

Abstract
Selective autophagy of damaged organelles is important to maintain cellular homeostasis. The mechanisms how autophagy selects specific targets is often poorly understood. Rabaptin5 was previously known as a major regulator of early endosome identity and maturation. Here, we identify two novel Rabaptin5 interactors: FIP200, a subunit of the ULK1 autophagy initiator complex, and ATG16L1, a central component of the E3‐like enzyme in LC3 lipidation. Autophagy of early endosomes damaged by chloroquine or monensin treatment requires Rabaptin5 and particularly a short sequence motif that binds to the WD domain of ATG16L1. Rabaptin5 and its interaction with ATG16L1 further contributes to the autophagic elimination of Salmonella enterica early after infection, when it resides in phagosomes with early endosomal characteristics. Our results demonstrate a novel function of Rabaptin5 in quality control of early endosomes in the selective targeting of autophagy to damaged early endosomes and phagosomes.
Keywords: ATG16L1, autophagy, early endosomes, Rabaptin5, Salmonella‐containing vacuoles
Subject Categories: Autophagy & Cell Death; Membranes & Trafficking; Microbiology, Virology & Host Pathogen Interaction
The endosomal regulator Rabaptin5 selectively targets autophagy to endosomes damaged by chloroquine or Salmonella infection via interaction with FIP200 and ATG16L1.
Introduction
Endosomes are dynamic organelles that receive endocytic cargo from the plasma membrane and exocytic material from the trans‐Golgi for sorting to late endosomes and lysosomes, to the cell surface via recycling endosomes, or back to the Golgi (Naslavsky & Caplan, 2018). Endosomal identities are defined by specific Rab GTPases, their effectors, and characteristic phosphoinositides. At early endosomes, Rab5 is the hallmark GTPase that activates VPS34/p150 to produce phosphatidylinositol‐3‐phosphate (PI3P) and recruits early endosome antigen 1 (EEA1) and Rabenosyn‐5, two multivalent PI3P‐binding proteins that act as membrane tethers to mediate homotypic endosome fusion. Rab5·GTP and PI3P are also responsible for recruitment of the Mon1/Ccz1 complex to activate Rab7 and deactivate Rab5 in the process of Rab conversion during maturation from early to late endosomes (Poteryaev et al, 2010; Huotari & Helenius, 2011).
Rab5 activity is regulated by a complex of Rabaptin5 and Rabex5, the GDP/GTP exchange factor of Rab5. Rabaptin5 binds to Rab4·GTP and Rab5·GTP, and Rabex5 binds to ubiquitin to mediate membrane recruitment and to activate Rab5 on early endosomes (Mattera et al, 2006; Mattera & Bonifacino, 2008; Kälin et al, 2015, 2016).
Searching for new interactors of Rabaptin5, we performed a yeast two‐hybrid screen and discovered as a novel binding partner FIP200, a component of the ULK1–FIP200–ATG13–ATG101 autophagy initiator complex. Autophagy is a self‐degradative survival mechanism of eukaryotic cells important to preserve cellular homeostasis in response to stress conditions such as lack of nutrients, accumulation of misfolded or aggregated proteins, damaged organelles, and pathogen infection (Bento et al, 2016; Dikic & Elazar, 2018; Mercer et al, 2018; Morishita & Mizushima, 2019). Clearance of damaged organelles is an important process to preserve overall organelle function. Defects in its mechanisms have been shown to contribute to some forms of cancer, to neurodegeneration, and inflammatory diseases (Anding & Baehrecke, 2017; Leidal et al, 2018). Selective autophagy has been described for many organelles including mitochondria, peroxisomes, endoplasmic reticulum, and lysosomes (mitophagy, pexophagy, ER‐phagy, and lysophagy; Kirkin & Rogov, 2019).
Typically, autophagy is set in motion by the activation of the ULK1–FIP200 complex under the control of mTOR. The ULK complex initiates a dynamic interactome of autophagy components by recruiting and activating the class III PI‐3‐kinase complex VPS34–VPS15–Beclin1–ATG14 to produce PI3P, further recruiting PI3P‐binding proteins, such as WIPI (WD‐repeat phosphoinositide‐interacting protein) family members. Together, these complexes and PI3P bind ATG16L1 with its partners ATG12–ATG5 acting as an E3‐ubiquitin ligase‐like enzyme for the lipidation of LC3 family proteins for elongation and closure of the phagophore membrane and fusion with lysosomes.
Selective autophagy of organelles destined for degradation differs from starvation‐induced autophagy in its mechanism of initiation, which is independent of the nutrient situation, and in its targeting to specific cargo (Kirkin & Rogov, 2019). Autophagy is often targeted to damaged organelles after they are marked for degradation by ubiquitination of lumenal proteins or by galectins binding to exposed glycans (Randow & Youle, 2014). Galectins 3, 8, and 9 have been implicated in the recognition of damaged lysosomes, endosomes, or salmonella‐containing vacuoles together with galectin‐interacting autophagy receptors NDP52 or TRIM16 (Thurston et al, 2012; Chauhan et al, 2016; Jia et al, 2018, 2020; Fraser et al, 2019).
ATG16L1 emerges as an important hub for specific cargo to connect to the autophagy machinery (reviewed by Gammoh, 2020). Besides its role in LC3 lipidation, it has binding sites for ubiquitin, FIP200 and WIPI2, phosphoinositides, and generally for membranes. It was further shown to interact with TRIM16 on damaged lysosomes (Chauhan et al, 2016) and with TMEM59 involved in autophagic degradation of late endosomes/lysosomes and Staphylococcus aureus‐containing phagosomes (Boada‐Romero et al, 2013).
ATG16L1 is also involved in LC3‐associated phagocytosis (LAP), where the LC3 conjugation system is recruited directly to a single‐membrane phagosome for fusion with lysosomes (Martinez et al, 2011, 2015; Heckmann et al, 2017). LAP has been described mainly in phagocytes such as macrophages, microglia, and dendritic cells that actively take up bacteria, opsonized particles, or dead cells by specific cargo receptors (such as Toll‐like receptors, immunoglobulin receptors, and the phosphatidylserine receptor TIM4, respectively). LAP is independent of the ULK–FIP200–ATG13–ATG101 complex and of WIPI proteins.
So far, little is known about selective autophagy of damaged early endosomes. Autophagy of endosomes disrupted upon uptake of transfection reagent‐coated latex beads showed a requirement for ubiquitination and ATG16L1 binding to ubiquitin (Fujita et al, 2013). As an alternative method of damaging endosomes, lysosomotropic agents such as chloroquine and monensin have been used, which cause swelling of acidic compartments due to osmotic imbalance (Florey et al, 2015; Jacquin et al, 2017; Mauthe et al, 2018). They induce autophagy of endosomal compartments, while simultaneously blocking autophagic flux by inhibiting fusion of autophagosomes with lysosomes (Mauthe et al, 2018). Monensin treatment induced localization of Gal8, ATG16L1, and LC3 at early endosomes in an ATG13‐dependent manner (Fraser et al, 2019), suggesting similarities to lysophagy.
As a morphologically distinctive model of an endosomal compartment, entotic phagosomes or latex bead‐containing phagosomes had been analyzed (Florey et al, 2015; Jacquin et al, 2017). Treatment with chloroquine or monensin was found to induce an LAP‐like process of direct LC3 recruitment to the intact single phagosomal membrane, without ubiquitination, PI3P production, or WIPI involvement, and independent of ATG13 and of the FIP200‐binding domain of ATG16L1.
In the present study, we analyzed the connection between Rabaptin5 and autophagy. We found Rabaptin5 to interact with FIP200 and with the WD domain of ATG16L1 to selectively target autophagy to early endosomes damaged by chloroquine or monensin treatment. In addition, we show that Rabaptin5 initiates autophagy of early Salmonella‐containing vacuoles and is thus responsible for killing a significant fraction of bacteria early upon infection.
Results
FIP200 is a novel interactor of Rabaptin5 on early endosomes
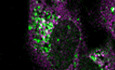
To identify new interaction partners of Rabaptin5, we used human Rabaptin5 as a bait for a yeast two‐hybrid screen with a HeLa cell prey library. The screen reproduced the known interactions of Rabaptin5 with itself and with Rabex5, but also revealed new candidates (Appendix Table S1). Very interestingly, the screen detected FIP200 (Fig 1A and B), a component of the ULK1–ATG13 autophagy initiator complex, as a high confidence interactor of Rabaptin5. The smallest isolated interacting fragment encompassed residues 281–439 of FIP200, a segment outside the coiled‐coil regions of the protein (Fig 1C). Importantly, an interaction of FIP200 with Rabaptin5 could be confirmed in HeLa and HEK293A cells by co‐immunoprecipitation from cell lysates (Fig 1D).
Figure 1. Interaction of Rabaptin5 with FIP200 as identified by two‐hybrid analysis in yeast and by co‐immunoprecipitation in HeLa and HEK293A cells.
-
ASchematic representation of the sequence of Rabaptin5. Coiled‐coil (CC) segments are shown in yellow. Colored backgrounds highlight the segments shown to interact with Rab4, Rab5, Rabex5, and the GAE and GAT domains of GGAs (Golgi‐localizing, γ‐adaptin ear homology domain, ARF‐binding proteins). Below, the segments used to test yeast two‐hybrid interaction with residues 257–444 of FIP200 are shown with their number (#) and the observed interaction (+ or –).
-
BYeast two‐hybrid analysis for interaction between the above‐shown Rabaptin5 segments (Rbpt5#, fused to LexA on the bait plasmid) and residues 257–444 of FIP200 (FIP, fused to the Gal4 activation domain on the prey plasmid) to drive HIS3 expression. Three different clones each were replica‐plated on medium with His or without His, but containing 3‐amino‐1,2,4‐triazole (3AT; an inhibitor of His synthesis to increase stringency) and grown in the absence of Trp and leucine as a control. As negative controls, empty bait or prey plasmids were used. The asterisk indicates a clone invalidated by recombination.
-
CSchematic representation of the sequence of FIP200 with its coiled‐coil segments in yellow. Residues 281–439 (gray) indicate the minimal sequence identified to interact with Rabaptin5 in the yeast two‐hybrid screen.
-
DFIP200 was immunoprecipitated (IP) from lysates of HeLa or HEK293A cells and probed for FIP200, Rabaptin5 (Rbpt5), and EEA1 (early endosome antigen 1) by immunoblotting. Input lysate (10%) was immunoblotted blotted parallel. As a negative control, the immunoprecipitation was performed using an anti‐GAPDH antibody.
-
E–HLysates of HeLa cells transiently transfected with full‐length FIP200‐mCherry (FIP200‐mCh) or a deletion mutant lacking the segment interacting with Rabaptin5 (∆280–440) were immunoprecipitated with anti‐mCherry (IP FIP200‐mCh) or, as a control, with anti‐FLAG antibodies (IP FLAG). Immunoprecipitates and input lysates (10%) were immunoblotted for mCherry and Rabaptin5 (E), ATG13 (F), or ULK1 (G). Co‐immunoprecipitation of Rabaptin5, ATG13, and ULK1 with FIP200∆280–440 (FIP∆) was quantified in comparison with that with wild‐type FIP200 (H; signals normalized to that of the immunoprecipitated protein; mean ± SD of three independent experiments each).
By yeast two‐hybrid testing of different protein fragments, the interacting segment in Rabaptin5 was identified to be the coiled‐coil domain CC2‐1 (Fig 1A and B), where also Rabex5 and the GAT domain of GGA proteins have been shown to bind (Zhu et al, 2004; Zhang et al, 2014). Since this domain and binding of Rabex5 are important for Rabaptin5 recruitment to early endosomes (Kälin et al, 2015), specific disruption of FIP200‐binding by its deletion is not possible. Deletion of the segment 280–440 of FIP200 that is sufficient for interaction with Rabaptin5 in the two‐hybrid assay was found to be necessary for co‐immunoprecipitation of Rabaptin5 (Fig 1E). Unfortunately, deletion of this segment also strongly reduced co‐immunoprecipitation of ATG13 (Fig 1F) and to a lesser extent of ULK1 with FIP200 (Fig 1G and H), indicating that it did not specifically abrogate binding to Rabaptin5, but also destabilized the incorporation of FIP200 into the ULK1 initiator complex. This is in line with recent structural data indicating that overlapping residues 435–442 are important for protein stability and that residues 443–450 of FIP200, right next to the Rabaptin5‐binding region, binds ATG13 (Shi et al, 2020). The FIP200∆280–440 mutant thus cannot help to define the role of Rabaptin5 in autophagy.
