

Abstract
A genome editing tool targeting the high-risk human papillomavirus (HPV) oncogene is a promising therapeutic strategy to treat HPV-related cervical cancer. To improve gene knockout efficiency, we developed a gene knockout chain reaction (GKCR) method for continually generating mutagenic disruptions and used this method to disrupt the HPV18 E6 and E7 genes. We verified that the GKCR Cas9/guide RNA (gRNA) cassettes could integrated into the targeted loci via homology-independent targeted insertion (HITI). The qPCR results revealed that the GKCR method enabled a relatively higher Cas9/gRNA cassette insertion rate than a control method (the common CRISPR-Cas9 strategy). Tracking of Indels by DEcomposition (TIDE) assay results showed that the GKCR method produced a significantly higher percentage of insertions or deletions (indels) in the HPV18 E6 and E7 genes. Furthermore, by targeting the HPV18 E6/E7 oncogenes, we found that the GKCR method significantly upregulated the P53/RB proteins and inhibited the proliferation and motility of HeLa cells. The GKCR method significantly improved the gene knockout efficiency of the HPV18 E6/E7 oncogenes, which might provide new insights into treatment of HPV infection and related cervical cancer.
Keywords: CRISPR-Cas9, gene knockout chain reaction (GKCR), HITI, HPV, cervical cancer
Graphical abstract
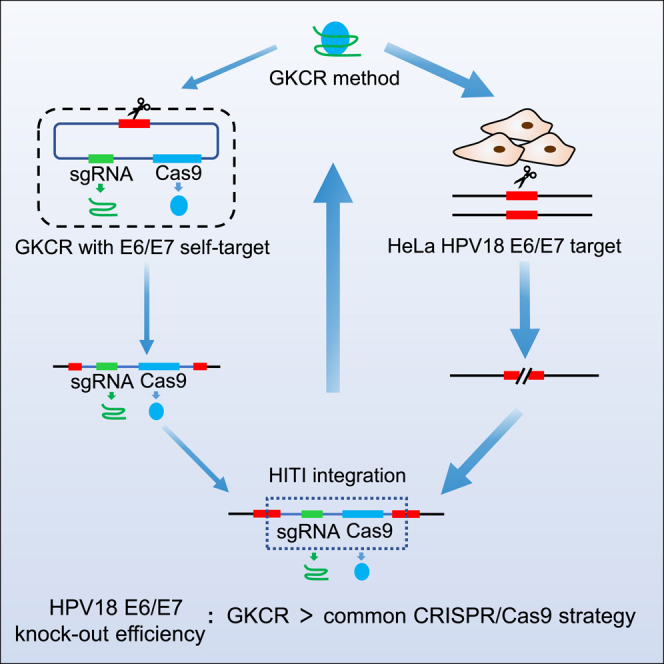
We developed a gene knockout chain reaction (GKCR) method by inserting CRISPR-Cas9 into the edited allele and generating sustained disruptions. Compared with the traditional CRISPR-Cas9 strategy, the GKCR method could result in higher disruption of the HPV18 E6/E7 oncogenes, which provides new insights into treatment of HPV infection and related cervical cancer.
Introduction
High-risk human papillomavirus (Hr-HPV), especially type 16 and 18, is recognized as the main cause of cervical cancer.1 The HPV E6 and E7 viral genes are major oncogenes that participate in carcinogenesis of cervical epithelial cells by inhibiting the P53 and RB proteins, respectively.2,3 Therefore, the E6 and E7 genes are considered critical targets for prevention and treatment of cervical cancer.
The clustered regularly interspaced short palindromic repeats-associated 9 (CRISPR-Cas9) system is a specific defense mechanism against foreign nucleic acids in bacteria and archaea that has been repurposed by researchers and is used widely in the gene editing field.4,5 In the CRISPR-Cas9 system, the single guide RNA (sgRNA) combines with the targeted DNA, leads the Cas9 endonuclease to the targeted DNA sequence, and produces sequence-specific double-strand breaks.6 Then mammal cells are repaired mainly by the error-prone non-homologous end joining (NHEJ) mechanism, which introduces random insertions or deletions (indels) at the targeted sites and causes gene disruption.7, 8, 9
We and other researcher have used the CRISPR-Cas9 system to target the E6 and E7 genes to reduce E6 and E7 protein expression, up-regulate P53 and RB, and inhibit the proliferation of cervical cancer cells.10, 11, 12, 13 Use of CRISPR-Cas9 to knock out the E6/E7 oncogenes also enhances the anticancer effect of chemotherapeutic or targeted drugs.14,15 However, in most cases, CRISPR-Cas9 plasmids are transiently transfected into the cells and are expressed instantaneously in the cells. The editing period is short, which leads to low efficiency.16,17 How to improve editing efficiency is an urgent problem to be solved.
