

Abstract
Pyrrolizidine alkaloids (PAs) are the most common phytotoxins with documented human hepatotoxicity. PAs require metabolic activation by cytochromes P450 to generate toxic intermediates which bind to proteins and form protein adducts, thereby causing cytotoxicity. This study investigated the role of the gut–liver axis in PA intoxication and the underlying mechanisms. We exposed mice to retrorsine (RTS), a representative PA, and for the first time found RTS-induced intestinal epithelium damage and disruption to intestinal barrier function. Using mice with tissue-selective ablation of P450 activity, we found that hepatic P450s, but not intestinal P450s, were essential for PA bioactivation. Besides, in RTS-exposed, bile duct-cannulated rats, we found the liver-derived reactive PA metabolites were transported by bile into the intestine to exert enterotoxicity. The impact of gut-derived pathogenic factors in RTS-induced hepatotoxicity was further studied in mice with dextran sulfate sodium (DSS)-induced chronic colitis. DSS treatment increased the hepatic endotoxin level and depleted hepatic reduced glutathione, thereby suppressing the PA detoxification pathway. Compared to RTS-exposed normal mice, the colitic mice displayed more severe RTS-induced hepatic vasculature damage, fibrosis, and steatosis. Overall, our findings provide the first mode-of-action evidence of PA-induced enterotoxicity and highlight the importance of gut barrier function in PA-induced liver injury.
KEY WORDS: Pyrrolizidine alkaloid, Cytochrome P450, Inflammatory bowel disease, Intestinal injury, Liver injury, Gut–liver axis
Graphical abstract
Pyrrolizidine alkaloids (PAs) are common phytotoxins. We provide the first mode-of-action evidence of PA-induced enterotoxicity. A disrupted gut barrier in turn exacerbates PA-induced liver injury via depleting the hepatic glutathione.

1. Introduction
Pyrrolizidine alkaloids (PAs) are secondary metabolites produced by more than 6000 plant species worldwide1. Approximately 600 PAs and PA N-oxides have been identified, and over half of them have been reported to be hepatotoxic or pneumotoxic2. Because of their ubiquitous distribution, people are readily exposed to PAs via the consumption of PA-containing herbal remedies or PA-contaminated food products. Ingestion of high doses of PAs causes acute hepatotoxicity, particularly with severe liver damage known as hepatic sinusoidal obstruction syndrome (HSOS), which is characterized by hepatomegaly, hemorrhagic necrosis, ascites, jaundice, and hepatic veno-occlusion3,4. Compared with the acute hepatotoxic manifestations, chronic PA intoxication is less studied. However, in more common situations, people are exposed to PAs via ingestion of PA-contaminated food or dietary supplements, and such exposure level is too low to cause acute liver injury. Instead, the more commonly occurring long-term dietary exposure to PAs causes chronic liver diseases such as liver fibrosis, cirrhosis, and cancer5. Recently, we reported the first indication between PA exposure and advanced human liver cancer6. However, progressive intoxication due to chronic PA exposure needed further evaluation.
PAs require metabolic activation to exert toxicity. After ingestion, PAs are metabolized by cytochrome P450 (CYP) enzymes, especially CYP3A4 isozyme, to generate reactive PA metabolites, namely dehydropyrrolizidine alkaloids (DHPAs)7. DHPAs can be hydrolyzed to (±)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine (DHP), and both are reactive PA metabolites that can bind to cellular proteins to form pyrrole–protein adducts, thereby causing hepatotoxicity8. DHPAs can also conjugate with reduced glutathione (GSH) to generate pyrrole–GSH conjugates which are then excreted from the body9,10. Therefore, the conjugation of DHPAs with GSH is generally regarded as the PA detoxification pathway. Apart from exerting toxicity in the liver, PAs can also cause toxicities in other organs such as the lungs11,12. However, the toxic effects of PAs in the intestine, which is the first-line defense against orally ingested chemicals and coordinates with the liver in various biological processes via the gut–liver axis, were unexplored.
The intestine plays an important role in liver homeostasis, and gut-derived pathogens contribute to liver pathogenesis13. One of the most devastating chronic intestinal disorders is inflammatory bowel disease (IBD), which encompasses Crohnʹs disease and ulcerative colitis14. As an epidemic intestine disease with increasing incidence over the recent decades, IBD could have a significant promoting effect on the susceptibility of the liver to other insults15. However, the toxicological outcomes of PA exposure in IBD patients remain unknown.
In the present study, we have investigated the intestinal injury induced by liver-derived reactive PA metabolites and the effects of gut-derived bacterial antigens on PA-induced liver injury. We first characterized the toxicological outcome of chronic PA exposure in mouse intestines. The roles of hepatic and intestinal P450 enzymes in PA bioactivation and the resulting formation of pyrrole–protein adducts were then determined using mice with tissue-selective ablation of P450 activity. Transport of liver-derived reactive PA metabolites into the intestine was confirmed by examining the bile of PA-exposed rats. Finally, we investigated the effects of chronic colitis on PA-induced hepatotoxicity in mice and the underlying factors affecting PA intoxication and detoxification in the liver.
2. Materials and methods
2.1. Animal models of chronic colitis and PA exposure
Male C3H mice (18–20 g, 8–12 weeks old) were supplied by the Laboratory Animal Service Center, the Chinese University of Hong Kong (Hong Kong, China). The colitis model was established by intermittent administration of dextran sulfate sodium (DSS, 36,000–40,000 Da, MP Biomedicals, Solon, OH, USA) according to previous studies16,17 with minor modification. The chronic PA exposure model was established by treatment with a representative PA, retrorsine (RTS, Sigma–Aldrich, St. Louis, MO, USA), according to our previous report6. Mice were divided randomly into four age-matched groups (n = 5 per group): control, RTS, DSS, RTS + DSS. DSS and RTS + DSS mice received multi-cycle administration of DSS (3% w/v DSS in drinking water). One DSS dosing cycle consisted of seven days of DSS-added drinking water followed by 14 days of normal drinking water. Control and RTS mice received normal drinking water. For PA exposure, RTS and RTS + DSS mice were orally administered with RTS at a non-acute toxic dose (Supporting Information Figs. S1 and S2) of 20 mg/kg body weight (b.w.)/day for 14 weeks, while control and DSS mice were orally administered with normal saline (0.1 mL/10 g b.w.). DSS treatment began at the same time with RTS administration (Fig. 1A). Procedures involving the care and handling of mice were reviewed and approved by the Animal Experimental Ethics Committee, the Chinese University of Hong Kong (Hong Kong, China).
Figure 1.

Long-term RTS exposure and intermittent DSS administration induced intestinal injury. (A) C3H mice (4-month old, male) receiving intermittent administration of DSS in drinking water 3% (w/v) or water alone, and oral administration of RTS (20 mg/kg/day) or saline for 14 weeks were divided into control, DSS, RTS, and RTS + DSS groups. (B) Representative histological changes indicated by H&E staining of paraffin-embedded mouse jejunum (upper panel) and colon (lower panel) sections. Arrowhead indicates inflammatory neutrophil infiltration. Star (∗) indicates villus or epithelial brush boarder erosion. Plus (+) indicates crypt or epithelium distortion. Scale bar: 50 μm. (C and D) Length of mouse small intestines (C) and colons (D). Data plotted as mean ± SD (n = 5). One-way ANOVA with Tukey's post hoc test was used. #P < 0.05, ##P < 0.01, ####P < 0.0001 versus control; ∗∗P < 0.01. ns, not significant.
2.2. Intestine histology
Mice were euthanized at the end of the 14-week treatment period, and intestine samples were collected. The lengths of the small intestines (from 1 cm below the pyloric ring to the cecum) and colon were measured. The luminal contents were removed by flushing with normal saline at room temperature. The tissues were prepared for histological examination using the Swiss roll method as previously described18. Briefly, each of the small and large intestines was gently inflated with ice-cold formalin fixative (4% formaldehyde in phosphate-buffered saline) by intraluminal injection. The pre-fixed specimens were opened along the mesenteric border and unfolded, and the residual contents were cleared by gently rinsing the mucosa with normal saline. Then the intestines were rolled longitudinally using the Swiss roll method, with the mucosa side inwards and the distal end of the intestine in the center of the roll, followed by formalin fixation for 12 h. The rolled tissues were embedded horizontally in paraffin and sectioned vertically across all layers in the roll. Tissue sections (5 μm thick) were stained with hematoxylin and eosin (H&E). The degree of pathological changes that reflect intestinal injury was scored on five cross sections of small and large intestines for each mouse according to a modified method as described previously19. The assessment of histological changes was done in a blinded fashion by two independent observers.
