

Abstract
Introduction
Young African women bear a disproportionately high risk for HIV acquisition. HIV technologies that empower women to protect themselves are needed. Safe, potent antiretroviral agents such as tenofovir alafenamide (TAF), formulated as long-acting subdermal implants, offer an innovative solution.
Methods and analysis
CAPRISA 018 is a phase I/II trial to evaluate the safety, acceptability, tolerability and pharmacokinetics (PKs) of a TAF free base subdermal silicone implant containing 110 mg of TAF with an anticipated 0.25 mg/day release rate.
The phase I trial (n=60) will assess the safety of one implant inserted in six participants (Group 1), followed by dose escalation components (Groups 2 and 3) assessing the safety, tolerability and PK of one to four TAF 110 mg implants releasing between 0.25 mg and 1 mg daily in 54 healthy women at low risk for HIV infection. Data from this phase I trial will be used to determine the dosing, implant location and implant replacement interval for the phase II trial.
The phase II component (Group 4) will assess extended safety, PK, tolerability and acceptability of the implant in 490 at risk women, randomised in a 1:1 ratio to the TAF implant and placebo tablet or to the placebo implant and an oral pre-exposure prophylaxis tablet. Safety will be assessed by calculating the percentage change in creatinine clearance from baseline at weeks 4, 12, 24, 36, 72, 96 and 120, compared with the percentage change in the control group.
Ethics and dissemination
The South African Health Products Regulatory Authority and the University of KwaZulu-Natal’s Biomedical Research Ethics Committee have approved the trial. Results will be disseminated through open access peer reviewed publications, conference presentations, public stakeholder engagement and upload of data into the clinical trials registry.
Trial registration number
PACTR201809520959443.
Keywords: HIV & AIDS, preventive medicine, public health, clinical trials
Strengths and limitations of this study.
CAPRISA 018 is a first-in-human trial assessing the safety, acceptability, tolerability and pharmacokinetics (PKs) of a tenofovir alafenamide (TAF) free-base subdermal implant formulation.
This trial adopts a master protocol design, where phase I and phase II studies are conducted sequentially under a single protocol.
Optimal dosing selected for the phase II trial is generated from phase I safety and PK data.
The phase II part of the trial is powered on a safety endpoint using a randomised, double-dummy design.
The study is not powered to show efficacy of the TAF implant against HIV.
Introduction
Despite the global decline in HIV infections by 23% since 2010, the number of people who acquire HIV each year remains unacceptably high.1 Adolescent girls and young women in sub-Saharan Africa account for approximately 25% of all new HIV infections globally.1 Young women in this region are particularly vulnerable and acquire HIV infection 3–5 years earlier than their male peers.2 3 Despite their greater vulnerability, young women have limited access to non-user-dependent prevention options to reduce their risk for HIV acquisition.
Since antiretrovirals (ARVs) were first shown by the CAPRISA 004 trial4 in 2010 to prevent sexual transmission of HIV, the HIV prevention landscape has been transformed, principally through oral tenofovir (TFV)-containing pre-exposure prophylaxis (PrEP)5–10 or through early ARV therapy initiation in HIV-positive individuals (treatment as prevention).11 Although daily oral PrEP has been shown to be consistently effective in men who have sex with men and transgender women globally,5 6 results have been inconsistent in African women, most likely due to varying adherence.7–10 Other novel long-acting PrEP agents and innovative delivery systems such as ARV containing intravaginal rings, viz. the Dapivirine ring12 and possibly long-acting injectable ARVs,13 are poised to be accessible soon. These formulations along with the implant under study offer specific adherence advantages over daily oral PrEP.
Pharmacological models suggest that while an individual only needs 2–3 doses per week of oral TDF/FTC to successfully prevent HIV infection via receptive anal intercourse, 6–7 doses per week are needed to successfully prevent HIV infection via receptive vaginal intercourse, because drug concentrations in the lower female genital tract after oral administration are 10 times lower than those found in the colorectal mucosa.14 Unless adherence improves, many women and men using oral TDF/FTC will remain unprotected. An effective, low-cost, method of HIV prevention that overcomes adherence challenges is needed. Furthermore, products that have fewer renal and bone mineral density side effects are also desirable given that TDF/FTC oral PrEP has been shown to decrease creatinine clearance and lower bone density.15–18
This protocol focuses on the study of tenofovir alafenamide (TAF), a phosphonamidate prodrug of the HIV-1 nucleotide reverse transcriptase inhibitor TAF.19 TAF enters the cell via OATP1B1 and OATP1B3-mediated transport and is subject to ester hydrolysis. In peripheral blood mononuclear cells (PBMCs), this is performed by the serine protease cathepsin A and in hepatocytes, by carboxyesterase 1.20 Following penetration of TAF into the cells and hydrolysis of the isopropyl ester, the TFV-Ala conjugate is formed eventually releasing free TFV. TFV is then phosphorylated by intracellular kinases to the active metabolite tenofovir diphosphate (TFV-DP). Residual TFV is slowly released from cells into plasma for renal elimination by a combination of glomerular filtration and active tubular secretion.21 22 TFV-DP inhibits HIV-1 replication through incorporation into viral DNA by HIV reverse transcriptase, resulting in DNA chain-termination.