In our study, we envisage further editing efficiency improvements by exploring the gene knockout chain reaction (GKCR) based on the CRISPR-Cas9 system. We aim to use the GKCR method to significantly improve the knockout efficiency of the HPV18 E6/E7 genes, which may provide new insight into treatment of cervical cancer.
Results
Outline of the GKCR
The HPV18 E6 and E7 oncogenes integrated into the host genome are multi-copied. Effective disruption of the integrated viral genes is desirable and challenging. Taking advantage of the CRISPR-Cas9 genome editing method, we developed a strategy to improve gene knockout efficiency and refer to this method as GKCR. An outline of the method is presented in Figure 1. We reasoned that an autocatalytic knockout effect could be generated with a construct having three components: (1) a Cas9 gene (expressed in somatic and germline cells), (2) a gRNA targeted to a genomic sequence of interest and the construct itself, and (3) the 23-bp target sequence including -NGG protospacer adjacent motif (PAM) sequence, which is the same as the targeted genomic sequence.18 We named this tripartite construct the GKCR plasmid. In such a tripartite construct, Cas9 should cleave the genomic target at the site directed by the gRNA and then insert the Cas9/gRNA cassette into that locus via homology-independent targeted insertion (HITI).18 Cas9 and the gRNA produced from the insertion allele should then cleave the other alleles. In this study, we called the plasmid carrying the sgRNA and Cas9 cassettes without the 23-bp target sequence the control plasmid (which is actually a common CRISPR-Cas9 plasmid). The only transfection of the GKCR plasmid into cells was called the GKCR method, the only transfection of the control plasmid into cells was called the control method, and co-transfection of GKCR with control plasmids into cells was the GKCR + Ccntrol method. The Cas9/gRNA cassette derived from the linearized GKCR plasmid is nondirectional. It can integrate into the targeted genomic locus as a forward insertion or reverse insertion. When the Cas9/gRNA cassette is integrated, the Cas9/gRNA cassette is expressed continually and cuts other alleles like a chain reaction regardless of the insertion direction. Because linearized plasmids can be degraded faster than circular plasmids, we performed the GKCR + control method to compare it with the GKCR method and control method.
Figure 1.

Schematic outlining the GKCR
A GKCR donor could be integrated into the AAVS1 locus
We first chose the AAVS1 locus, the safe harbor in the human genome, to test our GKCR methods in the HEK293T cell line. The corresponding GKCR plasmids were constructed according to the approaches mentioned above, with the sgRNA sequences targeting the AAVS1 locus (Table 1). We called these AAVS1-related plasmids AAVS1-GKCR and AAVS1-control, respectively. Theoretically, in the GKCR and GKCR + control groups, a specific primer pair could detect three different integration modes at the target locus: (1) primer genome 1 and Primer insert 1 for AAVS1-GKCR plasmid donor forward insertion, (2) primer genome 1 and primer insert 2 for donor reverse insertion, and (3) primer genome 1 and primer genome 2 for indels without donor insertion. In the control group, primer genome 1 and primer genome2 could detect indels without insertion into the target locus (Figure 2A).
Table 1.
The sgRNA sequences and their corresponding PAM sequences
| Targeted gene | sgRNA sequence (5′-3′) | PAM sequence (5′-3′) |
|---|---|---|
| AAVS1 | GGGGCCACTAGGGACAGGAT | TGG |
| HPV18 E6 | CACAGATCAGGTAGCTTGTA | GGG |
| HPV18 E7 | GGTCAACCGGAATTTCATTT | TGG |
Figure 2.

The Cas9/gRNA cassette could be integrated into the AAVS1 locus by the GKCR method in HEK293T cells
(A) Schematic of the GKCR methods at the AAVS1 locus. (B and C) Agarose gel electrophoresis (B) and Sanger sequencing (C) of PCR products to verify Cas9/gRNA cassette integration into the targeted AAVS1 locus. (D) PCR and Sanger sequencing verified that the inserted Cas9/gRNA cassette was fully retained.
