captured once a week and quantified for analysis. At the treatment endpoint, all mice were euthanized (one mouse in the control group died before the endpoint). Lungs containing tumor nodules were harvested for bioluminescent imaging and signal quantification. Average luminescence signals were plotted, and results presented as mean ± standard deviation (SD). All mice were handled according to protocols approved by the Institutional Animal Care and Use Committee of Houston Methodist Research Institute.
Tumor nodule quantification in lungs
At experimental endpoint, lungs were removed and fixed with 4% formalin (Thermo Fisher Scientific, Waltham, MA, USA). Metastatic tumor nodules were counted using a stereomicroscope (10X, Olympus, Tokyo, Japan). The number of non-calcified lung nodules present was recorded. All values were presented as mean ± SD. A two-tailed Student’s t-test was used to determine the statistical significance of the in vivo assay between the control and ApEn-NK-treated groups and the NK-92-treated and ApEn-NK-treated groups. P < 0.05 was considered to be statistically significant.
Histological examination of tumor and normal tissues
The lung tissues of xenograft mice were fixed overnight in 4% formalin and embedded with paraffin. Entire fixed lungs (five lobes per mouse) were examined under a 10X stereomicroscope (Nikon Inc., Melville, NY, USA) and visible lung tumor nodules were manually counted. Total tumor nodules of lung metastasis per mouse were calculated and statistically analyzed. In addition, tissue sections of the lung were prepared with hematoxylin and eosin (H&E) staining. Tumor cells of lung metastasis were then morphologically investigated under microscope to confirm TNBC cell origin.
To rule out potential side effect of treatments, major organs of the treated mice were collected at the experimental endpoint, including heart, liver, kidney, and spleen. Tissues were fixed overnight in 4% formalin and embedded with paraffin and prepared with H&E staining. Tissue sections were then examined under a microscope to determine pathology changes due to side toxicity of treatments.
Statistical analysis
Statistical significance was assessed using a two-tailed Student’s t-test for each in vitro assay. Each assay was performed in triplicate and representative results are presented in all figures. All values are presented as mean ± standard deviation (SD). For the in vivo experiments (n = 8 mice /group), tumor signals from lungs and whole body are shown as mean ± standard error of the mean (SEM). The nonparametric Mann–Whitney test was used to determine the statistical significance of the numbers of the tumor nodules between the control and ApEn-NK treated groups or between NK-92-treated and ApEn-NK-treated groups. P <0.05 was considered to be statistically significant in all assays.
Results
Aptamer PDGC21-T selectively binds to TNBC cells
To explore cell binding capacity, cultured TNBC cells were treated with Cy3-labeled aptamer PDGC21-T (Fig. S1) and the resultant cell binding was analyzed by flow cytometry. The PDGC21-T aptamer selectively bound 9 of 10 TNBC cell lines (MDA-MB-468, MDA-MB-436, HCC70, HCC1937, MDA-MB-157, SUM159PT, BT-549, Hs 578T, MDA-MB-231, and HCC38) (Fig. 1A). In contrast, the aptamer PDGC21-T did not react with T-47D and MCF-7 cells, luminal A (LA) breast cancer subtypes that express ER and PR but not HER2. Aptamer PDGC21-T binding to breast cancer cells is summarized in Table 1. Binding affinities exceeding two-folds of the mean background were classified as positive (+) binding and those exceeding five-folds of the mean background were classified as strongly positive (++) binding. To validate binding specificity, cell-binding assays were performed using different types of non-breast tumor cells, including lymphoma/leukemia cells (U-937, Jurkat, L-428, CA46, KM-H2, Karpas 299, Maver-1, SUDHL-1, HDLM-2, Mino, Jeko-1) (Fig. 1B) and solid tumor cells (HeLa, AsPC-1, LNCaP, VCaP, NCI-H1666, and NIH/3T3, a mouse embryonic fibroblast cell line. The PDGC21-T aptamer did not react with these non-breast tumor cells. Notably, aptamer PDGC21-T did not bind to NK-92 cells, a cultured NK cell line widely used for immunotherapy research[25–27]. Thus, aptamer PDGC21-T selectively binds to the majority of TNBC cells based on their poorly differentiated pathology, suggesting it may have utility as a targeted TNBC therapy.
Figure 1. Aptamer PDGC21-T binds most cultured TNBC cells.
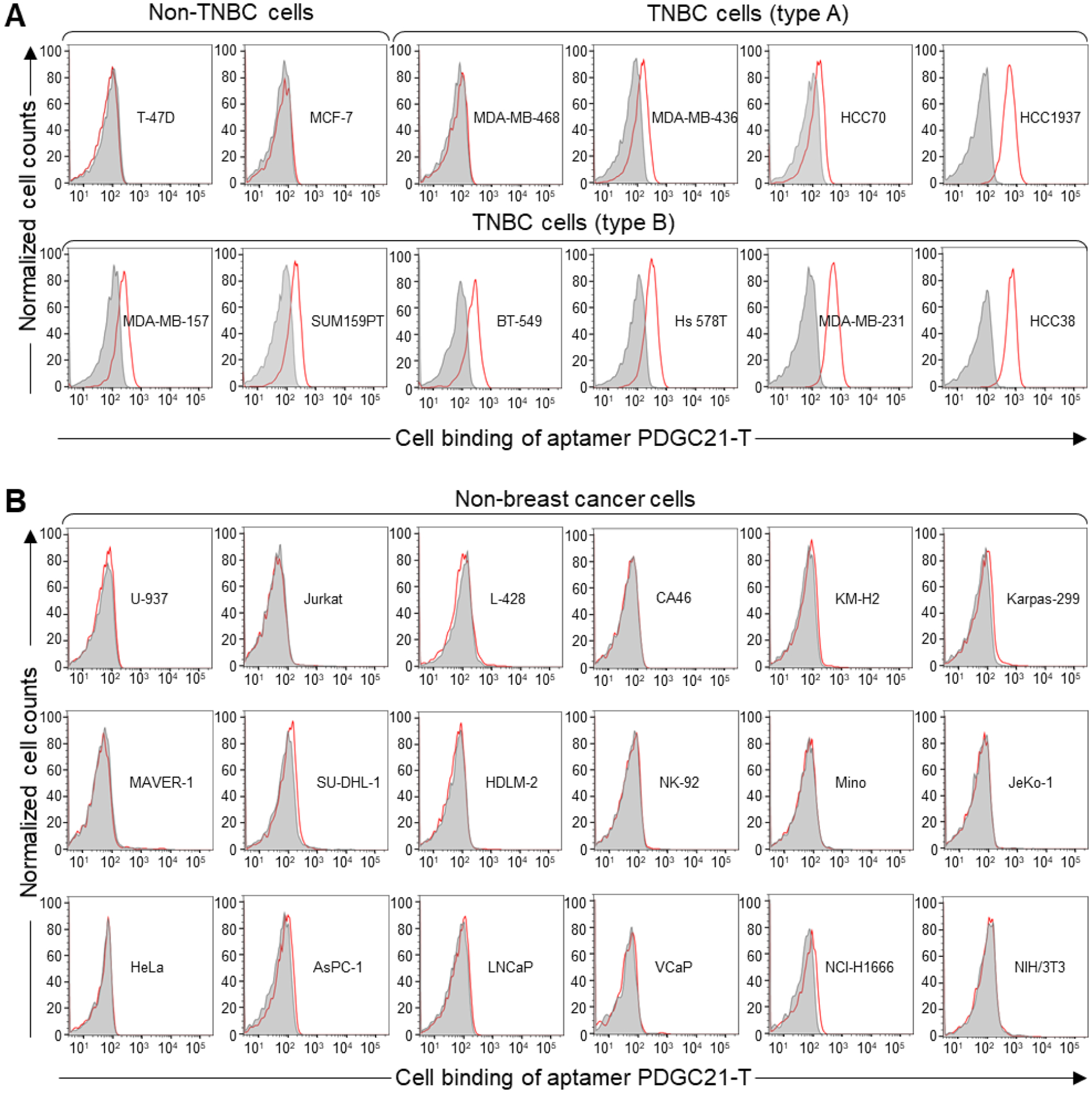
(A) Flow cytometry analysis of aptamer cell binding. The synthetic aptamer probe PDGC21-T bound 9 out of 10 TNBC cell lines but did not react with non-TNBC breast cancer cells. Note: T-47D and MCF-7 cell lines (non-TNBC) are ER+/PR+/Her2−; all TNBC cell lines are ER−/PR−/HER2−. (B) Aptamer PDGC21-T did not bind to lymphoma/leukemia or other types of solid tumor cells.