While oral emtricitabine and TAF hemifumarate (F/TAF) in combination with other ARVs has long been established to be safe and effective for HIV treatment,23 the DISCOVER trial reported that daily oral F/TAF was similarly effective in reducing incident HIV infections compared with TFV disoproxil fumarate and emtricitabine (TDF/FTC) in 5387 men and transgender women.24 While the number of adverse events for both regimens was low, F/TAF had favourable outcomes on bone mineral density and biomarkers of renal safety.25
In a study of four Beagle dogs, an early prototype of the current implant under study, was assessed over 40 days. The implant delivered TAF free-base at a rate of 1.07±0.02 mg/day. TFV-DP was observed in PBMCs at levels over 30 times higher than those associated with HIV-1 PrEP efficacy in humans. No adverse treatment-related events or clinical evidence of inflammation at the implantation site was reported. Importantly, there was no evidence of toxicity or poor tolerability. In addition, the incision sites appeared healthy on days 2–9 following surgery, with staples/sutures removed on day 8.26 In contrast, a reservoir polyurethane implant delivering TAF hemifumarate demonstrated local inflammation and in some instances, severe necrosis around the active implants in white rabbits and rhesus macaques.27
The CAPRISA 018 implant consists of TAF free-base micro-tablets encased in a cylindrical medical grade silicone elastomer sheath, with two delivery channels mechanically punched perpendicular to the longitudinal axis of the sheath.28 Each implant is approximately 40–45 mm in length with an inner diameter of 2.01±0.051 mm and a wall thickness of 0.19+0.051/–0.25 mm.28 The rationale for the 0.25 mg daily release was extrapolated from the earlier described Beagle dog study in which median PBMC TFV-DP levels of 512 fmol/106 cells were achieved and maintained over the first 35 days.26 This concentration is 11–32 times higher than the protective target from iPrEX (corresponding to a TFV-DP concentration range of 16–48 fmol/106 cells).29 Simple allometric scaling (exponent, 0.75) from Beagle dogs (mean weight, 10.8 kg) to humans (70 kg) affords a preliminary, lower target daily TAF release rate of 0.14 mg/day in humans to maintain a median TFV-DP PBMC concentration of 16 fmol/106 cells. The concentration of PBMCs in Beagle dog whole blood (mean, 1.6×106 cells/mL; SD, 0.7×106 cells/mL) was comparable to typical values for HIV-negative humans. Since 0.14 mg TAF per day in humans is estimated to yield TFV-DP PBMC concentrations of 16 fmol/106 cells (lower end of expected efficacy), the planned clinical study will evaluate a target of 0.25 mg TAF per day per implant, ranging from 1 to 4 implants (0.25 mg/day to 1 mg/day).
Recent PK modelling simulations of a potential TAF implant have estimated that multiple implants deliver a total of 1.4 mg/day of TAF subcutaneously and predict protection against HIV for approximately 6 months to 1 year.30 Innovative research into biodegradable, reservoir style TAF implants31 has also shown promise for future application. The use of subdermal implants as the drug delivery mechanism in this trial is supported by several studies showing that the contraceptive implant is highly acceptable to young women,32 with continuation rates of ~80% after 1 year, including in studies from sub-Saharan Africa.33 34
TAF is promising as a subdermal implant for PrEP due to its track record for improved safety compared with TDF, high potency and prolonged intracellular activity.16 35 The CAPRISA 018 trial will assess a novel sustained-release implant technology containing 110 mg of TAF for the prevention of HIV infection. The implant combines two well-established elements; (a) TAF, which is a licenced AFV drug widely used in HIV treatment and (b) a subdermal implant, which is widely used as a route of administration for contraception.
Methods and analysis
Trial setting
The phase I (Groups 1–3) component of the trial will be conducted at the urban CAPRISA eThekwini Clinical Research Site in Durban, South Africa. The phase II, which is a randomised controlled trial (Group 4), will continue at this urban site and include the rural CAPRISA Vulindlela Clinical Research Site in uMgungundlovu district, South Africa.
Trial population
The phase I study will enrol 60 healthy, HIV negative women at low risk for HIV into Groups 1–3, while phase II, Group 4 comprises 490 healthy, HIV-negative women from the general population. Potential study participants who consent for screening to assess for eligibility and subsequently participants who consent for enrolment will be enrolled in the study within 56 days of providing informed consent for screening. Enrolment into the trial is contingent on strict eligibility criteria being met (table 1).
Table 1.
Eligibility criteria
| Inclusion criteria | Exclusion criteria |
|
|
LDL, low-density lipoprotein; PrEP, pre-exposure prophylaxis; STI, sexually transmitted infection.
Trial design
The trial comprises an initial safety assessment in six participants (Group 1) followed by a dose escalation component (Groups 2 and 3) assessing the safety and PK of TAF 110 mg implants releasing a daily dose of 0.25 mg (1 implant), 0.5 mg (2 implants), 0.75 mg (3 implants) and 1 mg (4 implants) in 54 healthy, low risk, HIV-negative women. Comparator drugs include TAF 25 mg oral tablets and the placebo implant. Once data from Groups 1 to 3 are available, the phase II component (Group 4) of the trial will be initiated. Atotal of 490 HIV-negative women will be randomised in a double-blinded, double-placebo controlled trial to assess safety, acceptability, and PK of the TAF implant (table 2 and figure 1). Study progression from one group to the next is dependent on the approval of the Data Safety and Monitoring Board (DSMB) and Protocol Safety Review Team (PSRT).
Table 2.
