


Abstract
Here, we reported the compendium of protein lysine modifications (CPLM 4.0, http://cplm.biocuckoo.cn/), a data resource for various post-translational modifications (PTMs) specifically occurred at the side-chain amino group of lysine residues in proteins. From the literature and public databases, we collected 450 378 protein lysine modification (PLM) events, and combined them with the existing data of our previously developed protein lysine modification database (PLMD 3.0). In total, CPLM 4.0 contained 592 606 experimentally identified modification events on 463 156 unique lysine residues of 105 673 proteins for up to 29 types of PLMs across 219 species. Furthermore, we carefully annotated the data using the knowledge from 102 additional resources that covered 13 aspects, including variation and mutation, disease-associated information, protein-protein interaction, protein functional annotation, DNA & RNA element, protein structure, chemical-target relation, mRNA expression, protein expression/proteomics, subcellular localization, biological pathway annotation, functional domain annotation, and physicochemical property. Compared to PLMD 3.0 and other existing resources, CPLM 4.0 achieved a >2-fold increase in collection of PLM events, with a data volume of ∼45GB. We anticipate that CPLM 4.0 can serve as a more useful database for further study of PLMs.
INTRODUCTION
Protein lysine modifications (PLMs) belong to a sub-class of post-translational modifications (PTMs) that covalently attach small-molecule moieties or small proteins to positively charged ϵ-amino groups of specific lysine residues in protein sequences, in a reversible manner (1–3). In 1964, two types of PLMs, acetylation and methylation, were first discovered to modify histone lysine residues by Vincent Allfrey and his colleagues (4). These ‘relatively minor modifications’ were generally regarded as constitutive and inert marks in histones (4), until the discovery of GCN5 as the first histone acetyltransferase (HAT) from Tetrahymena thermophile, a ciliated protozoan, by C. David Allis and James E. Brownell in 1996 (5,6). Later, it was demonstrated that the lysine residue is particularly active and can be regulated by a variety of PLMs, including acylation modifications such as acetylation (7), crotonylation (8), succinylation (9) and lactylation (10), ubiquitin and ubiquitin-like (Ub/Ubl) conjugations such as ubiquitination (11) and sumoylation (12), and other types of PLMs such as methylation (13) and lipoylation (14). PLMs occurred at histone lysine residues constitute the ‘histone code’ together with other histone modifications, and play a critical role in transcriptional regulation and gene expression (15,16). PLMs also extensively occur at non-histone proteins, change their activity, stability and trafficking (17), and regulate a variety of biological processes such as signal transduction, metabolism, and autophagy (7,18,19). PLMs are precisely regulated in vivo, and the dysregulation of PLMs is highly associated with human diseases such as cancer, neurodegenerative diseases and metabolic disorders (20,21).
With the rapid progress of mass spectrometry-based technology and development of PLM-specific antibodies, a large number of protein substrates and sites have been identified for both well-characterized and newly discovered PLMs, in a high-throughput manner (22–25). For example, using a diglycine (diGly, K-ϵ-GG) antibody and a pan-acetyl antibody, Elia et al. quantified the changes of 33 500 ubiquitination and 16 740 acetylation sites in HeLa cells responding to ultraviolet or ionizing radiation, and discovered many new PLM substrates in regulation of DNA damage response (DDR) (22). By developing a pan-benzoyl antibody, Yingming Zhao's group discovered a novel PLM type, lysine benzoylation (Kbz), and identified 22 Kbz sites in core histone proteins from mammalian cells (23). They also developed a pan-lactyl antibody and discovered a novel lactate-derived PLM by identifying 28 lysine lactylation (Kla) sites on histones in human and mouse cells (10). More recently, Zhao's group developed a highly efficient pan-β-hydroxybutyryl antibody, and identified 3248 lysine β-hydroxybutyrylation (Kbhb) sites on 1397 proteins (24,25). Due to the biological importance of PLMs, collection, curation, integration and annotation of experimentally identified PLM events will provide a highly useful data resource for further experimental consideration and biomedical design.
In 2011, we developed the compendium of protein lysine acetylation (CPLA, v1.0), containing 7151 known acetylation sites in 3311 proteins (26). Later, we made a considerable improvement and released the compendium of protein lysine modifications (CPLM, v2.0), containing 203 972 modification events on 189 919 sites of 45 748 proteins for 12 PLM types across 122 species (27). In 2017, the v3.0 contained 284 780 modification events in 53 501 proteins across 176 species for 20 PLM types, with a 39.6% increase of the PLM events (28). During the past years, we continuously maintained and updated the database, and CPLM 4.0 contained 592 606 modification events on 463 156 unique lysine residues of 105 673 proteins for 29 types of PLMs across 219 species, with a >2-fold increase in PLM events. Also, we carefully annotated the data by integrating the knowledge from 102 additional resources. Compared to v3.0 (∼150MB), CPLM 4.0 (∼45GB) has a ∼300-fold increase in the data volume. We believe that CPLM 4.0 can be more helpful for the scientific community.