HEK293T cells were transfected with (1) only the AAVS1-GKCR plasmid, (2) only the AAVS1-control plasmid, and (3) the AAVS1-GKCR plasmid and AAVS1-control plasmid. 72 h after transfection, we extracted the genomic DNA of the HEK293T cells and used the primers listed in Table 2 to perform PCR and amplify the target sequences. We observed that the AAVS1-GKCR donor could be inserted into the target site in the forward and reverse directions when using the AAVS1-GKCR and AAVS1-GKCR + control methods (Figure 2B). Then we Sanger sequenced these PCR products and verified that the AAVS1-GKCR donor was precisely inserted into the AAVS1 locus (Figure 2C). Moreover, we designed specific primers to amplify the 5' and 3' integration terminal and verified by PCR and Sanger sequencing that the inserted Cas9/gRNA cassette was fully retained (Figure 2D). The results are representative of three independent experiments.
Table 2.
The sequences of the primers used in PCR amplification to verify insertion
| Targeted gene | Edited categories | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|---|
| AAVS1 | forward insertion | AAAACTGACGCACGGAGG | ACGATACAAGGCTGTTAGAGAGA |
| reverse insertion | AAAACTGACGCACGGAGG | AGAGTGAAGCAGAACGTGGG | |
| no insertion | AAAACTGACGCACGGAGG | CATCTCTCCTCCCTCACCCA | |
| HPV18 E6 | forward insertion | GAAAGGACGAAACACCGCAC | ATTCAACGGTTTCTGGCACC |
| reverse insertion | GGGGCGTACTTGGCATATGA | ATTCAACGGTTTCTGGCACC | |
| no insertion | CGAAATAGGTTGGGCAGCAC | ATTCAACGGTTTCTGGCACC | |
| HPV18 E7 | forward insertion | AAGGACGAAACACCGGTCAA | CCTCCCCGTCTGTACCTTCT |
| reverse insertion | GGGGCGTACTTGGCATATGA | CCTCCCCGTCTGTACCTTCT | |
| no insertion | TAATAAGGTGCCTGCGGTGC | CCTCCCCGTCTGTACCTTCT |
The GKCR method highly knocked out AAVS1 alleles in HEK293T cells
Because there are two copies of the AAVS1 locus in the genome, it is easy to confirm the editing results of the two alleles by PCR and Sanger sequencing. To further confirm the allele editing results of the GKCR and GKCR + control methods at the AAVS1 locus, we picked single clones of the transfected HEK293T cells and verified the editing types of the two alleles in each clone by PCR and Sanger sequencing. There were 10 kinds of edited types at the AAVS1 locus (Figure 3A). We picked 10 clones from GKCR- and GKCR + control-transfected HEK293T cells, respectively. Details of two clones in the AAVS1-GKCR group are shown in Figure 3B. We verified that two alleles of clone 1 were induced indels, whereas in clone 2, one allele exhibited the AAVS1-GKCR donor forward insertion, and the other allele was induced indels (Figure 3B). Among 10 monoclonals of the AAVS1-GKCR group, seven clones had the AAVS1-GKCR donor forward integrated in allele 1 and indels without donor integration in allele 2; the other three clones had indels in both alleles (Table S1). Among 10 monoclonals of the AAVS1-GKCR + control group, four clones had the AAVS1-GKCR donor forward integrated in allele 1 and indels without plasmid integration in allele 2, five clones had indels in both alleles, one clone had indels in one allele, and the other allele was unedited (Table S2).
Figure 3.

GKCR method significantly knocked out AAVS1 alleles in HEK293T cells
(A) Schematic of the possible editing results of the two AAVS1 alleles in one HEK293T cell. (B) Editing results of two monoclonals in the AAVS1-GKCR group, verified by agarose gel electrophoresis and Sanger sequencing. (C) TIDE assays revealed that the GKCR method induced a significantly higher indel percentage than the control method at the AAVS1 locus. Data are presented as means ± SD (n = 3 per group). ∗∗p < 0.01; n.s., not significant; analyzed by one-way ANOVA and Tukey’s post hoc test.
We also performed a TIDE assay of polyclonal cells before monoclonal cultivation to analyze the indel percentage on the AAVS1 locus. The results showed that the knockout efficiency, on average, was 21.47% in the AAVS1-GKCR group, 14.20% in the AAVS1-control group, and 17.8% in the AAVS1-GKCR + control group. The knockout efficiency of the AAVS1-GKCR group was significantly higher than that of the AAVS1-control group (p < 0.01). The knockout efficiency was not significantly enhanced in the AAVS1-GKCR + control group (Figure 3C). The results are representative of three independent experiments.