2.3. Alcian blue and periodic acid-Schiff staining
To detect mucin-secreting goblet cells, the prepared mouse intestine sections were stained with alcian blue and periodic acid-Schiff (AB-PAS). For AB staining, sections were deparaffinized in xylene, rehydrated in ethanol, and brought to distilled water for 5 min. AB 8GX (Biocare Medical, Concord, CA, USA) was applied to the sections for 15 min, followed by counterstained with Nuclear Fast Red. For PAS staining, deparaffinized and rehydrated sections were treated with periodic acid for 5 min. Slides were washed in distilled water then stained with Schiff's reagent for 15 min, followed by a 5-min wash in running tap water. The sections were then counterstained with hematoxylin, washed in running tap water for 2 min, followed by dehydration (twice in 95% ethanol and twice in 100% ethanol) and cover-slipped.
2.4. Intestinal tight junction analysis
Immunohistochemistry (IHC) analysis was performed on formalin-fixed mouse intestine sections to investigate the integrity of intestinal epithelium, which was stained with anti-zonula occludens-1 (ZO-1) and anti-occludin antibodies (Abcam, Cambridge, MA, USA), and counterstained with hematoxylin.
2.5. Measurement of intestinal permeability
Intestinal permeability was assessed according to the method previously described20. Fluorescein isothiocyanate-conjugated dextran of molecular weight 4000 Da (FD4000), a non-metabolizable macromolecule, was used as the permeability probe. Mice (n = 5 per group) were orally administered with FD4000 at 500 mg/kg b.w. 4 h before sacrifice. After 4 h, mice were sacrificed, and blood samples were collected. The concentration of FD4000 was determined by measuring the fluorescence intensity using a BioTeK Synergy H1 spectrophotometer (Winooski, VT, USA) with excitation wavelength at 485 nm and emission wavelength at 535 nm.
2.6. Endotoxin assay
The level of hepatic exposure to endotoxin (lipopolysaccharides) was determined by measuring endotoxin levels in 20 μL of blood collected from the mouse portal vein using the ToxinSenso Chromogenic limulus amebocyte lysate Endotoxin Assay Kit (GenScript, Piscataway, PA, USA). Detailed methods are available in Supporting Information.
2.7. Toxicokinetic analysis using mice with tissue-specific P450 abrogation
To demonstrate the source of reactive PA metabolites responsible for PA-induced intestinal injury, we investigated the respective roles of hepatic and intestinal P450 enzymes in PA metabolic activation by using cytochrome P450 reductase (Cpr) gene knockout mice. A functional microsomal P450 system relies on the electron donor, CPR, and deletion of the Cpr gene abrogates the activities of all microsomal P450 enzymes21. Here we used the liver-specific Cpr-null (LCN) mice with abrogated hepatic P450 activity21 and the intestinal epithelial-specific Cpr-null (IECN) mice with abolished intestinal P450 activity22,23. LCN and IECN mice (both on C57BL/6J background), as well as wild type (WT) C57BL/6J mice, were obtained from breeding stocks maintained at the University of Arizona (Tucson, AZ, USA). Procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Arizona (Tucson, AZ, USA). Blood samples (20–25 μL) were collected from the tail vein of each mouse (n = 3 per group) at multiple time points (0, 5, 15, 30, 60, 120, 240, 480, and 1440 min) after the oral administration of 120 mg/kg b.w. of monocrotaline (MCT, Sigma–Aldrich), a representative PA. Liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis was performed to determine the serum level of MCT with detailed methods available in Supporting Information.
2.8. Quantitation of pyrrole–protein adducts
Pyrrole–protein adducts are specific markers for PA exposure3. Using a derivatization method established previously24, we determined pyrrole–protein adducts, the biomarker for PA intoxication, in mouse serum, liver, and small intestine samples. Detailed methods for pyrrole–protein adducts quantitation are provided in Supporting Information.
2.9. Detection of reactive PA metabolites in bile
To investigate the transfer of reactive PA metabolites (e.g., DHPAs and DHP) from the liver to the gut, we performed the bile excretion assay to detect the presence of reactive RTS metabolite in bile. Adult (4-month-old) male Sprague–Dawley rats were supplied by the Laboratory Animal Service Center, the Chinese University of Hong Kong (Hong Kong, China). Rats were anesthetized by an intraperitoneal injection of a mixed solution containing 37.5 mg/mL of ketamine and 5 mg/mL of xylazine at 0.2 mL per 100 g b.w. Bile duct cannulation was performed in anesthetized rats, and 70 mg/kg b.w. RTS or normal saline was intravenously injected into the rats (n = 3 per group). Bile samples were collected at designated sampling intervals (0–15, 15–30, 30–45, 45–60, 60–75, 75–90, 90–105, and 105–120 min) after RTS or saline administration. After collection, we immediately divided each of the bile samples into two equal parts. GSH (0.1 mol/L) and paired saline vehicle were added into each part of bile samples followed by incubation for 2 min at 37 °C. The stable products, namely pyrrole–GSH conjugates, were determined by LC–MS/MS analysis with detailed methods described in Supporting Information. The rat handling protocols were approved by the Animal Experimental Ethics Committee, the Chinese University of Hong Kong.
2.10. Biochemical assay
Alanine aminotransferase (ALT) activity was measured in 50 μL serum using a kit obtained from Sigma (St. Louis, MO, USA) following the manufacturer's instruction. The total bilirubin level was determined in 200 μL serum using the QuantiChrom™ Bilirubin Assay Kit purchased from BioAssay Systems (Hayward, CA, USA) following the manufacturer's protocol.
2.11. Liver histology
Formalin-fixed, paraffin-embedded mouse livers were stained with H&E. Detailed information of histological examinations is available in Supporting Information. The degrees of hemorrhage, endothelial damage, sinusoidal dilatation, mixed cellular infiltration, and steatotic hepatocytes of the liver sections were scored on five cross liver sections of each mouse according to a modified method described previously25. The assessment of histological changes was done in a blinded fashion by two independent observers.
2.12. Hepatic vasculature imaging
To visualize the vasculature damage, scanning electron microscopy was performed to inspect ultrastructural changes of mouse hepatic sinusoids. Detailed methods are provided in Supporting Information.
2.13. Liver fibrosis assay
The paraffin-embedded mouse liver sections were stained with Sirius red to visualize fibrotic collagen with details provided in Supporting Information. The Sirius red positive area was measured on five cross liver sections of each mouse. Staining intensity was then determined by measuring the integrated density and subtracting the background intensity using ImageJ software (NIH Image, Bethesda, MA, USA) with details provided in Supporting Information. The fibrosis severity was scored according to a previous report26. The values (expressed as area%) from each measurement were averaged and expressed as mean ± SD.
2.14. Steatosis assay
Oil red O staining was performed to visualize lipid droplets in cryosections of mouse liver with details provided in Supporting Information. The steatosis level was quantified by determining oil red O positive area. The percent area occupied by oil red O-stained fat droplets was estimated on five cross liver sections of each mouse using ImageJ software with details provided in Supporting Information.
2.15. Metabolic study in mouse liver S9 fraction
The activity of mouse hepatic CYP3A11 (an ortholog of human CYP3A4) which predominately mediates PA bioactivation7 was determined by incubating liver S9 fraction prepared from C3H mice after the 14-week study with 50 μmol/L nifedipine (Sigma–Aldrich). In addition, to test the enzymatic activity toward PA metabolism, RTS (100 μmol/L) was also incubated with mouse liver S9. Detailed methods are provided in Supporting Information.
2.16. Hepatic GSH determination
GSH and glutathione disulfide (GSSG) levels in mouse liver samples were quantified following a modified reductase recycling assay27. Detailed methods are provided in Supporting Information.