Study drug administration in the CAPRISA 018 trial assessing initial safety and dose escalation followed by an extended safety assessment
| Study group (n) | Study drug | Estimated TAF implant daily drug release rate (mg/day) | Insertion site or oral | Duration of study drug exposure* |
| Group 1 (n=6) | ||||
| 1 (6) | TAF 110 mg implant | 0.25 | Arm | Up to 28 days |
| Group 2 (n=30) | ||||
| 2a (12) | TAF 110 mg implant | 0.25 | Arm | Approximately 24–48 weeks |
| 2b (3) | Placebo implant | 0 | Arm | Approximately 24–48 weeks |
| 2c (12) | 2 TAF 110 mg implants | 0.50 mg | One arm | Approximately 24–48 weeks |
| 2d (3) | 2 placebo implants | 0 | One arm | Approximately 24–48 weeks |
| Group 3 (n=24) | ||||
| 3a (6) | 2 TAF 110 mg implants | 0.50 | One implant per arm | Up to 24 weeks |
| 3b (6) | 3 TAF 110 mg implants | 0.75 | One arm | Approximately 24–48 weeks |
| 3c (6) | TAF 25 mg tablet | 25 | Oral | Up to 24 weeks |
| 3d (6) | 4 TAF 110 mg implants | 1.0 | One arm | Approximately 24–48 weeks |
| Group 4 (n=490) | ||||
| 4a (245) | TAF implant(s) plus placebo oral tablet | 0.50 mg† | †Two TAF implants per arm plus oral placebo tablets | Approximately 48–120 weeks |
| 4b (245) | TDF 300mg/FTC 200 mg oral tablet+placebo implant/s | 0 | †Two placebo implants per arm plus oral TDF/FTC tablets | Approximately 48–120 weeks |
*Follow-up extended based on safety review of the adverse events occurring during the first 4 weeks after insertion in Groups 1–3.
†Based on the PK and safety assessment in dog models but is subject to change after PK data become available from Groups 1–3 in the trial.
FTC, emtricitabine; TAF, tenofovir alafenamide fumarate; TDF, tenofovir disoproxil fumarate.
Figure 1.
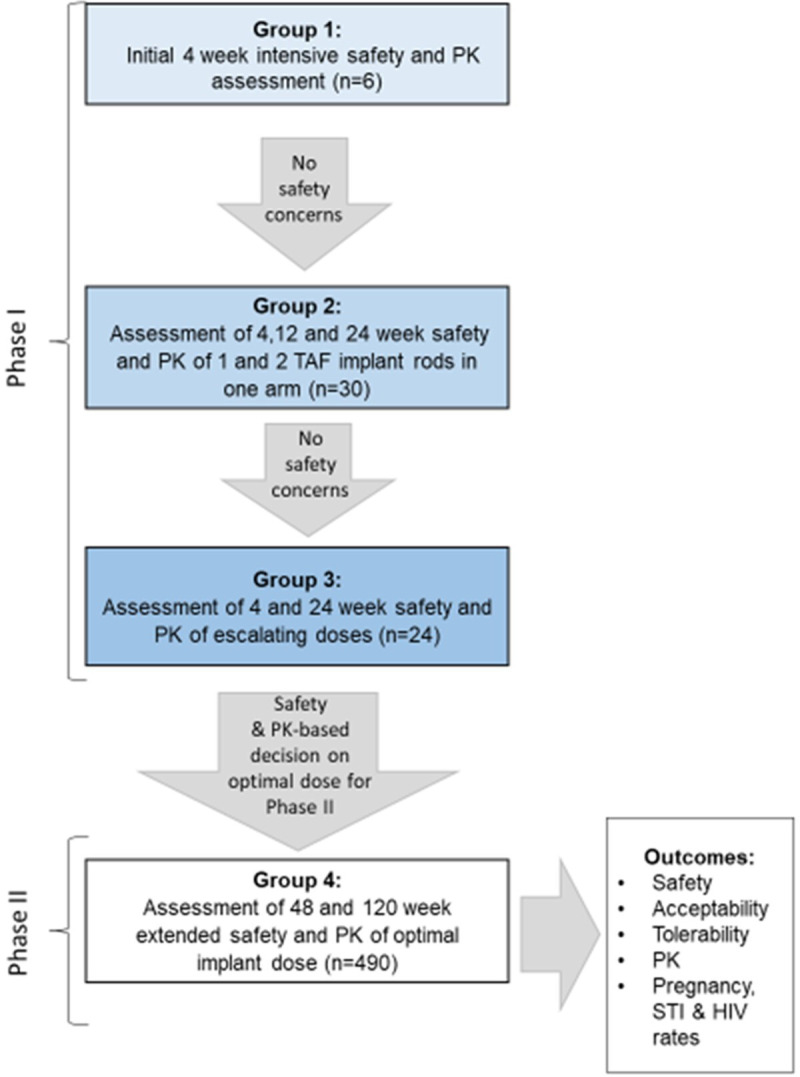
CAPRISA 018, phase I/II trial design summary graphic (extracted from the study protocol version 2.0, 12 August 2019). PK, pharmacokinetics; STI, sexually transmitted infection; TAF, tenofovir alafenamide.
Implants will be inserted subdermally in the upper arm/s, similar to the contraceptive implant, in a controlled sequence as follows (also see figure 1 for a graphical representation).
Phase I
The first six participants in Group 1 (open-label) will have one active implant inserted and will be enrolled sequentially on separate days. Participants will be followed up daily for the first 3 days following insertion and weekly thereafter. At the day 28 visit, the implant will be removed, and participant will be followed up weekly for a further 4 weeks.