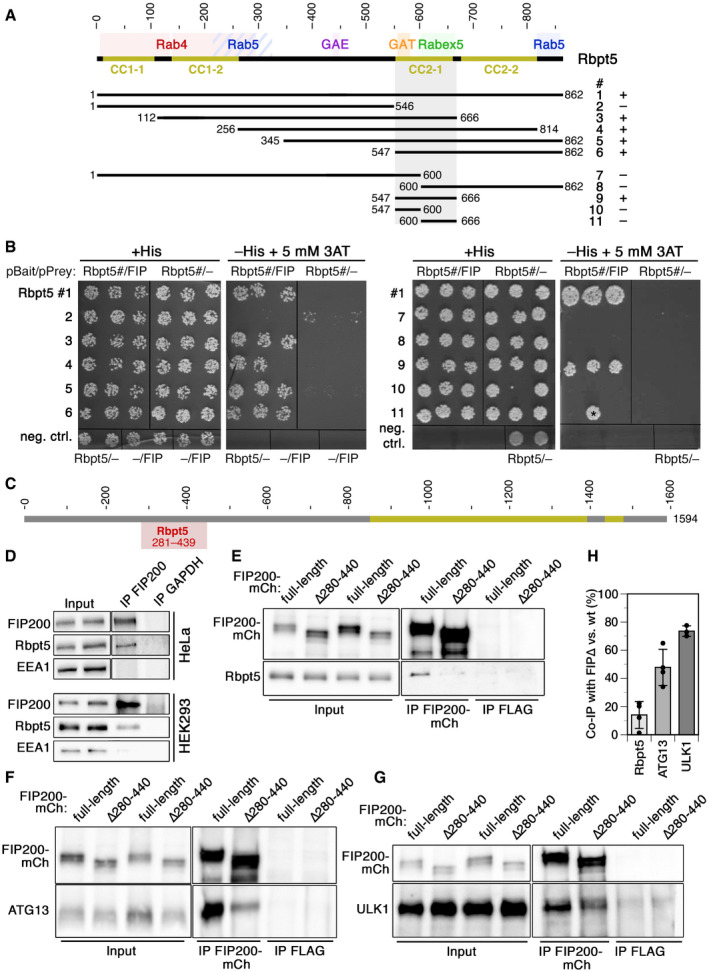
FIP200 transfected alone in HeLa cells localized to small intracellular puncta throughout the cell (Fig EV1A), corresponding to steady‐state autophagosomes (Hara et al, 2008). When expressed together with wild‐type Rabaptin5, they colocalized in slightly larger structures (Fig EV1B). Similarly, FIP200 colocalized with the co‐transfected early endosomal markers Rab4 and Rab5, but not with the late endosomal marker Rab7 (Fig EV1, EV2, EV3, EV4, EV5). This observation is consistent with an interaction of FIP200 with early Rabaptin5‐positive endosomes.
Figure EV1. FIP200 colocalizes with endosomal markers.
-
A–EHeLa cells were transfected with FLAG‐tagged FIP200 alone (A) or together with Rabaptin5 (Rbpt5‐wt) (B), GFP‐Rab4 (C), RFP‐Rab5 (D), or Citrine‐Rab7 (E), fixed after 24 h, and subjected to immunofluorescence microscopy. Scale bar, 10 µm.
Figure EV2. Chloroquine damages transferrin receptor‐ and Galectin 3‐positive early endosomes without recruiting ESCRT components.
- HEK+Rbpt5 cells transfected with mCherry‐galectin 3 (mCh‐Gal3) were treated with 60 µM chloroquine for 30 min and analyzed by immunofluorescence microscopy for Rabaptin5, transferrin receptor (TfR), and mCherry‐galectin 3. Arrowheads point out swollen Rabaptin5‐positive endosomes. Scale bar, 10 µm.
- Cells from experiments as in panel A were quantified for costaining of Rabaptin5‐positive rings with transferrin receptor and galectin 3 (mean ± SD of three independent experiments counting > 50 rings each).
- HEK+Rbpt5 cells were treated without (–) or with 60 µM chloroquine (+CQ) or 1 mM LLOMe (+LLOMe) for 30 min and analyzed by immunofluorescence microscopy for Rabaptin5 and ALIX. Arrowheads point out swollen Rabaptin5‐positive endosomes. Scale bar, 10 µm.
- HeLa cells stably expressing CHMP4B‐GFP were treated as in panel C and analyzed for Rabaptin5, CHMP4B‐GFP, and ALIX. Scale bar, 10 µm.
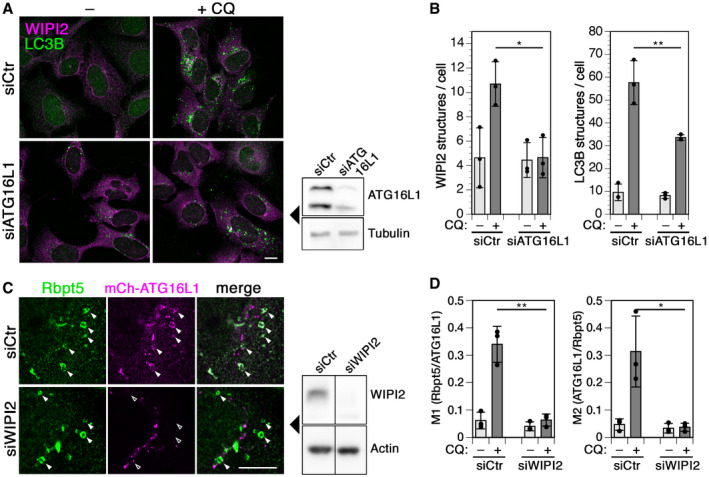
Figure EV3. Interdependence of WIPI2 and ATG16L1 recruitment to chloroquine‐damaged endosomes.
-
A, BCells were transfected with control siRNA (siCtr) or siRNA silencing ATG16L1, treated without (–) or with 60 µM chloroquine (+CQ) for 150 min, fixed, and stained for WIPI2 and LC3B (A). Scale bar, 10 µm. Efficiency of ATG16L1 knockdown was assayed by immunoblotting on the right. WIPI2 of LC3B puncta per cell was quantified for each condition (mean ± SD of three independent experiments; two‐tailed Student’s t‐test: *P < 0.05, **P < 0.01).
-
CHEK+Rbpt5 cells were transfected with nontargeting siRNA (siCtr) or siRNAs silencing WIPI2 (siWIPI2) for 72 h and with mCherry‐ATG16L1 for 24 h. Cells were treated without or with 60 µM chloroquine (+CQ) for 30 min and stained for Rabaptin5 and mCherry‐ATG16L1. Fluorescence micrographs of chloroquine‐treated cells are shown (left panel). Scale bar, 10 µm. Arrowheads point out chloroquine‐induced enlarged early endosomes. The efficiency of WIPI2 knockdown was assayed by immunoblotting using actin as a loading control (middle panel).
-
DManders’ colocalization coefficients were determined, M1 showing the fraction of Rabaptin5‐positive structures also positive for mCherry‐ATG16L1 and M2 showing the inverse. Mean ± SD of three independent experiments; ANOVA: *P < 0.05, **P < 0.01.
Figure EV4. Recruitment of full‐length ATG16L1 and ATG16L1α to damaged endosomes depends on the interaction motif in Rabaptin5.
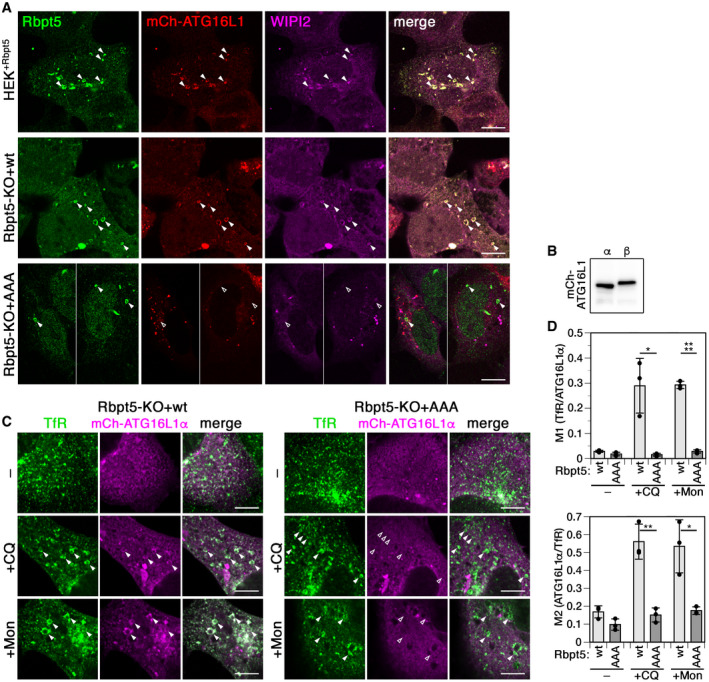
- HEK+Rbpt5 cells and Rabaptin5‐knockout cells stably re‐expressing wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA) were transfected with mCherry‐ATG16L1, treated with 60 µM chloroquine (CQ) for 30 min, and analyzed by immunofluorescence microscopy for Rabaptin5, mCherry‐ATG16L1, and WIPI2. Manders’ colocalization coefficients determined from these experiments are shown in Fig 6F and G. Scale bar, 10 µm. Arrowheads point out enlarged Rabaptin5‐positive endosomes (empty arrowheads when negative for the stained protein).
- HEK293A cells were transfected with the mCherry‐tagged full‐length b isoform of ATG16L1 or the a isoform lacking residues 266–284 and analyzed by immunoblot analysis.
- Rabaptin5‐knockout cells stably re‐expressing wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA) were transfected with mCherry‐ATG16L1α, treated without (–) or with 60 µM chloroquine (+CQ) or 100 µM monensin (+Mon) for 30 min, and analyzed by immunofluorescence microscopy for the transferrin receptor (TfR) and mCherry‐ATG16L1α. Arrowheads point out chloroquine‐induced enlarged early endosomes. Scale bar, 10 µm. Arrowheads point out enlarged Rabaptin5‐positive endosomes (empty arrowheads when negative for the stained protein).
- From experiments as in panel C, Manders’ colocalization coefficients were determined, M1 showing the fraction of transferrin receptor‐positive structures also positive for mCherry‐ATG16L1α and M2 showing the inverse (mean ± SD of three independent experiments; two‐tailed Student’s t‐test: *P < 0.05, **P < 0.01, ****P < 0.0001).
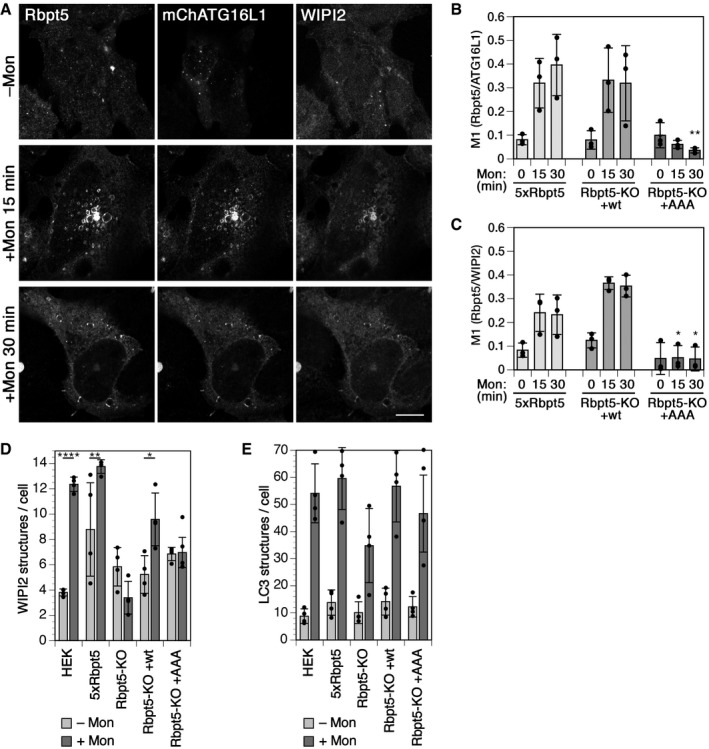
Figure EV5. Monensin induces endosomal autophagy dependent on Rabaptin5 and ATG16L1‐binding just like chloroquine.
-
AHEK+Rbpt5 cells, 24 h after transfection with mCherry‐ATG16L1, were treated with 100 µM monensin for 0, 15, and 30 min and stained for Rabaptin5, WIPI2, and mCherry‐ATG16L1 to assess their colocalization on swollen early endosomes. Scale bar, 10 µm.
-
B, CManders’ colocalization coefficients were determined, showing the fraction of Rabaptin5‐positive structures also positive for mCherry‐ATG16L1 (B) or for WIPI2 (C) (mean ± SD of three independent experiments, quantifying ~40 cells for each sample; ANOVA for Rbpt5‐KO+AAA versus HEK+Rbpt5 cells : *P < 0.05, **P < 0.01).
-
D, EWild‐type HEK293A cells, HEK+Rbpt5 cells, and Rabaptin5‐knockout cells without (Rbpt5‐KO) or with stable re‐expression of wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA) were treated without (–Mon) or with 100 µM monensin for 150 min (+Mon), and analyzed by immunofluorescence microscopy for WIPI2 or LC3B. WIPI2 (D) of LC3B (E) puncta per cell were quantified for each condition (mean ± SD of four independent experiments; ANOVA: *P < 0.05, **P < 0.01, ****P < 0.0001).