2.17. Reverse transcription-quantitative PCR analysis
Total RNA was extracted from mouse livers using the RNeasy mini kit (Qiagen, Valencia, CA, USA). The relative levels of specific mRNAs in the liver were assessed by real-time quantitative PCR on the Applied Biosystems Plus Real-Time PCR System using the SYBER premix ExTaq kit (TaKaRa, Dalian, China). Expression levels of target genes were calculated relative to the level of Gapdh. The PCR primers were obtained from Tech Dragon Ltd. (Hong Kong, China). Gene expression was analyzed using the following pairs of primers: tumor necrosis factor-α (Tnfa: forward, 5ʹ-TATGGCTCAGGGTCCAACTC-3ʹ; reverse, 5ʹ-AATGGACATTCGAGGCTCCA-3ʹ); interleukin-6 (Il6: forward, 5ʹ-CAGAAGGAGTGGCTAAGGA-3ʹ; reverse, 5ʹ-CGTAGAGAACAACATAAGTCAG-3ʹ); interferon-γ (Ifng: forward, 5ʹ-ACCTTTACTTCACTGACCAA-3ʹ; reverse, 5ʹ-GTTACTACCTGACACATTCG-3ʹ); and Gapdh (forward, 5ʹ-TCTCCTGCGACTTCAACA-3ʹ; reverse, 5ʹ-CTGTAGCCGTATTCATTGTC-3ʹ). The relative levels of target genes were quantitated using 2−ΔΔCt method.
2.18. Statistics and data analysis
Kinetic parameters were calculated with WinNonlin 4.0 (Pharsight Corporation, Mountain View, CA, USA), using a non-compartmental model. Statistical analyses were performed using Student's t-test for comparison of two variables, and one-way ANOVA or two-way ANOVA with Tukey's post hoc test for comparison of multiple variables. Analyses were performed using GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA). P values < 0.05 were considered statistically significant.
3. Results
3.1. Long-term RTS exposure causes intestinal injury
After the intermittent DSS treatment, DSS mice demonstrated marked intestinal epithelium disruption, shortened and adhesive microvilli, inflammatory cell infiltration, and loss of crypts in small intestine segments: jejunum (Fig. 1B), duodenum, and ileum (Supporting Information Fig. S3). RTS mice also showed pathological changes similar to those observed in DSS mice. However, compared to DSS mice, which served as the positive colitis control and exhibited apparent chronic colitic lesions manifested by shortening and collapse of crypts, loss of brush boarder epithelial cells and infiltration with inflammatory cells, RTS mice had less severe colon lesions (Fig. 1B). Furthermore, compared with RTS and DSS mice, RTS + DSS mice showed remarkably exacerbated intestinal lesions, indicated by increased epithelial cell layer erosions, cellular infiltration into the lamina propria, and extensive distortion of the crypts, indicating that RTS did exacerbate the DSS-induced colitic lesions in RTS + DSS mice (Fig. 1B). The histopathologic assessment demonstrated that the degree of intestinal injury in RTS mice was comparable to that in DSS mice, whereas RTS + DSS mice had the highest score, indicating apparently additive toxic effects of RTS and DSS on the intestine (Table 1). Consistent with the histological observations, the small intestine was shortened in DSS, RTS, and RTS + DSS mice (Fig. 1C), indicating pathological changes in the intestines after DSS or RTS treatment. For the large intestine, a trend of colon shortening was also observed in all three treated groups, though a statistically significant shortening was only demonstrated in the DSS and RTS + DSS groups (Fig. 1D).
Table 1.
Histological scoring of intestinal injury.
| Group | Score indexa |
|||||
|---|---|---|---|---|---|---|
| Epitheliumb | Villus shapec | Villus tipd | Stromae | Crypt statusf | Infiltrationg | |
| Control |  |
|
 |
|
|
|
| DSS |  |
 |
 |
|
 |
 |
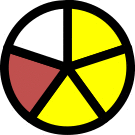
| RTS | |
 |
|
 |
|
 |
| RTS + DSS |  |
 |
|
|
|
 |
Each score index is presented as histology pies based on individual mouse intestine sections. Each pie is divided into slices equal to the number of mice inspected (n = 5 per group).
Graded on: normal (white), flattened or loss of epithelial continuity (yellow), and severe denudation (orange).
Graded on: normal (white), short and rounded (yellow), and extremely short and thick (orange).
Graded on: normal (white), mildly damaged (yellow), and severely damaged (orange).
Graded on: normal (white), retracted (yellow), and severely retracted or denudation (orange).
Graded on: normal (white), amorphic or elongated (yellow), and crypt loss (orange).
Graded on: normal (white), mild infiltration (yellow), and severe infiltration (orange).
3.2. DSS administration increases the gut permeability and endotoxin level in portal blood
With the observation of injured intestines in mice treated with DSS and/or RTS, we then examined whether the DSS and/or RTS treatment caused an increase in intestinal permeability, which may result in exposure of the liver to the gut-derived bacterial antigens. We investigated the effect of DSS and RTS on the quantity and function of mouse intestinal goblet cells. The mucins, stained by AB-PAS, are produced by goblet cells and provide a frontline of intestinal defense against enteric pathogens, while changes in goblet cells and mucin levels influence epithelial barrier function28. Compared with control mice, DSS, RTS, and RTS + DSS mice demonstrated reduced density of goblet cells in mouse small intestines (jejunum, upper panel of Fig. 2A). In mouse colons, DSS and/or RTS treatment reduced the mucin levels compared with vehicle control (lower panel of Fig. 2A). We next compared the tight junction proteins in mouse intestines. Compared with control mice, DSS, RTS, and RTS + DSS mice showed significantly decreased ZO-1 and occludin in both small intestines and large intestines (Fig. 2B and C). These data indicated the loss of intestinal epithelial barrier integrity. Consistently, the intestinal permeability, as measured by the recovery of orally administered FD4000 in serum, was significantly increased in DSS, RTS, and RTS + DSS mice, although the increased level induced by RTS-treated mice was not as high as that in DSS mice (Fig. 2D). This result indicated that long-term treatment with RTS increased the intestinal permeability. However, DSS mice and RTS + DSS mice had similar levels of FD4000 in serum, indicating absence of additive effect between RTS and DSS. To assess whether increased intestinal permeability directly led to hepatic exposure of gut-derived bacterial products, endotoxin levels were further determined in portal blood samples. Both DSS and RTS mice had significantly increased portal endotoxin levels compared with control mice, and RTS + DSS mice had the highest level of portal endotoxin among all groups (Fig. 2E).
Figure 2.

Long-term RTS exposure and intermittent DSS administration disrupted intestinal barrier integrity, increased intestinal permeability and hepatic endotoxin level. Intestine samples from C3H mice in control, DSS, RTS, and RTS + DSS groups were used. (A) The intestinal mucin indicated by AB-PAS staining in mouse jejunum (upper panel) and colon (lower panel) sections. Goblet cells in the jejunum samples are indicated by the green arrowheads, and mucin in the colon is stained blue. Scale bar: 25 μm. (B and C) IHC analysis of tight junction proteins ZO-1 (B) and occludin (C) in mouse jejunum (upper panels) and colon (lower panels) sections. Scale bar: 50 μm. (D) FD4000 concentration in mouse serum samples collected from mouse peripheral blood. (E) Endotoxin concentration in serum samples collected from mouse portal blood. EU, endotoxin units. Data plotted as mean ± SD (n = 5). One-way ANOVA with Tukey's post hoc test was used. #P < 0.05, ####P < 0.0001 versus control; ∗∗P < 0.01, ∗∗∗∗P < 0.0001.