CONSTRUCTION AND CONTENT
Data collection, curation and integration
In this update, we mainly focused on expanding new PLM events and types from the literature and public PTM databases (Figure 1A). First, we individually used multiple keywords to search PubMed, and manually curated the abstract or full text of each returned paper published after 1 January 2017. These keywords included but were not limited to ‘acetylation’, ‘lactylation’, ‘β-hydroxybutyrylation’, ‘succinylation’, ‘crotonylation’, ‘benzoylation’, ‘2-hydroxyisobutyrylation’, ‘malonylation’, ‘butyrylation’, ‘propionylation’, ‘glutarylation’, ‘formylation’, ‘3-hydroxyl-3-methylglutarylation OR HMGylation’, ‘3-methylglutaconylation OR MGcylation’, ‘3-methylglutarylation OR MGylation’, ‘ubiquitination’, ‘sumoylation’, ‘neddylation’, ‘pupylation’, ‘methylation’, ‘diethylphosphorylation’, ‘carboxymethylation’, ‘carboxyethylation’, ‘carboxylation’, ‘phosphoglycerylation’, ‘glycation’, ‘hydroxylation’, ‘lipoylation’ and ‘biotinylation’. To avoid missing any important studies, we also used additional keyword combinations, such as ‘acetyl AND lysine’, ‘acetylated AND lysine’, ‘ubiquitinated AND lysine’, ‘SUMO AND lysine’ and other similar terms, to find more related studies on PLMs.
Figure 1.

The procedure for development of CPLM 4.0. (A) First, we manually collected experimentally identified PLM substrates and sites from PubMed. We also integrated the existing data of 10 public databases, including PLMD 3.0 (28), dbPTM (30), ProteomeScout (31), iPTMnet (32), BioGRID (33), PhosphoSitePlus (34), mUbiSiDa (35), HPRD (36), ActiveDriverDB (37) and UniProt (29) (Supplementary Table S1). Furthermore, we annotated the PLM proteins and sites, using the knowledge from 102 additional databases that covered 13 aspects: (i) variation and mutation; (ii) disease-associated information; (iii) protein–protein interaction; (iv) protein function; (v) DNA & RNA element; (vi) chemical–target relation; (vii) protein structure; (viii) mRNA expression; (ix) physicochemical property; (x) protein expression/proteomics; (xi) subcellular localization; (xii) biological pathway; (xiii) domain annotation (Supplementary Table S2). Kla, lysine lactylation; Kcr, lysine crotonylation; Kmal, lysine malonylation; Kbhb, lysine β-hydroxybutyrylation; Kub: lysine ubiquitination; Kac, lysine acetylation; Ksucc, lysine succinylation. (B) A comparison of PLM events and proteins between CPLM 4.0 and other existing resources.
To obtain the position information, all collected PLM peptides were mapped to a merged file of uniprot_sprot.fasta, uniprot_trembl.fasta and uniprot_sprot_varsplic.fasta downloaded from UniProt (Benchmark sequences, release version 2020-05, ftp://ftp.uniprot.org/) (29), according to the species information. In UniProt, the accession numbers of proteins are organized in a chronological order with the initial character of O, P, Q, A, B and so on. For a PLM site mapped with multiple identical sequences, we only reserved the most anterior sequence to avoid the redundancy. In total, we collected 236,885 non-redundant PLM events on 200 723 lysine residues of 60 896 protein substrates from the literature (Supplementary Table S1).
Next, we integrated these newly reported PLM events with the existing data in PLMD 3.0 (28) and nine additional public databases, including dbPTM (30), ProteomeScout (31), iPTMnet (32), BioGRID (33), PhosphoSitePlus (34), mUbiSiDa (35), HPRD (36), ActiveDriverDB (37) and UniProt (29) (Figure 1A). Again, all PLM sites were re-mapped to UniProt for redundancy clearance. Finally, we obtained 592 606 non-redundant modification events on 463 156 lysine residues of 105 673 protein substrates for 29 PLM types across 219 species (Figure 1B, Supplementary Table S1). Compared to PLMD 3.0 and other existing databases, CPLM 4.0 achieved a >2-fold increase in collection of PLM events. Another public resource, BioGRID (33), only contained 226 210 ubiquitination sites in 15 478 proteins.