Chloroquine treatment induces autophagy of Rabaptin5‐positive early endosomes
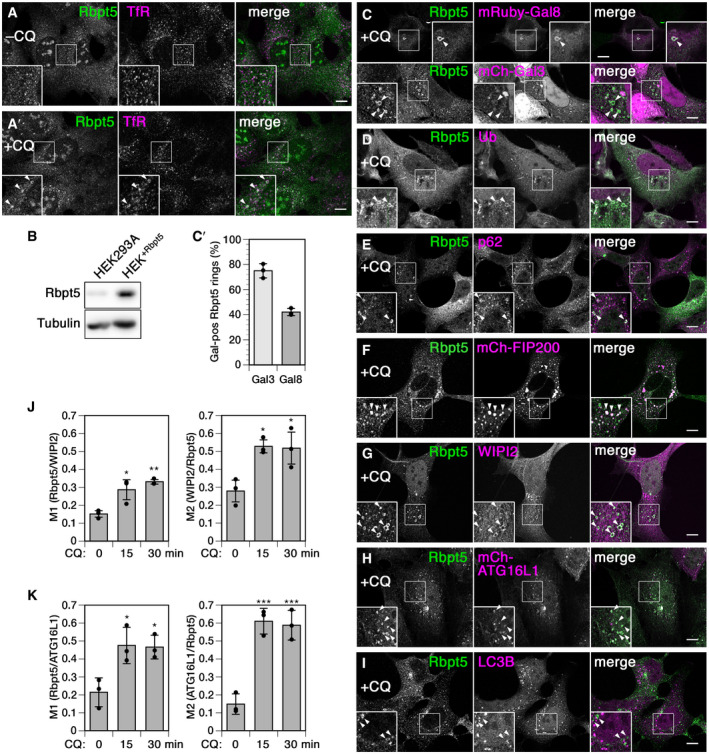
To assess a potential involvement of Rabaptin5 in autophagy at early endosomes, we employed chloroquine treatment as in previous studies (e.g., Mauthe et al, 2018). To be able to easily detect Rabaptin5‐positive early endosomes by immunofluorescence without transient transfection, we generated a stable HEK293A cell line moderately overexpressing Rabaptin5 approximately 5 times (HEK+Rbpt5; Fig 2A and B). Incubation of these cells with 60 µM chloroquine for 30 min indeed produced swollen structures appearing as small rings positive for Rabaptin5 and transferrin receptor and thus identifiable as early endosomes (Fig 2A'). In transfected cells expressing moderate levels of mCherry‐galectin 3 or mRuby3‐galectin 8, these lectins were found to be recruited to the Rabaptin5‐positive endosomes at high frequency (Fig 2C, quantified in C'), indicating membrane rupture. The endosomal identity of Rabaptin5‐ and galectin 3‐positive enlarged ring‐like structures was further confirmed by triple‐labeling with transferrin receptor (Fig EV2A and B). The structures also stained positive for ubiquitin (Fig 2D) and p62 (Fig 2E), a selective autophagy receptor connecting ubiquitin to LC3, consistent with exposure of lumenal protein domains to the cytosol. Membrane damage of Rabaptin5‐positive endosomes is distinct from the effect of chloroquine on phagophores or entotic vacuoles where the membranes remain intact and LAP‐like autophagy is initiated (Florey et al, 2015).
Figure 2. Autophagy proteins are targeted to Rabaptin5‐positive endosomes upon chloroquine treatment.
-
A–ITo more easily visualize Rabaptin5, a stable HEK293A cell line overexpressing Rabaptin5 (HEK+Rbpt5) was generated. Immunofluorescence microscopy of Rabaptin5 and transferrin receptor (TfR) showed swelling of early endosomes upon treatment with 60 µM chloroquine for 30 min (+CQ, panel A) compared with untreated cells (–CQ, A'). Rabaptin5 levels were analyzed by immunoblotting in comparison with parental HEK293A cells (panel B). HEK+Rbpt5 cells, untransfected or 24 h after transfection with mCherry‐galectin 3 (mCh‐Gal3), mRuby3‐galectin 8 (mRuby‐Gal8), mCherry‐FIP200, or mCherry‐ATG16L1 were analyzed upon chloroquine treatment by immunofluorescence microscopy for Rabaptin5 and mCherry‐galectin 3 or mRuby3‐galectin 8 (C), ubiquitin (Ub; D), p62 (E), mCherry‐FIP200 (F), WIPI2 (G), mCherry‐ATG16L1 (H), or LC3B (I). Scale bar, 10 µm. In the enlarged insets, arrowheads point out chloroquine‐induced enlarged, ring‐like early endosomes. Rabaptin5‐positive enlarged endosomes positive for mCherry‐galectin 3 (Gal3) or mRuby3‐galectin 8 (Gal8) were quantified (C'; mean ± SD and individual values of three independent experiments counting > 45 structures each).
-
J, KHEK+Rbpt5 cells, untransfected or 24 h after transfection with mCherry‐ATG16L1, were treated with 60 µM chloroquine for 0, 15, and 30 min and stained for Rabaptin5 and either WIPI2 or mCherry‐ATG16L1. Manders’ colocalization coefficients were determined, M1 showing the fraction of Rabaptin5‐positive structures also positive for WIPI2 (J) or mCherry‐ATG16L1 (K), and M2 showing the respective inverse (mean ± SD of three independent experiments; ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001).
The rings further stained positive for early autophagy components. Transfected mCherry‐FIP200 strongly colocalized with Rabaptin5‐positive structures (Fig 2F), which appeared enlarged most likely due to the overexpression of FIP200. WIPI2 and ATG16L1 (Fig 2G and H) also localized to swollen early endosomes. At this early time point of 30 min, the late component LC3B had not accumulated yet (Fig 2I).
Colocalization of WIPI2 and ATG16L1 was not only observed qualitatively on large Rabaptin5‐positive rings, but also globally as quantified using Manders' colocalization coefficients (Fig 2J and K): The fraction of Rabaptin5 on WIPI2‐ or ATG16L1‐positive structures (M1) and the fractions of these two proteins on Rabaptin5‐positive structures (M2) were significantly increased already after 15‐min chloroquine treatment and similarly after 30 min.
A rapid effect of endomembrane damage has been reported to be membrane repair by an ESCRT‐mediated mechanism involving ALIX and the ESCRT‐III effector CHMP4B on damaged lysosomes (Radulovic et al, 2018; Skowyra et al, 2018; Jia et al, 2020). Lysosome disruption and subsequent lysophagy can be specifically induced by l‐leucyl‐l‐leucine methyl ester (LLOMe) that is condensed into a membranolytic polymer by cathepsin C in lysosomes (Maejima et al, 2013; Chauhan et al, 2016). While 30‐min incubation with LLOMe strongly recruited CHMP4B and ALIX to damaged lysosomes, chloroquine treatment caused hardly any activation of CHMP4B or ALIX (Fig EV2C and D). No recruitment of either ESCRT protein was observed to enlarged Rabaptin5‐positive endosomes.
Chloroquine‐induced autophagy, but not Torin1‐induced autophagy or lysophagy, depends on Rabaptin5
Upon longer exposure of 150 min to chloroquine or to Torin1, an inhibitor of mTOR to induce autophagy mimicking starvation, control cells showed a strong increase in the numbers of both early WIPI2‐positive and late LC3B‐positive autophagosomal puncta (Fig 3A and B). Silencing of FIP200 expression by RNA interference blocked this increase in WIPI2 autophagosomes completely and of LC3B autophagosomes to a large extent for both treatments. Silencing of Rabaptin5 similarly inhibited specifically the formation of chloroquine‐induced WIPI2 and LC3B structures, but did not affect autophagy induction by Torin1. This result indicates that Rabaptin5 is specifically involved in autophagy triggered by chloroquine, but not in starvation‐induced autophagy regulated by mTOR, while FIP200 is required for both. The observation that knockdown of Rabaptin5 as well as of FIP200 appears less effective in reducing LC3B structures with chloroquine than WIPI2 puncta is likely due to stabilization of late autophagosomes by inhibition of autophagic flux (e.g., Mauthe et al, 2018).
Figure 3. Chloroquine‐ but not Torin1‐ or LLOMe‐induced autophagy depends on Rabaptin5.
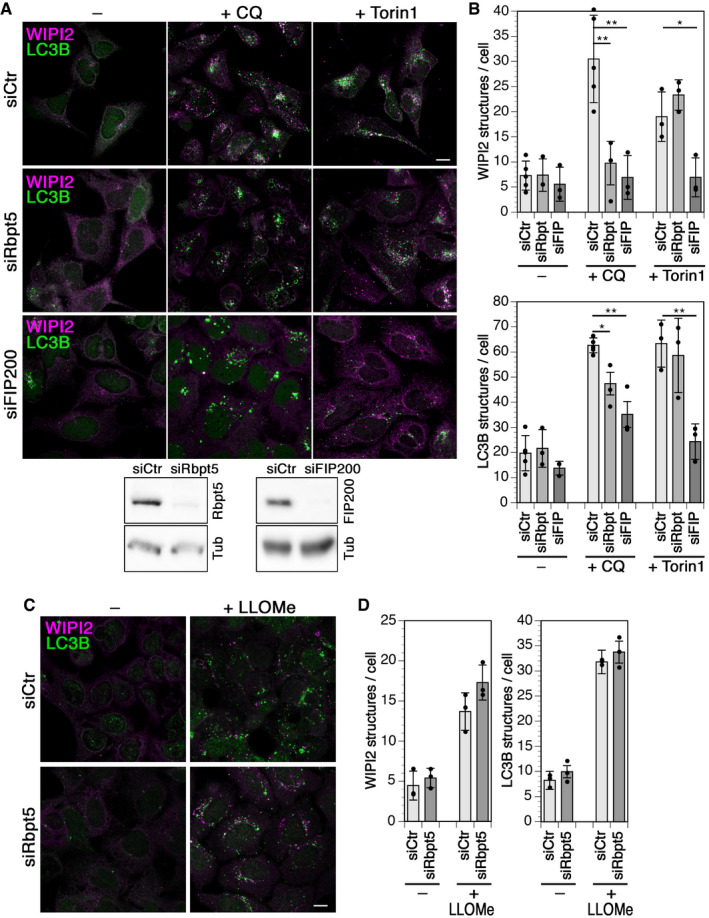
- HEK293A cells were transfected with nontargeting siRNA (siCtr) or siRNAs silencing Rabaptin5 (siRbpt5) or FIP200 (siFIP200) for 72 h and treated without (–) or with 60 µM chloroquine (+CQ) or 250 nM Torin1 for 150 min. Scale bar, 10 µm. Below, the efficiency of Rabaptin5 and FIP200 knockdown was assayed by immunoblotting using tubulin (Tub) as a loading control.
- WIPI2 or LC3B puncta per cell were quantified for each condition (mean ± SD of three independent experiments; ANOVA: *P < 0.05, **P < 0.01).
- HEK293A cells were transfected with siCtr or siRbpt5 as in A and incubated without or with 280 µM LLOMe for 150 min to induce lysophagy. Cells were fixed and immunostained for endogenous WIPI2 and LC3B. Scale bar, 10 µm.
- WIPI2 or LC3B puncta per cell were quantified (mean ± SD of three independent experiments).
Since chloroquine more generally affects acidified organelles, we attempted to differentiate between autophagy of lysosomes (lysophagy) and of endosomes. Incubation with LLOMe for 150 min induced the formation of WIPI2 and LC3B puncta that was not affected by silencing of Rabaptin5 (Fig 3C and D), indicating a specific role of Rabaptin5 in autophagy of damaged endosomes.
Chloroquine‐induced autophagy requires the FIP200 complex and ATG16L1
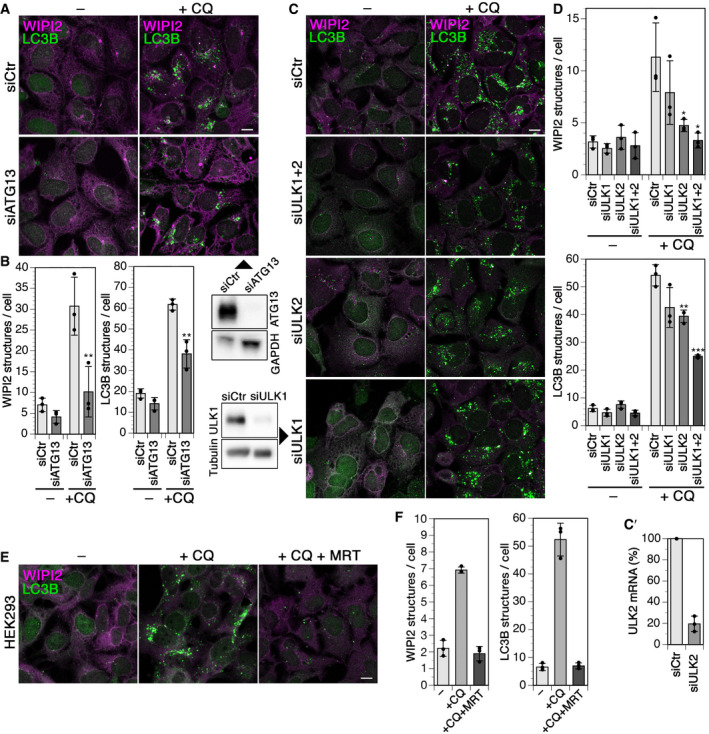
We further tested for an involvement of ATG13 and ULK1 and 2, the main partners of FIP200 in the classical autophagy initiation complex, in chloroquine‐induced autophagy. Silencing of ATG13 essentially blocked an increase in WIPI2 puncta and strongly reduced LC3B structures, just like silencing of FIP200 or Rabaptin5 did (Fig 4A and B). In contrast, silencing of ULK1 did not significantly reduce chloroquine induction of WIPI2‐ or LC3B‐positive autophagosomes (Fig 4C and D), even though it inhibited starvation‐ and Torin1‐induced autophagy (Appendix Fig S1A and B). Knockdown of its homolog ULK2 showed a significant reduction of chloroquine‐induced WIPI2 and LC3B puncta, while silencing of both ULKs was even more effective (Fig 4C and D). Incubation of cells with the general ULK inhibitor MRT68921 (Petherick et al, 2015; Chaikuad et al, 2019) entirely blocked induction of WIPI2 or LC3B autophagosomes by chloroquine (Fig 4E and F). The results suggest that mainly ULK2 is required for this function. Upon knockdown of ATG13 or inhibition of ULK1 and ULK2 by MRT68921, recruitment of mCherry‐ATG16L1 to chloroquine‐induced swollen endosomes and colocalization with Rabaptin5 was blocked (Appendix Fig S2A and B). FIP200 thus acts in chloroquine induction of autophagy together with its complex partners.
