
Figure 1.
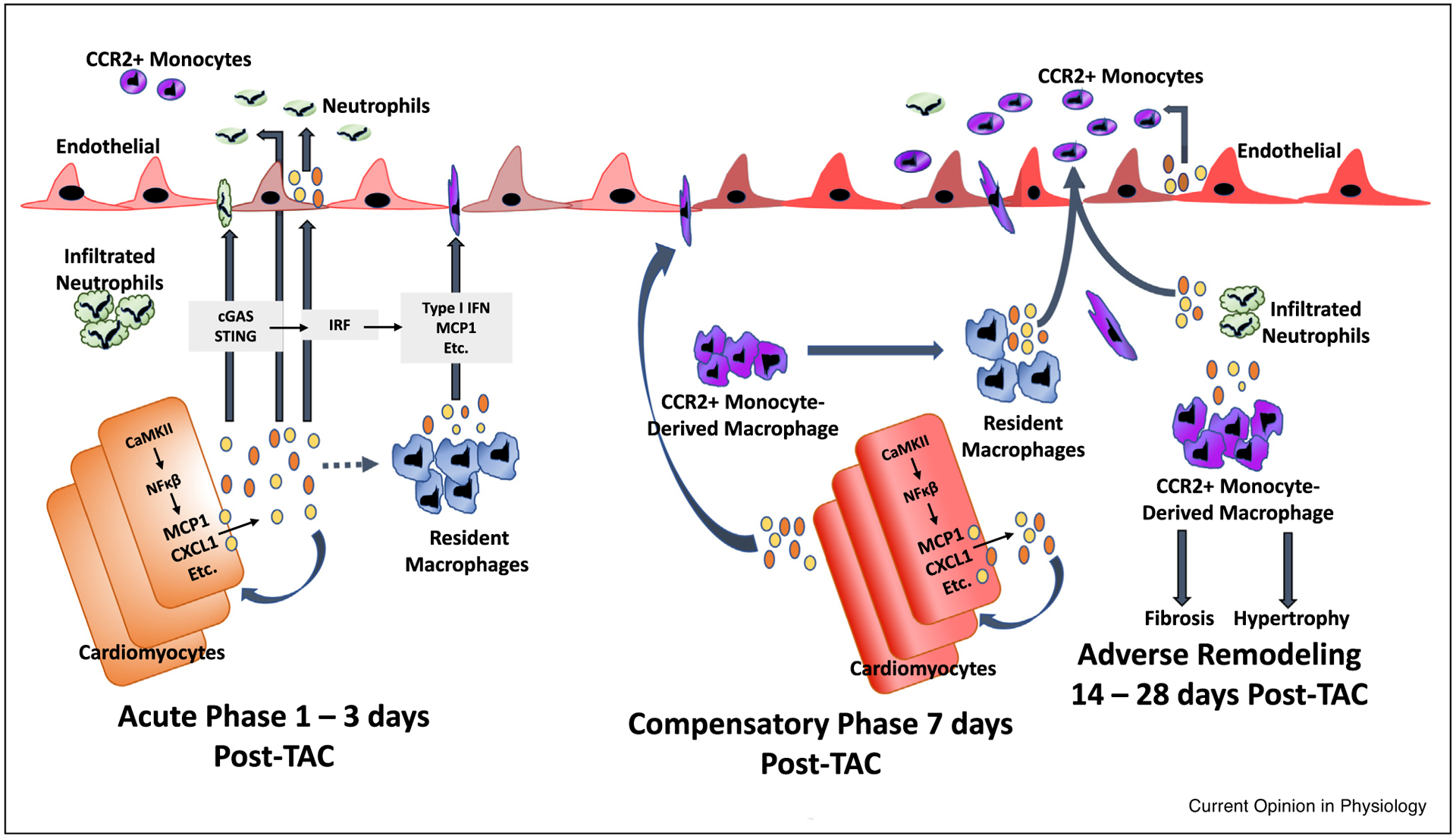
Inflammatory cascade initiated by cardiomyocytes in pressure overload (TAC) induced injury - the role of the cardiomyocyte as a major initiator of inflammation may be of paramount importance in the development of heart failure. During the acute phase (1–3 days post-TAC), cardiomyocytes express cytokines such as MCP1 and CXCL1 in response to CaMKII-mediated NFkB activation. Likewise, the presence of cytosolic DNA may activate the cGAS/STING pathway to induce type I interferon expression [16]. Cardiomyocyte-generated chemokines and other cytokines may be released into the circulation to mediate, feedback in a juxtacrine or autocrine manner, recruitment of monocytes and neutrophils, and locally activate tissue resident macrophages and endothelial cells [52]. Bajpai et al. demonstrated that distinct subsets of tissue-resident cardiac macrophages are crucial in the extravasation of bone-marrow derived neutrophils and monocytes after injury [46••,53••]. At approximately seven days post-TAC, cardiomyocyte-specific inflammatory processes mediate a significant influx of CCR2+ monocyte-derived macrophages into the myocardium [9]. The progression to adverse remodeling and heart failure, characterized by excess fibrosis and hypertrophy, requires infiltration of CCR2+ monocyte-derived macrophages, which replace the resident macrophage population; these results validate the widespread appreciation for the role of CCR2+ monocytes in TAC induced heart failure [43••]. TAC-induced adverse remodeling and heart failure has also been shown by Wang et al. to require neutrophil infiltration, and neutrophil depletion reduces the absolute number of recruited monocytes [44•]. Since infiltrated neutrophils appear earlier than recruited CCR2+ monocytes, it is possible that resident macrophages and infiltrated neutrophils synergistically recruit circulating CCR2+ monocyte-derived macrophages; disruption of any of these processes prevents downstream responses that lead to adverse remodeling.