The GKCR method significantly knocked out the HPV18 E6 and E7 oncogenes in HeLa cells
Cervical cancer is caused by Hr-HPV persistence. HPV18 is one of the most common pathogenic subtypes. Furthermore, the E6 and E7 viral genes play critical roles in carcinogenesis. There are multiple copies of HPV genes in HeLa cells.19 Because the integrated GKCR donor can express sgRNA and Cas9 to continuously target other copies, we assume that the GKCR strategy is feasible for significantly disrupting HPV oncogenes to treat cervical cancer.
First we designed sgRNA targeting the HPV18 E6 and E7 genes, called HPV18-E6-31 and HPV18-E7-51, respectively. The sgRNA sequences are listed in Table 1. The corresponding GKCR, control (traditional CRISPR-Cas9), GKCR + control (simultaneous transfection of GKCR and traditional CRISPR-Cas9) and vector (gRNA empty vector) plasmids were then constructed as mentioned above.
To disrupt the HPV18 E6 gene, we transfected HeLa cells with HPV18-E6-31-GKCR, HPV18-E6-31-control, HPV18-E6-31-GKCR + control, or vector plasmids. We also disrupted the HPV18 E7 gene by using the related HPV18-E7-GKCR plasmids. 72 h after transfection, the cells were harvested to extract the genomic DNA. We performed PCR and Sanger sequencing to verify that the GKCR donor integrated into the target sites. As shown in Figure 4A, we observed that the HPV18-E6-31-GKCR donor could be inserted into the E6 target site in forward and reverse directions when using the HPV18-E6-31-GKCR and HPV18-E6-31-GKCR + control methods. The same insertion patterns were found in the HPV18-E7-51-GKCR and HPV18-E7-51-GKCR + control groups. Sanger sequencing proved the PCR products’ specific sequences (Figure 4B). The results are representative of three independent experiments.
Figure 4.

GKCR method highly knocked out HPV18 E6 and E7 oncogenes in the HeLa cells
(A and B) Agarose gel electrophoresis (A) and Sanger sequencing (B) of PCR products to verify that the Cas9/gRNA cassette integrated into the targeted HPV18 E6 and E7 locus. (C) Fold change of forward and reverse integration of the corresponding Cas9/gRNA cassettes into the respective target sites, analyzed by qPCR. Data are presented as mean ± SD (n = 3 per group). ∗∗p < 0.01, ∗∗∗p < 0.001; analyzed by two-tailed Students’ t test. (D) TIDE assays revealed that the GKCR method induced a significantly higher indel percentage than the control method at the HPV18 E6 and E7 locus. Data are presented as mean ± SD (n = 3 per group). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; analyzed by one-way ANOVA and Tukey’s post hoc test.
Next we compared the editing efficiency on the HPV18 E6/E7 locus of each group. Because HPV18 E6 and E7 are multi-copied genes in HeLa cells, PCR and Sanger sequencing to identify allele editing results is not feasible to determine which strategy is better. The editing results at the target site are of four types: (1) donor forward integration, (2) donor reverse integration, (3) indels without donor integration, and (4) unedited. Except for the unedited type, the other three types of editing results could destroy the DNA sequence of the HPV18 E6/E7 genes. Therefore, to compare the donor insertion efficiency of each group, we performed qPCR by using donor insertion-specific primers to identify which method is more likely to insert the donor into the target sites. Then relative insertion fold change (FC) was determined according to FC = 2−ΔΔCT (ΔΔCT = ΔCT – ΔCTvector). The forward insertion efficiency of the HPV18-E6-31-GKCR donor in the GKCR group was, on average, 24.21 times that of the control group (p < 0.001). The value of the reverse insertion efficiency was, on average, 13.46 times that of the control group (p < 0.001). The forward and reverse insertion efficiency of the HPV18-E7-51-GKCR donor in the GKCR group was, on average, 18.29 times and 5.83 times that of the control group (p < 0.001). The GKCR method generally performed better regarding donor insertion efficiency compared with the GKCR + control and control methods (Figure 4C). Moreover, we performed a TIDE assays to observe the indel efficiency (without donor integration) of the target sites in each group. At the HPV18-E6-31 site, the indel percentage of the GKCR method was, on average, 16.97%, and that of GKCR + control was 13.03% compared with the 7.93% of the control method. The indel percentage of the GKCR method was significantly higher than that of the control method (p < 0.001). At the HPV18-E7-51 site, the indel percentage of the GKCR strategy was, on average, 21.45%, whereas that of GKCR + control was 10.6% compared with the 8.1% of the control method. The indel percentage of the GKCR method was significantly higher than that of the control method (p < 0.01) (Figure 4D). The results are representative of three independent experiments.