Figure 4. Chloroquine‐induced autophagy requires the FIP200–ULK1/2–ATG13 complex.
-
AHEK293A cells were transfected with nontargeting siRNA (siCtr) or siRNAs silencing ATG13 (siATG13) for 72 h, treated without (–) or with 60 µM chloroquine (+CQ) for 150 min, and fixed, and immunostained for endogenous WIPI2 and LC3B. Scale bar, 10 µm. Efficiency of ATG13 knockdown was assayed by immunoblotting below on the right using GAPDH as loading controls.
-
BWIPI2 of LC3B puncta per cell was quantified for each condition (mean ± SD of three independent experiments; ANOVA: **P < 0.01).
-
C, C′HEK293A cells were transfected with nontargeting siRNA (siCtr) or siRNAs silencing ULK1 (siULK1), ULK2 (siULK2), or both (siULK1+2) for 72 h, treated without (–) or with 60 µM chloroquine (+CQ) for 150 min, and fixed and immunostained for endogenous WIPI2 and LC3B. Scale bar, 10 µm. Efficiency of ULK1 knockdown was assayed by immunoblotting on the left and its effect on autophagy induced by starvation and Torin1 treatment in Appendix Fig S1. The efficiency of ULK2 knockdown was assayed by quantitative RT–PCR, and the result is shown in panel C'.
-
DWIPI2 of LC3B puncta per cell was quantified for each condition (mean ± SD of three independent experiments; ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001).
-
EHEK293A cells were incubated without (–), with 60 µM chloroquine (+CQ), or with chloroquine and 5 µM MRT68921 (+CQ +MRT) for 150 min and fixed and stained for WIPI2 and LC3B. Scale bar, 10 µm.
-
FWIPI2 of LC3B puncta per cell was quantified for each condition as in panel B (mean ± SD of three independent experiments).
Silencing of ATG16L1, which recruits the ATG5/12 E3‐like enzyme complex for LC3 lipidation, not only reduced LC3B autophagosomes induced by chloroquine as expected, but also blocked formation of WIPI2 puncta (Fig EV3A and B). ATG16L1 thus appears to be required to recruit or stabilize WIPI2. It has already been reported that WIPI2 puncta were reduced in cells lacking ATG16L1 or expressing ATG16L1 mutated in its PI3P‐binding motif (Dudley et al, 2019). But as observed in previous studies (Dooley et al, 2014; Bakula et al, 2017; Dudley et al, 2019), the opposite was also the case: Recruitment of mCherry‐ATG16L1 to chloroquine‐induced enlarged Rabaptin5‐positive endosomes as well as colocalization in general was inhibited upon siRNA silencing of WIPI2 (Fig EV3C and D). This indicates mutual stabilization of WIPI2 and ATG16L1 on early endosomes.
In addition to the involvement of the FIP200 complex and WIPI2 in chloroquine‐induced autophagy of endosomes, also the recruitment of galectins and p62, indicating membrane rupture, suggested canonical autophagy rather than an LAP‐like process. Silencing galectin 3 or p62 confirmed this notion, since chloroquine‐induced WIPI2‐positive autophagosomes were clearly reduced, while—as expected—Torin1‐induced autophagy was not affected (Appendix Fig S3A and B). LC3B structures were not significantly reduced. Since chloroquine inhibits autophagic flux, accumulating LC3B‐positive structures are likely less sensitive to incomplete inhibition by knockdowns.
A further distinction between canonical and LAP‐like autophagy is sensitivity to bafilomycin A1 (BafA1) (Florey et al, 2015; Jacquin et al, 2017), an inhibitor of the V‐type H+‐ATPase that acidifies endosomes and lysosomes. Resulting neutralization of lysosomes also blocks autophagic flux. In contrast, chloroquine was shown not to neutralize acidic compartments and instead to inhibit autophagosome–lysosome fusion (Mauthe et al, 2018). We found chloroquine to induce LC3B lipidation, formation of WIPI2 and LC3B puncta, and enhancement of LC3B–Rabaptin5 colocalization independently of BafA1 (Appendix Fig S3C–E), consistent with induction of canonical autophagy.
Rabaptin5 binds to the WD domain of ATG16L1 via a conserved interaction motif
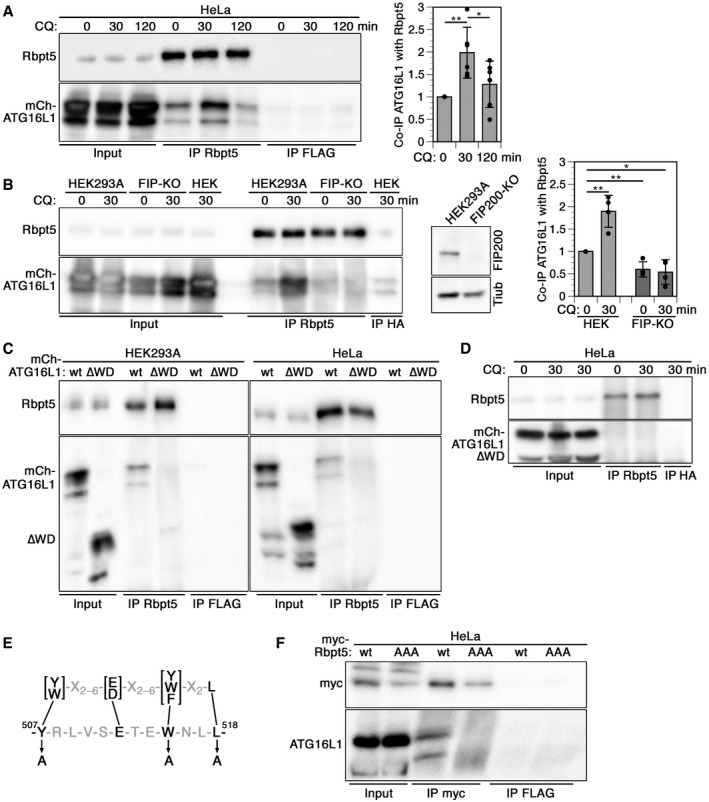
To test whether Rabaptin5 interacts with ATG16L1 on early endosomes, similar to TRIM16 or TMEM59 in autophagy of lysosomes or bacteria‐containing phagosomes (Boada‐Romero et al, 2013; Chauhan et al, 2016; Jia et al, 2020), we performed co‐immunoprecipitation experiments. Indeed, ATG16L1 was co‐immunoprecipitated with Rabaptin5 at low levels already from untreated HeLa cells and more extensively after 30 min of chloroquine treatment (Fig 5A). The interaction decreased after 2 h with chloroquine. Co‐immunoprecipitation was also observed in HEK293A cells after a 30‐min incubation with chloroquine (Fig 5B). However, in a cell line lacking FIP200 generated by CRISPR/Cas9 gene inactivation, no specific co‐isolation with immunoprecipitated Rabaptin5 was detected (Fig 5B). In FIP200‐knockout cells, mCherry‐ATG16L1 recruitment to chloroquine‐induced enlarged Rabaptin5‐positive endosomes and colocalization between Rabaptin5 and mCherry‐ATG16L1 were inhibited (Appendix Fig S4A and B). Endosomal autophagy and interaction of Rabaptin5 with ATG16L1 thus depend on FIP200.
Figure 5. Rabaptin5 binds to the WD domain of ATG16L1 via a conserved interaction motif† .
-
AHeLa cells transiently transfected with full‐length mCherry‐ATG16L1 were treated with 60 µM chloroquine (CQ) for 0, 30, or 120 min, lysed, and immunoprecipitated with anti‐Rabaptin5 (IP: Rbpt5) or, as a control, with anti‐FLAG antibodies (IP FLAG). Immunoprecipitates and input lysates (10%) were immunoblotted for Rabaptin5 and ATG16L1. Signals were quantified and the ratios of mCherry‐ATG16L1/Rabaptin5 normalized to that without (0 min) chloroquine treatment (mean ± S of six independent experiments; two‐tailed Student’s t‐test: *P < 0.05, **P < 0.001).
-
BCo‐immunoprecipitation was performed as in panel A using parental HEK293A cells and CRISPR/Cas9 knockout cells lacking FIP200 (FIP‐KO). Anti‐HA antibodies were used as a control (IP HA). On the right, HEK293A‐ and FIP200‐knockout cells were immunoblotted for FIP200 and as a loading control of tubulin (Tub). Signals were quantified and the ratios of mCherry‐ATG16L1/Rabaptin5 normalized to that of HEK293A cells without chloroquine treatment (mean ± SD of four independent experiments; two‐tailed Student’s t‐test: *P < 0.05, **P < 0.001).
-
C, DLysates of HEK293A or HeLa cells transiently transfected with full‐length mCherry‐ATG16L1 (wt) or a mutant lacking the WD domain (∆WD; residues 1–319 of ATG16L1, precisely deleting only the WD40 repeats residues 320–607) were immunoprecipitated with anti‐Rabaptin5 or anti‐FLAG antibodies, and immunoblotted for Rabaptin5 and mCherry‐ATG16L1 (C), or immunoprecipitated with anti‐Rabaptin5 or anti‐HA antibodies, and immunoblotted for Rabaptin5 and mCherry‐ATG16L1 (D). In panel D, cells were incubated with or without 60 µM chloroquine for 30 min before analysis. Co‐immunoprecipitation of ATG16L1∆WD with Rabaptin5 (C) was reduced to 6.6 ± 2.1 and 3.4 ± 2.1% in HEK293A and HeLa cells, respectively, relative to that of full‐length ATG16L1 (signals normalized to the immunoprecipitated protein; mean ± SD deviation of three independent experiments each).
-
EThe consensus sequence of the ATG16L1 interaction motifs of TMEM59, NOD2, and TLR2 (above; Boada‐Romero et al, 2013) is shown together with the matching sequence in Rabaptin5 (below). The three point mutations to alanine to produce the AAA mutant of Rabaptin5 are indicated.
-
FLysates of HeLa cells transiently transfected with myc‐tagged wild‐type Rabaptin5 (wt) or triple‐alanine mutant (AAA) were immunoprecipitated with anti‐myc (IP myc) or anti‐FLAG antibodies (IP FLAG), and immunoblotted for myc and ATG16L1. Co‐immunoprecipitation of ATG16L1 with Rabaptin5‐AAA triple mutant was reduced to 1.5 ± 1.2% relative to that with wild‐type Rabaptin5 (signals normalized to that of the immunoprecipitated protein; mean ± SD of three independent experiments).
Co‐immunoprecipitation of ATG16L1 with Rabaptin5 was lost upon deletion of the WD‐repeat domain (residues 320–607) in ATG16L1 (Fig 5C) and not rescued by treatment with chloroquine (Fig 5D). As expected, the interaction of ATG16L1∆WD with FIP200 was retained (Fig 5E). Rabaptin5 thus interacts differently with ATG16L1 than TRIM16, which was shown to bind to the coiled‐coil domain (Chauhan et al, 2016). In contrast, the membrane protein TMEM59, a promoter of selective autophagy of Staphylococcus aureus‐containing phagosomes, had previously been shown to bind to the WD domain (Boada‐Romero et al, 2013). A 19‐amino acid peptide motif containing four essential residues had been identified to mediate this interaction. Similar largely conserved motifs were also found in NOD2, a protein known to recruit ATG16L1 at bacterial entry sites, and in TLR2 that promotes LC3 lipidation at phagosomes. Rabaptin5 also contains a sequence (residues 507–518) that complies with this consensus motif (Fig 5F). Mutation of the three most important residues of this motif to alanines (Y507A/W515A/L518A in Rabaptin5‐AAA), which in TMEM59 completely inactivated ATG16L1 binding, also eliminated co‐immunoprecipitation of ATG16L1 with mutant Rabaptin5 (Fig 5G). The mutant Rabaptin5‐AAA was also co‐immunoprecipitated with FIP200 confirming that the mutations did not interfere with FIP200 binding (Fig 5H).