Following Data Safety Monitoring Board (DSMB) approval to proceed with the trial, the next 30 eligible low risk HIV-negative women will be enrolled into Group 2 (double-blinded). Group 2 participants will be randomised to one of four subgroups where they could receive either one or two active implants or placebo implants in a 4:1, active to placebo ratio. Participants will attend study visits weekly in the first 4 weeks postactive/placebo implant insertion and thereafter study visits will be conducted every 4 weeks through week 24 or 48 if safety reviews conducted at weeks 4, 12 and 24 permit. Participants will be followed up weekly for a further 4 weeks after implant removal.
Contingent on DSMB approval to proceed with the study after the review of the week 4 safety data from participants in Group 2, the dose escalation component (Group 3) of the study will proceed in 24 eligible low-risk HIV-negative women. Group 3 participants will be enrolled in parallel in three subgroups (one implant in each arm, three implants in one arm, and an oral TAF 25 mg control group) while the maximum dose group (four implants in one arm) will be enrolled sequentially. Participants follow a similar visit schedule and safety review time points to Group 2 participants.
Phase II
Enrolment into phase II (Group 4) may proceed with two implant rods in one arm provided that the DSMB review of the 4-week safety data in Group 2 recommended study continuation and the Protocol Safety Review Team (PSRT) review of the safety and PK data from Group 3 recommended progression to Group 4 without change.
-
Group 4 participants (double-blinded, double-dummy design) will be randomised in a 1:1 ratio and could receive either two active implants+daily placebo tablets or two placebo implants+TDF 300 mg/FTC 200 mg tablets. While the trial protocol allows for insertion of two active/placebo implants in this group, the actual number of implants for insertion will be determined from the PK and safety data that emerge from the Group 1–3 experience.
Participants enrolled in Group 4 will attend a study visit 1 week after implant insertion and thereafter from week 4 the study visits will be conducted monthly. The minimum follow-up period for Group 4 is 48 weeks. Implants will be removed at week 48 and replacement implants will be inserted. These participants will have implants removed at week 116 and will be exited from the study at week 120. Implants may be removed without replacement at any time; however, in accordance with study visits, they will be scheduled to be removed 4 weeks before study exit.
Trial objectives
Primary objective
To evaluate the safety and tolerability of sustained-release TAF 110 mg subdermal implant/s in HIV uninfected women.
Secondary objectives
To assess systemic and genital compartment PK of single and multiple TAF 110 mg implant/s to determine in-human release rate characteristics.
To compare the PK profiles of insertion of two implants in one arm vs insertion of one implant in each arm.
To assess participant acceptability of implant technology after insertion of one or more TAF implants.
To assess the incidence of HIV infection, as well as other sexually transmitted infections (STIs), including (but not limited to) herpes simplex virus type 2 (HSV-2), human papillomavirus (HPV), gonorrhoea, chlamydia and trichomonas infections.
To assess viral load and frequency of resistance mutations in HIV seroconverters.
To assess pregnancy rates and outcomes.
Trial endpoints
Primary endpoint
To evaluate the safety of the TAF 110 mg implant.
Secondary endpoints
Adverse event rates by grade (according to the National Institutes of Health Division of AIDS (DAIDS) table for grading adverse events)
Adverse event rates by degree of association with study product
Number of early implant removals (prior to scheduled removal) and the reasons for removal
Systemic PK profile
Genital compartment PK profile
Acceptability of the insertion of 1, 2, 3 and 4 implants.
Incidence rates of STIs, including HIV, HSV-2, HPV, gonorrhoea, chlamydia and trichomonas
Pregnancy rates and outcomes
TAF resistance in HIV seroconverters
Viral load in HIV seroconverters
Sample size calculation
Phase I (Groups 1–3)
The goal of the phase I study is to identify safety concerns associated with product administration during dose escalation. No formal sample size calculation is needed. However, given the chosen sample size per group, the ability of the study to detect serious adverse events (SAEs) for different group sizes is shown in table 3. Sample sizes were selected by calculating the probabilities of experiencing 0, ≥1 or ≥2 events under different possible true event rates33 36 as shown in table 3. For each of the groups with n=6 (ie, participants in Group 1, Groups 3a, 3b and 3d), there is a 26% chance of observing at least one event, if the true event rate is 4.8%. However, when the true event rate is doubled or sixfold higher, this probability rises to 47% and 88%, respectively. When we consider the groups that are doubled in size (n=12), who will receive one or two TAF implants (ie, Groups 2a and 2c), the probabilities of detecting at least one event are also increased. They are 45%, 72% and 95% when the event rate is 4.8%, 10% and 30%, respectively. The probability of observing 0, 1+ and 2+ events for a range of true event rates among different groups, including all 54 participants who will be receiving active treatment is provided in table 3.
Table 3.
Probability of observing 0 events, 1 or more events and 2 or more events, for a range of hypothetical true event rates
| True event rate (%) | Number of participants | 0 events | 1+ events | 2+ events |
| 1 | 6 | 0.94 | 0.06 | <0.01 |
| 12 | 0.89 | 0.11 | <0.01 | |
| 24 | 0.79 | 0.21 | 0.02 | |
| 54 | 0.58 | 0.42 | 0.1 | |
| 4.8 | 6 | 0.74 | 0.26 | 0.03 |
| 12 | 0.55 | 0.45 | 0.11 | |
| 24 | 0.31 | 0.69 | 0.32 | |
| 54 | 0.07 | 0.93 | 0.74 | |
| 6 | 6 | 0.69 | 0.31 | 0.05 |
| 12 | 0.48 | 0.52 | 0.16 | |
| 24 | 0.23 | 0.77 | 0.43 | |
| 54 | 0.04 | 0.96 | 0.84 | |
| 10 | 6 | 0.53 | 0.47 | 0.11 |
| 12 | 0.28 | 0.72 | 0.34 | |
| 24 | 0.08 | 0.92 | 0.71 | |
| 54 | <0.01 | >0.99 | 0.98 | |
| 30 | 6 | 0.12 | 0.88 | 0.58 |
| 12 | 0.01 | 0.99 | 0.91 | |
| 24 | <0.01 | >0.99 | >0.99 | |
| 54 | <0.01 | >0.99 | >0.99 |
Since the phase I trial will help identify the maximally tolerated dose, the chances of detecting rare events will vary depending on the dosing strategy and how big or small the sample size is.
