

Abstract
The novel coronavirus strain SARS-CoV-2 is the virus responsible for the recent global health crisis, as it causes the coronavirus disease-19 (COVID-19) in humans. Due to its high rate of spreading and significant fatality rates, the situation has escalated to a pandemic, which is the cause of immense disruption in daily life. In this study, we have taken a docking-based virtual screening approach to select natural molecules (from plants) with possible therapeutic potential. For this purpose, AUTODOCK Vina-based determination of binding affinity values (blind and active-site oriented) was obtained to short-list molecules with possible inhibitory potential against the main Mpro in SARS-CoV-2 (PDB ID 6Y2F -the monomeric form). The 4 molecules selected were Chebuloside (−8.2; −8.2), Acetoside (−8.0; −8.0), Corilagin (−8.1; −7.7) and Arjunolic Acid (−8.0; −7.6) (blind and active-site oriented docking scores (Kcal/mol) in parenthesis, respectively). Further, a comparative search, with FDA-approved drugs, has shown that Ouabain was comparable to Chebuloside with a similarity score of 0.227. This in silico finding with respect to Ouabain is significant, since this polycyclic glycoside has been shown to treat COVID-19 positive patients with a cardiovascular disease. Hydrocortisone was similar to Arjunolic acid with a score of 0.539. Again, this likeness is worthy of mention, since hydrocortisone has been used earlier for the treatment of SARS-CoV1 and MERS. However, further experimentation and validation of the results, in suitable biological model systems, are necessary to gain more insight and relevance as well as provide corroborative evidence for our in-silico findings.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40011-021-01292-5.
Keywords: COVID-19, Docking, Drug likeliness screening, Medicinal plant, Bioinformatics
Introduction
Under the electron microscope, the CoVs (RNA viruses with a positive strand) appear to have a crown-like appearance. This molecular feature is attributable to be due to the spike glycoproteins on the viral envelope. The viruses in the Coronaviridae family belong to the order Nidovirales. Several viruses from this family have been known to cause many enteric, neurological, hepatic and respiratory diseases in diverse species (camels, cats, bats, etc.), while a select few are capable of infecting humans. [1] The current global pandemic, due to significant increases in the occurrence and rapid transmission of the novel coronavirus—SARS-CoV-2, has resulted in infections in over 213 countries. This outbreak has been thought to have originated in Hubei and Wuhan City in China in December of 2019. Due to the high rates of spreading and significant fatality rates, the World Health Organization convened an emergency committee and declared it a global health emergency in January 2020. It was dubbed a pandemic in March 2020, with several countries going into lockdown in an effort to contain its spread [2]. Also, this infection has posed an enormous economic and psychological burden on the public health maintenance systems globally as well as being a major factor contributing to the decline in productivity levels.
The SARS-CoV-2, referred to as COVID-19, belongs to the genera Betacoronavirus (betaCoV), under the subfamily Orthocoronavirinae. The CoVs belonging to this genera predominantly manifest as respiratory and extra-respiratory pathogenesis with variable severity. The recent genomic studies have revealed that the SARS-CoV-2 may have evolved from the betaCoV RaTG13 strain present in bats, showing approximately 96% similarity. [3]
SARS-CoV-2 has a diameter of 60–140 nm, and its form is spherical and often pleomorphic. The number of nucleotides and amino acids in the single-stranded RNA genome of this corona virus is 29891 and 9860, respectively [1]. The number of open reading frames (ORFs) in the genome of CoVs is generally around 6–11. The first ORF (ORF1a/b)) corresponds to two polyproteins, namely—pp1a and pp1ab, as well as the 16 non-structural proteins (NSPs) and is encoded by about 67% of the viral RNA genome. The rest of the ORFs code for the remaining accessory and structural proteins. The envelope (E), spike (S), membrane (M) and nucleocapsid (N) represent four structural proteins, along with the other accessory proteins [4]. The spike and membrane proteins play a key role in the assembly of the virus during replication, while the accessory proteins are crucial as triggers to initiate and facilitate entry processes.