Binding of Rabaptin5 to ATG16L1 is essential for endosomal autophagy
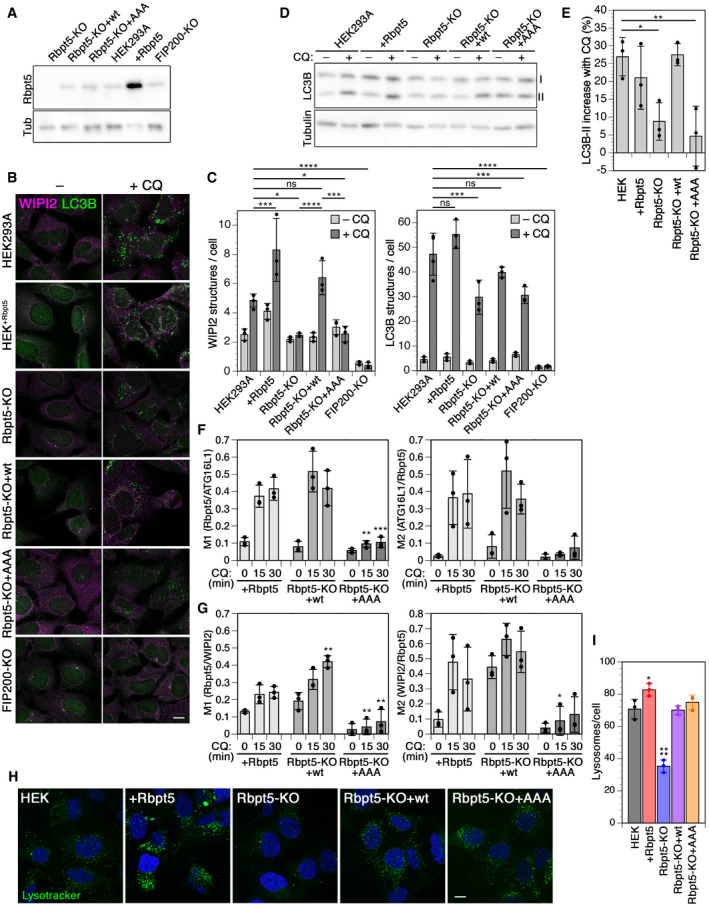
To assess the importance of the interaction between Rabaptin5 and ATG16L1, we produced a Rabaptin5‐knockout HEK293A cell line (Rbpt5‐KO) by CRISPR/Cas9 gene inactivation. By transfecting wild‐type Rabaptin5 or the triple mutant Rabaptin5‐AAA, we produced the stable cell lines Rbpt5‐KO+wt and Rbpt5‐KO+AAA that produced similar Rabaptin5 levels as the original HEK293A cell line (Fig 6A). Again, we analyzed WIPI2‐ and LC3‐positive autophagosomes before and after chloroquine treatment for 150 min (Fig 6B and C). While overexpression of Rabaptin5 resulted in a slight increase in steady‐state and chloroquine‐induced WIPI2 puncta, Rabaptin5 knockout left the steady‐state levels unchanged and eliminated chloroquine‐induced formation of WIPI2 autophagosomes (Fig 6C, left). Re‐expression of wild‐type Rabaptin5 rescued WIPI2 autophagosome induction by chloroquine, while Rabaptin5‐AAA did not. This result suggests that the interaction of Rabaptin5 with ATG16L1 is essential for early endosomal autophagy upon chloroquine damage.
Figure 6. Chloroquine‐induced endosomal autophagy depends on Rabaptin5 and its ATG16L1‐binding motif.
-
ABy immunoblot analysis, the levels of Rabaptin5 and as a loading control of tubulin (Tub) were assessed in wild‐type HEK293A cells, HEK+Rbpt5 cells stably overexpressing Rabaptin5, and Rabaptin5‐knockout cells without (Rbpt5‐KO) or with stable re‐expression of wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA).
-
BThe same stable HEK293A‐derived cell lines were treated without (–CQ) or with 60 µM chloroquine for 150 min (+CQ) and analyzed by immunofluorescence microscopy for WIPI2 or LC3B. Scale bar, 10 µm.
-
CWIPI2 and LC3B puncta per cell were quantified for each condition (mean ± SD of three independent experiments; ANOVA: *P < 0.05, ***P < 0.001, ****P < 0.0001).
-
D, EThe HEK293A‐derived cell lines were treated with 60 µM chloroquine for 30 min, and non‐lipidated and lipidated LC3B (I and II, resp.) were assayed by immunoblot analysis (D). The increase in the LC3B‐II fraction of total LC3B upon chloroquine treatment was quantified (mean ± SD of three independent experiments; ANOVA: *P < 0.05, **P < 0.01).
-
F, GHEK+Rbpt5 cells and Rabaptin5‐knockout cells stably re‐expressing wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA) were transfected with mCherry‐ATG16L1, treated with 60 µM chloroquine (CQ) for 0, 15, and 30 min, and analyzed by immunofluorescence microscopy for Rabaptin5, mCherry‐ATG16L1, and WIPI2. Manders’ colocalization coefficients were determined, M1 showing the fraction of Rabaptin5‐positive structures also positive for mCherry‐ATG16L1 (F) or WIPI2 (G) and M2 showing the inverse (mean ± SD of three independent experiments; ANOVA for Rbpt5‐KO+wt or +AAA versus HEK+Rbpt5 cells: *P < 0.05, **P < 0.01, ***P < 0.001).
-
HWild‐type HEK293A cells, HEK+Rbpt5 cells, Rabaptin5‐knockout cells without (Rbpt5‐KO) or with stable re‐expression of wild‐type (Rbpt5‐KO+wt) or AAA mutant Rabaptin5 (Rbpt5‐KO+AAA) were stained with LysoTracker and with DAPI for nuclei. Scale bar, 10 µm.
-
IThe number of lysosomes (LysoTracker‐positive structures) per cell was quantified from cells as in panel C (mean ± SD of three independent experiments). Images for at least 15 cells per sample were quantified. The value for HEK+Rbpt5 cells is an underestimate, because the density of puncta makes them difficult to distinguish as separate structures. ANOVA: *P < 0.05, ****P < 0.0001.
Rabaptin5 knockout also reduced the chloroquine‐dependent increase in LC3B‐positive puncta (Fig 6C, right), consistent with inhibition of endosomal autophagy, but did not block it. Again, re‐expression of wild‐type Rabaptin5, but not Rabaptin5‐AAA, rescued formation of LC3B‐positive structures. FIP200 knockout strongly reduced both steady‐state and chloroquine‐induced autophagosomes positive for either WIPI2 or LC3B, consistent with an essential role for both steady‐state autophagy and endosomal autophagy.
These results with stable cell lines confirm those with siRNA knockdown of Rabaptin5 or FIP200 of Fig 3. (It should be noted that the numbers of WIPI2 and LC3B puncta were consistently higher under knockdown conditions even at steady state, which is nonspecifically caused by siRNA transfection.)
Immunoblot analysis of LC3B lipidation upon chloroquine treatment (Fig 6D and E) showed the expected increase in LC3B‐II in wild‐type and Rabaptin5‐overexpressing cells already after 30 min, which was reduced in Rabaptin5‐knockout cells and rescued by re‐expression of wild‐type Rabaptin5, but not of the AAA mutant.
We furthermore quantified colocalization of Rabaptin5 and ATG16L1 or WIPI2 upon chloroquine treatment for 0, 15, or 30 min in Rabaptin5‐overexpressing cells and in knockout cells re‐expressing wild‐type or AAA mutant Rabaptin5 (Figs 6F and G, and EV4A). Colocalization with either autophagy protein was lower and hardly induced in knockout cells expressing the Rabaptin5‐AAA mutant unable to bind ATG16L1, demonstrating that this interaction is required to induce endosomal autophagy.
ATG16L1 exists in two isoforms, the full‐length β isoform and the α isoform lacking residues 266–284. This β‐isoform‐specific segment was shown by Lystad et al (2019) to be part of a membrane‐binding region that is dispensable for canonical autophagy, but is required for membrane recruitment of ATG16L1 and LC3B lipidation upon monensin treatment when canonical autophagy was blocked by inhibition of VPS34 and/or ULK1/2. To test the requirement of the β segment for the recruitment of ATG16L1 to chloroquine‐damaged early endosomes, we expressed the α form of mCherry‐ATG16L1 in HEK293A cells (Fig EV4B). In cells containing wild‐type Rabaptin5, but not in cells expressing the AAA mutant, ATG16L1α was recruited to chloroquine‐enlarged transferrin receptor‐positive endosomes resulting in increased localization of ATG16L1α to early endosomes (Fig EV4C and D), just like the β isoform did (Figs EV4A and 2H and K), in support of canonical autophagy at endosomes.
There are a number of interconnections between autophagy and endosomes (Birgisdottir & Johansen, 2020). To address the possibility that absence or mutation of Rabaptin5 might alter endosome properties in a way that indirectly affects chloroquine‐induced autophagy, we confirmed that chloroquine induces early endosome swelling also in Rabaptin5‐KO cells by immunofluorescence microscopy of EEA1 (Appendix Fig S5A). Rabaptin5 deletion did not affect autophagic flux, since the expression of tandem fluorescent‐tagged RFP‐GFP‐LC3 containing an acid‐sensitive GFP and a pH‐insensitive RFP (Kimura et al, 2007) showed an indistinguishable fraction of autophagosomes (red/green double‐positive LC3 puncta) and autolysosomes (red‐only LC3 puncta) in HEK293A and Rabaptin5‐KO cells, without treatment or upon induction of autophagy by starvation or Torin1 treatment (Appendix Fig S5B and C).
To further exclude the possibility that the triple AAA mutation inactivated the function of Rabaptin5 in endosomal maturation and thus perhaps affected endosomal autophagy indirectly, we analyzed the number of lysosomal structures in the different HEK cell lines by LysoTracker staining (Fig 6H). The marked reduction of lysosomes upon Rabaptin5 knockout was completely rescued by re‐expression of wild‐type Rabaptin5 and also by Rabaptin5‐AAA, demonstrating that the endosomal function of the mutant protein was intact (Fig 6I).
To test for degradation of damaged endosomes, transferrin receptor levels were analyzed with and without chloroquine treatment. We found a significant reduction of ~15 and ~30% after overnight incubation with chloroquine in parental HEK293A cells and in Rabaptin5‐KO cells re‐expressing wild‐type Rabaptin5, resp., but no reduction in Rabaptin5‐KO cells expressing the Rabaptin5‐AAA mutant defective in binding to ATG16L1 (Appendix Fig S5D and E).
Monensin‐induced endosomal autophagy also depends on Rabaptin5 and its interaction with ATG16L1
Chloroquine is protonated and trapped within acidified endosomes and lysosomes, osmotically promoting compartment swelling by water influx. Monensin promotes the exchange of protons for osmotically active monovalent cations such as Na+ and thus results in osmotic swelling of acidified compartments in a different way. Monensin treatment of HEK+Rbpt5 cells rapidly produced swollen Rabaptin5‐positive early endosomes that stained positive for ATG16L1 and WIPI2 (Fig EV5A), just like chloroquine treatment (Fig 2F and G). Similarly, overall colocalization of ATG16L1 and WIPI2 with Rabaptin5‐positive structures increased in Rabaptin5‐overexpressing HEK+Rbpt5 cells and in Rabaptin5‐knockout cells re‐expressing wild‐type Rabaptin5, but not in knockout cells expressing the Rabaptin5‐AAA mutant that is unable to bind to ATG16L1 (Fig EV5B and C). The same was observed for the α isoform of ATG16L1 upon monensin treatment (Fig EV4C and D). The increase in total WIPI2‐positive structures upon longer monensin treatment of 150 min was also dependent on Rabaptin5, since it was abolished upon knockout of Rabaptin5 and recovered upon re‐expression of wild‐type Rabaptin5, but not by the AAA mutant (Fig EV5D). The effect on the induction of LC3B‐positive structures was less clearly dependent on Rabaptin5 (Fig EV5E).
Rabaptin5 targets Salmonella‐containing vacuoles to autophagy
As Salmonella enterica enters host cells such as fibroblasts or epithelial cells, its surrounding phagosomal membrane quickly acquires the characteristics of early endosomes through fusion with Rab5‐positive endosomes (Steele‐Mortimer et al, 1999; LaRock et al, 2015; Levin et al, 2016). The Salmonella‐containing vacuole (SCV) is thus positive for Rab5, Rabaptin5/Rabex5, EEA1, transferrin receptor (TfR), and the PI‐3‐kinase VPS34. Perforation of the early SCV membrane by the type III secretion system 1 (T3SS‐1) results in binding of cytosolic galectins and ubiquitination to activate antibacterial autophagy to the SCV membrane (LaRock et al, 2015). Because of this similarity of the early SCV with damaged early endosomes, we tested the role of Rabaptin5 in autophagy of Salmonella early after infection.