Table 1.
Binding affinity of PDGC21-T aptamers to breast cancer cell lines
| BC cell lines | T-47D | MCF-7 | MDA-MB-468 | MDA-MB-436 | HCC70 | HCC1937 | MDA-MB-157 | SUM159PT | BT-549 | Hs 578T | MDA-MB-231 | HCC38 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biomarker expression | ER+/PR+/HER2− | ER−/PR−/HER2− | ||||||||||
| Aptamer (100 nM) | − | − | − | + | + | +++ | + | + | + | + | ++ | +++ |
Note: −: < 2 folds; +: 2~5 folds; ++: 5~10 folds; +++: > 10 folds (in binding affinity)
Production and characterization of aptamer-engineered NK cells (ApEn-NK)
To utilize aptamer PDGC21-T for targeted TNBC immunotherapy, we combined NK cells as a therapeutic agent and synthetic aptamer as a ligand to produce ApEn-NK. Aptamer-anchor structures were synthesized by conjugating a Cy3-labeled aptamer sequence to a lipophilic anchor (2 × C18), as reported previously (Fig. 2A)[21]. Aptamer-anchor structures were then incubated with NK-92 cells to form ApEn-NKs (Fig. 2B). The lipophilic anchor portions of aptamer-anchor structures were inserted into NK cell membranes, leaving the hydrophilic aptamer portions on the cell surface. In a validation study, confocal microscopy confirmed that ApEn-NK products were morphologically intact and revealed a fluorescent signal of surface-engineered aptamers surrounding ApEn-NK nuclei (Fig. 2C). To optimize reaction conditions for ApEn-NK cell production, reaction temperature, reaction time, and aptamer-anchor structure concentration were varied. The optimal condition for ApEn-NK cell production was reached with 1 μM aptamer-anchor structures at 37°C for 30 min (Fig. S2A-B). This condition was then used to produce ApEn-NK cells in the following experiments. Next, to validate cell surface aptamer engineering, ApEn-NKs were exposed to DNase treatment to digest cell surface aptamer sequences. Confocal microscopy and flow cytometry confirmed that DNase treatment digested surface-engineered aptamers on ApEn-NKs (Fig. S3A-B). To evaluate the stability of the aptamers anchoring on ApEn-NK cells, cellular fluorescent signals were monitored. The flow cytometry assay demonstrated that the fluorescent signals derived from surface anchored aptamers were stable up to 10 h post-production (Fig. 2D). Additional time course study revealed that ApEn-NK products were stable in culture medium containing 10% FBS at 37°C, with remaining of cellular aptamer signal 67.5% at 24 h, 48.4% at 48 h, and 33.5% at 72 h post-production (Fig. S4). Finally, cell proliferation assays demonstrated that neither the presence of the aptamers in media, nor the embedded aptamer-anchor structures adversely affected NK-92 cell growth (Fig. 2E). Together, these data demonstrated the successful generation of ApEn-NK cells.
Figure 2. Production and characterization of aptamer-engineered NK cells (ApEn-NKs).
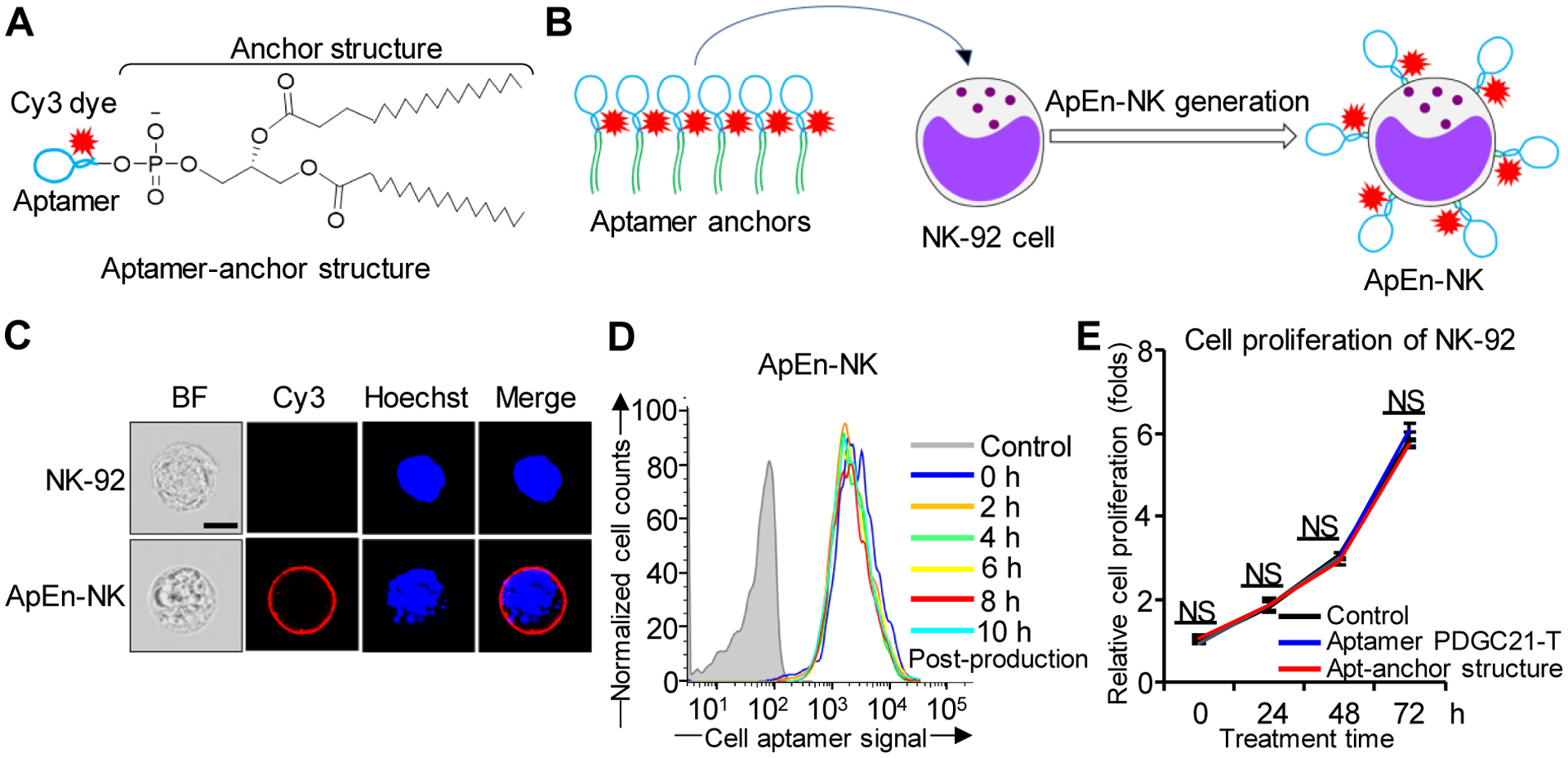
(A) Aptamer-anchor structures were formulated by conjugating the aptamer PDGC21-T sequence to two C18 anchor structures. (B) ApEn-NKs were produced by simple incubation of NK-92 cells with synthetic aptamer-anchor structures. (C) Confocal microscopy showed intact morphology of parental NK-92 cells (top) and fluorescent signal (red) derived from surface engineered aptamers on ApEn-NK (bottom). For tracking purposes, cell nuclei were stained with Hoechst dye (blue). Scale bar: 10 μm. (D) Stability assay of ApEn-NK products. No changes in fluorescent signals of surface-engineered aptamers on ApEn-NK were detected by flow cytometry analysis 10 h post-production. (E) Cell proliferation assay. The presence of aptamer alone or the aptamer-anchor structure to form ApEn-NK had no effect on NK-92 cell proliferation 72 h post incubation. NS: not significant.
