
Fig. 2.
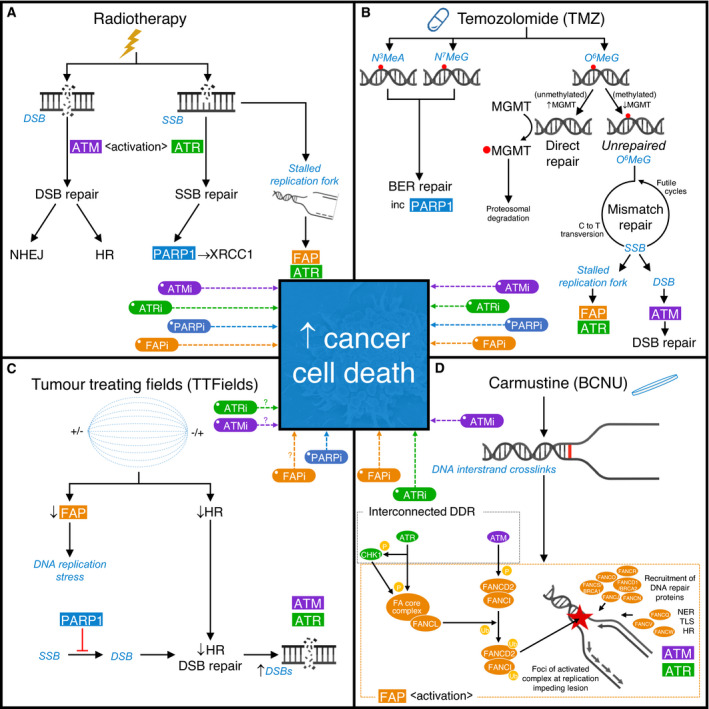
The effects of clinically approved therapies on the DNA damage response (DDR) and novel strategies to enhance efficacy of current standard‐of‐care treatments. Schematic representation of the main DNA damage lesions (in blue italic) induced by therapies approved for clinical use to treat glioblastoma and associated DDR mechanisms. For each approved treatment, putative strategies to enhance therapeutic efficacy through targeting relevant DDR mechanism(s) are indicated. (A) Radiotherapy: generates large amounts of DNA single‐strand breaks (SSBs) and double‐strand breaks (DSBs), which activate ATR and ATM, respectively. DSB repair is then predominantly undertaken by either nonhomologous end joining (NHEJ), which is available throughout the cell cycle but compromises fidelity, or homologous recombination (HR) DNA repair, which provides a high‐fidelity repair mechanism, but is only available during S and G2 phases of the cell cycle due to the requirement for a sister chromatid. SSB repair relies on PARP1 to detect SSBs and facilitate the recruitment of XRCC1. However, the presence of strand breaks also leads to stalling of DNA replication forks, which depend on the functions of ATR and proteins within the Fanconi anaemia pathway (FAP) for stability and replication restart. Consequently, a strong scientific rationale exists supporting inhibition of either ATM (ATMi), ATR (ATRi), PARP1 (PARPi) or the FAP (FAPi) to enhance the efficacy of radiotherapy. (B) Temozolomide: produces an array of methylation lesions including N3‐methyladenine (N3MeA) and N7‐methylguanine (N7MeG), which are substrates for effective removal via DNA base excision repair (BER), and O6‐methylguanine (O6MeG), which is removed directly by the enzyme MGMT in a suicide reaction. Hypermethylation of the MGMT gene promoter region leads to reduced MGMT expression, shifting the balance in favour of persistent O6MeG. O6MeG can act as a miscoding base during DNA replication, leading to a corresponding C‐to‐T transversion within the complementary DNA strand. If O6MeG is not successfully excised by the mismatch repair (MMR) DNA repair machinery, it endures as a perpetually miscoding base, instigating ‘futile cycles’ of MMR with consequent stalling of DNA replication forks or DSBs. (C) Tumour‐treating fields (TTFields): may negatively impact FAP and HR‐mediated DNA repair processes. TTFields‐induced ‘BRCAness’ (reflecting a relative HR deficiency) provides a compelling rationale to combine this therapeutic modality with PARPi, or potentially FAPi, ATRi or even ATMi. (D) Carmustine (BCNU) – Gliadel® wafers: provide local delivery of this bidirectional DNA alkylating agent, leading to the generation of DNA interstrand crosslinks which impede DNA replication during S phase. This leads to activation of the FAP, within which monoubiquitination of FANCD2 within the FANCD2‐I complex is a key quantifiable step. Activated FANCD2‐I coalesces as foci at sites of DNA damage and acts as a master regulator of downstream DNA repair, recruiting proteins involved in nucleotide excision repair (NER), translesion synthesis (TLS) and HR. Interplay with associated DDR mechanisms, for example ATM and ATR, leads to the phosphorylation of multiple FAP proteins (examples indicated), providing a rationale for the use of non‐FAP DDR inhibitors (e.g. ATRi or ATMi) to sensitise to crosslinking chemotherapy, and for the concept of combining multiple DDR inhibitors (including FAPi) to potentially maximise therapeutic enhancement.