We first infected HeLa cells with Salmonella enterica serovar Typhimurium with or without siRNA‐mediated silencing of Rabaptin5 or FIP200 and determined the number of phagocytosed live bacteria after 0, 1, 3, and 6 h (Fig 7A, left). For this, bacteria were gently centrifuged onto the cell layer for 5 min at 37°C and further incubated for 10 min. Bacteria that had not entered the cells were thoroughly washed away. For further incubation, the medium was supplemented with gentamycin to eliminate any remaining extracellular bacteria. Yet, even immediately after infection, confocal immunofluorescence microscopy confirmed that essentially all cell‐associated bacteria were internalized. The infection efficiency was not affected by silencing Rabaptin5 or FIP200 (~14% of cells infected; Fig 7A, right). At different time points, cells were lysed and plated out to determine the number of live bacteria.
Figure 7. Rabaptin5‐mediated autophagy contributes to early killing of Salmonella .
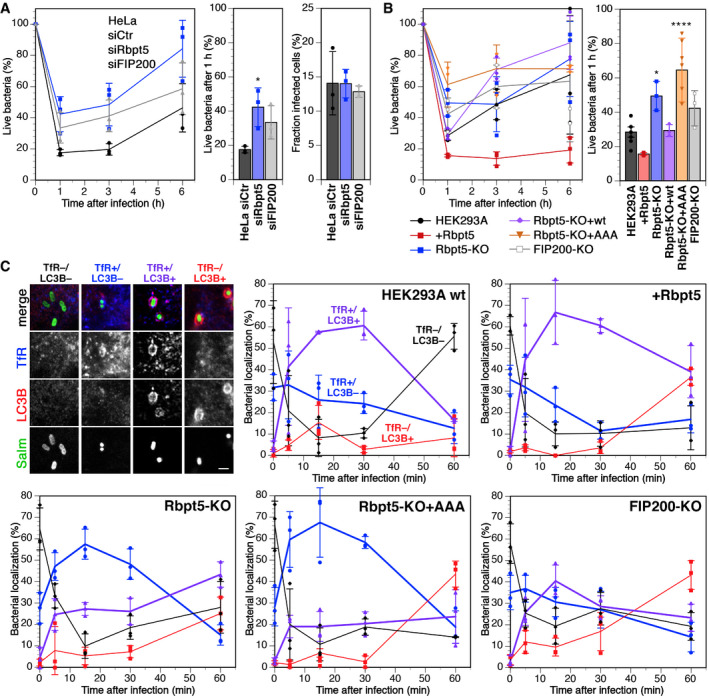
- HeLa cells were transfected with nontargeting siRNA (siCtr) or siRNAs silencing Rabaptin5 (siRbpt5) or FIP200 (siFIP200) for 72 h. The cells were infected with Salmonella by centrifugation at 500 × g for 5 min at 37°C and incubation for 10 min at 37°C, washed three times, and incubated in fresh culture medium containing gentamicin to prevent growth of extracellular bacteria for 0, 1, 3, or 6 h before lysis of the host cells and plating of the bacteria on LB agar plates at various dilutions to determine the number of live bacteria at the different time points, shown as a percentage of internalized cells after infection (mean ± SD of three independent experiments). The fractions of internalized bacteria alive 1 h after infection are shown separately in the middle (mean ± SD of three independent experiments; ANOVA: *P < 0.05). On the right, the fraction of infected cells was determined for HeLa cells transfected with siRNAs and infected as above with Salmonella expressing GFP, washed, and immediately fixed for fluorescence microscopy and stained with anti‐transferrin receptor and anti‐LC3B as cellular markers. Z‐stacks for > 5,000 cells/sample were acquired and analyzed in Fiji to determine the fraction of infected cells (mean ± SD of three independent experiments). The average number of bacteria per infected cell was identical (2.16, 2.11, and 2.12 bacteria per cell transfected with siCtr, siRbpt5, and siFIP200, respectively).
- Wild‐type HEK293A, HEK+Rbpt5, Rbpt5‐KO, Rbpt5‐KO+wt, Rbpt5‐KO+AAA, and FIP200‐KO cells were infected with Salmonella and treated and analyzed as in panels A (mean ± SD of three independent experiments; ANOVA: *P < 0.05, ****P < 0.0001).
- Wild‐type HEK293A, HEK+Rbpt5, Rbpt5‐KO, Rbpt5‐KO+AAA, and FIP200‐KO cells were infected with Salmonella expressing GFP as in panel B, incubated in fresh culture medium containing gentamicin for 0, 5, 15, 30, and 60 min, fixed with methanol, and immunostained for transferrin receptor (TfR) as a marker of early endosomes and for LC3B as a marker of autophagy. Salmonella were classified according to their association with a TfR‐ and/or LC3B‐positive compartment—as illustrated on the top left (scale bar, 2 µm)—during the first hour after infection. In the absence of Rabaptin5, LC3‐positive SCVs with early endosomal characteristics (containing TfR) were strongly reduced. (mean ± SD of three independent experiments, analyzing >50 bacteria for each time point.)
Within the first hour of infection, ~80% of initially internalized bacteria were killed, after which live cell numbers increased again. Part of the observed initial killing is due to autophagy, since knockdown of FIP200 almost doubled the number of live cells 1 h after infection (Fig 7A, middle). Silencing of Rabaptin5 expression was at least as effective, indicating a contribution of Rabaptin5 to early bacterial killing. These findings were confirmed by experiments using our stable HEK293A cell lines (Fig 7B). Only 25% of internalized Salmonella survived the first hour in parental HEK293A cells, whereas overexpression of Rabaptin5 in HEK+Rbpt5 cells reduced this fraction by half and knockout of Rabaptin5 doubled it. Re‐expression of the wild‐type protein in knockout cells recovered wild‐type levels of survival, while the expression of the mutant Rabaptin5‐AAA that is unable to bind ATG16L1 did not.
These results suggest that Rabaptin5 targets autophagy to the damaged membrane of SCVs and contributes to killing of phagocytosed Salmonella. To confirm this concept, bacteria infecting the different HEK293A cell lines were analyzed by immunofluorescence microscopy for colocalization with TfR as a marker for early endosomal membrane identity and for LC3B as an autophagy marker (Fig 7C). Right after infection, while virtually all bacteria were intracellular, more than half of them were negative for both markers. This fraction rapidly declined for the next 15 min and the fraction in a TfR‐positive environment with or without LC3B increased significantly, consistent with increasing acquisition of endosomal markers of the SCVs by fusion with early endosomes.
In wild‐type HEK293A cells and Rabaptin5‐overexpressing HEK+Rbpt5 cells, the predominant population 5–30 min after infection was that of Salmonella in double‐positive (TfR+/LC3B+) SCV–autophagosomes (Fig 7C). In Rabaptin5‐knockout cells, this population was reduced in favor of a majority of TfR single‐positive SCVs as a result of reduced autophagy initiation. The expression of Rabaptin5‐AAA did not change this situation, confirming that the interaction with ATG16L1 is required to initiate autophagy of SCVs. At the same time, it shows that the phenotype of the knockout cells is not just the result of reduced endosome maturation in the absence of Rabaptin5, since the AAA mutant is functional for its other endosomal functions. In FIP200‐knockout cells, TfR+/LC3B+ SCVs were also reduced, as expected.
In summary, Rabaptin5, in addition to its function as a regulator of early endosome identity and endosomal maturation, plays an unexpected role in initiating autophagy of damaged early endosomes and bacteria‐containing phagosomes via interaction with FIP200 and ATG16L1.
Discussion
Previously, Rabaptin5 had been characterized as a regulator of Rab5 activity by complex formation with Rabex5, the GDP/GTP exchange factor of Rab5. In addition, it had been shown to bind to Rab4·GTP and Rab5·GTP, the hallmark Rab proteins of early endosomes (Horiuchi et al, 1997; Lippé et al, 2001; Mattera et al, 2006; Kälin et al, 2015, 2016). Rabaptin5 thus was known to contribute to endosome identity and maturation (Huotari & Helenius, 2011). Silencing of Rabaptin5 has no effect on cell viability, most likely because Rabex5 can recruit independently in the absence of Rabaptin5 to endosomes and activate Rab5 (Mattera & Bonifacino, 2008). However, silencing Rabaptin5 or reduced expression of Rabaptin5 due to hypoxia‐activated hypoxia‐inducible factor (HIF) or overexpressed histone deacetylase 6 (HDAC6) in gastric cancer has been found to delay early endosome fusion, reduce EGF receptor degradation, and thereby prolong receptor signaling (Wang et al, 2009; Park et al, 2014). Consistent with these functions, we found here that the number of lysosomes, the endpoint organelles of the endosomal pathway, was increased by overexpression and reduced upon knockout of Rabaptin5. This phenotype was rescued by re‐expression of wild‐type as well as of Rabaptin5‐AAA that is unable to bind ATG16L1, indicating that the mutant protein performs its endosomal functions normally.
In this study, we identified FIP200 and ATG16L1 as novel interactors of Rabaptin5 and thus an additional unexpected role for Rabaptin5 in autophagy. Endosomes—specifically recycling endosomes—were previously implicated as a source of membranes and of ATG9 and ATG16L1 for growing phagophores (Knævelsrud et al, 2013; Puri et al, 2013; Popovic & Dikic, 2014; Søreng et al, 2018). Rab11A‐positive recycling endosomes were found to act as a platform for autophagosome assembly mediated by the interaction of WIPI2 with PI3P and Rab11A (Puri et al, 2018). These recycling endosomes were demonstrated to be distinct from Rab5‐ and thus also from Rabaptin5‐positive early sorting endosomes. While Rab11A was shown to be broadly important for starvation‐induced autophagy as well as mitophagy (Puri et al, 2018), silencing of Rabaptin5 did not affect Torin1‐induced autophagy or lysophagy, but specifically inhibited autophagy induced by chloroquine or monensin and of SCVs. This specificity argues against a defect in sorting endosomes due to silencing Rabaptin5 to generally inhibit autophagy.
On the contrary, there is evidence that defects in autophagy affect sorting endosome function: Cells lacking ATG7 or ATG16L1 were shown to perturb EGF receptor endocytic trafficking and to abrogate EGF receptor signaling (Fraser et al, 2019). Damaged, galectin 8‐positive early endosomes accumulated in the absence of autophagy, like during chemical disruption with monensin. These findings suggested autophagy of early endosomes (“endosome‐phagy”) to be a housekeeping activity to keep sorting endosomes functional. Rabaptin5 is a good candidate to target autophagy to this compartment.
We found Rabaptin5 to be essential to target the autophagy machinery to damaged endosomes by binding to FIP200 and ATG16L1. Rabaptin5 binds to the WD domain of ATG16L1 via an interaction motif 507 YRLVSETEWNLL 518 based on a conserved consensus initially identified in TMEM59, NOD2, and TLR2 to promote autophagy in the context of bacterial phagosomes (Boada‐Romero et al, 2013). Rabaptin5 with three of the key residues in this sequence mutated to alanines could not rescue chloroquine‐induced WIPI2 and LC3B puncta beyond the level of the Rabaptin5 knockout and did not induce colocalization with WIPI2 or ATG16L1. This suggests that, in the context of damaged early endosomes, Rabaptin5 makes an essential contribution to recruit ATG16L1. The importance of the interaction with FIP200 could not be assessed, since deletion of the interacting segment in Rabaptin5 (residues 547–666; CC2‐1) abolishes Rabex5‐binding and membrane recruitment (Kälin et al, 2015) and in FIP200 destabilized complex formation with ATG13 and ULK1.
Chloroquine treatment caused Rabaptin5‐positive endosomes to swell and break, recruiting galectins 3 and 8, ubiquitination, p62, FIP200, WIPI2, and ATG16L1 within 30 min. At longer time points, also LC3B‐positive autophagosomes accumulated. Induction of WIPI2 puncta was strongly reduced by silencing of Rabaptin5, FIP200, ATG13, or by inhibition of ULK1/2. LC3B autophagosomes/autolysosomes were also reduced, but in most cases to a lesser extent. The involvement of the ULK–FIP200 initiator complex and of WIPI2 and the insensitivity to BafA1 is in agreement with canonical selective autophagy upon chloroquine treatment, similar to lysophagy induced by LLOMe treatment (Maejima et al, 2013; Chauhan et al, 2016). However, it differs from a noncanonical LAP‐like mechanism that was observed for entotic vacuoles or latex bead‐containing phagosomes as model organelles (Florey et al, 2015; Jacquin et al, 2017; Fletcher et al, 2018). Upon chloroquine or monensin treatment, LC3 was recruited directly to the single‐membrane vacuoles without PI3P production or WIPI involvement, without ubiquitination, and independently of ATG13 and of the FIP200‐binding domain of ATG16L1. Chloroquine and monensin appear to trigger a different response on large phagosomal structures and entotic vacuoles than on regular early endosomes.