A multi-layer annotation of PLM substrates and sites
CPLM 4.0 was developed as a protein-centred database. To organize the database, a CPLM ID was automatically generated as the primary accession for each PLM substrate, e.g., CPLM017735 for a core histone protein H3C1 in Homo sapiens. For each protein entry, the basic annotations such as UniProt/RefSeq/Ensembl accession numbers, protein name/synonyms, gene name/synonyms, gene ID, NCBI Taxa ID, functional descriptions, protein sequence, nucleotide sequence, Gene Ontology (GO) terms and keywords were integrated from UniProt (29), and a typical protein 3D structure was selected from PDB (38) if available, for visualization of known PLM sites. For each PLM site, the modification position, a flanking peptide with a length of 15 aa around the middle residue, the modification type and the reference information with PMIDs were provided. For PLM sites identified from high-throughput studies, the original PLM peptides detected by mass spectrometry, cell or tissue sources, and software packages for data processing were reserved if provided. For some data processing tools such as MaxQuant, the localization probability (LP) score would be computationally assigned to each PLM site (39). The LP score ranges from 0 to 1, and a higher LP score denotes a higher probability of a site to be a real PLM site. Similar to the analysis of phosphorylation (39), here we classified the high-throughput PLM sites into four categories, including class I (LP > 0.75), class II (0.5 < LP ≤ 0.75), class III (0.25 ≤ LP ≤ 0.5) and class IV (LP < 0.25). In CPLM 4.0, there were 141,068 (99.25%) Class I sites, 659 (0.46%) Class II sites, and 405 (0.28%) Class III sites. To ensure the data quality, the Class IV sites were discarded.
In addition to the literature and basic annotations, we further compiled the knowledge from 102 additional databases and carefully annotated the PLM proteins and sites, especially human PLM substrates and sites. These public resources covered 13 aspects: (i) variation and mutation; (ii) disease-associated information; (iii) protein–protein interaction; (iv) protein function; (v) DNA & RNA element; (vi) protein structure; (vii) chemical–target relation; (viii) mRNA expression; (ix) protein expression/proteomics; (x) subcellular localization; (xi) biological pathway; (xii) domain annotation; (xiii) physicochemical property (Figure 1A, Supplementary Table S2). The distribution of numbers of annotation entries was counted, and it could be found that up to 217 species had at least one entry (Supplementary Table S3). Obviously, PLM substrates and sites in H. sapiens had the most annotations, with a total of 699 440 281 entries (94.12%). Details on processing the annotations of each resource were carefully described in Supplementary Methods. All data sets and annotations in CPLM 4.0 could be downloaded at http://cplm.biocuckoo.cn/Download.php.
The data statistics of CPLM 4.0
Previously in PLMD 3.0 (28), we classified the 20 PLM types into three categories: (i) acylation modifications, including acetylation, butyrulation, formylation, succinylation, propinoylation, crotonylation, glutarylation, malonylation and 2-hydroxyisobutyrylation; (ii) Ub/Ubl conjugations, including ubiquitination, sumoylation, pupylation, and neddylation; (iii) others, including methylation, lipoylation, glycation, carboxylation, hydroxylation, biotinylation and phosphoglycerylation. In CPLM 4.0, we added six new acylation modifications including lactylation, β-hydroxybutyrylation, benzoylation, HMGylation, MGcylation and MGylation, and three other PLMs, including carboxymethylation, carboxyethylation and diethylphosphorylation. The numbers of modification events and proteins were shown for each PLM type (Figure 2A). Obviously, ubiquitination had the most PLM events (235 888 events, 39.80%), whereas acetylation held the second position and had the most acylation events (208 299 events, 35.15%) (Figure 2A). Of note, sumoylation occupied the third position (53 483 events, 9.02%), mainly due to the advance in proteomic technology. In 2017, Hendriks et al. significantly improved the K0-SUMO strategy that used a lysine-deficient SUMO protein, and identified 40 765 sumoylation sites in 6747 human proteins (40). The distribution of different types of PLM sites and proteins was counted for each species (Supplementary Table S4), and the top 10 species with the most PLM proteins were visualized by Heat map Illustrator (HemI) (41) (Figure 2B). Compared to PLMD 3.0 (28), the numbers of reported PLM substrates were dramatically increased to a considerable number of species. For example, PLMD 3.0 only contained 2018 PLM events in Oryza sativa subsp. japonica, whereas this number was 22 349 with a >11-fold increase in CPLM 4.0. Also, there were only 62 PLM events in Sus scrofa maintained by PLM 3.0, while 2711 PLM events of 924 pig proteins were collected in CPLM 4.0.
Figure 2.