ApEn-NK selectively binds to TNBC cells
Circulating TNBC tumor cells play a critical role in cancer metastasis, a hallmark of TNBC tumor progression[28–32]. To determine the specific binding of ApEn-NK cells to circulating TNBC cells, we co-incubated parental NK cells and ApEn-NK cells with target cancer cell lines in suspension at ratios ranging from 1:3 to 3:1. A rotating incubator was used to keep the cells in suspension throughout testing to mimic blood circulation and minimize non-specific cell clotting. Cell clustering analysis under fluorescent microscopy was used to quantify the effect of aptamer-anchoring on NK-92 cell binding capacity. The tested tumor cells and parental NK-92 cells in control experiments were pre-stained with a green or red fluorescent dye, respectively. ApEn-NKs were tracked using Cy3 signals derived from aptamer-anchor structures. Cell binding was assessed using three different NK to tumor cell ratios (1:3, 1:1, and 3:1) and fluorescent microscopy. Compared to parental NK-92 cell baseline binding, ApEn-NKs bound more TNBC cells (HCC1937 and MDA-MB-231) and formed more cell clusters than that observed in cell mixtures of non-TNBC cells (MCF-7) under the same conditions (Fig. 3).
Figure 3. ApEn-NK specifically target suspended TNBC cells.
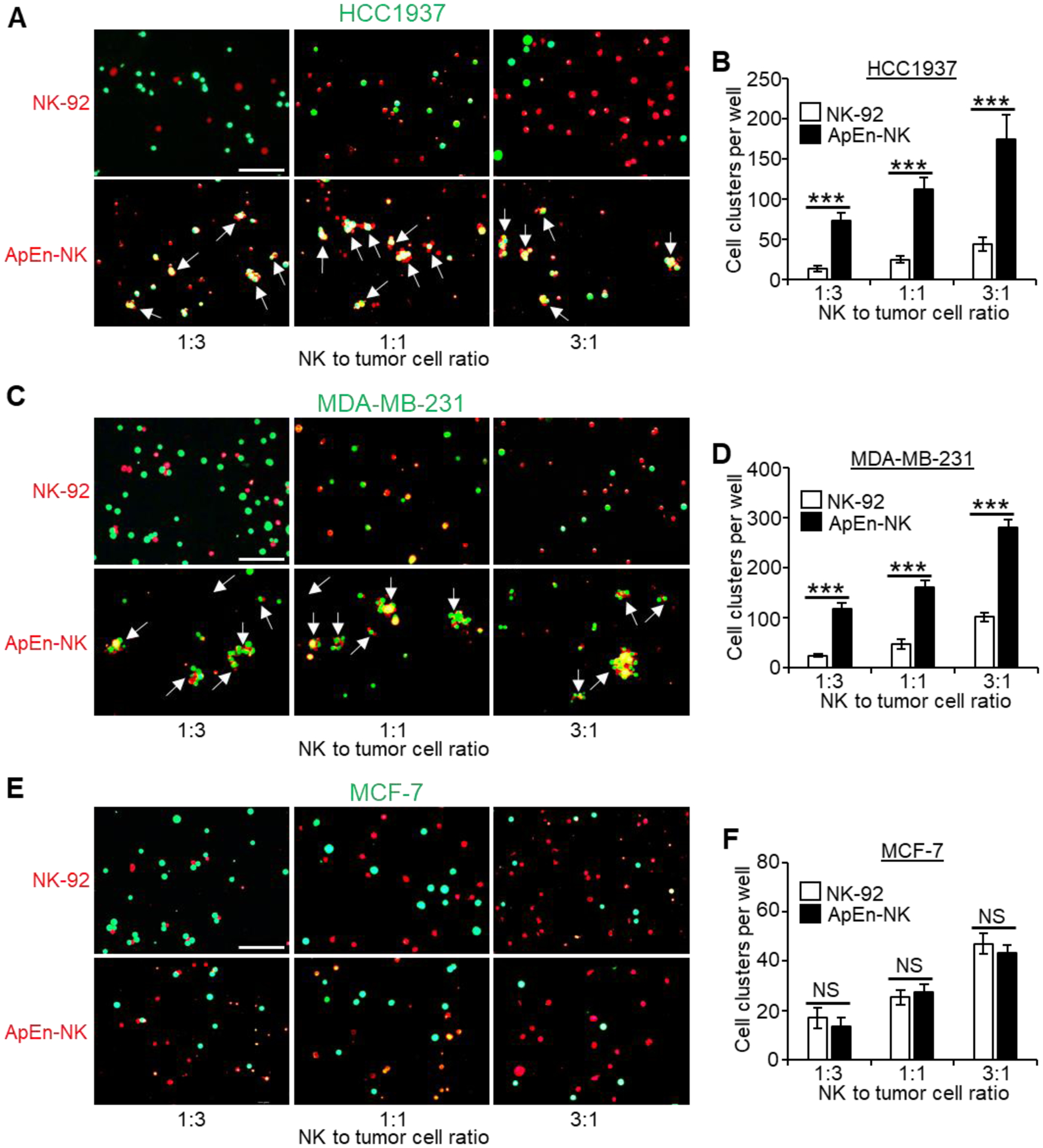
(A-D) ApEn-NK specifically bound and formed more clusters (pointed by arrows) with TNBC cells (HCC1937 and MDA-MB-231) in comparing to baseline controls observed in mixtures with NK92 cells. (E, F) ApEn-NK and parental NK-92 cells showed similar background non-specific binding to non-TNBC cells (MCF-7). Quantitative analyses of cell cluster formation under individual conditions are shown to the right of corresponding microscopic images. Scale bar: 100 μm. All experiments were performed ≥ 3 times with similar results. Representative results are shown. Data are presented as mean ± S.D. ***: P<0.001 (Student’s t-test, two-tailed). NS: not significant.