Phase II (Group 4)
In the phase II study, the primary safety endpoint is a change in creatinine clearance from baseline to week 12 postrandomisation. The sample size calculation was based on data from the iPrEx study,15 17 which showed a mean creatinine clearance decline of 5% from baseline to week 12. Preliminary clinical data37 regarding TAF oral use suggest minimal creatinine clearance alterations. Assumptions in calculating sample size include a mean decline of 5% from baseline in the TDF/FTC group and a mean decline of 1% in the TAF group, with a common SD of 13%, using a two-group t-test with 0.05 2-sided significance. Loss to follow-up was set at 10%. A sample size of 245 in each arm will have 90% power to detect a fivefold difference in the mean decline in creatinine clearance from baseline to 12 weeks between the two groups.
In table 4, the statistical power for varying declines in mean creatinine clearance in both TAF and TDF/FTC groups is presented when overall sample size is fixed at 490. These estimates are subject to differences in adherence to daily oral TDF/FTC in the control group.
Table 4.
Power calculation at a constant sample size of 490, allowing for varying percentage declines in creatinine clearance (CrCl) estimates in the TAF implant and TDF/FTC oral groups
| Mean CrCl % decline in TDF/FTC group | ||||||
| 3 | 4 | 5 | 6 | 7 | ||
| Mean CrCl % decline in the TAF group | 0.5 | 56 | 84 | >95 | >95 | >95 |
| 1 | 39 | 72 | 90 | >95 | >95 | |
| 1.5 | 24 | 56 | 84 | >95 | >95 | |
TAF, tenofovir alafenamide; TDF/FTC, tenofovir disoproxil fumarate and emtricitabine.
Trial procedures
Informed consent
Written informed consent will be obtained from each study participant in English or isiZulu prior to screening and enrolment, in accordance with South African Good Clinical Practice (GCP) guidelines, 21 CFR Part 50 and ICH GCP guidelines. Separate written informed consent will be obtained for trial screening, specimen storage and possible future testing, enrolment into the trial and permissions for off-site visits. Study participation will be permitted even if consent for long-term specimen storage or off-site study visits is declined by study participants.
Recruitment, screening and enrolment
Study staff will conduct targeted recruitment, by focusing study outreach on women likely to be between 18 and 40 years of age for Groups 1–3 and are between 18 and 30 years for Group 4. Participants may be recruited from sexual reproductive/family planning health services or directly from the community. Walk-in participants, who may have heard about the trial during community outreach activities, may also be screened for participation.
To prevent deliberate or inadvertent co-enrolment in multiple trials, each participant’s identification will be verified against the Biometric Co-Enrolment Prevention System database at the screening visit and at each subsequent contact visit. Screening is completed in a stepwise manner. The first step is to provide introductory study information and obtain written informed consent. A unique participant identification number is assigned. HIV testing and counselling, using two rapid antibody test kits and/or antibody/antigen, one of which must be a fourth generation test, is conducted at the outset. HIV-infected participants or those with discordant results will be linked to immediate care and treatment. Only HIV-negative participants will continue with screening. A complete medical and contraceptive use history will be recorded along with a full physical examination, urine collection and phlebotomy to assess laboratory test results for further participation. These tests include urinalysis, urine pregnancy, pap smear, sexually transmitted and reproductive tract infection testing, haematology, and serum chemistries.
If all screening parameters conform to the trial inclusion criteria, enrolment into the trial, defined as implant insertion, must take place within 56 days of the first screening attempt. A separate enrolment informed consent is conducted prior to implant insertion along with a physical examination and review of contraceptive and medical history. Additional testing includes an assessment of bone densitometry, genital specimen collection and bloods for PK assessments. The implant insertion procedure is conducted under local anaesthetic, by a trained study clinician or professional nurse.
Randomisation
Group 1 participants will not be randomised but are enrolled sequentially until targeted numbers are reached. In Group 2, participants will be randomised in a 4:1 ratio, stratified by whether participants will be receiving one or two TAF implants. Group 3 participants are not randomised but will be enrolled in parallel for groups 3a, 3b and 3c with Group 3d enrolling sequentially to Group 3b until targeted numbers are reached. In Group 4, participants will be assigned at random to one of the two study arms in equal proportions to receive active implants and placebo tablets or placebo implants and active tablets.
A statistician who is not involved in the study will produce a computer-generated randomisation list for Groups 2 and 4, which will then be provided to the unblinded study pharmacist. For Group 4, the statistician will use a randomly permuted block design, with two or more prespecified block sizes. The study pharmacist will also receive sealed, sequentially numbered opaque randomisation envelopes. These envelopes will be assigned in sequence to eligible study participants by the study pharmacist. Electronic copies of the randomisation schedule and the programmes used to generate the randomisation schedule will be access controlled and password protected.