The data statistics and analysis of the data in CPLM 4.0. (A) The 29 PLM types were classified into three categories, including acylation, Ub/Ubl conjugation and others. The numbers of PLM sites and proteins were shown for each PLM type. (B) The distribution of different types of PLM protein substrates for the top 10 abundant species. More details on the data statistics were shown in Supplementary Table S4. (C) The proteins with at least one PLM site to be multiply regulated by acetylation, succinylation, malonylation, 2-hydroxyisobutyrylation and crotonylation. More details on the potential crosstalks among different types of PLMs were shown in Supplementary Table S5. (D) The GO-based enrichment analysis of the 687 multiply regulated PLM proteins (P-value < 1E–18).
Next, we analyzed the sequence preference around the modification sites for each of the 22 PLM types with enough data, using a sequence logo generator pLogo (https://plogo.uconn.edu/) (42). From the results, it could be found that acidic amino acids (D/E) preferred to locate at the −1 position of 2-hydroxyisobutyrylation, acetylation, and crotonylation sites, at the −2 position of sumoylation sites, and at +1 and +2 positions of succinylation sites (Supplementary Figure S1A). Also, the basic amino acid K preferentially occurred at the +4 position of benzoylation and lactylation sites (Supplementary Figure S1A). The motif analysis demonstrated that distinct sequence profiles were recognized by different PLM types.
Moreover, the co-occurrence of PLM events on the same lysine residues were pairwisely analyzed. In total, we identified 87 608 PLM sites (18.92%) could be regulated in situ by at least two PLM types, indicating a widespread existence of PTM crosstalks (Supplementary Figure S1B, Supplementary Table S5). From the results, we found 687 proteins containing at least one site to be regulated by the top 5 abundant acylations, including acetylation, succinylation, malonylation, 2-hydroxyisobutyrylation and crotonylation (Figure 2C). A hypergeometric test-based analysis detected that biological processes related to translation, transcription and metabolism were highly enriched (Figure 2D, P-value < 1E–18), supporting the functional importance of these multiply modified substrates (43,44). In addition, we used IUPred (https://iupred.elte.hu/) (45), an online service to predict disordered regions in proteins, and the disorder propensity scores were calculated for sites of the top 5 abundant acylations. From the results, it could be found that all the five types of acylation sites preferred to locate in disordered regions (Supplementary Figure S1C, >70%).
USAGE
The online service of CPLM 4.0 was developed in an easy-to-use manner. Here, we took the human histone protein H3C1, newly discovered to be regulated by lactylation (10), as an example to describe the usage of CPLM 4.0. In the browse page, we implemented two types of browsing options, including ‘Browse by Modification Types’ and ‘Browse by species’. In the former option, users could click on ‘Lactylation’ and then choose ‘Homo sapiens’ to access a tabular list of all human lactylation substrates (Figure 3A). For the latter, users could first click on ‘Homo sapiens’ and then ‘Lactylation’ to return the same result (Figure 3B). By clicking on the CPLM ID ‘CPLM017735’, the protein page of H3C1 could be viewed (Figure 3C). An ‘Enlarging’ button was implemented below the structure window, and allowed the protein 3D structure of human H3C1 to be viewed in a larger window. Besides the basic information and details on known PLM sites, additional annotations could be accessed either by clicking the ‘Annotation’ button in the left bar, or by clicking the ‘Integrated Annotation’ icon adjacent to the CPLM ID (Figure 3C). Then, each type of additional annotations could be presented by clicking on its corresponding name. For example, users could click on ‘ICGC’ under the variation & mutation section to view all cancer missense mutations identified in H3C1 gene, from International Cancer Genome Consortium (ICGC) (46) (Figure 3D).
Figure 3.

The browse options of CPLM 4.0. (A) Browse by modification types. (B) Browse by species. (C) The tabular list and the protein page of human H3C1. Besides the basic information and details on PLM sites, additional annotations could be accessed. By clicking on the ‘Enlarging’ button under the structure window, the protein 3D structure of human H3C1 could be viewed in a larger window. (D) The annotation page of H3C1. As an example, ICGC cancer missense mutations that change PLM sites of H3C1 were shown (46).
Also, multiple search options were implemented. In the search page (http://cplm.biocuckoo.cn/Search.php), there were four options including ‘Substrate Search’, ‘Advanced Search’, ‘Batch Search’ and ‘BLAST Search’ provided for searching the data in CPLM 4.0 (Supplementary Figure S2). The ‘Substrate Search’ option was also present at the home page, and users could click on ‘Example’ and then ‘Submit’ to search human H3C1 and its analogs in other species (Supplementary Figure S2A).