3.3. Hepatic P450 enzymes are essential for PA bioactivation
Because the toxic effects of PAs depend on P450-mediated bioactivation and the formation of reactive PA metabolites, unraveling the site of PA bioactivation is essential for delineating the mode of intoxicating action of PAs in the target organs. In our previous report11, liver P450 enzymes were shown to be essential for PA bioactivation, and the liver-derived reactive PA metabolites were found to be transported into the lungs, where they bind to pulmonary proteins and exert pneumotoxicity. Conversely, suppression of extrahepatic P450 activities had little effect on PA intoxication in the lungs. In this study, we further investigated the roles of hepatic and intestinal P450 enzymes in PA bioactivation. First, a kinetic study was performed to investigate the effect of hepatic P450 deficiency on circulating levels of MCT in MCT-exposed WT, LCN, and IECN mice. After oral administration, MCT was rapidly absorbed in all mouse groups to reach the maximum serum concentration at approximately 30 min (Fig. 3A and Table 2). The kinetic data of WT and LCN mice have been published in our previous study11. In brief, LCN mice had significantly higher Cmax and area under the curve (AUC) values compared with WT mice, indicating a large decrease in hepatic MCT metabolism in LCN mice. However, compared with IECN mice, LCN mice also showed significantly higher Cmax and AUC values, whereas IECN mice had no significant difference in any kinetic parameters compared with WT mice. The data indicated that dysfunctional hepatic P450s largely decreased MCT metabolism, while diminished enteric P450 activity did not alter MCT metabolism. Furthermore, LCN mice also had lower CL/F values compared with IECN mice (by > 2-fold in both males and females), further confirming that deficiency of hepatic P450s resulted in lower efficiency of metabolic clearance of MCT.
Figure 3.

Enterotoxic PA metabolites were generated by hepatic P450 metabolism. (A) Toxicokinetic profiles of serum MCT in WT, LCN, and IECN mice. Adult male and female mice of various genotypes were treated with a single oral administration of MCT via gavage at 120 mg/kg. Blood (20–25 μL) was collected via the tail vein at various times (0–24 h) for the analysis of serum MCT levels. Data plotted as mean ± SD (n = 3). (B) Pyrrole–protein adducts levels in serum, liver, and intestine samples collected from WT, LCN, and IECN mice at 48 h after oral treatment with MCT at 120 mg/kg or saline (vehicle control). Data plotted as mean ± SD (n = 5). <LOQ, below limit of quantification. Two-way ANOVA with Tukey's post hoc test was used. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. ns, not significant. Data of toxicokinetic parameter, and serum, and liver pyrrole–protein adducts of WT and LCN mice have been published11.
Table 2.
Toxicokinetic parameters of mouse serum MCT after a single oral administration of MCT at 120 mg/kg. Cmax, maximum peak concentration; AUC0–t, the area under the concentration versus time curve from zero to the last time point tested; t1/2, elimination half-life of serum MCT concentration; CL/F, apparent clearance rate. Data from Fig. 3A are used to calculate toxicokinetic parameters. Data are presented as mean ± SD (n = 3). Two-way ANOVA followed by Tukey's post hoc test was used.
| Toxicokinetic parameter | Male |
Female |
||||
|---|---|---|---|---|---|---|
| WT∗ | LCN∗ | IECN | WT∗ | LCNa | IECN | |
| Cmax (μg/mL) | 4.06 ± 0.87 | 11.2 ± 3.1a,b | 5.31 ± 0.61 | 4.24 ± 0.40 | 10.1 ± 1.7c,d | 4.45 ± 1.53 |
| AUC0–t (min × μg/mL) | 189 ± 23 | 746 ± 258e,f | 217 ± 52 | 207 ± 41 | 526 ± 46d,g | 196 ± 29 |
| t1/2 (min) | 56.5 ± 38.4 | 60.5 ± 16.0 | 60.4 ± 39.3 | 55.1 ± 36.4 | 48.2 ± 23.1 | 49.2 ± 30.0 |
| CL/F (mL/min) | 24.2 ± 2.1 | 6.62 ± 1.9b,e | 17.6 ± 5.2 | 17.4 ± 3.2 | 6.92 ± 0.29g,h | 18.3 ± 3.3 |
∗Kinetic data of WT and LCN mice have been published11.
aP<0.01, eP < 0.001 versus WT male mice; bP < 0.01, fP < 0.001 versus IECN male mice; gP < 0.05, cP < 0.01 versus WT female mice; dP < 0.05, hP < 0.01 versus IECN female mice.
The formation of pyrrole–protein adducts is an indicator of P450-mediated metabolic activation of PAs and a direct cause of PA-induced cytotoxicity8. The respective roles of the hepatic and intestinal P450s in the conversion of MCT into the reactive metabolites were determined by comparing levels of pyrrole–protein adducts in WT, LCN, and IECN mice. After MCT exposure, slightly lower levels of pyrrole–protein adducts were found in serum, liver, and intestine samples from IECN mice compared with those from WT mice (Fig. 3B), while no significant difference was observed among these samples except for male IECN mice which showed significantly lower hepatic pyrrole–protein adducts level than male WT mice. The data indicated that the substantial decrease in intestinal P450 activity in IECN mice did not have a major effect on the generation of reactive metabolites of MCT. In contrast, compared with WT mice and IECN mice, LCN mice had significantly lower levels (by > 60%) of pyrrole–protein adducts in all (serum, liver, and intestine) samples, a result indicating that liver Cpr deletion markedly inhibited the metabolic activation of MCT. Of note, IECN mice, with comparable hepatic P450 activity but much absence of intestinal P450 activity to WT mice, had only a slightly decreased level of pyrrole–protein adducts detected in the liver (~1.9 pmol/mg protein in IECN versus ~2.4 pmol/mg protein in WT) and the intestine (~1.1 pmol/mg protein in IECN versus ~1.4 pmol/mg protein in WT). Collectively, these data demonstrated that the production of the reactive MCT metabolite was predominantly mediated by hepatic P450s. Furthermore, the finding of comparable pyrrole–protein adducts levels in the intestine of WT and IECN mice suggested that the reactive PA metabolites generated in the liver could be transported to the intestine to exert toxicity.
3.4. Reactive PA metabolites generated in the livers are transferred into the intestine via bile excretion
We further explored the pathway through which liver-derived reactive PA metabolites were transported from the liver into the intestine. To directly prove this, we exposed rats to RTS. Rats were used, instead of mice, to facilitate surgical collection of bile secreted from the liver. The reactive metabolites of PAs are very unstable and nearly impossible to directly detect29. We measured the reactive metabolites of RTS by adding GSH, as a trapping reagent30, to freshly collected bile to form stable pyrrole–GSH conjugates (Fig. 4A). LC–MS/MS detection of pyrrole–GSH conjugates confirms the presence of reactive RTS metabolites. Over the 120-min sampling period, pyrrole–GSH conjugates were detected in both saline-added bile and GSH-added bile collected at all time intervals, indicating the formation of pyrrole–GSH conjugates after RTS exposure. More importantly, compared with the saline-added counterparts, the addition of GSH to the bile of RTS-exposed rats led to significant increases in biliary concentrations of pyrrole–GSH conjugates (Fig. 4B) especially within 0–60 min, a result confirming the presence of the reactive RTS metabolites in bile.
Figure 4.

Liver-derived PA metabolites were transported via bile from the liver into the gut. (A) Sprague–Dawley rats (4-month old, male) received a single intravenous injection of 40 mg/kg RTS, and bile samples were collected before RTS administration (at 0 min) and during 0–15, 15–30, 30–45, 45–60, 60–75, 75–90, 90–105, and 105–120 min after RTS administration and processed as illustrated. (B) The concentrations of pyrrole–GSH conjugates were determined in saline-added or paired GSH-added bile samples. Data plotted as mean ± SD (n = 3). Student's t-test was used. ∗P < 0.05, ∗∗P < 0.01. <LOQ, below limit of quantification.
3.5. DSS administration exacerbates the chronic liver injury induced by RTS
3.5.1. Biochemical index
We next assessed the toxicological effects of long-term exposure to RTS and/or DSS in mice. For both serum ALT activity and bilirubin level, there were no differences between control and DSS mice. After the 14-week RTS treatment, the serum ALT activity of RTS mice was elevated compared to control mice, although the difference between these groups did not reach a statistical significance. A significant ALT activity elevation was observed in RTS + DSS mice compared with control mice (Fig. 5A), suggesting that DSS exacerbated RTS-induced hepatotoxicity. In addition, compared to control mice, the serum levels of bilirubin were significantly increased in RTS and RTS + DSS mice but not in DSS mice, and RTS + DSS mice did not differ from RTS mice in the bilirubin level (Fig. 5B). The results indicate that RTS exposure mainly contributes to cholestasis in mice.
Figure 5.