Blinding
Both study staff (except for the study pharmacists) and participants will be blinded to active or placebo treatment assignments for Groups 2 and 4. However, it will not be possible to blind the number of implants received in Group 2. Blinding will be maintained until the last participant reaches study exit within their assigned group.
If knowledge of the received study product is necessary to protect a participant’s safety, the principal investigator and/or designee will give permission for emergency unblinding.
Safety monitoring
Clinical safety and adverse events
While clinical safety will be assessed by evaluating vital signs, weight, physical examination and clinical laboratory results, the main safety indicators are implant insertion site reactions (local) and changes in creatinine clearance (systemic). Each safety assessment will include a review of adverse events (AEs) at grade 2 or higher for local site reactions and serum chemistry. Product hold or discontinuation will be based on assessment of grade 3 or higher AEs that are deemed to be probably or definitely related to study product.
All participants reporting an AE will be followed clinically, until the AE resolves (returns to baseline/non-gradable range). Each AE will be graded for severity using the DAIDS Adverse Event Grading Tables, V.2.1, dated March 2017 (or latest version). Laboratory values meeting grade 1 and above will be reported as AEs. AEs related to implant insertion or removal will be graded using a study modified interpretation of the DAIDS AE grading table for site reactions to injections and infusions for insertion site pain, insertion site erythema, insertion site swelling or insertion site pruritus. All AE reports will be captured regardless of the association to the study product and will contain at least the date the AE occurred, a brief description of the event, the relationship to study drug, any treatment given, the outcome, date resolved and the seriousness of the event. AEs and SAEs will be coded using the Medical Dictionary for Regulatory Activities (MedDRA, V.21.1) terminology, that is, system organ class (SOC) and preferred terms.
PSRT and DSMB
Protocol Safety Review Team
Participant safety will be closely monitored both internally by the PSRT (designated study staff will be responsible for continuous close safety monitoring of all study participants) and externally by the DSMB. PSRT members will meet in-person and/or via teleconference regularly throughout the period of study implementation.
Data Safety Monitoring Board
An independent DSMB will be established before the clinical trial begins to monitor the safety of the trial participants. The DSMB will convene regularly to review cumulative safety data prior to opening enrolment into each of the four study groups. Following periodic review of the trial data, the DSMB may recommend that the study proceed as designed, proceed with design modifications, or be discontinued. A recommendation to stop the trial may be made by the DSMB at any such time that the board agrees an unacceptable type and/or frequency of AEs has been observed.
Data management and statistical analysis
Data management
Data will be collected on paper-based case report forms (CRFs) that have been developed by the study team. If data entered on the CRFs are taken from an external source (eg, laboratory reports, patient records), the source documents will be maintained in the participant’s medical chart or study file at the site and will be available for review. The CRFs will be faxed into the central CAPRISA database management system (DataFax Discover database) running on SuSe Linux V.11. Data Encoders will verify all data by cross-checking the faxed version with what is entered into the database. Queries arising during validation of the data will be recorded in quality control reports sent to the sites on a regular basis. Database files will be password-protected and access to the files will be limited to authorised study staff. All data will be backed up at regular intervals. On completion, the close-out site monitoring visit and finalisation of the database for analysis, the original forms will be bound and kept for long-term storage.
Statistical analysis
Demographic data of all participants enrolled in the study will be summarised using descriptive statistics. These will be reported by treatment assignment, study group and overall. The primary and secondary analyses will be performed on an intention to treat basis.
Laboratory test results will be summarised by study arm, group and time-point postenrolment. Creatinine clearance is an important laboratory marker in this trial. For Group 4, the mean percentage change in creatinine clearance will be calculated from baseline to week 4, 12, 24, 36, 48, 72, 96 and 120. The percentage change at week 12 will be compared between the two treatment groups using a t-test for independent groups. In addition, linear mixed models or generalised estimating equations, accounting for repeated measurements will be used to assess changes in creatinine clearance over time. These models will be adjusted for baseline prognostic covariates.
Summaries of AEs by treatment arm (active or placebo) and group will show number and percentage of participants experiencing AEs within each of the SOC and preferred terms. Moreover, number and percentages of participants experiencing each specific AE will be tabulated by severity and relationship to study product.
To assess the efficacy of TAF implant, the cumulative probability of HIV infection will be calculated for each treatment group using the Kaplan-Meier method and the curves will be compared using the log-rank test. The overall HIV incidence rates will be calculated for each treatment group and compared using a z-test. TAF implant efficacy will be calculated as 1 minus (HIV incidence rate in the TAF implant group/HIV incidence rate in the placebo group).
PK analysis
The PK analysis will involve analysis of TAF, TFV and TFV-DP concentrations at predetermined timepoints post-insertion in plasma, PBMCs, subdermal fluid on the removed implant and in the genital tract in both phase I and II of the trial. These data will be used to calculate PK parameters (AUC, half-life, clearance and volume of distribution) for the TAF implant using a non-compartmental PK model analysis. The TFV-DP active intracellular metabolite assayed in the PBMCs and genital tract cells along with TFV assayed from the genital fluid will be assessed to evaluate protection against HIV infection.