The role of the main protease in the pathogenesis of SARS-CoV-2 is highly significant. Its function is to proteolytically (enzymatically) cleave pp1a and pp1ab polyproteins, which subsequently makes them functional. Without the occurrence of this important cleavage event, important viral replication-associated proteins like RdRp and nsp13 will not fully function. Along with its essential role in viral replication, the Mpro is highly conserved/expressed selectively in SARS-CoV-2 and lacks any similar structures in humans making it an ideal antiviral drug target [5].
SARS-CoV-2 also dubbed as the Wuhan-Hu-1 coronavirus (WHCV), exhibits marked similarity to the human SARS-CoV (approximately 82%), at the phylogenetic and genomic levels [6]. This aspect indicates its capability of direct human to human transmission, especially due to the high similarity seen in the S-glycoprotein and receptor-binding domain (RBD). Based on the epidemiological investigations, COVID-19 has an incubation period of 1–14 days. However, it is contagious even during the latency period.
With the exponential spreading of SARS-CoV-2, there is an increasing demand for drugs to treat these infections. Compounds from traditional medicinal plants have proved to have antiviral properties in the past. AYUSH (Ayurveda, Yoga and Naturopathy, Unani, Siddha and Homoeopathy), Government of India, has recommended a formulation made up of ingredients from 15 plants [7]. Since this treatment was based on empirical findings, we decided to screen and identify key bioactive components (possibly major contributors) present in the extracts of the respective medicinal plants in the aforesaid Ayurvedic formulation, Hence, an extensive literature study was performed to obtain compounds from plants and a few other traditional medicinal plants as mentioned in Table S1 [8–25]. We have then used virtual screening as a tool to select plant compounds that can serve as possible leads for the development of anti-SARS-CoV-2 drugs.
Material and Methods
Ligand Selection, Retrieval and Preparation
A detailed literature survey for the identification and listing of active compounds in the plants was conducted (Table S1). The study enabled selection of ligands for docking against the main protease. 175 compounds were selected and their respective chemical structures and Ids were retrieved from PubChem [29]. The ligands were then imported into the OpenBabel suite of PyRx (http://%28https//pyrx.sourceforge.io/). Energy minimization, under default parameters, was done for all the ligands. They were further converted to the appropriate docking format using the make ligand option available in PyRx.
Protein Retrieval and Preparation
The monomeric form of the structure of the main Mpro of SARS-CoV-2 with PDB ID 6Y2F [26] was retrieved from RCSB-PDB. The protein was prepared by removing water and nonspecific molecules using the online USCF Chimera tool. The protein molecule was further prepared by energy minimization in USCF Chimera. It was then loaded to PyRx and was prepared for docking using the make macromolecule option in PyRx.
Protein Binding Pocket Identification
Before undertaking docking analysis, CASTp (Computed Atlas of Surface Topography of proteins) [27] was used to identify the major binding pockets in the protein- Mpro. Data obtained thereby were corroborated with published experimental findings to confirm, validate and lend more credence to our in-silico approach.
Virtual Screening
The ligands were virtually screened by docking of each of them with the Mpro in the Autodock Vina (http://www3.interscience.wiley.com/journal/122439542/abstract) suite of PyRx. Two docking runs were undertaken. In the first one, grid dimensions were maximized (X:50.7889,Y:66.6742,55.5860) with centre (X:4.7290,Y:0.7953,Z:1.0522) such that the ligand was allowed to bind to any part of the protein. For the second docking run, the grid box was minimized (Dimensions-X:25.9265,Y:25.1807,Z:38.0944 and Centre-X:11.20939,Y:8.9589,Z:15.0892) to cover only the active sites of the protein molecule-HIS41, CYS145, GLY143 and SER144 [28]. This “blind-docking” versus “targeted approach” was taken to better obtain a more precise binding affinity value based on active site-based interactions. The exhaustivenss was set at 12 and 8, respectively, for the two runs, and the docking of plant-derived active compounds with the SARS-CoV-2 Mpro was performed.
Drug Likeliness Screening
MolSoft (https://molsoft.com/mprop/), an in-silico tool, was used to check the drug likeliness score. Molsoft is a tool which calculates the drug likeliness of a compound using parameters such as logP and logS. This was done to further the assess the ability of selected natural compounds to act as drug lead compounds.