DISCUSSION
In recent years, great attention has been paid on the identification of PLM substrates and sites, as well as the discovery of new PLM types, mainly due to the functional importance of various PLMs in regulation of gene expression outputs and metabolic reprogramming in response to changes of the cellular or external environment, while abnormal PLM regulations are frequently associated with human diseases (1–3,7,15–21). The continuous collection, integration, biocuration and annotation of experimentally identified PLM events will provide a fundamental resource for further analysis of molecular mechanisms and regulatory roles of PLMs. Previously, we developed CPLA 1.0 (26), CPLM 2.0 (27) and PLMD 3.0 (28) for this purpose. In this study, the latest release CPLM 4.0 contained 592 606 PLM events occurred on 463 156 unique lysine residues of 105 673 proteins across 219 species, and expanded the PLM types from 20 to 29. Compared to PLMD 3.0 and other existing databases (28–37), CPLM 4.0 had a >2-fold increase in collection of PLM events, with a data volume of ∼45GB.
Besides the basic information and details on modification sites of each protein entry, additional annotations from 102 additional resources were integrated to cover 13 aspects. For example, from the Catalogue Of Somatic Mutations In Cancer (COSMIC) (47), there were 124 records of missense mutations that changed known PLM sites of human H3C1 protein (Figure 4). Six of the seven known lactylation sites including K9, K14, K18, K23, K27 and K56 could be changed by COSMIC mutations, whereas a K79N mutation was found in ICGC uterine corpus endometrial carcinoma (UCEC) (46). From the disease-associated information, acetylation and methylation of H3C1 at K27 have been associated with a number of diseases, including bladder cancer, pancreatic cancer, lung cancer and breast cancer, and obesity (48–53). Human H3C1 acts as a client protein of liquid-liquid phase separation (LLPS) in nucleolus (54), interacts with 2790 other proteins, is post-transcriptionally targeted by 14 miRNAs according to the annotations in miRTarBase (55). With 221 resolved 3D structures in PDB (38), H3C1 could be targeted by 65 chemicals, such as calcitriol (56), berberine (57) and coumestrol (58). According to the annotations of The Cancer Genome Atlas (TCGA) (59), the mRNA of H3C1 was highly expressed in thymoma (THYM), UCEC, and uterine carcinosarcoma (UCS).
Figure 4.
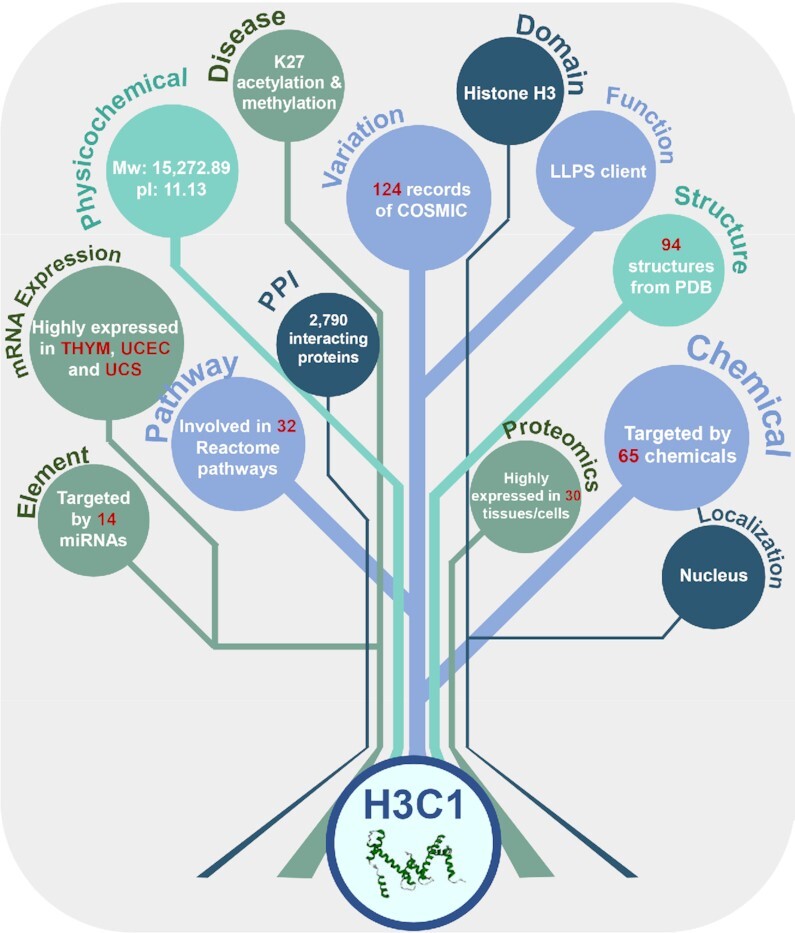
The overview of integrated annotations for human H3C1. A brief summary of all the 111 data resources used in this study is shown in Supplementary Table S2. The details on processing each of the resources were present in Supplementary methods.
