
Extended Data Fig. 1. Genome statistics of the prokaryotic skin MAGs.
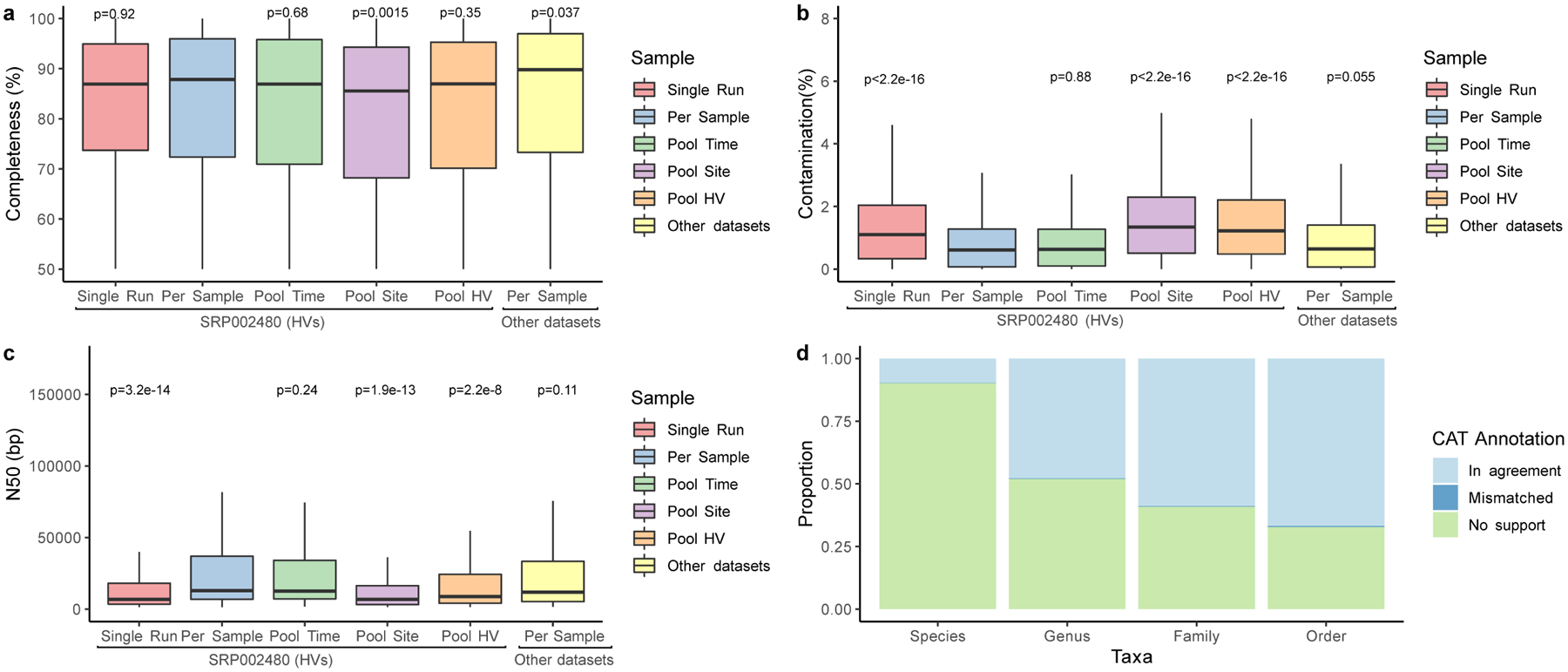
a, The completeness and b, contamination estimates for genomes (Single Run, n=2,389; Per Sample, n=1,206; Pool Time, n=973; Pool Site, n=1,171; Pool HV, n=1,054, Other datasets, n=1,099) recovered from different metagenomic samples as determined by CheckM. ‘Other datasets’ refers to skin metagenomes excluding the healthy volunteer dataset SRP002480. c, N50 of these MAGs as determined through BBMap. Significance for a-c was determined using the two tailed t-test relative to Per Sample, with ns representing not significant. d, The mean proportion of these genomes classified as taxonomically mismatched by comparing the annotation of the bin to the annotation of each contig via the contig annotation tool (CAT). ‘No support’ indicates that no taxonomic annotation was available at the respective rank. In panels a, b and c, box lengths represent the IQR of the data, with whiskers depicting the lowest and highest values within 1.5 times the IQR of the first and third quartiles, respectively.