DSS administration exacerbated the chronic liver injury induced by long-term RTS exposure in mice. Serum and liver samples from C3H mice in control, DSS, RTS, and RTS + DSS groups were used. (A) ALT activity and (B) bilirubin level in mouse serum samples. Data plotted as mean ± SD (n = 5). One-way ANOVA with Tukey's post hoc test was used. ##P < 0.01, ###P < 0.001 versus control. ns, not significant. (C) Representative histological changes indicated by H&E staining of paraffin-embedded mouse liver sections. Arrowhead indicates inflammatory neutrophil infiltration. Star (∗) indicates cellular dysplasia. Plus (+) indicates hemorrhage. Scale bar: 200 μm. (D) Representative scanning electron microscopy images of the mouse hepatic sinusoids. Arrows (→) indicate fenestration gaps. Scale bar: 3.00 μm.
3.5.2. Liver histological changes
Significant liver lesions were observed in mice exposed to RTS for 14 weeks (Fig. 5C). Compared with control mice, the major pathological finding in RTS mice was cytologic dysplasia of hepatocytes, represented by cytomegaly (increased cytoplasmic volume) and karyomegaly (enlarged nuclei with marginated chromatin). Cellular changes resulted in disruption of the hepatic cord architecture, manifested by distended hepatocytes with vacuolated cytoplasm, which further induced the mild to moderate proliferative lesions and bile duct hyperplasia in RTS mice. Additionally, necrotic foci were observed in RTS mice manifested by inflammatory neutrophil infiltration localized in the centrilobular region. Moreover, focal hemorrhage combined with the coagulative lesion was observed along with dilated centrilobular sinusoids in RTS mice. Compared with control mice, DSS-treated mice showed slight hepatic pathological changes. However, RTS + DSS mice demonstrated more severe liver lesions manifested by pronounced necrotic changes and cytologic alterations in comparison to RTS mice. The degrees of representative pathological lesions of the mouse liver sections were assessed and scored as previously described25,31. The scoring results are summarized in Table 3. RTS resulted in significant liver injury in most treated mice, while DSS treatment alone induced only mild cellular infiltration in mouse livers. Of note, DSS significantly enhanced the chronic liver lesions induced by RTS.
Table 3.
Histological scoring of liver injury.
| Group | Score indexa |
||||
|---|---|---|---|---|---|
| Endothelial cell damage | Focal hemorrhage | Mixed cellular infiltration | Hepatocyte hyperplasia | Steatotic hepatocyte | |
| Control | |
|
|
|
|
| DSS | |
|
 |
|
|
| RTS |  |
 |
 |
 |
|
| RTS + DSS | |
|
|
|
|
Each score index is presented as histology pies based on individual mouse liver sections. Each pie is divided into slices equal to the number of mice inspected (n = 5 per group) and scored based on the severity of individual liver lesions: normal (white), moderate lesion (yellow), and severe lesion (orange). Detailed scoring method is available in Supporting Information Table S1.
3.5.3. Changes in liver sinusoidal endothelium
Because hepatic microvascular injury derived from sinusoidal endothelial cell damage has been reported to be the typical intoxication outcome of PAs25,32, we next assessed hepatic microvascular changes after RTS and DSS treatments. The normal sinusoidal endothelium in control mice was characterized by scattered fenestrae-organized sieve plates with diameters of 100–500 nm, which are formed by lining of sinusoidal endothelial cells (Fig. 5D). Compared to control mice, RTS mice exhibited a reduced number of fenestrae in liver sinusoids. Additionally, small holes or gaps developed among sinusoidal endothelium in RTS mice. The data indicated that long-term RTS exposure caused sinusoidal endothelial cell damage and sinusoidal wall destruction. Conversely, the DSS treatment alone did not cause remarkable sinusoid endothelium damage. However, compared with RTS mice, RTS + DSS mice demonstrated significantly larger gaps in sinusoidal endothelium. The results reveal that although DSS treatment alone could not induce hepatic vasculature damage, co-exposure to DSS and RTS causes more extensive hepatic sinusoid injury than RTS exposure alone.
3.5.4. Liver fibrosis
Control mice exhibited Sirius red-positive collagen only around large hepatic vessels, whereas RTS mice developed disseminated collagen accumulation in peri-vascular areas (Fig. 6A). DSS mice also showed no significant fibrotic collagen. However, the livers of RTS + DSS mice demonstrated diffusive collagen deposition particularly in sub-endothelial and adventitial areas (Fig. 6A). The quantitative results of collagen-stained area indicated mild to moderate fibrosis (1.2%–1.5%) in the livers of RTS mice, but severe fibrosis (1.8%–2.4%) in the livers of RTS + DSS mice (Fig. 6B).
Figure 6.

DSS administration exacerbated liver fibrosis and steatosis induced by long-term RTS exposure in mice. Liver samples from C3H mice in control, DSS, RTS, RTS + DSS groups were used. (A) Representative images of paraffin-embedded mouse liver sections in which fibrotic collagen was stained by Sirius red. Scale bar: 200 μm. (B) Percentage of Sirius red stained area in mouse liver sections. (C) Representative images of cryosections of mouse liver in which lipid and fatty acids were stained by oil red O. Scale bar: 200 μm. (D) Percentage of oil red O stained area in mouse liver sections. Data plotted as mean ± SD (n = 5). One-way ANOVA with Tukey's post hoc test was used. ####P < 0.0001 versus control; ∗∗∗∗P < 0.0001.
3.5.5. Steatosis
The H&E stained liver sections demonstrated ballooned hepatocytes with vacuolated appearance in RTS mice and RTS + DSS mice (Fig. 5C), suggesting the presence of steatotic lesions. Hence, we performed oil red O staining to confirm and assess the lipid accumulation in hepatocytes with representative images shown in Fig. 6C. In contrast to control mice, RTS mice demonstrated hepatic lipid droplet accumulation in the liver, indicating that steatosis has developed after the 14-week RTS treatment. Notably, the steatotic hepatocytes were more prominent in RTS + DSS mice, which had both microvesicular and macrovesicular appearance, compared to RTS mice. The quantitative results of lipid accumulation demonstrated that the area occupied by fat was significantly increased in RTS mice compared to control mice (averaging 17% versus 5%). Whereas, compared to the mild degree of steatosis in RTS mouse livers, lipid accumulation was substantially increased in the livers of RTS + DSS mice (averaging 29% versus 17%) (Fig. 6D).
3.6. Chronic DSS administration does not further reduce hepatic CYP3A activity or PA bioactivation in RTS-exposed mice
Because hepatic CYP3A4 (CYP3A11 isoform in mice) is essential for PA bioactivation, we next examined whether long-term RTS or DSS administration altered the mouse hepatic CYP3A11 activity. Mouse liver S9 fractions, which were prepared from different groups of mice, had significantly higher activity in the generation of oxidized nifedipine (known metabolite of nifedipine by mouse CYP3A11) in control mice compared to all three treated groups of mice, while there was no difference between RTS mice and RTS + DSS mice (Fig. 7A). Consistently, the levels of generated RTS N-oxide (known metabolite of RTS by mouse CYP3A11) and pyrrole–protein adducts (products of PA bioactivation by mouse CYP3A11) were also lower in DSS- and/or RTS-treated mouse liver S9 fractions compared to control mice, but were the same between RTS mice and RTS + DSS mice (Fig. 7B and C). Collectively, the data indicated that CYP3A11 activity was significantly reduced by the long-term DSS treatment, however the degree of CYP3A11 reduction caused by DSS was not affected by concurrent exposure of RTS.
Figure 7.

Intermittent DSS administration did not change the hepatic P450 activity in RTS-exposed mice. Liver S9 fractions were prepared from C3H mice in control, DSS, RTS, RTS + DSS groups after the 14-week study. S9 fractions (equivalent of 2 mg protein/mL) were incubated with nifedipine (50 μmol/L) or RTS (100 μmol/L) in reaction mixtures containing 100 mmol/L potassium phosphate buffer (pH 7.4), 1 mmol/L nicotinamide adenine dinucleotide phosphate hydride, and 5 mmol/L magnesium chloride, in a final volume of 400 μL, at 37 °C for 40 min with nifedipine or 60 min with RTS. The reactions were terminated by adding 800 μL of ice-cold acetonitrile. The levels of (A) oxidized nifedipine and (B) RTS N-oxide were measured in the supernatants of the reaction mixture. The pyrrole–protein adducts (C) were measured in the precipitated protein pellets. De-S9 denotes denatured mouse liver S9. Data plotted as mean ± SD (n = 3 independent duplicates). One-way ANOVA with Tukey's post hoc test was used. #P < 0.05, ##P < 0.01, ###P < 0.001 versus the S9 fraction from control mice. ns, not significant. <LOQ, below limit of quantitation.