Gene knockout efficiency comprises three parts: donor forward insertion, donor reverse insertion, and indels without donor insertion, and the GKCR method performed better regarding the donor insertion rate and indel percentage compared with the control and GKCR + control method. These results are consistent with the results observed at the AAVS1 locus.
The GKCR method upregulated P53/RB proteins and inhibited the proliferation and motility of HeLa cells by targeting the HPV18 E6/E7 oncogenes
Western blot analyses of the P53/RB proteins were performed in HeLa cells transfected with the corresponding GKCR, control, GKCR + control, or vector plasmids. Compared with the control group, the expression levels of P53 proteins were upregulated in the HPV18-E6-31-GKCR-treated group (p < 0.01). Similarly, upon treatment of HPV18-E7-51-GKCR, the expression levels of RB proteins were upregulated (p < 0.05) (Figure 5A). The GKCR method targeting HPV18 E6/E7 sites could effectively disrupt the oncogenes and upregulate the expression levels of the P53/RB proteins.
Figure 5.

GKCR method upregulated the P53/RB proteins and inhibited the proliferation and motility of the HeLa cells by targeting the HPV18 E6/E7 oncogenes
(A) Western blot analysis. ∗p < 0.05, ∗∗p < 0.01; analyzed by two-tailed Students’ t test. (B) CCK-8 assay. ∗p < 0.05, analyzed by two-way RM ANOVA with Sidak’s multiple comparisons test. (C) Wound healing assay; scale bars, 250 μm. ∗p < 0.05, ∗∗p < 0.01; analyzed by two-tailed Students’ t test.
To examine whether the GKCR method could inhibit the growth of HeLa cells, the corresponding plasmids were transfected into HeLa cells, and then cell viability was measured using a Cell Counting Kit-8 (CCK-8) assay. Compared with the control group, proliferation activity of HeLa cells treated with HPV18-E6-31-GKCR or HPV18-E7-51-GKCR method was decreased significantly (p < 0.05) (Figure 5B).
A wound healing assay was performed to evaluate the motility of HeLa cells treated with related plasmids as described above. Cell migration rates were calculated for comparing the motility of the HeLa cells in different groups. Compared with control groups, the GKCR method targeting the HPV18 E6/E7 oncogenes specifically inhibited the motility of HeLa cells (HPV18-E6-31-GKCR, p < 0.05; HPV18-E7-51-GKCR, p < 0.01) (Figure 5C).
Discussion
With the increasing incidence and mortality rate of cervical cancer, there is a growing need for effective strategies to treat this disease. Previous research has revealed that using programmable gene-editing tools to disrupt HPV viral genes could inhibit their expression levels and treat cervical cancer.10, 11, 12,15 Integrated HPV viral genes in cervical cancer cells are multi-copied.19 However, transient transfection of editing tools targeting HPV viral genes might only cut some of the copies and not others. Editing efficiency needs to be improved.
Here we used GKCR strategies to disrupt the HPV18 E6 and E7 genes in HeLa cervical cancer cells. The control method is the regular gene knockout strategy; it just transiently expresses Cas9 protein and sgRNAs without donor insertion. The GKCR method uses the same Cas9 and sgRNAs with the same target sequence as the genomic locus to linearize and insert into the genome target locus. The GKCR + control method is a mixture of the GKCR and control methods (Figure 1). We speculated that GKCR could serve as the donor and be integrated into the target sites, where it continually expresses Cas9 protein and the sgRNA and disrupts the other copies. Therefore, the GKCR approach enables us to significantly disrupt the HPV18 viral genes. The multi-copied E6 and E7 viral genes would eventually be disrupted if sgRNA-Cas9 could be expressed continuously to perform the cleavage function.
We observed that the GKCR method could significantly improve the Cas9/gRNA cassette integration rate compared with the GKCR + control method (Tables S1 and S2; Figure 4C) and produce a significantly higher indel rate than the common CRISPR-Cas9 strategy (Figures 3C and 4D). This phenomenon might due to GKCR inducing a relatively high level of linearized donor insertion followed by continuous gene disruption, which is superior to the GKCR + control and control methods. Of note, the Cas9/gRNA cassette forward insertion rate is much higher than the reverse insertion rate among the picked monoclonals (Tables S1 and S2). We reasoned that the reverse insertion pattern could re-form a sgRNA target sequence that might induce re-editing until the targeted sequences are destroyed.18
The GKCR method could insert the Cas9/gRNA cassette into the targeted genome sites and activate a GKCR, which achieves a higher knockout efficiency than the common CRISPR-Cas9 strategy. This chain reaction may serve as an effective treatment approach to robustly disrupt multi-copied HPV viral genes to treat cervical cancer.