To determine whether ApEn-NKs target solid TNBC tumors, ApEn-NK was added into cultures of adherent tumor cells that mimic a solid tumor surface (Fig. 4A). Different ratios of ApEn-NK to tumor cells (1:3, 1:1, and 3:1) were tested and then imaged using the Incucyte S3 Live-Cell Imaging and Analysis System. Tumor cells were subjected to bright field imaging and the fluorescent signal derived from surface-engineered aptamers was used to detect ApEn-NKs. After incubating at 37°C for 1 h, cell cultures were gently washed twice to remove unbound ApEn-NK and re-imaged. ApEn-NKs and tumor cells from the same fields of individual cultures were quantified before and after washing using the Incucyte system. Cell binding capacity was calculated: ApEn-NK or parental NK-92 cells that bound to tumor cells vs. total tumor cells in the same cultures. Cell binding of parental NK-92 was used as non-specific baseline control. Increased tumor cell binding of ApEn-NK (folds) was normalized to baseline of parental NK-92 cells under the same conditions. As showed in Fig. 4B–E, ApEn-NK bound TNBC cells (HCC1937 and MDA-MB-231) with higher capacity, 3–6 folds of that in parental NK-92 cells. In contrast, ApEn-NKs and parental NK-92 cells exhibited similar background binding to non-TNBC cells (MFC-7) (Fig. 4F–G). Taken together, these findings demonstrate that ApEn-NKs can specifically target and bind suspended and adherent TNBC cells.
Figure 4. ApEn-NK specifically binds adherent TNBC cells with high capacity.
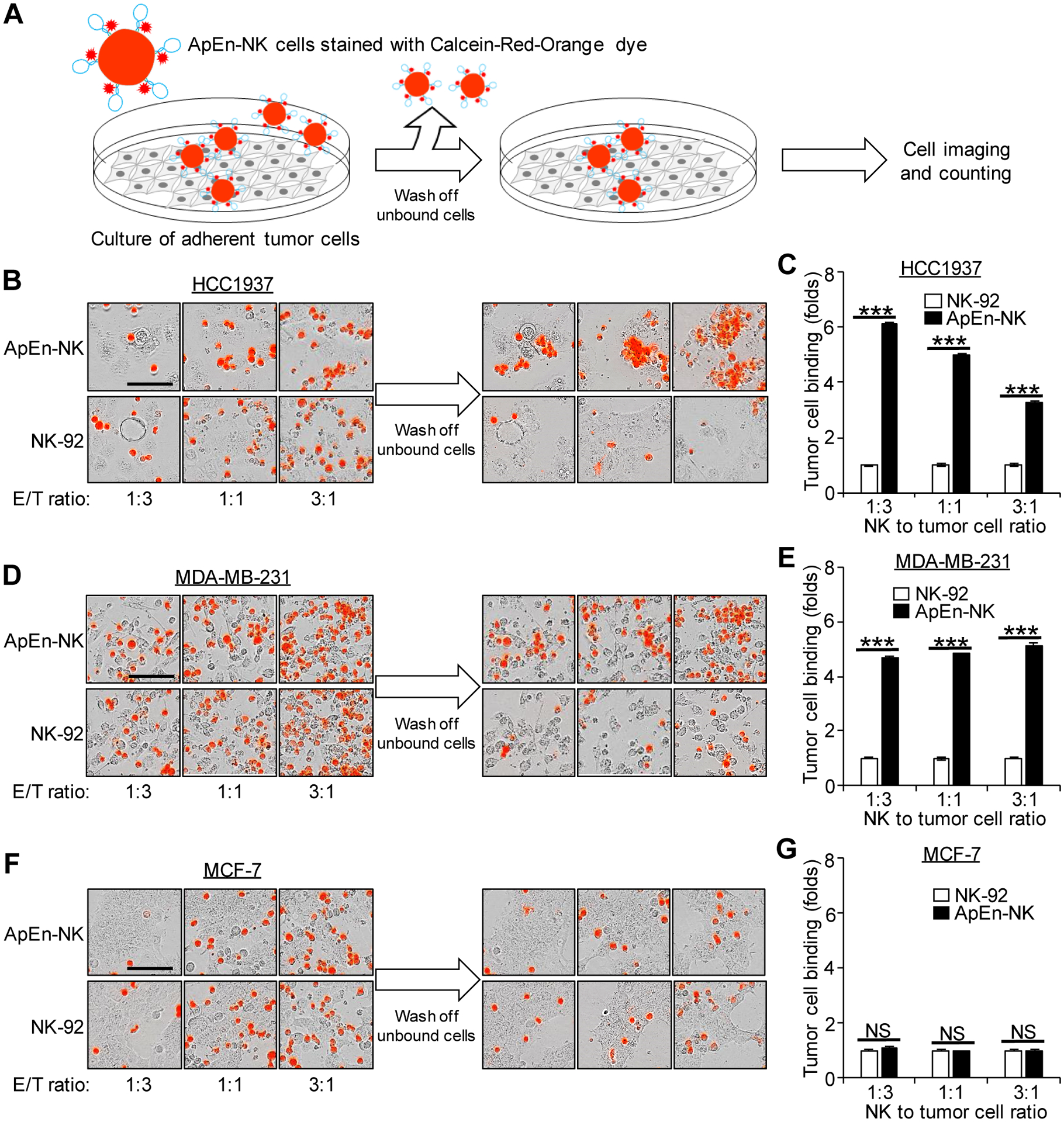
(A) Scheme of cell binding assays. (B-E) ApEn-NK specifically bound TNBC cells (HCC1937 and MDA-MB-231) with higher affinity compared to parental NK-92 cells. (F, G) Similar background baseline binding to off-target non-TNBC cells (MCF-7) was detected in cultures exposed to ApEn-NK or parental NK-92 cells. Scale bar: 100 μm. All experiments were repeated ≥ 3 times. Representative results are presented. Data are shown as mean ± S.D. ***: P<0.001 (Student’s t-test, two-tailed). NS: not significant.
ApEn-NK efficiently attacks TNBC cells
As a component of the innate immune system, NK cells can attack tumor cells without pre-activation[33–35], making them an ideal living drug for immunotherapy. However, a lack of cell receptors specific for tumor cells limits the therapeutic potential of NK cells. Cell-binding assays revealed that under aptamer guidance, ApEn-NK specifically bound TNBC cells with high affinity (Fig. 3 and 4). To assess the therapeutic potential of ApEn-NK to treat TNBC, adherent TNBC tumor cells were treated with ApEn-NKs at different ratios for 24 h as described under Materials and Methods. Cells were then stained, and the apoptotic or dead tumor cells were gated and quantified using flow cytometry based on the cellular signals from FITC Annexin V and eFluor 450, respectively. Apoptosis and death rates of tumor cells were calculated as percentage of total tumor cells (%). Fig. 5A–D showed that ApEn-NK treatment induced significantly higher TNBC cell (HCC1937 and MDA-MB-231) apoptosis/death than parental NK-92 cell treatment. In contrast, ApEn-NK treatment had minimal effects on off-target breast tumor cells (MCF-7), similar to the treatment with parental NK-92 cells (Fig. 5E–F). To rule out potential cellular effect of aptamer PDGC21-T, the same set of tumor cells were treated with aptamers alone as described above. As showed in Fig. S5, aptamer treatment alone had no effect on apoptosis and death rates of TNBC tumor cells or off-target non-TNBC tumor cells, in comparing to random sequence treatment or non-treatment controls. Taken together, these findings demonstrate therapeutic potential of ApEn-NKs for targeted TNBC treatment.