Visualization of Ligand -Protein Bonds
The docked complexes of the plant-derived compounds with Mpro were visualized using Discovery Studio 2020 (Dassault Systèmes BIOVIA, [Discovery Studio], [20.1.0.19295], San Diego: Dassault Systèmes, [2020]). The 2D interaction plot of bonds between compound and Mpro active sites was obtained.
Similarity Screening
The compounds with high binding affinities and drug likeliness scores were then checked for to select drugs that have already been approved by FDA using the SWISS similarity tool.
Results and Discussion
From the docking run results, the docked pose with best binding affinity was chosen for each ligand. The 30 ligands with the highest binding affinities from docking run 2 (active site-oriented) are summarized in Table 1 and chosen for further analysis. The drug likeliness scores of chosen compounds are also given in Table 1.
Table 1.
Consolidated virtual screening results
| Compound name | Pubchem ID | Binding affinity from docking run 1 | Binding affinity from docking run 2 (active site oriented) | Drug likeliness score |
|---|---|---|---|---|
| Chebuloside II | 44,567,150 | − 8.2 | − 8.2 | 0.66 |
| Acteoside | 5,281,800 | − 8 | − 8 | 0.51 |
| Ursolic acid | 64,945 | − 8 | − 7.8 | 0.66 |
| Luteolin | 5,280,445 | − 7.8 | − 7.8 | 0.38 |
| Arjunglucoside I | 14,658,050 | − 8.2 | − 7.7 | 0.63 |
| Corilagin | 73,568 | − 8.1 | − 7.7 | 0.64 |
| Daucosterol | 5,742,590 | − 7.9 | − 7.7 | 0.5 |
| Eucalyptin | 76,573 | − 7.6 | − 7.7 | − 0.13 |
| Arjunolic acid | 73,641 | − 8 | − 7.6 | 0.64 |
| Oleanolic acid | 10,494 | − 8.4 | − 7.6 | 0.37 |
| Arjunetin | 21,152,828 | − 8.6 | − 7.5 | 0.58 |
| Kaempferol-3-rutinoside | 5,318,767 | − 7.6 | − 7.5 | 0.9 |
| Coleon U Quinone | 9,974,772 | − 7.5 | − 7.5 | − 0.23 |
| Beta-sitosterol | 222,284 | − 8.1 | − 7.4 | 0.78 |
| Vitexin | 5,280,441 | − 7.5 | − 7.4 | 0.6 |
| Apigenin | 5,280,443 | − 7.3 | − 7.3 | 0.39 |
| Horminone | 2,751,795 | − 7.3 | − 7.3 | 0.04 |
| Cordioside | 101,915,817 | − 8.2 | − 7.2 | 0.37 |
| Pelargonidin | 440,832 | − 7.1 | − 7.2 | − 0.57 |
| 24-Ethylcholesta-5,24-dien-3beta-ol | 5,283,643 | − 7.8 | − 7.1 | 0.96 |
| Ecdysterone | 5,459,840 | − 7.5 | − 7.1 | 1.37 |
| Dehydrocostus lactone | 73,174 | − 7.1 | − 7.1 | − 1.37 |
| Ergosterol Peroxide | 5,351,516 | − 8.6 | − 7.1 | − 0.5 |
| Berberine | 2353 | − 8 | − 7 | 0.77 |