3.7. DSS administration exacerbates oxidative stress and enhances inflammation in RTS-exposed mice
Previous studies demonstrated hepatic GSH as an important detoxification molecule for not only PAs32, but also endotoxin33. To assess the hepatic detoxification capability, we measured hepatic GSH level. Compared with control mice, DSS treatment did not affect the hepatic GSH level in mice; whereas, RTS mice and RTS + DSS mice had significantly lower hepatic GSH level compared with control mice, but no significant difference was observed between these two groups (Fig. 8A). GSH acts as a potent anti-oxidant and is converted to its oxidized GSSG form upon sequestering free radicals34. RTS mice demonstrated a trend of higher GSSG:GSH ratio compared to control mice, and RTS + DSS mice showed significantly elevated GSSG:GSH ratio compared to both control and RTS mice, indicating exacerbated hepatic oxidative stress by DSS (Fig. 8B). Consistently, RTS + DSS mice also demonstrated significantly higher expression of proinflammatory cytokines (Tnfa, Il6, and Ifng) compared to RTS mice (Fig. 8C). Collectively, these data indicated that DSS administration exacerbated the hepatic oxidative damage and inflammation in RTS-exposed mice.
Figure 8.

Intermittent DSS administration markedly decreased the hepatic GSH level and increased oxidative stress. Liver samples from C3H mice in control, DSS, RTS, RTS + DSS groups were used. The contents of reduced GSH (A) and ratio of GSSG:GSH (B) in mice livers. (C) Relative mRNA levels of inflammatory cytokines (Il6, Ifng, and Tnfa) in mice livers. Data plotted as mean ± SD (n = 5). One-way ANOVA with Tukey's post hoc test was used. #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus control. ∗P < 0.05, ∗∗∗P < 0.001. ns, not significant. <LOQ, below limit of quantitation.
4. Discussion
Humans are frequently exposed to dietary PAs via the intake of PA-contaminated foods such as honey and milk, which are derived from the carry-over PAs in PA-exposed livestock, and this type of intermittent, low-level dietary exposure is likely to have occurred in the past and seems increasingly to continue to the present day to varying degrees in many regions worldwide5,35. Delineation of the chronic toxicological behavior of PAs is essential for risk management of these widely available phytotoxins. In this study, we have provided the first mode-of-action evidence on the enterotoxicity of liver-derived reactive PA metabolites and the hepatotoxicity-enhancing effect of gut-derived endotoxins.
We characterized PA-induced mouse intestinal injury and found that the lesions were similar to the DSS-induced intestinal injury which resembles the pathological lesions, though not the etiology, of human ulcerative colitis36. This result suggests that PA-induced intestinal injury may contribute to the development of IBD, of which the etiology is not fully understood. Notably, apart from the DSS model, other animal models of IBD exist, such as the interleukin-10 knockout mouse, which may present additional pathological features as well as etiological factors of human IBD37. Future studies incorporating PA exposure in these animal models are warranted to fully explore the role of PA exposure in IBD pathogenesis.
Considering that P450-mediated metabolic activation of PAs is required for their toxic effect7, we further delineated the metabolic mechanisms responsible for PA-induced enterotoxicity. Although P450 enzymes are most abundant in the liver, some extrahepatic tissues, such as the lungs and small intestine, have been reported to be highly efficient in target-tissue bioactivation of xenobiotics23,38,39. Cpr-null mouse models that are devoid of P450 activity in an organ-specific fashion enable the delineation of the roles of organ-specific P450s in PA metabolism21,22. Previously we have demonstrated that extrahepatic P450 enzymes play a minor role in PA bioactivation11. In this study, we further compared the influences of tissue-specific suppression of hepatic and intestinal P450 activity on PA metabolism. Compared with WT and IECN mice, hepatic P450 deficiency in LCN mice led to dramatically lower clearance and higher serum concentrations of PA, while the suppression of intestinal P450 activity in IECN mice did not significantly influence PA metabolism, as indicated by the essentially comparable toxicokinetic parameters between IECN and WT mice. The pyrrole–protein adducts analysis confirmed the decreased PA bioactivation in LCN mice. Comparable levels of pyrrole–protein adducts were found in WT and IECN mice, a result suggesting that intestinal P450 activity does not have a dominant role in the overall PA bioactivation in vivo. Notably, the reason for a lack of contribution of intestinal P450, particularly CYP3A, may be at least in part due to the rapid absorption of PA in the intestine40, which results in only limited opportunity for intestinal CYP3A to act. Compared to WT and IECN mice, LCN mice showed a significant decrease in the generation of pyrrole–protein adducts, a result indicating that hepatic P450 ablation resulted in remarkable inhibition of the metabolic activation of PAs. Notably, because the pyrrole–protein adducts are the basis of PA-induced cytotoxicity8, their presence in the intestines of IECN mice supports the concept that liver-produced reactive metabolites are responsible for the PA-induced intestinal injury. For the liver-derived reactive PA metabolites, our previous report has demonstrated that circulating blood can carry them to other organs (e.g., the lungs) to exert toxicity11. In this study, we further revealed the presence of reactive PA metabolites in bile efflux from the livers of PA-exposed rats by exogenously adding GSH to trap the reactive PA metabolites to form detectable pyrrole–GSH conjugates. The finding indicated that the bile served as the carrier for transport of the liver-derived reactive PA metabolites to the intestine. The finding is consistent with the histological findings in mouse intestines. Because the small intestine receives these bile-borne reactive PA metabolites ahead of the large intestine, it can be extrapolated that the small intestine proteins sequester these reactive metabolites in the first place, the large intestine thus receives less PA metabolites and thereby demonstrates weaker intoxication outcomes compared to the small intestine. Consistently, compared to DSS mice, RTS mice showed comparable lesions in small intestines but less severe damage in large intestines. Furthermore, with the same dose of two PA compounds (0.2 mmol/kg RTS or MCT) in rats, the concentration of pyrrole–GSH conjugates found in blood in our previous work11 or in bile in this study are similar, which indicates that the reactive PA metabolites formed in the livers are diffused into blood and bile without directional preference. Collectively, apart from the reported blood transport, the bile transport of reactive PA metabolites found in this study reveals another source for PA-induced enterotoxicity. To this end, our data suggest that PA-induced intestinal injury is due to hepatic P450-mediated metabolic activation to reactive PA metabolites, which are transported by blood and bile into the intestine and exert enterotoxicity. Future development of analytical approaches for direct measurement of reactive PA metabolites in vivo (e.g., in blood or bile samples) is needed to further support this conclusion.