Materials and methods
Plasmid construction
The pX330-U6-Chimeric_BB-CBh-hSpCas9 vector was a gift from Feng Zhang20 (Addgene, catalog number 42230). We used the website http://crispor.tefor.net/ to design sgRNA targeting the AAVS1 site and the HPV18 E6 and E7 genes. The sgRNAs sequences are listed in Table 1. Then the sgRNA oligos were synthesized and cloned into the pX330-U6-Chimeric_BB-CBh-hSpCas9 vector by Genewiz (Suzhou, China) to obtain the control plasmid. Then the target sequences (including the -NGG PAM) were cloned into the control vector to obtain the GKCR plasmid.
Mammalian cell culture
The human embryonic kidney 293T (HEK293T) cell line and HeLa cervical cancer cell line were purchased from the ATCC. These cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, catalog number C11995500BT) supplemented with 10% fetal bovine serum (FBS; Gibco, catalog number 10270-106) and incubated at 37°C and 5% CO2 in a constant-temperature incubator. The medium was changed every other day. Cell passaging was performed at a 1:3 split ratio when the cells reached 90% confluence.
Plasmid transient transfection
Plasmid transient transfection was carried out using X-tremeGENE HP DNA transfection reagent (Roche, catalog number 6366244001) according to the manufacturer’s protocol. HEK293T or HeLa cells were seeded at density of 3 × 105 cells per well with 2 mL 10% FBS-supplemented DMEM in 6-well culture plates and transfected after 24 h with a total amount of 2 μg plasmid per well. Specifically, for the GKCR method, 2 μg GKCR plasmid was used; for the control method, 2 μg control plasmid was used; and for the GKCR + control method, 1 μg GKCR and 1 μg control plasmids were used to transfect the cells. For the vector method, 2 μg empty sgRNA pX330-U6-Chimeric_BB-CBh-hSpCas9 vector plasmid was transfected as an additional control. Transfected cells were cultured for 3 days and then harvested for genomic DNA extraction using the EasyPure Genomic DNA Kit (Transgen, catalog number TEE101-01) according to the manufacturer’s instructions.
Donor insertion verification by PCR and sanger sequencing
Donor insertion was verified by PCR and Sanger sequencing. There are three types of editing categories: GKCR donor forward insertion, GKCR donor reverse insertion, and no GKCR donor insertion. After transfection, genomic DNA was extracted and amplified by PCR using the primer pairs listed in Table 2. We used 2×EasyTaq PCR SuperMix (Transgen, catalog number AS111-01) with 200 ng genomic DNA as the template and 10 μM forward and reverse primers in a final volume of 50 μL on a thermal cycler (Bio-Rad, C1000). The PCR program was as follows: 94°C for 3 min and then 35 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and elongation at 72°C for 1 min, final extension at 72°C for 5 min, and hold at 4°C. The PCR products were determined by gel electrophoresis and Sanger sequencing. The Sanger sequencing was consigned to Sangon Biotech.
Editing efficiency analysis by TIDE assay
A TIDE assay was used to assess editing efficiency as in a previous study.21 Genomic DNA was extracted with the EasyPure Genomic DNA Kit (Transgen, catalog number TEE101-01) according to the manufacturer’s instructions. Then 100 ng of genomic DNA was used in a PCR reaction using 2×EasyTaq PCR SuperMix (Transgen, catalog number AS111-01) and primers surrounding the sgRNA target region. The primer sequences used for amplification of the edited locus are listed in Table 3.
Table 3.
The sequences of the primers used in the TIDE assay
| Targeted gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| AAVS1 | AAAACTGACGCACGGAGG | CATCTCTCCTCCCTCACCCA |
| HPV18 E6 | CGAAATAGGTTGGGCAGCAC | ATTCAACGGTTTCTGGCACC |
| HPV18 E7 | TAATAAGGTGCCTGCGGTGC | CCTCCCCGTCTGTACCTTCT |
Relative donor insertion rate measurement by qPCR
Transfected HeLa cells were harvested to extract the genomic DNA and perform qPCR by using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific; catalog number A25742) and on a real-time PCR apparatus (Bio-Rad, CFX96). qPCR was conducted on 100 ng genomic DNA per reaction, with GAPDH as a reference gene. Primers used for relative donor insertion rate determination are listed in Table 4. The following program was used: one step at 50°C for 2 min and then 95°C for 2 min, 40 cycles of denaturation at 95°C for 15 s, and annealing/elongation at 60°C for 1 min. The amplification specificity was evaluated by determining the product melting curve. The Δ-Δ method was used for target sequence quantification normalized to GAPDH gene amplification comparative threshold (CT) value average. Relative doner insertion copy number was normalized using the formula ΔCT = CT (donor insertion) – CT (GAPDH) and determined according to the following formula ΔΔCT = ΔCT – ΔCTVector. The value used to plot relative donor insertion fold change was determined using FC = 2−ΔΔCT. Three technical replicates were used for each reaction.