Figure 5. ApEn-NK treatment induces TNBC cells apoptosis and death.
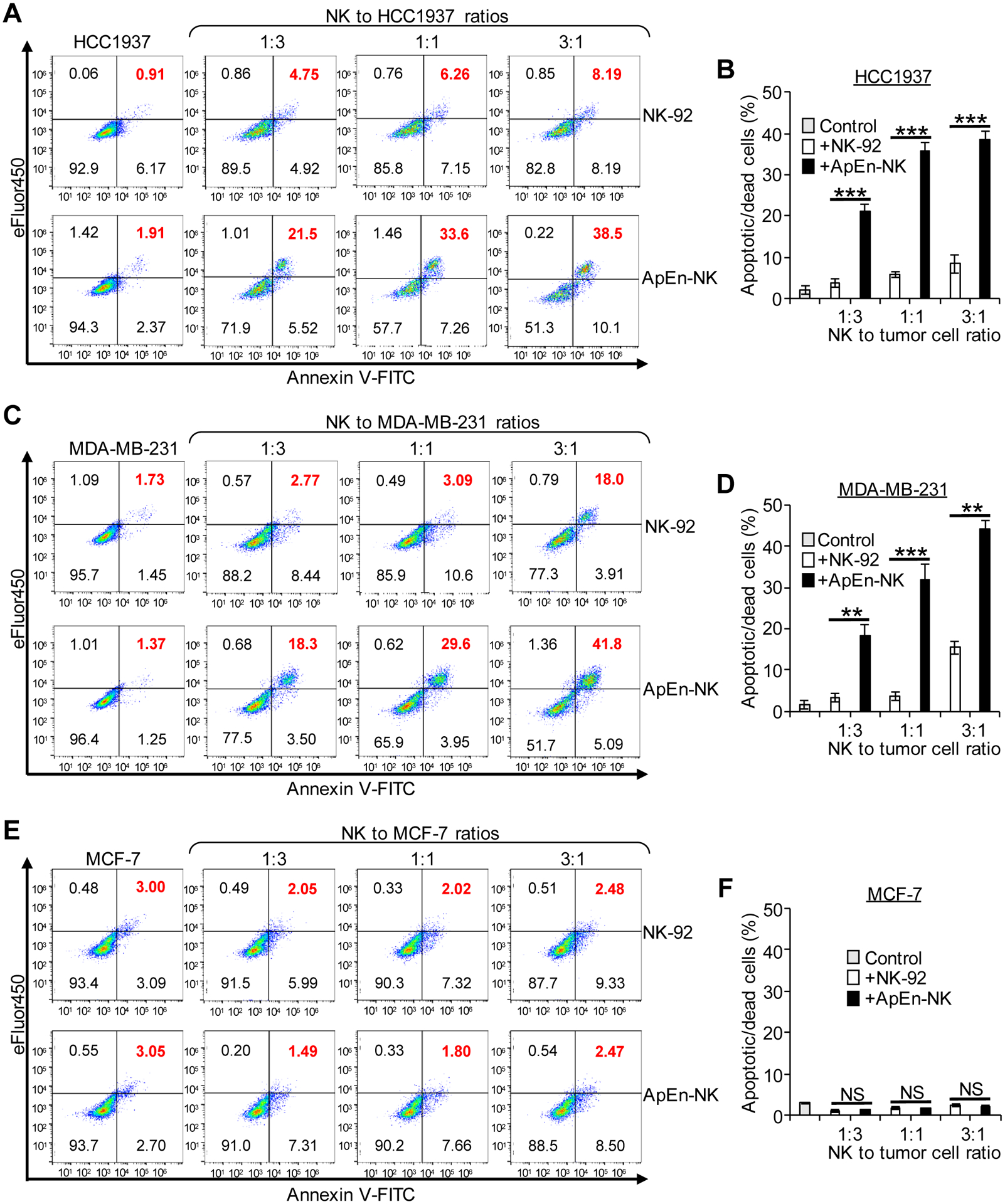
(A-D) ApEn-NK treatment triggered apoptosis/death of TNBC cells (HCC1937 and MDA-MB-231). (E, F) Similar background baseline effects on off-target breast cancer cells (MCF-7) were detected with treatments of ApEn-NK or parental NK-92 cells. All experiments were repeated ≥ 3 times. Data are shown as mean ± S.D. **: P<0.01; ***: P<0.001 (Student’s t-test, two-tailed). NS: not significant.
ApEn-NK treatment inhibits lung metastasis in mice harboring TNBC xenografts
To assess the clinical value of ApEn-NKs for targeted TNBC immunotherapy, a mouse model of lung metastasis was established by intravenously infusing traceable TNBC cells (MDA-MB-231) that stably express luciferase. Two weeks post tumor cell infusion, xenograft mice were treated with ApEn-NK, parental NK-92 cells, or vehicle alone control for a total of eight doses as showed in Fig. 6A. Lung metastasis formation was monitored weekly by bioluminescent imaging and quantified using total flux signal. Imaging results prior to treatment and at the study endpoint were shown in Fig. 6B and 6C, respectively.
Figure 6. ApEn-NK treatment inhibits lung metastasis of TNBC in mice without affecting body weight.