| Stigmasterol | 5,280,794 | − 7.8 | − 7 | 0.62 |
| TinosporinB | 122,206,356 | − 8.4 | − 6.9 | − 0.99 |
| Tinosporinone | 42,607,646 | − 6.8 | − 6.9 | − 0.05 |
| Moupinamide | 5,280,537 | − 6.9 | − 6.9 | − 0.23 |
| Palmatoside G | 184,515 | − 7.9 | − 6.8 | 0.12 |
| Constunolide | 5,380,858 | − 6.8 | − 6.8 | − 1.82 |
| Rutin | 5,280,805 | − 7.9 | − 6.8 | 0.91 |
| Ellagic Acid | 5,281,855 | − 7.3 | − 6.8 | − 1.11 |
| Betulin | 72,326 | − 7.8 | − 6.7 | − 0.13 |
| palmatine | 19,009 | − 7.5 | − 6.7 | 0.69 |
| Jatrorrhizine | 72,323 | − 7.4 | − 6.7 | − 0.47 |
| Tamarixetin | 5,281,699 | − 7.2 | − 6.7 | 0.29 |
| Tinocordiside | 102,504,931 | − 6.9 | − 6.7 | 0.49 |
| Sweroside | 161,036 | − 6.8 | − 6.7 | 0.63 |
| Piperolactam A | 3,081,016 | − 8.3 | − 6.7 | − 0.74 |
| Isocolumbin | 24,721,165 | − 8.4 | − 6.5 | − 0.52 |
Five compounds were then selected based on the highest binding affinities (active site oriented) and their bonds, visualized using Discovery Studio visualizer. The 2D images of bonds between the active sites and the selected compounds, namely Chebuloside, Acetoside, Corilagin, Arjunolic Acid and Beta Sitosterol are provided in Figs. 1, 2, 3, 4 and 5, respectively (depicting receptor-ligand interactions in terms of the chemical bonds as well in the form of a ribbon diagram). Chebuloside has a hydrogen bond with CYS145, ASN142, GLU166, Pi-Sigma bond with HIS41. Acetoside is connected to CYS145 by Pi-Alkyl and HIS41 by Pi-Cation bonding, GLU166, ASN142 by hydrogen bonding. Corilagin is connected to HIS41 by Pi-Pi T-shaped bonding, to GLU166 and VAL168 by hydrogen bonding, whereas the arjunolic acid has hydrogen bonding with HIS41, THR26. Beta Sitosterol has alkyl binding with CYS145, MET165. Hence, all 5 compounds have bonding with the Mpro active sites HIS41, CYS145. These results are consistent with those reported in other in-silico docking studies, wherein Acetoside [29] and Corilagin [30] have been reported to have good binding affinities with Mpro.
Fig. 1.

a Interactions between Main Protease and Chebuloside, b ribbon format—MPro and chebuloside
Fig. 2.

a Interactions between Main Protease and Acetoside, b ribbon format—MPro and acetoside
Fig. 3.
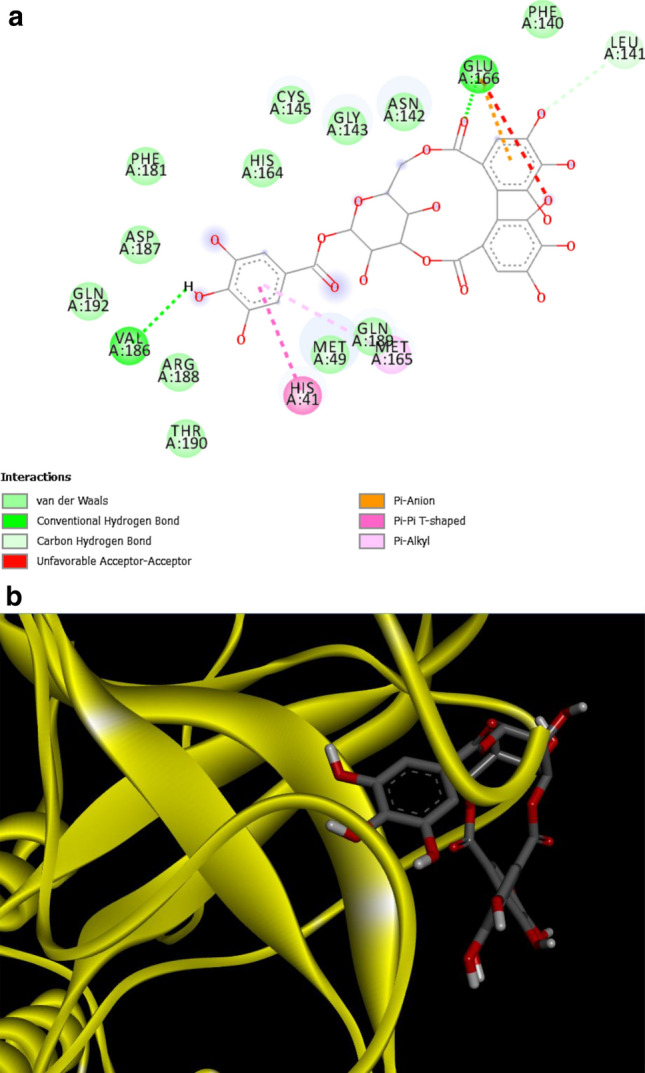
a Interactions between Main Protease and Corilagin, b ribbon format—MPro and corilagin
Fig. 4.