To test whether mass spectrometry-derived PLM substrates and sites tend to be quantified from proteins with high expression levels, here we re-analyzed the data sets from 4 published studies. To study how microbial metabolite biosynthesis is regulated by lysine acylation, Xu et al. quantified the proteomes and acetylomes in wild-type (WT) and high-yield E3 strains of Saccharopolyspora erythraea, a model actinomycete that produces erythromycin (60). Also, Li et al. performed a proteomic and ubiquitylomic quantification using a pair of gefitinib-resistant and sensitive non-small cell lung cancer (NSCLC) cell lines (61). Sap et al. quantified proteomes and ubiquitylomes of brain lysates of Huntington's disease mice and normal mice (62), whereas Karoutas et al. conducted a proteomic and acetylomic profiling of mouse embryonic fibroblasts (MEFs) and embryonic stem cells (ESCs) with or without Mof, a major lysine acetyltransferase responsible for histone H4K16 acetylation (63). From each study, PLM substrates mutually quantified by proteomics and PLMomics were reserved, and the scatter plot of PLM site intensities vs. protein intensities was illustrated by the R package ggplot2 (64) (Supplementary Figure S3). The marginal distribution of PLM site intensities or protein intensities was illustrated by the ggMarginal function of the R package ggExtra (version 0.9). From the results, it could be found that PLM substrates in the first and last studies tended to be quantified from proteins with higher expressions (Supplementary Figure S3A, D). However, PLM sites did not have such a tendency.
For the future plan, we will continuously maintain and update the database, when newly reported PLM substrates and sites are available from the literature. More PLM types and species will be included. Also, more annotations will be integrated from additional public resources. We believe that CPLM 4.0 can serve as a highly useful resource for further analysis of PLMs.
Supplementary Material
Contributor Information
Weizhi Zhang, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Xiaodan Tan, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Shaofeng Lin, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Yujie Gou, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Cheng Han, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Chi Zhang, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Wanshan Ning, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Chenwei Wang, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China.
Yu Xue, MOE Key Laboratory of Molecular Biophysics, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China; Nanjing University Institute of Artificial Intelligence Biomedicine, Nanjing, Jiangsu 210031, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Natural Science Foundation of China [31930021 and 31970633]; Fundamental Research Funds for the Central Universities [2019kfyRCPY043]; Changjiang Scholars Program of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Verdin E., Ott M.. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015; 16:258–264. [DOI] [PubMed] [Google Scholar]
- 2. Rape M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018; 19:59–70. [DOI] [PubMed] [Google Scholar]
- 3. Menzies K.J., Zhang H., Katsyuba E., Auwerx J.. Protein acetylation in metabolism - metabolites and cofactors. Nat. Rev. Endocrinol. 2016; 12:43–60. [DOI] [PubMed] [Google Scholar]
- 4. Allfrey V.G., Faulkner R., Mirsky A.E.. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. PNAS. 1964; 51:786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brownell J.E., Zhou J., Ranalli T., Kobayashi R., Edmondson D.G., Roth S.Y., Allis C.D.. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996; 84:843–851. [DOI] [PubMed] [Google Scholar]
- 6. Brownell J.E., Allis C.D.. HAT discovery: Heading toward an elusive goal with a key biological assist. Biochim. Biophys. Acta Gene Regul. Mech. 2021; 1864:194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Narita T., Weinert B.T., Choudhary C.. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019; 20:156–174. [DOI] [PubMed] [Google Scholar]
- 8. Gowans G.J., Bridgers J.B., Zhang J., Dronamraju R., Burnetti A., King D.A., Thiengmany A.V., Shinsky S.A., Bhanu N.V., Garcia B.A.et al.. Recognition of histone crotonylation by Taf14 links metabolic state to gene expression. Mol. Cell. 2019; 76:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y., Guo Y.R., Liu K., Yin Z., Liu R., Xia Y., Tan L., Yang P., Lee J.H., Li X.J.et al.. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017; 552:273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M.et al.. Metabolic regulation of gene expression by histone lactylation. Nature. 2019; 574:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pohl C., Dikic I.. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science (New York, N.Y.). 2019; 366:818–822. [DOI] [PubMed] [Google Scholar]
- 12. Zhou H.J., Xu Z., Wang Z., Zhang H., Zhuang Z.W., Simons M., Min W.. SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis. Nat. Commun. 2018; 9:3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhat K.P., Ümit Kaniskan H., Jin J., Gozani O.. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021; 20:265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang Q., Guo Y., Meng L., Chen X.. Chemical tagging of protein lipoylation. Angew. Chem. Int. Ed. Engl. 2021; 60:4028–4033. [DOI] [PubMed] [Google Scholar]