The relative insensitivity of chloroquine‐induced LC3B puncta to knockdown of FIP200, ATG13, and Rabaptin5—compared to the strong and robust reduction of WIPI2 puncta—suggests the possibility of LAP‐like autophagy occurring in parallel, perhaps on endosomes that were not damaged. Alternatively, inhibition of autophagic flux specifically causes LC3 puncta to accumulate over time. In support of the latter interpretation, complete ULK inhibition by MRT68921 (Fig 4E and F) or FIP200 knockout (Fig 6B and C) abolished chloroquine‐induced LC3 structures, suggesting that—unlike on phagosomes or entotic vacuoles—there is little LAP‐like autophagy on damaged early endosomes. Nevertheless, Lystad et al (2019) detected LC3B lipidation upon monensin treatment in the presence of ULK and VPS34 inhibitors to block canonical autophagy. This lipidation was partially dependent on the β segment of full‐length ATG16L1 providing membrane‐binding affinity. Yet, in the absence of these inhibitors, we found both ATG16L1α and ATG16L1 β to be equally recruited to chloroquine‐ or monensin‐treated early endosomes dependent on the WD domain interaction motif in Rabaptin5 (Fig EV4C and D).
Salmonella that are phagocytosed by professional phagocytes such as macrophages are the target of LAP (Masud et al, 2019). However, Salmonella also has the ability to actively infect other cell types such as epithelial cells and fibroblasts, triggering uptake into SCVs that are the target of canonical autophagy (LaRock et al, 2015). Salmonella manipulates its host by injecting effectors into the host cytosol via two type III secretion systems (T3SS). T3SS‐1 is required for internalization into SCVs. As SCVs are further acidified, T3SS‐2 is induced and its effectors mediate maturation to Salmonella‐induced filaments, where bacteria proliferate. However, T3SS‐1 also damages the SCV membrane. This may result in complete release of bacteria into the cytosol where they hyperproliferate and become direct targets of ubiquitination and autophagy. In contrast, damaged SCVs are recognized by selective autophagy in a similar manner as damaged endosomes or lysosomes: Exposure of lumenal protein domains and glycans triggers the binding of galectin 3 and galectin 8 (Thurston et al, 2012; Fujita et al, 2013), poly‐ubiquitination, and p62 and NDP52 recruitment (Zheng et al, 2009; Thurston et al, 2012). SCVs are surrounded by phagophores in a process requiring FIP200 and ATG9L1 (Kageyama et al, 2011; Fujita et al, 2013; Huang & Brumell, 2014; Kishi‐Itakura et al, 2020).
Because of the similarities between autophagy of SCVs and of damaged endosomes, we tested a potential role of Rabaptin5 in the early phase of Salmonella infection. We found that autophagic elimination of Salmonella during the first hour postinfection, when the SCVs most resemble early endosomes, strongly depended on Rabaptin5 and its ability to bind ATG16L1. In wild‐type HeLa and HEK293A cells, 70–80% of internalized bacteria were killed within the first hour. Knockdown or knockout of Rabaptin5 reduced this number to approximately 50%, i.e., to the same extent as silencing of FIP200 did. The same effect was observed, when wild‐type Rabaptin5 was replaced by a triple mutant defective in its interaction with ATG16L1.
Interestingly, Kageyama et al (2011) observed that LC3 recruitment to SVCs was not blocked in the absence of the ULK1/FIP200 complex, ATG9L1, or ATG14L, or in the presence of wortmannin, suggesting a noncanonical mechanism of recruiting ATG16L1 to SCVs in the absence of the canonical machinery. This alternative mechanism may be responsible for the residual killing of bacteria we observed in the absence of Rabaptin5 or FIP200.
Our study shows that Rabaptin5 has dual functions: In addition to its role as a regulator of early endosome identity and endosome maturation, it contributes to initiate and promote autophagy, when endosomes are damaged spontaneously, by chemical agents, or by pathogens.
Materials and Methods
Antibodies and reagents
Mouse anti‐transferrin receptor (OKT8 monoclonal hybridoma, kind gift of Dr. H. Farhan, 1:1,000), mouse anti‐Rabaptin5 (610676, BD Transduction Laboratories, 1:1,000), rabbit anti‐Rabaptin5 antibodies (NBP1‐47285, Novus Biologicals, 1:500), mouse anti‐WIPI2 (ab105459, Abcam, 1:500), and rabbit anti‐LC3B (3868S, Cell Signaling, 1:1,000 and 1:400) were used for immunofluorescence microscopy and immunoblotting; mouse anti‐tubulin (kind gift of Dr. H. Farhan, 1:1,000) and rabbit anti‐ATG13 (#13468S, Cell Signaling, 1:1,000) for immunoblotting; rabbit anti‐FIP200 (12436S, Cell Signaling, 1:1,000) for immunoblotting and immunoprecipitation; rabbit anti‐Galectin 3 (Abcam ab31707, 1:1,000), rabbit anti‐ATG16L1 (8089, Cell Signaling, 1:1,000), rabbit anti‐myc (GTX29106, GeneTEX, 1:5,000), rabbit anti‐mCherry (PA5‐34974, Invitrogen, 1:1,000), and rabbit anti‐EEA1(Abcam Ab2900, 1:1,000) for immunoprecipitation; mouse anti‐ubiquitin (SC‐8017, Santa Cruz, 1:500), mouse anti‐ALIX (634502, BioLegend, 1:400), and mouse anti‐p62/SQSTM1 antibody (2C11) (H00008878‐M01, Novus Biologicals, 1:400) for immunofluorescence microscopy.
Alexa‐Fluor‐488‐ or Alexa‐Fluor‐568‐tagged donkey anti‐mouse‐ or anti‐rabbit‐immunoglobulin antibodies (A32766, A32790, A10042, A10037, Molecular Probes, 1:200) and Alexa‐Fluor‐633 goat anti‐mouse immunoglobulin antibodies (A‐21050, Molecular Probes, 1:200) were used as secondary antibodies for immunofluorescence, and horseradish‐peroxidase‐coupled goat anti‐mouse‐ or anti‐rabbit‐immunoglobulin antibodies (A0168, A0545, Sigma Immunochemicals, 1:5,000) in combination with the enhanced chemiluminescence reaction kit (Amersham Pharmacia Biotech).
Monensin, MRT68921, and LLOME were purchased from Sigma‐Aldrich (M5273, SML1644, and L7393, respectively), and chloroquine was purchased from Serva (16919).
Plasmids and mutagenesis
Plasmids of human Rabaptin5, RFP‐Rab5 (pSI‐AAR6_Rab5), and GFP‐Rab7 (pSI‐AAL6_Rab7) were described in Kälin et al (2015). The cDNA of human FIP200 was purchased from OriGene (SC114884), pmCherry_Gal3 was a gift from Hemmo Meyer (Addgene plasmid #85662) (Papadopoulos et al, 2017), mRuby3‐Gal8 (PB‐CAG‐mRuby3‐Gal8‐P2A‐Zeo) was obtained from Addgene (#150815), pSpCas9(BB)‐2A‐GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138) (Ran et al, 2013), and mCherry‐ATG16L1 was a kind gift from Sharon Tooze (Francis Crick Institute). Mutations were introduced by PCR mutagenesis. HeLa cells stably expressing GFP‐CHMP4B were a kind gift by A. Hyman (MPI Dresden).
Cell culture and transfection
HeLaα (from ATCC) and HEK293A cells (a gift by Sharon Tooze, Crick Institute) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, 100 units/ml streptomycin, and 2 mM l‐glutamine at 37°C at 7.5% CO2.
To generate Rabaptin5‐ and FIP200‐knockout cell lines in HEK293A cells, the gRNA sequences AGAGTGTGTACCTACAGTGC (Rabaptin5) and TGGGCGCCTCACCGTCAGGC (FIP200) extended with Bpi I overhangs were cloned into pSpCas9(BB)‐2A‐GFP, digested with Bpi I (Thermo Scientific, FD1014). 24 h after transfection, the plasmid containing cells were selected by fluorescence‐activated cell sorting based on GFP expression. After 10 days of culture, single cells were sorted again for loss of GFP expression and expanded. Gene inactivation was confirmed by immunoblotting.
Transient transfections were performed using Fugene (Promega) or JetPRIME (Polyplus) and cells analyzed after 2 days. For stable expression, cells were transfected with the respective pcDNA3 constructs and subsequently selected with 2 mg/ml G418 (Geneticin from Thermo Fisher Scientific; 3 mg/ml for 10 days). Surviving cells were expanded in culture medium containing G418.
For knockdown experiments, siRNAs were purchased from Dharmacon Thermo Fisher Scientific (nontargeting siControl, D‐001810‐10‐05; Rabaptin5 siRABEP1, L‐017645‐01; FIP200 siRB1CC1, M‐021117‐01‐0005; siATG16L1, J‐021033‐11‐0010; siATG13, L‐020765‐01; siULK1, L‐005049‐00; siULK2, J005396‐07‐0005; siWIPI2, J‐020521‐09‐0005). Cells were transfected with 20 nM siRNA and HiPerFect (Qiagen) and used after 3 days.
Immunofluorescence microscopy and quantitation
Cells on coverslips were fixed with 3% paraformaldehyde (PFA) for 10 min at room temperature, quenched for 5 min with 50 mM NH4Cl, washed with PBS, permeabilized with 0.1% Triton X‐100 for 10 min, blocked with 3% BSA in PBS for 15 min, incubated for 1 h with primary antibodies in PBS with BSA, washed, and stained for 30 min with fluorescently tagged secondary antibodies in PBS with BSA. Coverslips were mounted in Fluoromount‐G (Southern Biotech).
For LC3B and WIPI2 staining, fixation was performed with methanol or, when costaining for Rabaptin5, with PFA/methanol. In the first case, cells were fixed for 15 min in –20°C methanol, permeabilized for 10 min with 0.3% Triton X‐100, blocked for 30 min with 3% BSA in PBS, and incubated overnight with the primary antibody at 4°C. In the second case, cells were first fixed in 3% PFA for 1 min and then for 15 min in 100% –20°C methanol, permeabilized for 10 min with 0.1% Triton X‐100, blocked for 15 min with 3% BSA in PBS, and incubated overnight with the primary antibody at 4°C. Secondary antibody was incubated in 3% BSA for 30 min at room temperature. PFA/methanol fixation was also used to co‐stain Rabaptin5 and ALIX in HEK293A cells, HEK293A‐FIP200‐KO cells, and GFP‐CHMP4B HeLa cells.
Images were acquired using an LSM700 confocal microscope or an API Delta Vision Core microscope with a 63× lens. All experiments were repeated at least three times. Images were analyzed with Fiji, using the JACoP plug‐in to determine Manders’ colocalization coefficients. Quantitations were based on images of 11 fields typically containing 3–8 cells. To quantify colocalization between Rabaptin5 and mCherry‐ATG16L1, transfected cells were fixed with PFA/methanol as above and stained for Rabaptin5. Images of at least 20 mCherry‐ATG16L‐expressing cells were analyzed per experiment.
To estimate the number of lysosomes, cells were loaded overnight with 450 nM LysoTracker Green (from Molecular Probes), fixed in 3% PFA, and imaged. Images for at least 15 cells per sample were quantified using Fiji.
To evaluate autophagic flux, HEK293A and Rabaptin5‐KO cells were transduced with the Premo Autophagy Tandem Sensor RFP‐GFP‐LC3B Kit (Thermo Fisher Scientific) for 48 h and incubated with or without HBSS starvation media or with 250 nM Torin1 for 3 h before fixation. GFP/RFP double‐positive autophagosomes and GFP‐negative/RFP‐positive autolysosomes were quantified by fluorescence microscopy.
Statistical analysis
Student's t‐test (two‐tailed, unpaired) was used to compare data of two groups, while one‐way ANOVA was the test used to compare three or more groups.
Co‐immunoprecipitation
Cells were lysed with lysis buffer (0.5% Na‐deoxycholate, 1% Triton X‐100, 2 mM PMSF, protease inhibitor cocktail). Post‐nuclear supernatants were incubated with antibody overnight at 4°C. Antigen–antibody complexes were collected with protein A‐Sepharose for 2 h at 4°C, washed three times with lysis buffer, and subjected to immunoblot analyses.
Salmonella infection and immunofluorescence analysis
Wild‐type Salmonella enterica serovar Typhimurium strain SL1344 was cultured in Luria broth (LB) overnight at 37°C with shaking, followed by dilution into 10 ml of fresh LB (1:33), and continued to grow under the same conditions for 3 h. One milliliter of bacteria was then centrifuged at 8,000 ×g for 2 min and resuspended in 1 ml of PBS. This suspension was diluted into DMEM + 10% FCS (no antibiotics) and added directly to HeLa or HEK293A cell lines to a multiplicity of infection of 100. The infected cell monolayers were centrifuged at 500 ×g for 5 min at 37°C to synchronize the infection and incubated 10 min at 37°C. The monolayers were then washed three times in PBS and then incubated in fresh culture medium with 100 µg/ml gentamycin. At 1 h postinfection, the gentamycin concentration was reduced to 10 µg/ml. At designated time points, cells were washed three times with PBS and lysed in 500 µl of lysis buffer (PBS with 1% Triton X‐100). Serial dilutions were plated onto LB agar plates (with 90 µg/ml streptomycin) to determine bacterial live cell count.