aInteractions between Protease and Arjunolic Acid, b ribbon format—Mpro and arjunolic acid
Fig. 5.
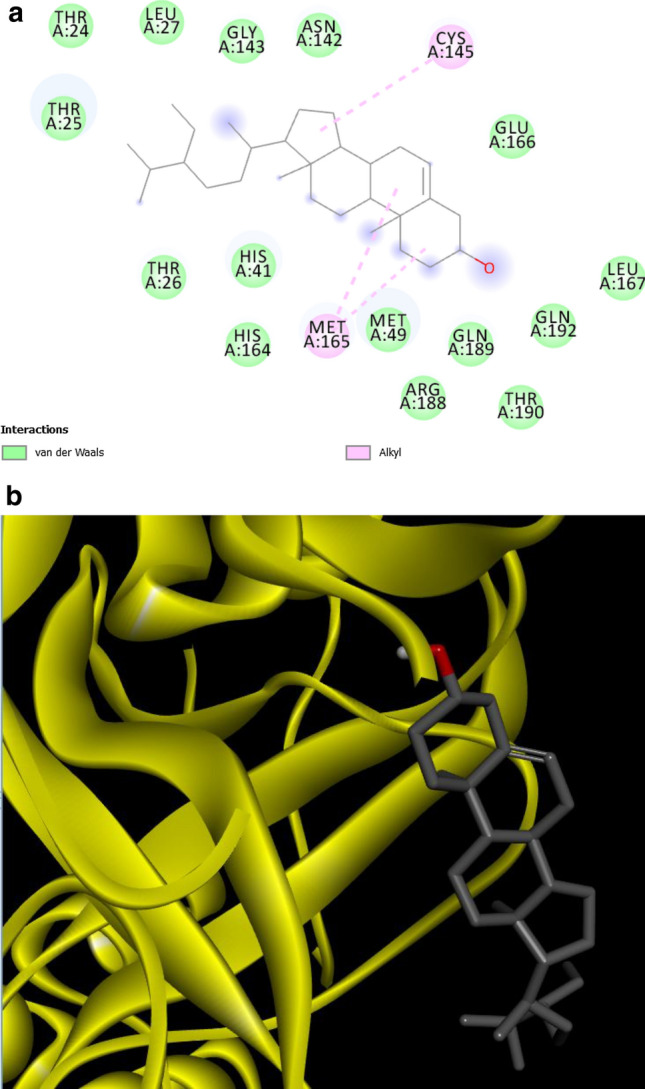
a Interactions between Protease and Beta Sitosterol, b ribbon format—MPro and beta sitosterol
The two main binding pockets were given by CASTp. The first pocket included the Mpro active sites HIS41, CYS145 around which the grid for docking run 2 was built. Based on data available in the literature, binding pocket 2, however, did not contain any active site. It is interesting to note that the results from docking run 1 showed a few compounds namely- Ergosterol Peroxide, Lupeol, Piperolactam A and Corilagin binding with Mpro in the second binding pocket strongly the with high binding affinities- −8.5, −8.4, −8.3, −8.1, respectively. Supplementary figure S1 shows bonding between Ergosterol Peroxide and Mpro at ILE200, VAL202, HIS246 (Binding pocket 2). However, due to absence of corroborating experimental data in the existing literature signifying the presence of active sites in binding pocket 2, it is not possible for these results to be considered as significant in the current study. However, conformational-related alterations in binding pocket 1, subsequent to the natural molecule interacting with the Mprotease at binding pocket 2, cannot be ruled out. Hence, our in-silico data provides a basis for more experimental work involving the targeting of binding pocket 2 by our natural molecules and evaluating the functional consequences thereafter.