- 15. Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N.et al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011; 146:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dutta A., Abmayr S.M., Workman J.L.. Diverse activities of histone acylations connect metabolism to chromatin function. Mol. Cell. 2016; 63:547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cornett E.M., Ferry L., Defossez P.A., Rothbart S.B.. Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell. 2019; 75:1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrico C., Meyer J.G., He W., Gibson B.W., Verdin E.. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018; 27:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim H.J., Kim S.Y., Kim D.H., Park J.S., Jeong S.H., Choi Y.W., Kim C.H.. Crosstalk between HSPA5 arginylation and sequential ubiquitination leads to AKT degradation through autophagy flux. Autophagy. 2021; 17:961–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cohen T.J., Guo J.L., Hurtado D.E., Kwong L.K., Mills I.P., Trojanowski J.Q., Lee V.M.. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011; 2:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun L., Zhang H., Gao P.. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2021; 10.1007/s13238-021-00846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elia A.E., Boardman A.P., Wang D.C., Huttlin E.L., Everley R.A., Dephoure N., Zhou C., Koren I., Gygi S.P., Elledge S.J.. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol. Cell. 2015; 59:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang H., Zhang D., Wang Y., Perez-Neut M., Han Z., Zheng Y.G., Hao Q., Zhao Y.. Lysine benzoylation is a histone mark regulated by SIRT2. Nat. Commun. 2018; 9:3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., Qi S., Li J., Colak G., Chen Y.et al.. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell. 2016; 62:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang H., Zhang D., Weng Y., Delaney K., Tang Z., Yan C., Qi S., Peng C., Cole P.A., Roeder R.G.et al.. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv. 2021; 7:eabe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Z., Cao J., Gao X., Zhou Y., Wen L., Yang X., Yao X., Ren J., Xue Y.. CPLA 1.0: an integrated database of protein lysine acetylation. Nucleic Acids Res. 2011; 39:D1029–D1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Z., Wang Y., Gao T., Pan Z., Cheng H., Yang Q., Cheng Z., Guo A., Ren J., Xue Y.. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014; 42:D531–D536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu H., Zhou J., Lin S., Deng W., Zhang Y., Xue Y.. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genomics. 2017; 44:243–250. [DOI] [PubMed] [Google Scholar]
- 29. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang K.Y., Su M.G., Kao H.J., Hsieh Y.C., Jhong J.H., Cheng K.H., Huang H.D., Lee T.Y.. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016; 44:D435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matlock M.K., Holehouse A.S., Naegle K.M.. ProteomeScout: a repository and analysis resource for post-translational modifications and proteins. Nucleic Acids Res. 2015; 43:D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang H., Arighi C.N., Ross K.E., Ren J., Li G., Chen S.C., Wang Q., Cowart J., Vijay-Shanker K., Wu C.H.. iPTMnet: an integrated resource for protein post-translational modification network discovery. Nucleic Acids Res. 2018; 46:D542–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R.et al.. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019; 47:D529–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hornbeck P.V., Kornhauser J.M., Latham V., Murray B., Nandhikonda V., Nord A., Skrzypek E., Wheeler T., Zhang B., Gnad F.. 15 years of PhosphoSitePlus®: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019; 47:D433–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen T., Zhou T., He B., Yu H., Guo X., Song X., Sha J.. mUbiSiDa: a comprehensive database for protein ubiquitination sites in mammals. PLoS One. 2014; 9:e85744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goel R., Harsha H.C., Pandey A., Prasad T.S.. Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012; 8:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krassowski M., Paczkowska M., Cullion K., Huang T., Dzneladze I., Ouellette B.F.F., Yamada J.T., Fradet-Turcotte A., Reimand J.. ActiveDriverDB: human disease mutations and genome variation in post-translational modification sites of proteins. Nucleic Acids Res. 2018; 46:D901–D910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Humphrey S.J., Yang G., Yang P., Fazakerley D.J., Stockli J., Yang J.Y., James D.E.. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013; 17:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hendriks I.A., Lyon D., Young C., Jensen L.J., Vertegaal A.C., Nielsen M.L.. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017; 24:325–336. [DOI] [PubMed] [Google Scholar]