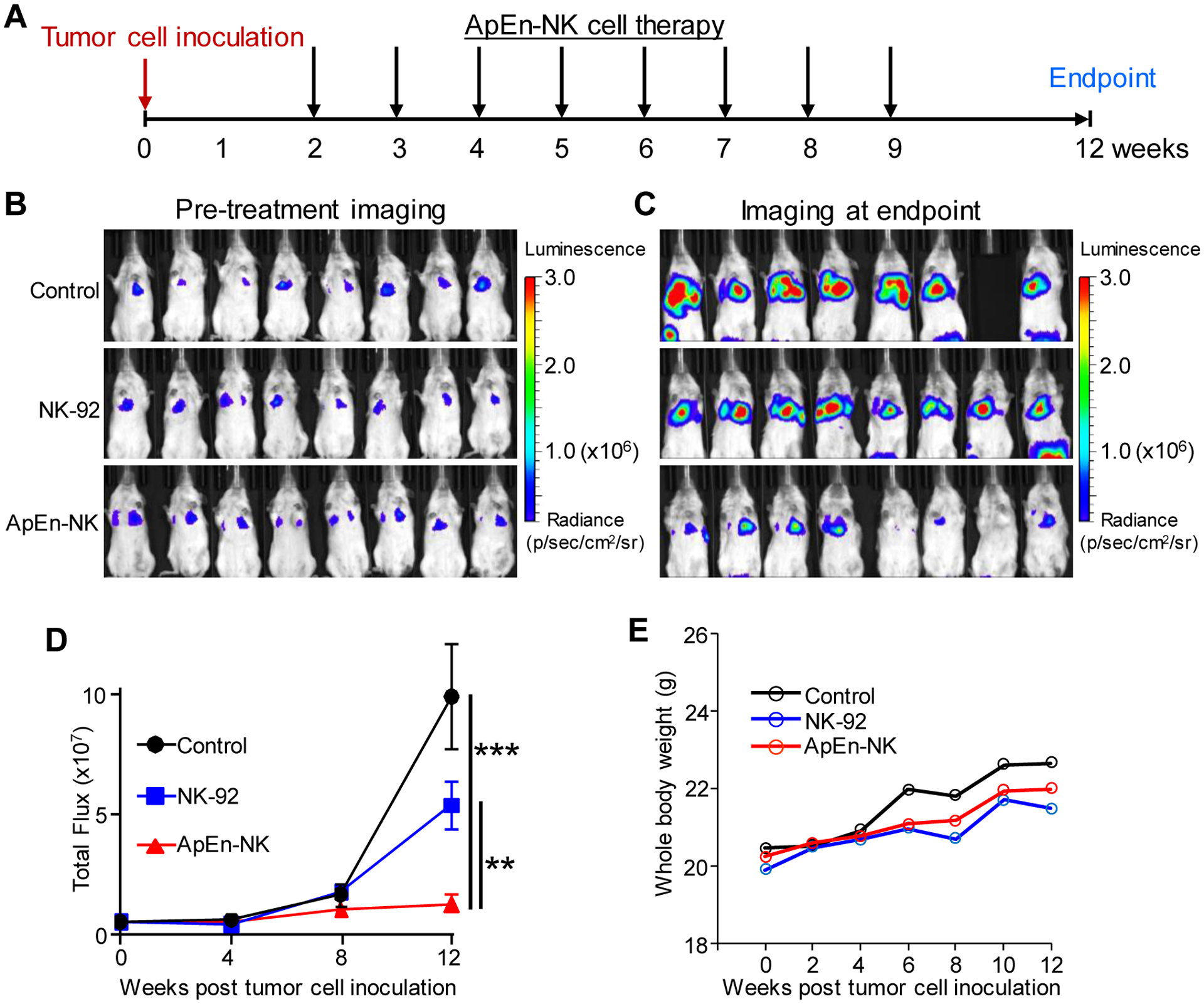
(A) Treatments were started two weeks post intravenous infusion of TNBC cells and repeated weekly for total eight doses. Lung metastasis formation was monitored weekly by whole-body imaging of mice. (B) Whole-body imaging pre-treatments. (C) Whole-body imaging at study endpoint. Luminescence radiance is displayed as photons/second/square centimeter/steradian. (D) Changes in lung metastasis signals derived from traceable TNBC cells over time. (E) Changes in body weight of treated mice over time. Data are presented as mean ± S.D. **: P<0.01; ***: P<0.001 (Student’s t-test, two-tailed). NS: not significant.
ApEn-NK treatment significantly inhibited TNBC cell lung metastasis at experimental endpoint compared to control group (1.28 × 107 vs. 9.91 × 107 in mean flux signals), and to parental NK-92 cell treatment group (1.28 × 107 vs. 5.38 × 107 in mean flux signals) (Fig. 6D). To rule out adverse treatment effects, body weight was monitored in treated mice. Treatment with ApEn-NK and parental NK-92 cells had no effect on body weight (Fig. 6E). Due to tumor growth, control group mice showed a slight increase in body weight.
To confirm our in vivo findings, entire lung tissues were removed from treated mice immediately after endpoint imaging (Fig. 7A). Ex vivo imaging confirmed that ApEn-NK treatment had higher inhibitory effect on TNBC cell lung metastasis than parental NK-92 cell treatment (Fig. 7B). Subsequently, intact lung tissues (five lobes per mouse) were fixed and visible tumor nodules in lung tissues were manually counted under a stereomicroscope. Total tumor nodules of lung metastasis per mouse were calculated and compared among mouse groups received different treatments (Fig. 7C). The number of tumor nodules was consistent with the intensity of tumor imaging signals detected both in vivo and ex vivo. Finally, lung tissue sections were stained with H&E. Histological examination revealed cytological and morphological features of tumor nodules and confirmed lung metastasis of TNBC cells (Fig. 7D). Finally, to rule out the side toxic effects of the treatments, mouse major organs were collected at the experimental endpoint, including heart, liver, kidney, and spleen. Tissue histology examination revealed that ApEn-NK treatment did not have side toxicity to normal tissues and thus, no morphological abnormalities were detected in mouse manor organs (Fig. 7E). Taken together, these findings demonstrate the potential utility of ApEn-NK for targeted TNBC immunotherapy with little side effects.
Figure 7. ApEn-NK treatment inhibits tumor nodule formation of lung metastasis in TNBC xenograft mice.
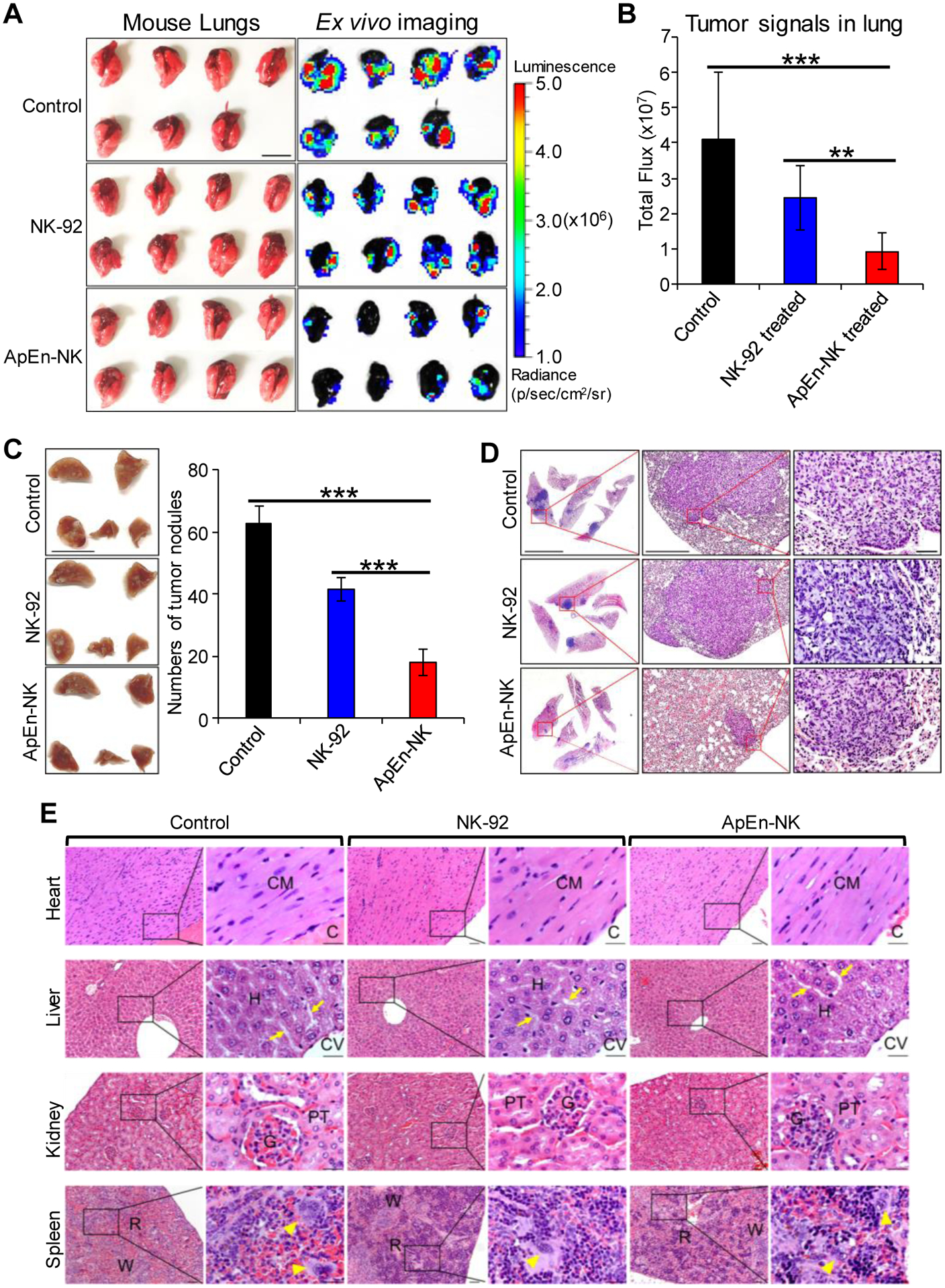
(A) Ex vivo imaging of lung tissues performed immediately following the whole-body imaging study at the study endpoint. Luminescence radiance is displayed as photons/second/square centimeter/steradian. Scale bar: 1 cm. (B) Tumor signals derived from lung metastasis in mice with different treatments. (C) Tumor nodules of metastasis in fixed lung tissues were counted using a stereomicroscope under 10X magnification. Scale bar: 1 cm. (D) Hematoxylin and Eosin staining of tumor nodules derived from TNBC cells at 10X (left), 40X (middle), and 200X magnification (right). Data are presented as mean ± S.D. **: P<0.01; ***: P<0.001 (Student’s t-test, two-tailed). NS: not significant. Scale bars from left to right: 5 mm, 0.5 mm, 0.05 mm. (E) ApEn-NK had no side toxicity to mouse normal tissues. Major organs (heart, liver, kidney, and spleen) were collected at experiment endpoint from mice different treatments as indicated. Histology examination was performed under a microscope with 100X and 400X magnification. No morphological abnormalities were detected in mouse major organs. (CM = Cardiac myocyte; C = Cardiac cavity; H =Hepatocyte; CV = Central vein; P = Portal area. Arrow = Hepatic sinusoid; G = Glomerulus; PT = Proximal tubule; R = Red pulp; W = White pulp; Arrowhead = Megakaryocyte. Scale bar of low and high magnification were 0.05 mm and 0.025 mm, respectively.