Patient and public involvement
CAPRISA, the study sponsor, has established a Community Advisory Board (CAB) at both trial sites, informed by Good Participatory Practice guidelines.38 CAB members consist of individuals who reside in the communities from where trial participants will be screened and recruited. They include community leaders, traditional leaders, previous trial participants, representatives of local HIV/AIDS organisations and people living with HIV from the community. The CAB meets at least bi-monthly to review concepts, protocols, provide input into study materials, alert researchers to concerns from the community, prepare messaging for the outcome of DSMB meetings, and plan for the dissemination of study results. Trial staff, designated as community liaison officers, work closely with the CAB and plan, with CAB support, participating in community-driven events within current COVID-19 restrictions.
Ethics and dissemination
Ethics and regulatory approval
Ethics approval was granted by the University of KwaZulu-Natal’s Biomedical Research Ethics Committee (UKZN BREC) (reference number: BFC107/18) on 16 October 2019 and regulatory approval by the South African Health Products Regulatory Authority (SAHPRA) (trial reference number: 20180523) on 19 September 2019 for the study protocol (V.2.0, dated 12 August 2019). Any future protocol modifications will be submitted to the relevant regulatory and ethics authorities for approval prior to implementation.
Trial results dissemination plan
Results from this research will be published in open access peer-reviewed journals. In addition, investigators will disseminate the results as broadly as possible to the scientific community by attending presenting the findings at local, national and international conferences and through presentations at public lectures, scientific institutions and stakeholder/partner meetings. The findings will be shared and discussed with the study participants, communities and lay persons. Summary results of the trial will also be made publicly available in a timely manner by posting to the results section of the clinical trial registry.
Standard Protocol Items: Recommendations for Interventional Trials guidelines
This protocol has been written in accordance with the Standard Protocol Items: Recommendations for Interventional Trials guidelines.
Trial status
Enrolment commenced on 4 August 2020 and is currently recruiting. The trial is anticipated to be completed in June 2024.
Supplementary Material
Footnotes
Contributors: SAK, TNG conceived the trial. SAK, QAK, TNG designed the trial. TNG, SAK, QAK, LM, NYZ and CB contributed to writing the study protocol. CH critically reviewed the protocol. NYZ performed sample size calculations and the statistical analysis strategy for the study protocol. PR, the study data manager, designed data collection tools and programmed the study database. MMB and JAM are the implant product developers, wrote the study dose rationale and provided the technical information on the study implant and generated the preclinical safety data. BP will assay the PK samples. LM is the trial project manager, IH is responsible for clinical oversight, NS is responsible for the setup of all laboratory procedures and BM is the study pharmacist. All authors contributed to the planning of the trial. TNG wrote the first draft of the protocol paper and SAK, QAK, CB, LM, NYZ, CH, NS, PR, IH, BM, JAM, MMB and BP contributed to critical edits to the manuscript. All authors reviewed the final version of this manuscript and consented to publication.
Funding: The study is funded by the European and Developing Countries Trial Partnership (EDCTP Grant No: SRIA2015-1061) which is a part of the EDCTP2 programme supported by the European Union Horizon 2020 framework. This research is also supported by a joint initiative between the South African National Department of Health, the South African Medical Research Council and the Centre for the AIDS Programme of Research in South Africa. Cipla Ltd (India) has donated the oral drugs being studied in the CAPRISA 018 trial and Gilead Sciences donated the tenofovir alafenamide free base active pharmaceutical ingredient (API) used to formulate the TAF implants.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1. Joint United Nations Programme on HIV/AIDS . UNAIDS data 2020 Geneva. Switzerland, 2020. Available: https://www.unaids.org/sites/default/files/media_asset/2020_aids-data-book_en.pdf
- 2. UNAIDS . The gap report, 2014. Available: http://www.who.int/tb/publications/global_report/en/
- 3. Abdool Karim Q, Baxter C, Birx D. Prevention of HIV in adolescent girls and young women: key to an AIDS-Free generation. J Acquir Immune Defic Syndr 2017;75 Suppl 1:S17–26. 10.1097/QAI.0000000000001316 [DOI] [PubMed] [Google Scholar]
- 4. Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010;329:1168–74. 10.1126/science.1193748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010;363:2587–99. 10.1056/NEJMoa1011205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCormack S, et al. Pragmatic open-label randomised trial of preexposure prophylaxis: the PROUD study Abstract 22LB. Conference on retroviruses and opportunistic infections (CROI); Seattle, USA 2015.
- 7. Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med 2012;367:423–34. 10.1056/NEJMoa1110711 [DOI] [PubMed] [Google Scholar]
- 8. Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 2012;367:399–410. 10.1056/NEJMoa1108524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med Overseas Ed 2012;367:411–22. 10.1056/NEJMoa1202614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med 2015;372:509–18. 10.1056/NEJMoa1402269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011;365:493–505. 10.1056/NEJMoa1105243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. PrEPWatch an initiative of AVAC . About the Dapivirine ring, 2021. Available: https://www.prepwatch.org/about-prep/dapivirine-ring/
- 13. Landovitz RJ, Donnell D, Clement ME, et al. Cabotegravir for HIV prevention in Cisgender men and transgender women. N Engl J Med 2021;385:595–608. 10.1056/NEJMoa2101016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cottrell ML, Yang KH, Prince HMA, et al. A translational pharmacology approach to predicting outcomes of preexposure prophylaxis against HIV in men and women using tenofovir disoproxil fumarate with or without emtricitabine. J Infect Dis 2016;214:55–64. 10.1093/infdis/jiw077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang EC, Vittinghoff E, Anderson PL, et al. Changes in kidney function associated with daily tenofovir disoproxil Fumarate/Emtricitabine for HIV preexposure prophylaxis use in the United States demonstration project. J Acquir Immune Defic Syndr 2018;77:193–8. 10.1097/QAI.0000000000001566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Markowitz M, Zolopa A, Squires K, et al. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J Antimicrob Chemother 2014;69:1362–9. 10.1093/jac/dkt532 [DOI] [PubMed] [Google Scholar]
- 17. Liu AY, Vittinghoff E, Anderson PL, et al. Changes in renal function associated with TDF/FTC PreP use in the US Demo project. Abstract number: 867. Conference on retroviruses and opportunistic infections; February 22–25; Boston, Massachusetts 2016.
