

Abstract
The MED13L-related intellectual disability or MRFACD syndrome (Mental retardation and distinctive facial features with or without cardiac defects; MIM # 616789) is one of the most common forms of syndromic intellectual disability with about a hundred cases reported so far. Affected individuals share overlapping features comprising intellectual disability, hypotonia, motor delay, remarkable speech delay, and a recognizable facial gestalt. De novo disruption of the MED13L gene by deletions, duplications, or sequence variants has been identified as deleterious. Siblings affected by intragenic deletion transmitted from a mosaic parent have been reported once in the literature. We now present the first case of paternal germinal mosaicism for a missense MED13L variant causing MRFACD syndrome in one of the father's children and being the likely cause of intellectual disability and facial dysmorphism in the other. As part of the Mediator complex, the MED proteins have an essential role in regulating transcription. Thirty-two subunits of the Mediator complex genes have been linked to congenital malformations that are now acknowledged as transcriptomopathies. The MRFACD syndrome has been suggested to represent a recognizable phenotype.
Keywords: abnormality of the maxilla; delayed gross motor development; intellectual disability, moderate; secundum atrial septal defect
INTRODUCTION
Disruption of the Mediator complex subunit 13-like gene (MED13L) is increasingly recognized as the cause of an intellectual disability syndrome with associated facial dysmorphism (Mental retardation and distinctive facial features with or without cardiac defects [MRFACD]; MIM # 616789) (Gordon et al. 2018). The gene, initially termed PROSIT240/THRAP2 (Protein Similar to TRAP240/Thyroid hormone receptor-associated protein 2) was first linked to intellectual disability and congenital heart disease in 2003 by Muncke et al. (2003), but congenital heart disease was later shown not to be a consistent feature of the MED13L-related syndrome (Adegbola et al. 2015). MED13L encodes a component of the Mediator complex, which regulates transcription via interactions between upstream transcriptional factors and the basal RNA polymerase II initiation machinery (Malik and Roeder 2010), and, as such, plays a role in the control of cell growth, repression of cell cycle target genes, and cell cycle inhibition (Angus and Nevins 2012). Thirty-two subunits of the Mediator complex have been linked to congenital malformations (Asadollahi et al. 2017). According to a recently proposed concept, the class of disorders that reflect a global disturbance in transcriptional regulation may be referred to as “transcriptomopathies” (Yuan et al. 2015; Caro-Llopis et al. 2016).
Both small-scale (missense and protein truncating mutations) and large-scale (intragenic copy-number variations) alterations can cause MRFACD syndrome (Smol et al. 2018). Tørring et al. reviewed the clinical presentation and genotype of 69 patients in 2019 (Tørring et al. 2019). They disclosed that all cases arose de novo; large deletions or large intragenic duplications of the MED13L were responsible for approximately one-third of the cases (possibly overrepresented because of array comparative genomic hybridization [CGH] being the first-tier diagnostic tool in the investigation of intellectual disability), whereas small sequence variants (predominantly frameshift and nonsense mutations and less frequently missense variants) account for the rest of the cases. The latter seem to cluster in exons 15–17 and 25–31 and typically result in a more severe phenotype. One familial case with variable, mild phenotype was described as having a 3-Mb duplication encompassing the MED13L gene by Adegbola et al. (2015).
Yamamoto et al. reported the case of siblings having a large deletion of the MED13L gene encompassing exons 3–14 inherited from their mother, who was mosaic for the deletion (Yamamoto et al. 2017).
Motor delay, intellectual disability, and speech delay are consistent features in the MED13L–intellectual disability syndrome; a congenital heart defect may or may not accompany the symptoms; and a recognizable facial dysmorphism consisting of macrostomia, macroglossia, an open mouth appearance, broad or depressed nasal root, and ear anomalies are frequent findings (Tørring et al. 2019).
We now present the first case of paternal germinal mosaicism for a missense MED13L variant causing MRFACD syndrome in one of the father's children and being the likely cause of intellectual disability and facial dysmorphism in his other child from a previous marriage.