- 41. Deng W., Wang Y., Liu Z., Cheng H., Xue Y.. HemI: a toolkit for illustrating heatmaps. PLoS One. 2014; 9:e111988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Shea J.P., Chou M.F., Quader S.A., Ryan J.K., Church G.M., Schwartz D. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods. 2013; 10:1211–1212. [DOI] [PubMed] [Google Scholar]
- 43. Sabari B.R., Tang Z., Huang H., Yong-Gonzalez V., Molina H., Kong H.E., Dai L., Shimada M., Cross J.R., Zhao Y.et al.. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell. 2015; 58:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H.et al.. Regulation of cellular metabolism by protein lysine acetylation. Science (New York, N.Y.). 2010; 327:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mészáros B., Erdos G., Dosztányi Z.. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018; 46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J., Baran J., Cros A., Guberman J.M., Haider S., Hsu J., Liang Y., Rivkin E., Wang J., Whitty B.et al.. International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database. 2011; 2011:bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S.et al.. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ellinger J., Bachmann A., Göke F., Behbahani T.E., Baumann C., Heukamp L.C., Rogenhofer S., Müller S.C.. Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urol. Int. 2014; 93:113–118. [DOI] [PubMed] [Google Scholar]
- 49. Woo J., Kim H.Y., Byun B.J., Chae C.H., Lee J.Y., Ryu S.Y., Park W.K., Cho H., Choi G.. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2014; 24:2486–2492. [DOI] [PubMed] [Google Scholar]
- 50. Yamada N., Hamada T., Goto M., Tsutsumida H., Higashi M., Nomoto M., Yonezawa S.. MUC2 expression is regulated by histone H3 modification and DNA methylation in pancreatic cancer. Int. J. Cancer. 2006; 119:1850–1857. [DOI] [PubMed] [Google Scholar]
- 51. Xu C., Hou Z., Zhan P., Zhao W., Chang C., Zou J., Hu H., Zhang Y., Yao X., Yu L.et al.. EZH2 regulates cancer cell migration through repressing TIMP-3 in non-small cell lung cancer. Med. Oncol. 2013; 30:713. [DOI] [PubMed] [Google Scholar]
- 52. Ko H.W., Lee H.H., Huo L., Xia W., Yang C.C., Hsu J.L., Li L.Y., Lai C.C., Chan L.C., Cheng C.C.et al.. GSK3β inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget. 2016; 7:57131–57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yi S.A., Um S.H., Lee J., Yoo J.H., Bang S.Y., Park E.K., Lee M.G., Nam K.H., Jeon Y.J., Park J.W.et al.. S6K1 phosphorylation of H2B mediates EZH2 trimethylation of H3: a determinant of early adipogenesis. Mol. Cell. 2016; 62:443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scherl A., Couté Y., Déon C., Callé A., Kindbeiter K., Sanchez J.C., Greco A., Hochstrasser D., Diaz J.J.. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell. 2002; 13:4100–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chou C.H., Shrestha S., Yang C.D., Chang N.W., Lin Y.L., Liao K.W., Huang W.C., Sun T.H., Tu S.J., Lee W.H.et al.. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018; 46:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang W.L., Chatterjee N., Chittur S.V., Welsh J., Tenniswood M.P.. Effects of 1α,25 dihydroxyvitamin D3 and testosterone on miRNA and mRNA expression in LNCaP cells. Mol. Cancer. 2011; 10:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Z., Liu Y., Xue Y., Hu H., Ye J., Li X., Lu Z., Meng F., Liang S.. Berberine acts as a putative epigenetic modulator by affecting the histone code. Toxicology In Vitro. 2016; 36:10–17. [DOI] [PubMed] [Google Scholar]
- 58. Dip R., Lenz S., Gmuender H., Naegeli H.. Pleiotropic combinatorial transcriptomes of human breast cancer cells exposed to mixtures of dietary phytoestrogens. Food Chem. Toxicol. 2009; 47:787–795. [DOI] [PubMed] [Google Scholar]
- 59. Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017; 169:1327–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu J.Y., Xu Y., Xu Z., Zhai L.H., Ye Y., Zhao Y., Chu X., Tan M., Ye B.C.. Protein acylation is a general regulatory mechanism in biosynthetic pathway of acyl-CoA-derived natural products. Cell Chem. Biol. 2018; 25:984–995. [DOI] [PubMed] [Google Scholar]
- 61. Li W., Wang H., Yang Y., Zhao T., Zhang Z., Tian Y., Shi Z., Peng X., Li F., Feng Y.et al.. Integrative analysis of proteome and biquitylome reveals unique features of lysosomal and endocytic pathways in gefitinib-resistant non-small cell lung cancer cells. Proteomics. 2018; 18:e1700388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sap K.A., Guler A.T., Bezstarosti K., Bury A.E., Juenemann K., Demmers J.A., Reits E.A.. Global proteome and ubiquitinome changes in the soluble and insoluble fractions of Q175 Huntington mice brains. Mol. Cell. Proteomics: MCP. 2019; 18:1705–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karoutas A., Szymanski W., Rausch T., Guhathakurta S., Rog-Zielinska E.A., Peyronnet R., Seyfferth J., Chen H.R., de Leeuw R., Herquel B.et al.. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat. Cell Biol. 2019; 21:1248–1260. [DOI] [PubMed] [Google Scholar]
- 64. Villanueva R.A.M., Chen Z.J.. ggplot2: elegant graphics for data analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 2019; 17:160–167. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.