- 18. Mulligan K, Glidden DV, Anderson PL, et al. Effects of Emtricitabine/Tenofovir on bone mineral density in HIV-negative persons in a randomized, double-blind, placebo-controlled trial. Clin Infect Dis 2015;61:572–80. 10.1093/cid/civ324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suo Z, Johnson KA. Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-Phosphonylmethoxypropyl)adenine. J Biol Chem 1998;273:27250–8. 10.1074/jbc.273.42.27250 [DOI] [PubMed] [Google Scholar]
- 20. Birkus G, Wang R, Liu X, et al. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother 2007;51:543–50. 10.1128/AAC.00968-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ray AS, Fordyce MW, Hitchcock MJM. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res 2016;125:63–70. 10.1016/j.antiviral.2015.11.009 [DOI] [PubMed] [Google Scholar]
- 22. Birkus G, Bam RA, Willkom M, et al. Intracellular activation of tenofovir Alafenamide and the effect of viral and host protease inhibitors. Antimicrob Agents Chemother 2016;60:316–22. 10.1128/AAC.01834-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Clercq E. Role of tenofovir alafenamide (TAF) in the treatment and prophylaxis of HIV and HBV infections. Biochem Pharmacol 2018;153:2–11. 10.1016/j.bcp.2017.11.023 [DOI] [PubMed] [Google Scholar]
- 24. Abbasi J. Long-Acting Cabotegravir shot prevents HIV among women. JAMA 2020;324:2247. 10.1001/jama.2020.23330 [DOI] [PubMed] [Google Scholar]
- 25. Mayer KH, Molina J-M, Thompson MA, et al. Emtricitabine and tenofovir alafenamide vs emtricitabine and tenofovir disoproxil fumarate for HIV pre-exposure prophylaxis (discover): primary results from a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet 2020;396:239–54. 10.1016/S0140-6736(20)31065-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gunawardana M, Remedios-Chan M, Miller CS, et al. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob Agents Chemother 2015;59:3913–9. 10.1128/AAC.00656-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su JT, Simpson SM, Sung S, et al. A subcutaneous implant of tenofovir Alafenamide fumarate causes local inflammation and tissue necrosis in rabbits and macaques. Antimicrob Agents Chemother 2020;64. 10.1128/AAC.01893-19. [Epub ahead of print: 21 02 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oak Crest Institute of Science . Investigators brochure: OCIS-001 tenofovir alafenamide (TAF) implant. Second edition, 2019. [Google Scholar]
- 29. Anderson PL, Glidden DV, Liu A, et al. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med 2012;4:151ra25. 10.1126/scitranslmed.3004006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rajoli RKR, Demkovich ZR, Flexner C, et al. Predicting pharmacokinetics of a tenofovir Alafenamide subcutaneous implant using physiologically based pharmacokinetic modelling. Antimicrob Agents Chemother 2020;64. 10.1128/AAC.00155-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li L, Johnson LM, Krovi SA, et al. Performance and stability of tenofovir Alafenamide formulations within subcutaneous biodegradable implants for HIV pre-exposure prophylaxis (PreP). Pharmaceutics 2020;12:1057. 10.3390/pharmaceutics12111057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diedrich JT, Klein DA, Peipert JF. Long-acting reversible contraception in adolescents: a systematic review and meta-analysis. Am J Obstet Gynecol 2017;216:364.e1–364.e12. 10.1016/j.ajog.2016.12.024 [DOI] [PubMed] [Google Scholar]
- 33. Pillay D, Chersich M, Morroni C, et al. User perspectives on Implanon NXT in South Africa: a survey of 12 public-sector facilities. S Afr Med J 2017;107:815–21. 10.7196/SAMJ.2017.v107i10.12833 [DOI] [PubMed] [Google Scholar]
- 34. O'Neill E, Tang J, Garrett J, et al. Characteristics of Kenyan women in a prospective cohort study who continue using subdermal contraceptive implants at 12 months. Contraception 2014;89:204–8. 10.1016/j.contraception.2013.11.016 [DOI] [PubMed] [Google Scholar]
- 35. Babusis D, Phan TK, Lee WA, et al. Mechanism for effective lymphoid cell and tissue loading following oral administration of nucleotide prodrug GS-7340. Mol Pharm 2013;10:459–66. 10.1021/mp3002045 [DOI] [PubMed] [Google Scholar]
- 36. Power J, French R, Cowan F. Subdermal implantable contraceptives versus other forms of reversible contraceptives or other implants as effective methods of preventing pregnancy. Cochrane Database Syst Rev 2007;3:CD001326. 10.1002/14651858.CD001326.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015;385:2606–15. 10.1016/S0140-6736(15)60616-X [DOI] [PubMed] [Google Scholar]
- 38. UNAIDS . Good participatory practice: guidelines for biomedical HIV prevention trials, 2011. Available: https://www.unaids.org/en/resources/documents/2011/20110629_JC1853_GPP_Guidelines_2011%20OK
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.