Table 4.
The sequences of the primers used in qPCR
| Targeted gene | Category | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|---|
| HPV18 E6 | forward insertion | CGGGACCGAAAACGGTGTAT | TATTGACGTCAATGGGCGGG |
| reverse insertion | CGGGACCGAAAACGGTGTAT | GCTAGTCCGTTTTTAGCGCG | |
| HPV18 E7 | forward insertion | TCCAACGACGCAGAGAAACA | TGACGTCAATGGAAAGTCCCT |
| reverse insertion | TCCAACGACGCAGAGAAACA | GAAAAAGTGGCACCGAGTCG | |
| GAPDH | reference gene | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
Western blot analysis
Cells were lysed in modified radio immunoprecipitation assay (RIPA) buffer (P0013B, Beyotime), and protein concentration was calculated by the bicinchoninic acid (BCA) protein quantification kit (P0012S, Beyotime). Target proteins and internal reference protein were analyzed by western blot assay, using the following antibodies: mouse anti-GAPDH antibody (97166T, CST, USA), Mouse anti-P53 antibody (2524T, Cell Signaling Technology), mouse anti-RB antibody (9309T, CST), and horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (SA00001-1, Proteintech). Equal amounts of 30 μg of total protein were separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes. All PVDF membranes were blocked in skim milk for 1 h at room temperature and then incubated with primary antibodies at 4°C overnight. The next day, the membranes were washed with tris buffered saline with tween (TBST) and incubated with an HRP-conjugated secondary antibody at room temperature for 1 h. An enhanced chemiluminescence (ECL) system was used to visualize the immunoreactive bands.
Cell viability assay
Cell viability was evaluated using CCK-8 (CK04, Dojindo). HeLa cells were seeded into 96-well plates with 5 × 103 cells per well and transfected with the corresponding plasmids as described previously after 16–20 h. 0, 24, 48, 72, and 96 h after transfection, 10 μL CCK-8 solution was added to each well. Immediately after 2 h incubation at 37°C, absorbance values of cells were measured at 450 nm using a microplate reader. Each experiment was repeated three times.
Wound healing assay
After 24 h of corresponding plasmid transfection, cells were seeded in 6-well plates at a density of 1 × 105 cells. A linear scratch was created in the monolayer of cells with a sterile 10-μL micropipette tip. The detached cells were rinsed three times using PBS and then incubated in DMEM. After 24-h and 48-h cell culture, the distance between the two sides of the scratch was captured, and we measured the dynamic change of width and analyzed cell migration using ImageJ.
Statistical analyses
We present the data as mean ± SD. Two-tailed Students’ t test, one-way ANOVA with Tukey’s post hoc test or two-way RM ANOVA with Sidak’s multiple comparisons test was used to analyze the differences between groups. GraphPad Prism 8 software (GraphPad) was used to perform statistical analyses. A p value of less than 0.05 was defined to be statistically significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 32171465 and 82102392), the National Science and Technology Major Project of the Ministry of Science and Technology of China (grant 2018ZX10301402), the General Program of the Natural Science Foundation of Guangdong Province of China (grant 2021A1515012438), the National Postdoctoral Program for Innovative Talent (grant BX20200398), the China Postdoctoral Science Foundation (grant 2020M672995), the Guangdong Basic and Applied Basic Research Foundation (grant 2020A1515110170), the Characteristic Innovation Research Project of University Teachers (grant 2020SWYY07), the National Ten Thousand Plan-Young Top Talents of China, major projects funded by Hubei Provincial Health Commission (grant WJ2019H312) major projects of Wuhan Municipal Health Commission (grant WX19M02), the Foundation of Health Commission of Hubei Province of China (grant WJ2019Q008), and the Dongguan Science and Technology of Social Development Program (grant 202050715007221).