In support of the gut–liver axis concept, we have proved the toxic effect of liver-derived reactive PA metabolites on the intestine, and we also delineated the impact of gut-derived bacterial antigens on PA-induced liver injury. Dietary PA exposure in prolonged periods is unavoidably accompanied with other diet-related pathologies, among which IBD is one of the most common diseases in the digestive system14. The utility of a DSS mouse model helps to reveal the impact of gut-derived pathological factors, such as a compromised gut barrier and the increases in endotoxin exposure, on PA-induced hepatotoxicity. In this study, though we found no significant liver injury in mice with DSS-induced chronic colitis, we discovered that DSS treatment substantially enhanced the RTS-induced liver injury manifested by extensive loss of sinusoidal fenestrae, increased fibrotic collagen and lipid deposition. We then investigated the mechanisms underlying the impact of disrupted gut homeostasis on PA intoxication and detoxification. Among all gut-derived pathological factors, endotoxins produced by Gram-negative bacteria have been reported to induce hepatocyte death and aggravate inflammatory liver injury41. Because the gut epithelium acts as the first barrier against the translocation of gut microbiota and bacterial products, normally only a few gut-derived pathogens can reach the liver in a healthy state42. However, the gut barrier can be disrupted by various pathological conditions such as IBD. The leaky gut hypothesis of IBD links the translocation of microbial products with the onset and progression of liver disease20. Endotoxins are considered as one of the critical co-factors of hepatopathogenesis43. Here we observed reduced goblet cell density and impaired intestinal tight junction proteins after DSS and/or RTS treatment, which indicated that the goblet cell-derived mucous membrane in intestinal epithelial surface was disrupted28. As a consequence of this impaired mechanical protective barrier, the intestinal permeability was significantly increased, and hepatic endotoxin level was elevated in DSS-treated mice. Moreover, the gut-derived bacterial antigens, typically endotoxins, enter the liver wherein they are in immediate contact with hepatic sinusoids, while the fenestrae of sinusoidal endothelium permit the substance interaction with parenchymal hepatocytes and hepatic immune cells44. Hepatic sinusoidal endothelial cells and Kupffer cells, which reside in hepatic sinusoids, act as scavengers for gut-derived pathogens by forming the reticulo-endothelial system45,46. Hence in physiological conditions, no obvious liver injury is induced by gut-derived microbial products transferred into the liver. Here we found that RTS treatment caused hepatic sinusoidal endothelium damage represented by fenestrae loss and gap formation. Although DSS treatment alone did not induce marked sinusoid injury, it synergistically enhanced the sinusoidal endothelium damage caused by RTS, which indicated deficient cleansing function of the reticulo-endothelial system in RTS + DSS mice. This explains that RTS increased the mouse gut permeability to a lesser extent than DSS, but RTS increased the hepatic endotoxin level in comparable level with DSS, which could be ascribed to the compromised sinusoidal endotoxin clearance in RTS mice. Collectively, the first pathway responsible for the exacerbated liver injury in RTS + DSS mice is likely due to i) the elevated gut-derived endotoxins caused by increased intestinal permeability and ii) the compromised endotoxin clearance capability caused by damage in hepatic sinusoids.
Furthermore, as being reported in our previous work11 and validated in this study, hepatic P450 enzymes are critical for PA intoxication, and it has also been contemplated that altered CYP3A4 activity could affect the PA bioactivation and resulting intoxication outcomes7. Previous studies implicated that DSS-induced colitis could downregulate the expression of P450s and inhibit the activity of CYP3A4 in the acute context23,47. RTS has been reported to inhibit CYP3A11 activity48. In chronic situations, as in this study, the CYP3A11 (CYP3A4 isoform in humans) activity was reduced in RTS, DSS, and RTS + DSS mice to a similar extent. Although the reason for no significant enhancement on CYP3A11 inhibitory effect in the concurrent exposure to DSS and RTS is unknown and needs to be further investigated, we can at least exclude the possibility that the hepatotoxicity difference between these two groups was derived from varied PA bioactivation rate. On the other hand, although DSS did not affect the hepatic generation of reactive PA metabolites, it significantly compromised the hepatic GSH-mediated PA detoxification32. Hepatotoxins, such as PAs and endotoxins, could induce the imbalance of normal redox state in liver microenvironment, thereby exerting toxicity by producing free radicals, and GSH plays an important role in counteracting such oxidative stress49. In this study, DSS treatment synergistically reduced the GSH-mediated detoxifying capability and enhanced hepatic oxidative stress in mouse livers, thereby increasing their susceptibility to PA intoxication.
5. Conclusions
This study highlights the importance of the gut–liver axis in PA-induced liver and intestinal injury. We provide the first mode-of-action evidence for a major role of hepatic P450 enzymes, and bile transport of liver-derived reactive metabolites in PA-induced enterotoxicity. Considering the prevalence of PA-contaminated food, our finding warrants further investigation into the possible roles of PA exposure in human gut diseases, especially IBD with unknown or unexplained causes. In addition, this study reveals that enterogenous pathological factors, in turn, exacerbate PA-induced liver injury via depleting GSH and increasing endotoxin level in the liver. These findings suggest reducing dietary PA exposure and maintaining gut homeostasis as preventive interventions against chronic PA intoxication.
Acknowledgments
This work was supported by Research Grants Council of Hong Kong Special Administrative Region (GRF Project Nos. 14160817 and 14106318 to Ge Lin, China), and a grant from the National Institutes of Health (No. R01 GM082978 to Qing-Yu Zhang, USA). We thank Ms. Weizhu Yang for assistance with mouse breeding and genotyping analysis, Dr. Lei Yin and Dr. Xiangmeng Wu for assistance with analytical instruments, and Dr. Weiguo Han and Dr. Nataliia Kovalchuk for assistance with histological assays.
Author contributions
Yisheng He, Qing-Yu Zhang, and Ge Lin conceived the idea and designed the research studies. Yisheng He, Jiang Ma, Xiaoyu Fan, and Liang Ding conducted experiments and acquired data. Yisheng He and Jiang Ma performed data analysis and generated figures. Yisheng He, Jiang Ma, and Xinxin Ding wrote the manuscript. Xinxin Ding, Qing-Yu Zhang, and Ge Lin gave critical review of the manuscript.
Conflicts of interest
The authors report no conflicts of interest. The authors are responsible for the content and writing of this article.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.07.013.
Contributor Information
Qing-Yu Zhang, Email: qyzhang@pharmacy.arizona.edu.
Ge Lin, Email: linge@cuhk.edu.hk.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Jank B., Rath J. The risk of pyrrolizidine alkaloids in human food and animal feed. Trends Plant Sci. 2017;22:191–193. doi: 10.1016/j.tplants.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Robertson J., Stevens K. Pyrrolizidine alkaloids: occurrence, biology, and chemical synthesis. Nat Prod Rep. 2017;34:62–89. doi: 10.1039/c5np00076a. [DOI] [PubMed] [Google Scholar]
- 3.Lin G., Wang J.Y., Li N., Li M., Gao H., Ji Y.A., et al. Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J Hepatol. 2011;54:666–673. doi: 10.1016/j.jhep.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L., Zhang C.Y., Li D.P., Chen H.B., Ma J., Gao H., et al. Tu-San-Qi (Gynura japonica): the culprit behind pyrrolizidine alkaloid-induced liver injury in China. Acta Pharmacol Sin. 2021;42:1212–1222. doi: 10.1038/s41401-020-00553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prakash A.S., Pereira T.N., Reilly P.E.B., Seawright A.A. Pyrrolizidine alkaloids in human diet. Mutat Res Genet Toxicol. 1999;443:53–67. doi: 10.1016/s1383-5742(99)00010-1. [DOI] [PubMed] [Google Scholar]
- 6.He Y., Shi M., Wu X., Ma J., Ng K.T., Xia Q., et al. Mutational signature analysis reveals widespread contribution of pyrrolizidine alkaloid exposure to human liver cancer. Hepatology. 2021;74:264–280. doi: 10.1002/hep.31723. [DOI] [PubMed] [Google Scholar]