The compounds Chebuloside, Acetoside, Corilagin and Arjunolic acid were chosen to check for similarity with FDA-approved drugs, Beta sitosterol was exempted as it lacked strong bonding with active sites of Mpro. The results of the SWISS similarity tool as lists of FDA-approved drugs similar to Chebuloside and Arjunolic Acid are shown in Tables S2 and S3, respectively. Spinosad and Ouabain were the drugs similar to Chebuloside with similarity scores of 0.269 and 0.227, respectively. In the case of Ouabain, this significant in silico finding is consistent with certain reports, wherein Ouabain has been shown to treat COVID-19 positive patients with a cardiovascular disease. Hydrocortisone, Dinoprost Tromethamine was similar to Arjunolic acid with scores 0.539 and 0.523, respectively. Again, this similarity is significant since hydrocortisone has been used earlier for the treatment of SARS-CoV1 and MERS. The tool showed that there are no FDA-approved drugs similar to Acetoside and Corilagin.
Conclusion
This study, involving a preliminary in silico screening of known natural compounds from traditional medicine, has revealed that certain compounds such as Chebuloside, Acetoside, Corilagin and Arjunolic acid may serve as possible drug development leads against the SARS-CoV-2. The lists of FDA-approved drugs similar to Chebuloside and Arjunolic acid obtained in this study may help in drug repurposing against SARS-CoV-2, especially in individuals with certain co-morbid conditions. Further validation of our preliminary in silico findings with advanced computational tools, supplemented by in vitro and in vivo experimental data would be pivotal in increasing treatment options and fastening the drug development process against SARS-CoV-2.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary file1. Figure S1 Interactions Protease & Ergosterol Peroxide (PNG 86 kb)
Acknowledgements
The authors acknowledge the management of VIT, Vellore for their constant encouragement and support and for creating the necessary scientific ambience that made this in-silico study possible.
Declarations
Conflict of interest
The authors declare they have no conflict of interest.
Footnotes
Significance statement: The virtual screening performed examines the possibility of using compounds from traditional medicinal plants as possible drug leads for COVID-19.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R (2021) Features, evaluation and treatment coronavirus (COVID-19). https://www.ncbi.nlm.nih.gov/books/NBK554776/ [PubMed]
- 2.Kumar Poonam S, Rathi B. Coronavirus disease COVID-19: a new threat to public health. Curr Top Med Chem. 2020 doi: 10.2174/1568026620999200305144319. [DOI] [PubMed] [Google Scholar]
- 3.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020 doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019 doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan JFW, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020 doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasad A, Muthamilarasan M, Prasad M. Synergistic antiviral effects against SARS-CoV-2 by plant-based molecules. Plant Cell Rep. 2020 doi: 10.1007/s00299-020-02560-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh S, Saha S. Tinospora cordifolia: one plant, many roles. Anc Sci Life. 2012 doi: 10.4103/0257-7941.107344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhujbal, S. M. . Kewatkar, L. More, and M. Patil, “Antioxidant Effects of Roots of Clerodendrum serratum Linn,” Pharmacognosy Res., vol. 1, no. 5, p. 294, 2009, Accessed: Nov. 23, 2020. [Online]. Available: http://www.phcogres.com/article.asp?issn=0974-8490;year=2009;volume=1;issue=5;spage=294;epage=298;aulast=Bhujbal
- 10.Reddy BS, Rao NR, Vijeepallam K, Pandy V. Phytochemical, pharmacological and biological profiles of tragia species (Family: Euphorbiaceae) Afr J Tradit Complement Altern Med. 2017;14(3):105–112. doi: 10.21010/ajtcam.v14i3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali Z, Khan I. Chemical constituents of terminalia chebula. Planta Med. 2009 doi: 10.1055/s-2009-1216479. [DOI] [Google Scholar]