Author contributions
R.T., J.L., W.F., and Z. Hu conceptualized and designed the study. Z.C. and Z.J. performed pre-experiments. R.T., J.L., and W.F. performed the experiments with support from Zhaoyue Huang, R.L., H.X., L.L., and Zheying Huang. R.T., J.L., and R.L. acquired, analyzed, and interpreted data. R.T., J.L., R.L., and W.F. composed the figures and manuscript. X.T., P.Z., and Z. Hu critically revised and all authors approved the final version of the manuscript.
Declaration of interests
The authors declare no conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.12.011.
Contributor Information
Zheng Hu, Email: huzheng1998@163.com.
Ping Zhou, Email: 2045214580@qq.com.
Xun Tian, Email: tianxun@zxhospital.com.
Supplemental information
References
- 1.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Hu Z., Zhu D., Wang W., Li W., Jia W., Zeng X., Ding W., Yu L., Wang X., Wang L., et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015;47:158–163. doi: 10.1038/ng.3178. [DOI] [PubMed] [Google Scholar]
- 3.Hoppe-Seyler K., Bossler F., Braun J.A., Herrmann A.L., Hoppe-Seyler F. The HPV E6/E7 oncogenes: key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 2018;26:158–168. doi: 10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 6.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 8.Miyaoka Y., Berman J.R., Cooper S.B., Mayerl S.J., Chan A.H., Zhang B., Karlin-Neumann G.A., Conklin B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016;6:23549. doi: 10.1038/srep23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkman E.K., Chen T., de Haas M., Holland H.A., Akhtar W., van Steensel B. Kinetics and fidelity of the repair of cas9-induced double-strand DNA breaks. Mol. Cell. 2018;70:801–813.e6. doi: 10.1016/j.molcel.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling K., Yang L., Yang N., Chen M., Wang Y., Liang S., Li Y., Jiang L., Yan P., Liang Z. Gene targeting of HPV18 E6 and E7 synchronously by nonviral transfection of CRISPR/Cas9 system in cervical cancer. Hum. Gene Ther. 2020;31:297–308. doi: 10.1089/hum.2019.246. [DOI] [PubMed] [Google Scholar]
- 11.Hu Z., Yu L., Zhu D., Ding W., Wang X., Zhang C., Wang L., Jiang X., Shen H., He D., et al. Disruption of HPV16-E7 by CRISPR/Cas system induces apoptosis and growth inhibition in HPV16 positive human cervical cancer cells. Biomed. Res. Int. 2014;2014:612823. doi: 10.1155/2014/612823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshiba T., Saga Y., Urabe M., Uchibori R., Matsubara S., Fujiwara H., Mizukami H. CRISPR/Cas9-mediated cervical cancer treatment targeting human papillomavirus E6. Oncol. Lett. 2019;17:2197–2206. doi: 10.3892/ol.2018.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y., Jiang H., Wang T., He D., Tian R., Cui Z., Tian X., Gao Q., Ma X., Yang J., et al. In vitro and in vivo growth inhibition of human cervical cancer cells via human papillomavirus E6/E7 mRNAs' cleavage by CRISPR/Cas13a system. Antivir. Res. 2020;178:104794. doi: 10.1016/j.antiviral.2020.104794. [DOI] [PubMed] [Google Scholar]
- 14.Zhen S., Lu J., Liu Y.H., Chen W., Li X. Synergistic antitumor effect on cervical cancer by rational combination of PD1 blockade and CRISPR-Cas9-mediated HPV knockout. Cancer Gene Ther. 2020;27:168–178. doi: 10.1038/s41417-019-0131-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhen S., Lu J.J., Wang L.J., Sun X.M., Zhang J.Q., Li X., Luo W.J., Zhao L. In vitro and in vivo synergistic therapeutic effect of cisplatin with human Papillomavirus16 E6/E7 CRISPR/Cas9 on cervical cancer cell line. Transl. Oncol. 2016;9:498–504. doi: 10.1016/j.tranon.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siva N., Gupta S., Gupta A., Shukla J.N., Malik B., Shukla N. Genome-editing approaches and applications: a brief review on CRISPR technology and its role in cancer. 3 Biotech. 2021;11:146. doi: 10.1007/s13205-021-02680-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F., Wen Y., Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum. Mol. Genet. 2014;23:R40–R46. doi: 10.1093/hmg/ddu125. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adey A., Burton J.N., Kitzman J.O., Hiatt J.B., Lewis A.P., Martin B.K., Qiu R., Lee C., Shendure J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.