Discussion
TNBC cells lack expression of ER, PR, and HER2 and are not responsive to current hormonal therapy or HER2-targeted therapies. The absence of targetable antigens limits the development of antibody drug-conjugates and CAR-T/NK therapies for targeted TNBC treatment. Chemotherapy, the only established therapeutic option for TNBC, may cause severe off-target toxicity in patients. To address these clinical challenges, we used ApEn-NK technology to target TNBC based on its poorly differentiated pathology. Aptamer probes target TNBC cells and NK cells act as an immunotherapeutic agent that lacks off-target toxicity. Our in vitro and in vivo studies validated the potential utility of ApEn-NK for targeted TNBC immunotherapy (Fig. 4, 5, and 6).
Aptamer PDGC21-T was initially identified through cell-based SELEX and successfully targeted poorly differentiated gastric cancer through an unidentified binding target[18]. In addition, aptamer PDGC21-T recognizes poorly differentiated colorectal cancer cells[18]. Here, we reveal that PDGC21-T also recognizes poorly differentiated TNBC tumor cells (Fig. 1). Thus, aptamer PDGC21-T may target a common molecule related to poor differentiation in tumor cells. Identifying the target molecule(s) of aptamer PDGC21-T is a clinically relevant and necessary endeavor for developing targeted therapy for TNBC and other poorly differentiated cancers. To this end, we are conducting immunoprecipitation assays with aptamer PDGC21-T probes and subsequent mass spectrum identification. Our findings may provide a foundation with which to develop future antibody-based therapies, including CAR-T/NK.
ApEn-NKs inhibited TNBC lung metastasis formation without side effects on body weight of treated mice (Fig. 6 and 7). To enhance immunotherapeutic potential, a pre-activation step could be introduced using immunostimulatory adjuvants to stimulate innate NK cells prior to or post ApEn-NK production. In addition, co-administration of an immune checkpoint inhibitor may enhance ApEn-NK therapeutic efficacy by preventing negative effects from PD-1/PD-L1 signaling pathways in PD-1-expressing tumor cells. As the safety of allogeneic NK cells for adoptive transfer immunotherapy has been clinically validated, our ApEn-NK products may have potential as an off-the-shelf therapeutic to treat TNBC. Pre-clinical studies and clinical trials will be required before clinical use. In contrast to T cells, NK cells generally have a shorter circulating half-life and surface-engineered aptamers will become diluted as ApEn-NKs proliferate in vivo. Therefore, multiple doses of ApEn-NK may be needed in contrast to a single dose treatment of CAR-T therapy.
Conclusion
The study introduced a simple method to produce ApEn-NK for targeted immunotherapy and also demonstrated an unique approach to target TNBC by aptamers based on pathological features of tumor cells. Pre-clinical study demonstrated that targeted immunotherapy by ApEn-NK inhibited lung metastasis of TNBC cells without side toxicity to normal tissues in treated mice. Our findings shed light on the development of new targeted therapy to treat TNBC.
Supplementary Material
Highlights:
Pragmatically anchoring aptamers on NK cells to produce Aptamer-Engineered NK cells (ApEn-NK)
ApEn-NK selectively target triple-negative breast cancer (TNBC) based on poorly differentiated pathological features of tumor cells
ApEn-NK treatment induces apoptosis and death of TNBC cells with little effect on off-target cells
Targeted immunotherapy by ApEn-NK inhibits lung metastasis of TNBC cells without side toxicity to normal tissues in treated mice
Acknowledgements:
We want to thank Houston Methodist Research Institute’s Advanced Cellular and Tissue Microscopy Core Facility for the support. We also thank Drs. Heather McConnell and Sasha Pejerrey for their scientific editing of the manuscript.