For microscopy, cells were infected with GFP‐expressing Salmonella SL1344 as above, fixed after different times by the methanol fixation protocol, and incubated with anti‐TfR and anti‐LC3B antibodies in PBS with 3% BSA overnight at 4°C, followed by fluorescent secondary antibodies for 30 min at room temperature. Images were acquired using a Zeiss LSM700 confocal microscope and analyzed in Fiji. To determine the infection rate, HeLa cells infected with GFP‐expressing Salmonella SL1344 as above were fixed in methanol and stained for TfR and LC3B. Z‐stacks if images were acquired on an API Delta Vision Core microscope using the 20× lens and a spacing of 0.13 μm. A range of 5,000–18,000 cells/sample were imaged on 5–7 different areas on the coverslip, deconvoluted, and stitched together on a SoftWoRx Imaging Workstastion. The final 3D pictures were analyzed with Fiji.
Yeast two‐hybrid screen and interaction domain identification
The pB29‐Rbpt5(1–862)‐LexA bait plasmid was used to screen a random‐primed HeLa cells_RP1 cDNA library cloned into the pP6‐Gal4‐AD plasmid using a high‐throughput proprietary yeast two‐hybrid‐based technology (ULTImate Y2H Screen; Hybrigenics, Paris, France). To identify the FIP200 interaction segment in Rabaptin5, interaction domain mapping analysis of different fragments in pB29‐Rbpt5(#)‐LexA was performed with pP6‐Gal4AD‐FIP200(257–444) (Hybrigenics).
qPCR
Total RNA was isolated from HEK293A cells using RNeasy Mini Kit (QIAGEN). cDNA synthesis was performed with 50 ng RNA in GoScript Reverse Transcription Mix (Promega Corporation, A2790) and thermocycler settings as indicated by the manufacturer. Real‐time quantitative PCR was performed using the GoTaq DNA Polymerase Kit (Promega, M3001) and the qTower qPCR System. Two pairs of primers were used for ULK2 (TTTAAATACAGAACGACCAATGGA with GGAGGTGCCAGAACACCA and CTCCTCAGGTTCTCCAGT with TTGGTGGGAGAAGTTCCAAG). Amplifications for actin and GAPDH were performed for normalization.
Author contributions
VM, SS, and SK planned and performed experiments and analyzed data. MS planned experiments and analyzed data. VM and MS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
We thank Drs. Mauricio Rosas Ballina and Dirk Bumann (Biozentrum) for materials and assistance with the Salmonella experiments, Drs. Kai Schleicher, Alexia Loynton‐Ferrand, and the Biozentrum Imaging Core Facility, and Janine Bögli of the FACS Core Facility for their support. This work was supported by Grant 31003A‐182519 from the Swiss National Science Foundation. Open access funding provided by Universitat Basel.
EMBO reports (2022) 23: e53429.
Footnotes
Correction added on 5 January 2022, after first online publication: The legend has been corrected according to the displayed figure: Panel group C‐E has been corrected to C, D and FIP200 has been corrected to Rabaptin5. Panels F and G have been corrected to E and F, respectively. The text for panel H has been deleted.
Data availability
This study includes no data deposited in external repositories.
References
- Anding AL, Baehrecke EH (2017) Cleaning house: selective autophagy of organelles. Dev Cell 41: 10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakula D, Müller AJ, Zuleger T, Takacs Z, Franz‐Wachtel M, Thost A‐K, Brigger D, Tschan MP, Frickey T, Robenek H et al (2017) WIPI3 and WIPI4 β‐propellers are scaffolds for LKB1‐AMPK‐TSC signalling circuits in the control of autophagy. Nat Commun 8: 15637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC (2016) Mammalian autophagy: how does it work? Annu Rev Biochem 85: 685–713 [DOI] [PubMed] [Google Scholar]
- Birgisdottir ÅB, Johansen T (2020) Autophagy and endocytosis ‐ interconnections and interdependencies. J Cell Sci 133: jcs228114 [DOI] [PubMed] [Google Scholar]
- Boada‐Romero E, Letek M, Fleischer A, Pallauf K, Ramón‐Barros C, Pimentel‐Muiños FX (2013) TMEM59 defines a novel ATG16L1‐binding motif that promotes local activation of LC3. EMBO J 32: 566–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A, Koschade SE, Stolz A, Zivkovic K, Pohl C, Shaid S, Ren H, Lambert LJ, Cosford NDP, Brandts CH et al (2019) Conservation of structure, function and inhibitor binding in UNC‐51‐like kinase 1 and 2 (ULK1/2). Biochem J 476: 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd M, Kimura T, Choi S, Peters R, Mandell M, Bruun J‐A et al (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 Co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39: 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley LJ, Cabodevilla AG, Makar AN, Sztacho M, Michelberger T, Marsh JA, Houston DR, Martens S, Jiang X, Gammoh N (2019) Intrinsic lipid binding activity of ATG16L1 supports efficient membrane anchoring and autophagy. EMBO J 38: e100554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, Mayer U, Carding SR, Wileman T, Beale R et al (2018) The WD40 domain of ATG16L1 is required for its non‐canonical role in lipidation of LC3 at single membranes. EMBO J 37: e97840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M (2015) V‐ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11: 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J, Simpson J, Fontana R, Kishi‐Itakura C, Ktistakis NT, Gammoh N (2019) Targeting of early endosomes by autophagy facilitates EGFR recycling and signalling. EMBO Rep 20: e47734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S et al (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N (2020) The multifaceted functions of ATG16L1 in autophagy and related processes. J Cell Sci 133: jcs249227 [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S‐I, Natsume T, Guan J‐L, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Boada‐Romero E, Cunha LD, Magne J, Green DR (2017) LC3‐associated phagocytosis and inflammation. J Mol Biol 429: 3561–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi H, Lippé R, McBride HM, Rubino M, Woodman P, Stenmark H, Rybin V, Wilm M, Ashman K, Mann M et al (1997) A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin‐5 links nucleotide exchange to effector recruitment and function. Cell 90: 1149–1159 [DOI] [PubMed] [Google Scholar]
- Huang J, Brumell JH (2014) Bacteria‐autophagy interplay: a battle for survival. Nat Rev Microbiol 12: 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30: 3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquin E, Leclerc‐Mercier S, Judon C, Blanchard E, Fraitag S, Florey O (2017) Pharmacological modulators of autophagy activate a parallel noncanonical pathway driving unconventional LC3 lipidation. Autophagy 13: 854–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Abudu YP, Claude‐Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M et al (2018) Galectins control mTOR in response to endomembrane damage. Mol Cell 70: 120–135.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Claude‐Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA et al (2020) Galectin‐3 coordinates a cellular system for lysosomal repair and removal. Dev Cell 52: 69–87.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama S, Omori H, Saitoh T, Sone T, Guan J‐L, Akira S, Imamoto F, Noda T, Yoshimori T (2011) The LC3 recruitment mechanism is separate from Atg9L1‐dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell 22: 2290–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kälin S, Buser DP, Spiess M (2016) A fresh look at the function of Rabaptin5 on endosomes. Small GTPases 7: 34–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kälin S, Hirschmann DT, Buser DP, Spiess M (2015) Rabaptin5 is recruited to endosomes by Rab4 and Rabex5 to regulate endosome maturation. J Cell Sci 128: 4126–4137 [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Kirkin V, Rogov VV (2019) A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell 76: 268–285 [DOI] [PubMed] [Google Scholar]
- Kishi‐Itakura C, Ktistakis NT, Buss F (2020) Ultrastructural insights into pathogen clearance by autophagy. Traffic 21: 310–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knævelsrud H, Søreng K, Raiborg C, Håberg K, Rasmuson F, Brech A, Liestøl K, Rusten TE, Stenmark H, Neufeld TP et al (2013) Membrane remodeling by the PX‐BAR protein SNX18 promotes autophagosome formation. J Cell Biol 202: 331–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock DL, Chaudhary A, Miller SI (2015) Salmonellae interactions with host processes. Nat Rev Microbiol 13: 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidal AM, Levine B, Debnath J (2018) Autophagy and the cell biology of age‐related disease. Nat Cell Biol 20: 1338–1348 [DOI] [PubMed] [Google Scholar]
- Levin R, Grinstein S, Canton J (2016) The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev 273: 156–179 [DOI] [PubMed] [Google Scholar]
- Lippé R, Miaczynska M, Rybin V, Runge A, Zerial M (2001) Functional synergy between Rab5 effector Rabaptin‐5 and exchange factor Rabex‐5 when physically associated in a complex. Mol Biol Cell 12: 2219–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lystad AH, Carlsson SR, de la Ballina LR, Kauffman KJ, Nag S, Yoshimori T, Melia TJ, Simonsen A (2019) Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy‐related processes. Nat Cell Biol 21: 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y et al (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR (2011) Microtubule‐associated protein 1 light chain 3 alpha (LC3)‐associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA 108: 17396–17401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan J‐L, Tan H, Peng J et al (2015) Molecular characterization of LC3‐associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Masud S, Prajsnar TK, Torraca V, Lamers GEM, Benning M, Van Der Vaart M, Meijer AH (2019) Macrophages target Salmonella by Lc3‐associated phagocytosis in a systemic infection model. Autophagy 15: 796–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Bonifacino JS (2008) Ubiquitin binding and conjugation regulate the recruitment of Rabex‐5 to early endosomes. EMBO J 27: 2484–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Tsai YC, Weissman AM, Bonifacino JS (2006) The Rab5 guanine nucleotide exchange factor Rabex‐5 binds ubiquitin (Ub) and functions as a Ub ligase through an atypical Ub‐interacting motif and a zinc finger domain. J Biol Chem 281: 6874–6883 [DOI] [PubMed] [Google Scholar]
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K‐J, Coppes RP, Engedal N, Mari M, Reggiori F (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy 14: 1435–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TJ, Gubas A, Tooze SA (2018) A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 293: 5386–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, Mizushima N (2019) Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol 35: 453–475 [DOI] [PubMed] [Google Scholar]
- Naslavsky N, Caplan S (2018) The enigmatic endosome ‐ sorting the ins and outs of endocytic trafficking. J Cell Sci 131: jcs216499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K et al (2017) VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36: 135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Kim JK, Bae HJ, Eun JW, Shen Q, Kim HS, Shin WC, Yang HD, Lee EK, You JS et al (2014) HDAC6 sustains growth stimulation by prolonging the activation of EGF receptor through the inhibition of rabaptin‐5‐mediated early endosome fusion in gastric cancer. Cancer Lett 354: 97–106 [DOI] [PubMed] [Google Scholar]
- Petherick KJ, Conway OJL, Mpamhanga C, Osborne SA, Kamal A, Saxty B, Ganley IG (2015) Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)‐dependent autophagy. J Biol Chem 290: 11376–11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Dikic I (2014) TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep 15: 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D, Datta S, Ackema K, Zerial M, Spang A (2010) Identification of the switch in early‐to‐late endosome transition. Cell 141: 497–508 [DOI] [PubMed] [Google Scholar]
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2013) Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154: 1285–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri C, Vicinanza M, Ashkenazi A, Gratian MJ, Zhang Q, Bento CF, Renna M, Menzies FM, Rubinsztein DC (2018) The RAB11A‐positive compartment is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P‐RAB11A. Dev Cell 45: 114–131.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic M, Schink KO, Wenzel EM, Nähse V, Bongiovanni A, Lafont F, Stenmark H (2018) ESCRT‐mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37: e99753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, Youle RJ (2014) Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15: 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yokom AL, Wang C, Young LN, Youle RJ, Hurley JH (2020) ULK complex organization in autophagy by a C‐shaped FIP200 N‐terminal domain dimer. J Cell Biol 219: e201911047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360: eaar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søreng K, Munson MJ, Lamb CA, Bjørndal GT, Pankiv S, Carlsson SR, Tooze SA, Simonsen A (2018) SNX18 regulates ATG9A trafficking from recycling endosomes by recruiting Dynamin‐2. EMBO Rep 19: e44837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele‐Mortimer O, Méresse S, Gorvel JP, Toh BH, Finlay BB (1999) Biogenesis of Salmonella typhimurium‐containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell Microbiol 1: 33–49 [DOI] [PubMed] [Google Scholar]
- Thurston TLM, Wandel MP, von Muhlinen N, Foeglein A, Randow F (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YI, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, Hast BE, Hanna SC, Wondergem B, Furge KA et al (2009) Regulation of endocytosis via the oxygen‐sensing pathway. Nat Med 15: 319–324 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang T, Wang S, Gong Z, Tang C, Chen J, Ding J (2014) Molecular mechanism for Rabex‐5 GEF activation by Rabaptin‐5. Elife 3: e2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH (2009) The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183: 5909–5916 [DOI] [PubMed] [Google Scholar]
- Zhu G, Zhai P, He X, Wakeham N, Rodgers K, Li G, Tang J, Zhang XC (2004) Crystal structure of human GGA1 GAT domain complexed with the GAT‐binding domain of Rabaptin5. EMBO J 23: 3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]