- 7.Ruan J., Yang M., Fu P., Ye Y., Lin G. Metabolic activation of pyrrolizidine alkaloids: insights into the structural and enzymatic basis. Chem Res Toxicol. 2014;27:1030–1039. doi: 10.1021/tx500071q. [DOI] [PubMed] [Google Scholar]
- 8.Ma J., Ruan J., Chen X., Li D., Yao S., Fu P.P., et al. Pyrrole–hemoglobin adducts, a more feasible potential biomarker of pyrrolizidine alkaloid exposure. Chem Res Toxicol. 2019;32:1027–1039. doi: 10.1021/acs.chemrestox.8b00369. [DOI] [PubMed] [Google Scholar]
- 9.Lin G., Cui Y.Y., Hawes E.M. Microsomal formation of a pyrrolic alcohol glutathione conjugate of clivorine firm evidence for the formation of a pyrrolic metabolite of an otonecine-type pyrrolizidine alkaloid. Drug Metab Dispos. 1998;26:181–184. [PubMed] [Google Scholar]
- 10.Lame M.W., Jones A.D., Morin D., Segall H.J., Wilson D.W. Biliary excretion of pyrrolic metabolites of [14C]monocrotaline in the rat. Drug Metab Dispos. 1995;23:422–429. [PubMed] [Google Scholar]
- 11.He Y., Lian W., Ding L., Fan X., Ma J., Zhang Q.Y., et al. Lung injury induced by pyrrolizidine alkaloids depends on metabolism by hepatic cytochrome P450s and blood transport of reactive metabolites. Arch Toxicol. 2021;95:103–116. doi: 10.1007/s00204-020-02921-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Z., He Y., Ma J., Fu P.P., Lin G. Pulmonary toxicity is a common phenomenon of pyrrolizidine alkaloids. J Environ Sci Health C. 2020;38:124–140. doi: 10.1080/26896583.2020.1743608. [DOI] [PubMed] [Google Scholar]
- 13.Nolan J.P. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 14.Ng S.C., Shi H.Y., Hamidi N., Underwood F.E., Tang W., Benchimol E.I., et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 15.Mawdsley J.E., Rampton D.S. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut. 2005;54:1481–1491. doi: 10.1136/gut.2005.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dieleman L.A., Palmen M.J., Akol H., Bloemena E., Pena A.S., Meuwissen S.G., et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thaker A.I., Shaker A., Rao M.S., Ciorba M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS) 2012;67 doi: 10.3791/4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams J.M., Duckworth C.A., Vowell K., Burkitt M.D., Pritchard D.M. Intestinal preparation techniques for histological analysis in the mouse. Curr Protoc Mouse Biol. 2016;6:148–168. doi: 10.1002/cpmo.2. [DOI] [PubMed] [Google Scholar]
- 19.Nadatani Y., Watanabe T., Tanigawa T., Machida H., Okazaki H., Yamagami H., et al. High mobility group box 1 promotes small intestinal damage induced by nonsteroidal anti-inflammatory drugs through Toll-like receptor 4. Am J Pathol. 2012;181:98–110. doi: 10.1016/j.ajpath.2012.03.039. [DOI] [PubMed] [Google Scholar]
- 20.Cani P.D., Bibiloni R., Knauf C., Waget A., Neyrinck A.M., Delzenne N.M., et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 21.Gu J., Weng Y., Zhang Q.Y., Cui H.D., Behr M., Wu L., et al. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q.Y., Fang C., Zhang J., Dunbar D., Kaminsky L., Ding X. An intestinal epithelium-specific cytochrome P450 (P450) reductase-knockout mouse model: direct evidence for a role of intestinal p450s in first-pass clearance of oral nifedipine. Drug Metab Dispos. 2009;37:651–657. doi: 10.1124/dmd.108.025429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan X., Ding X., Zhang Q.Y. Hepatic and intestinal biotransformation gene expression and drug disposition in a dextran sulfate sodium-induced colitis mouse model. Acta Pharm Sin B. 2020;10:123–135. doi: 10.1016/j.apsb.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruan J., Gao H., Li N., Xue J., Chen J., Ke C., et al. Blood pyrrole–protein adducts--a biomarker of pyrrolizidine alkaloid-induced liver Injury in humans. J Environ Sci Health C. 2015;33:404–421. doi: 10.1080/10590501.2015.1096882. [DOI] [PubMed] [Google Scholar]
- 25.DeLeve L.D., McCuskey R.S., Wang X., Hu L., McCuskey M.K., Epstein R.B., et al. Characterization of a reproducible rat model of hepatic veno-occlusive disease. Hepatology. 1999;29:1779–1791. doi: 10.1002/hep.510290615. [DOI] [PubMed] [Google Scholar]
- 26.Angulo P., Hui J.M., Marchesini G., Bugianesi E., George J., Farrell G.C., et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–854. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 27.Rahman I., Kode A., Biswas S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 28.Linden S.K., Sutton P., Karlsson N.G., Korolik V., McGuckin M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008;1:183–197. doi: 10.1038/mi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huxtable R.J., Yan C.C., Wild S., Maxwell S., Cooper R. Physicochemical and metabolic basis for the differing neurotoxicity of the pyrrolizidine alkaloids, trichodesmine and monocrotaline. Neurochem Res. 1996;21:141–146. doi: 10.1007/BF02529131. [DOI] [PubMed] [Google Scholar]
- 30.Mattocks A.R., Jukes R. Trapping and measurement of short-lived alkylating-agents in a recirculating flow system. Chem Biol Interact. 1990;76:19–30. doi: 10.1016/0009-2797(90)90031-h. [DOI] [PubMed] [Google Scholar]
- 31.Chan P.C., Mahler J., Bucher J.R., Travlos G.S., Reid J.B. Toxicity and carcinogenicity of riddelliine following 13 weeks of treatment to rats and mice. Toxicon. 1994;32:891–908. doi: 10.1016/0041-0101(94)90368-9. [DOI] [PubMed] [Google Scholar]
- 32.Yang M., Ruan J., Fu P.P., Lin G. Cytotoxicity of pyrrolizidine alkaloid in human hepatic parenchymal and sinusoidal endothelial cells: firm evidence for the reactive metabolites mediated pyrrolizidine alkaloid-induced hepatotoxicity. Chem Biol Interact. 2016;243:119–126. doi: 10.1016/j.cbi.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 33.Spolarics Z. Endotoxin stimulates gene expression of ROS-eliminating pathways in rat hepatic endothelial and Kupffer cells. Am J Physiol. 1996;270:G660–G666. doi: 10.1152/ajpgi.1996.270.4.G660. [DOI] [PubMed] [Google Scholar]
- 34.Pompella A., Visvikis A., Paolicchi A., De Tata V., Casini A.F. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–1503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 35.He Y., Zhu L., Ma J., Wong L., Zhao Z., Ye Y., et al. Comprehensive investigation and risk study on pyrrolizidine alkaloid contamination in Chinese retail honey. Environ Pollut. 2020;267:115542. doi: 10.1016/j.envpol.2020.115542. [DOI] [PubMed] [Google Scholar]
- 36.Singh S., Stroud A.M., Holubar S.D., Sandborn W.J., Pardi D.S. Treatment and prevention of pouchitis after ileal pouch-anal anastomosis for chronic ulcerative colitis. Cochrane Database Syst Rev. 2015;11:CD001176. doi: 10.1002/14651858.CD001176.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Deventer S.J., Elson C.O., Fedorak R.N. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn's disease. Crohn's Disease Study Group. Gastroenterology. 1997;113:383–389. doi: 10.1053/gast.1997.v113.pm9247454. [DOI] [PubMed] [Google Scholar]
- 38.Ding X., Kaminsky L.S. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 39.Xie F., Ding X., Zhang Q.Y. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016;6:374–383. doi: 10.1016/j.apsb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang M., Ma J., Ruan J., Zhang C., Ye Y., Pi-Cheng Fu P., et al. Absorption difference between hepatotoxic pyrrolizidine alkaloids and their N-oxides - mechanism and its potential toxic impact. J Ethnopharmacol. 2020;249:112421. doi: 10.1016/j.jep.2019.112421. [DOI] [PubMed] [Google Scholar]
- 41.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altin J.G., Bygrave F.L. Non-parenchymal cells as mediators of physiological responses in liver. Mol Cell Biochem. 1988;83:3–14. doi: 10.1007/BF00223193. [DOI] [PubMed] [Google Scholar]
- 43.Crispe I.N. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 44.Racanelli V., Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 45.Knook D.L., Sleyster E.C. Isolated parenchymal, Kupffer and endothelial rat liver cells characterized by their lysosomal enzyme content. Biochem Biophys Res Commun. 1980;96:250–257. doi: 10.1016/0006-291x(80)91207-3. [DOI] [PubMed] [Google Scholar]
- 46.Smedsrod B., Melkko J., Araki N., Sano H., Horiuchi S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem J. 1997;322:567–573. doi: 10.1042/bj3220567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masubuchi Y., Enoki K., Horie T. Down-regulation of hepatic cytochrome P450 enzymes in rats with trinitrobenzene sulfonic acid-induced colitis. Drug Metab Dispos. 2008;36:597–603. doi: 10.1124/dmd.107.018754. [DOI] [PubMed] [Google Scholar]
- 48.Dai J., Zhang F., Zheng J. Retrorsine, but not monocrotaline, is a mechanism-based inactivator of P450 3A4. Chem Biol Interact. 2010;183:49–56. doi: 10.1016/j.cbi.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.