Funding:
This study was partially supported by an NIH grant R01CA224304 (Y.Z.), a department seed fund (Y.Z.), and Golfers Against Cancer grant (Y.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests: The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA Cancer J Clin 71(3) (2021) 209–249. [DOI] [PubMed] [Google Scholar]
- [2].Ryu DW, Jung MJ, Choi WS, Lee CH, Clinical significance of morphologic characteristics in triple negative breast cancer, J Korean Surg Soc 80(5) (2011) 301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Denkert C, Liedtke C, Tutt A, von Minckwitz G, Molecular alterations in triple-negative breast cancer-the road to new treatment strategies, Lancet 389(10087) (2017) 2430–2442. [DOI] [PubMed] [Google Scholar]
- [4].Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA, Triple-negative breast cancer: clinical features and patterns of recurrence, Clin Cancer Res 13(15 Pt 1) (2007) 4429–34. [DOI] [PubMed] [Google Scholar]
- [5].Lyons TG, Targeted Therapies for Triple-Negative Breast Cancer, Curr Treat Options Oncol 20(11) (2019) 82. [DOI] [PubMed] [Google Scholar]
- [6].Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L, Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease, Nat Rev Clin Oncol 13(11) (2016) 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shigdar S, Schrand B, Giangrande PH, de Franciscis V, Aptamers: Cutting edge of cancer therapies, Mol Ther (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou SF, Kong L, Li Y, Pu C, Duan W, Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine, Theranostics 5(1) (2015) 23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zamay TN, Zamay GS, Kolovskaya OS, Zukov RA, Petrova MM, Gargaun A, Berezovski MV, Kichkailo AS, Current and Prospective Protein Biomarkers of Lung Cancer, Cancers (Basel) 9(11) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ku TH, Zhang T, Luo H, Yen TM, Chen PW, Han Y, Lo YH, Nucleic Acid Aptamers: An Emerging Tool for Biotechnology and Biomedical Sensing, Sensors (Basel) 15(7) (2015) 16281–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ni S, Zhuo Z, Pan Y, Yu Y, Li F, Liu J, Wang L, Wu X, Li D, Wan Y, Zhang L, Yang Z, Zhang BT, Lu A, Zhang G, Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications, ACS Appl Mater Interfaces 13(8) (2021) 9500–9519. [DOI] [PubMed] [Google Scholar]
- [12].Zhu G, Ye M, Donovan MJ, Song E, Zhao Z, Tan W, Nucleic acid aptamers: an emerging frontier in cancer therapy, Chem Commun (Camb) 48(85) (2012) 10472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sefah K, Shangguan D, Xiong X, O’Donoghue MB, Tan W, Development of DNA aptamers using Cell-SELEX, Nat Protoc 5(6) (2010) 1169–85. [DOI] [PubMed] [Google Scholar]
- [14].Li L, Xu S, Yan H, Li X, Yazd HS, Li X, Huang T, Cui C, Jiang J, Tan W, Nucleic Acid Aptamers for Molecular Diagnostics and Therapeutics: Advances and Perspectives, Angew Chem Int Ed Engl 60(5) (2021) 2221–2231. [DOI] [PubMed] [Google Scholar]
- [15].Liu X, Wang YL, Wu J, Qi J, Zeng Z, Wan Q, Chen Z, Manandhar P, Cavener VS, Boyle NR, Fu X, Salazar E, Kuchipudi SV, Kapur V, Zhang X, Umetani M, Sen M, Willson RC, Chen SH, Zu Y, Neutralizing Aptamers Block S/RBD-ACE2 Interactions and Prevent Host Cell Infection, Angew Chem Int Ed Engl (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wan Q, Liu X, Zeng Z, Chen Z, Liu Y, Zu Y, Aptamer Cocktail to Detect Multiple Species of Mycoplasma in Cell Culture, Int J Mol Sci 21(11) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu Y, Jiang W, Yang S, Hu J, Lu H, Han W, Wen J, Zeng Z, Qi J, Xu L, Zhou H, Sun H, Zu Y, Rapid Detection of Mycoplasma-Infected Cells by an ssDNA Aptamer Probe, ACS Sens 4(8) (2019) 2028–2038. [DOI] [PubMed] [Google Scholar]
- [18].Li W, Wang S, Zhou L, Cheng Y, Fang J, An ssDNA aptamer selected by Cell-SELEX for the targeted imaging of poorly differentiated gastric cancer tissue, Talanta 199 (2019) 634–642. [DOI] [PubMed] [Google Scholar]
- [19].Waks AG, Winer EP, Breast Cancer Treatment: A Review, JAMA 321(3) (2019) 288–300. [DOI] [PubMed] [Google Scholar]
- [20].Yin L, Duan JJ, Bian XW, Yu SC, Triple-negative breast cancer molecular subtyping and treatment progress, Breast Cancer Res 22(1) (2020) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang S, Wen J, Li H, Xu L, Liu Y, Zhao N, Zeng Z, Qi J, Jiang W, Han W, Zu Y, Aptamer-Engineered Natural Killer Cells for Cell-Specific Adaptive Immunotherapy, Small 15(22) (2019) e1900903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J, Genes that mediate breast cancer metastasis to lung, Nature 436(7050) (2005) 518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fantozzi G Christofori, Mouse models of breast cancer metastasis, Breast Cancer Res 8(4) (2006) 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pillar N, Polsky AL, Weissglas-Volkov D, Shomron N, Comparison of breast cancer metastasis models reveals a possible mechanism of tumor aggressiveness, Cell Death Dis 9(10) (2018) 1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pieta A, Barbieri V, Zuccolotto G, Rosato A, Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer, Cells 9(6) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, Loureiro LR, Bergmann R, Mathe D, Hegedus N, Kovacs T, Zhang C, Oberoi P, Jager E, Seliger B, Rossig C, Temme A, Eitler J, Tonn T, Schmitz M, Hassel JC, Jager D, Wels WS, Bachmann M, “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells, Sci Rep 10(1) (2020) 2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang QM, Tang PM, Lian GY, Li C, Li J, Huang XR, To KF, Lan HY, Enhanced Cancer Immunotherapy with Smad3-Silenced NK-92 Cells, Cancer Immunol Res 6(8) (2018) 965–977. [DOI] [PubMed] [Google Scholar]
- [28].Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S, Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis, Cell 158(5) (2014) 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nole F, Munzone E, Zorzino L, Minchella I, Salvatici M, Botteri E, Medici M, Verri E, Adamoli L, Rotmensz N, Goldhirsch A, Sandri MT, Variation of circulating tumor cell levels during treatment of metastatic breast cancer: prognostic and therapeutic implications, Ann Oncol 19(5) (2008) 891–7. [DOI] [PubMed] [Google Scholar]
- [30].Shaw JA, Guttery DS, Hills A, Fernandez-Garcia D, Page K, Rosales BM, Goddard KS, Hastings RK, Luo J, Ogle O, Woodley L, Ali S, Stebbing J, Coombes RC, Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts, Clin Cancer Res 23(1) (2017) 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S, Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition, Science 339(6119) (2013) 580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF, Circulating tumor cells, disease progression, and survival in metastatic breast cancer, N Engl J Med 351(8) (2004) 781–91. [DOI] [PubMed] [Google Scholar]
- [33].Freund-Brown J, Chirino L, Kambayashi T, Strategies to enhance NK cell function for the treatment of tumors and infections, Crit Rev Immunol 38(2) (2018) 105–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J, NK cell-based cancer immunotherapy: from basic biology to clinical development, J Hematol Oncol 14(1) (2021) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu SY, Fu T, Jiang YZ, Shao ZM, Natural killer cells in cancer biology and therapy, Mol Cancer 19(1) (2020) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.