- 12.Sutradhar RK, Rahman AKMM, Ahmad MU, Bachar SC. Bioactive flavones of Sida cordifolia. Phytochem Lett. 2008 doi: 10.1016/j.phytol.2008.09.004. [DOI] [Google Scholar]
- 13.Rose K, Wan C, Thomas A, Seeram NP, Ma H. Phenolic compounds isolated and identified from amla (Phyllanthus emblica) juice powder and their antioxidant and neuroprotective activities. Nat Prod Commun. 2018 doi: 10.1177/1934578x1801301019. [DOI] [Google Scholar]
- 14.Kumar V, Van Staden J. A review of Swertia chirayita (Gentianaceae) as a traditional medicinal plant. Front Pharmacol. 2016 doi: 10.3389/fphar.2015.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corrêa GM, de Alcântara AFC. Chemical constituents and biological activities of species of justicia -a review. Braz J Pharmacogn. 2011 doi: 10.1590/S0102-695X2011005000196. [DOI] [Google Scholar]
- 16.Chao WW, Kuo YH, Lin BIF. Anti-inflammatory activity of new compounds from andrographis paniculata by nf-κb transactivation inhibition. J Agric Food Chem. 2010 doi: 10.1021/jf903629j. [DOI] [PubMed] [Google Scholar]
- 17.Al-Snafi A. Alhagi maurorum as a potential medicinal herb: an overview. Int J Pharm Rev Res. 2015;5:130–136. [Google Scholar]
- 18.Yulianto W, Andarwulan N, Giriwono PE, Pamungkas J. Bioactive compounds from torbangun [Plectranthus amboinicus (Lour.) Spreng] chloroform fraction induce apoptosis in breast cancer (MCF-7 Cells) in vitro. Maj Obat Tradis. 2017;22(1):37. doi: 10.22146/tradmedj.24312. [DOI] [Google Scholar]
- 19.Choudhary N, Singh V. A census of P. longum’s phytochemicals and their network pharmacological evaluation for identifying novel drug-like molecules against various diseases, with a special focus on neurological disorders. PLoS ONE. 2018 doi: 10.1371/journal.pone.0191006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pandey MM, Rastogi S, Rawat AKS. Saussurea costus: botanical, chemical and pharmacological review of an ayurvedic medicinal plant. J Ethnopharmacol. 2007 doi: 10.1016/j.jep.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 21.Ahn JH, et al. 6-Acetoxy cyperene, a patchoulane-type sesquiterpene isolated from cyperus rotundus rhizomes induces caspase-dependent apoptosis in human ovarian cancer cells. Phyther Res. 2015 doi: 10.1002/ptr.5385. [DOI] [PubMed] [Google Scholar]
- 22.Mao QQ, et al. Bioactive compounds and bioactivities of ginger (zingiber officinale roscoe) Foods. 2019 doi: 10.3390/foods8060185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nisar S, Lanza Júnior U, Waqar Azeem M (2019) Isolation of bioactive components of Carom: A Review. Accessed: Nov. 23, 2020. [Online]. Available: www.iscientific.org/Journal.html
- 24.Cortés-Rojas DF, de Souza CRF, Oliveira WP (2014) Clove (Syzygium aromaticum): A precious spice. Asian Pac J Trop Biomed 4(2):90–96. Asian Pacific Tropical Biomedicine Press. 10.1016/S2221-1691(14)60215-X [DOI] [PMC free article] [PubMed]
- 25.Usmani A, Khushtar M, Arif M, Siddiqui MA, Sing SP, Mujahid M. Pharmacognostic and phytopharmacology study of Anacyclus pyrethrum: an insight. J Appl Pharm Sci. 2016 doi: 10.7324/JAPS.2016.60325. [DOI] [Google Scholar]
- 26.Zhang L et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science (80- ). 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed]
- 27.Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018 doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar Y, Singh H, Patel CN. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health. 2020 doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincent S, Arokiyaraj S, Saravanan M, Dhanraj M. Molecular docking studies on the anti-viral effects of compounds from kabasura kudineer on SARS-CoV-2 3CLpro. Front Mol Biosci. 2020;7:613401. doi: 10.3389/fmolb.2020.613401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos-Filho OA. Identification of potential inhibitors of severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) main protease from non-natural and natural sources: a molecular docking study. J Braz Chem Soc. 2020;31(12):2638–2643. doi: 10.21577/0103-5053.20200139. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1. Figure S1 Interactions Protease & Ergosterol Peroxide (PNG 86 kb)