RESULTS
Clinical Presentation and Family History
The proband was a teenage patient born from nonconsanguineous healthy parents. He was the first child of the mother and second child of his father (the first was from a previous spouse). He was born to term with a weight of 25–50 percentile, his length and head circumference were not registered, his Apgar score was 7/9, and he needed short-term oxygen support in the perinatal period. Muscular hypotonia and dysmorphic facies were noted right after birth. A congenital cardiac defect was diagnosed consisting of transposition of the pulmonary veins and secundum-type atrial septal defect. Brain magnetic resonance imaging (MRI) was normal. Focal bilateral abnormal waves on electroencephalogram (EEG) necessitated the initiation of valproic acid. In his early teens he could speak a few words, and sphincter control was developing.
His multiple congenital malformations and developmental delay suggested a genetic origin of symptoms.
His longitudinal growth and weight were normal (height 10 pc, weight 10–25 pc, head circumference 10 percentile). Hypotrophy of the skeletal muscles was described. The facial dysmorphism was dominated by a long face, maxillary hypoplasia, prominent tongue, and mandibular prognathism. A more detailed phenotypic description is available via contact with the corresponding author. On the hands, palmar eminences were flattened and the fingers were slender, whereas on the lower limbs there were prominent calcanei, bilateral pes planus and hallux valgus, and overriding second and fourth toes. He was a cooperative, friendly child with moderate intellectual deficit, producing sounds during examination, understanding simple commands but without meaningful speech. A strong resemblance to his half-sibling's facial morphological features and intellectual status was seen, with special regards to maxillary hypoplasia and mandibular prognathism, open mouth, protruding tongue, large and prominent ears, and very little expressive speech.
His half-sibling, born from the father's previous partner, was an adult individual affected by intellectual disability and facial dysmorphism. This individual was born after term with normal parameters (weight at 25 pc, length at 50 pc, occipital frontal circumference [OFC] at 50 pc) as the first child of healthy, nonrelated, Caucasian parents. He had bilateral pes equinovarus, spastic limbs, cryptorchidism, kyphoscoliotic spine due to axial hypotonia, strabismus, and epilepsy. He was nonverbal, and his IQ was at the bottom border of moderate intellectual disability. Fragile X syndrome was suspected, and an FMR1 Southern blot was performed. Results of these tests were not available to us.
Genomic Analyses
In early childhood the proband underwent G-banding chromosome analysis and an FMR1 gene test, both of which showed normal results. His array CGH did not reveal a pathological copy-number variation.
Whole-exome sequencing of the proband revealed two variants in two genes associated with intellectual disability: a heterozygous c.5695G > A, p.(Gly1899Arg) missense variant in the MED13L gene (NM_015335.4) and a hemizygous c.638 + 2del (r.spl) splicing variant in the TMLHE gene (NM_018196.3) (Table 1). Pathogenic mutations in the MED13L gene have been described before as causative for mental retardation and distinctive facial features with or without cardiac defects (MIM # 616789).
Table 1.
Variant table
| Gene | Chromosome (GRCh37) | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP ID | Genotype |
|---|---|---|---|---|---|---|---|
| MED13L | Chr 12: g.116413012C > T | c.5695G > A | p.Gly1899Arg | Substitution | Missense | n/a | Hetero zygous |
| TMLHE | Chr X: g.154743645del | c.638 + 2del | p.? | Deletion | Exon skipping | n/a | Hemi zygous |
The heterozygous MED13L variant detected in our patient leads to the substitution of a strongly conserved nucleotide and amino acid residue, which is located in the Mediator complex, subunit Med13 domain of the MED13L protein. This specific variant has been reported before by Caro-Llopis et al. (2016) and McRae et al. (2017). It is not present in the gnomAD and ClinVar databases, and computational predictional tools unanimously support a deleterious effect on the MED13L gene. According to the American College of Medical Genetics and Genomics (ACMG) 2015 guideline, the variant can be classified as likely pathogenic (Richards et al. 2015) (evidences: PM2, PP3, PP5, PP1).
Pathogenic mutations in the TMLHE gene have been described before as causative for autism, susceptibility to X-linked 6 (MIM # 300872). The c.638 + 2del hemizygous variant concerns a deletion of a single nucleotide in the splice donor site of intron 4 of TMLHE. Prediction tools predict that this deletion will disrupt the functionality of this splice donor site. The consequence of this change is not predictable, but a skip of exon 4 is predicted to be likely. The variant has not been reported before in the literature, gnomAD database, and ClinVar database. It is classified as likely pathogenic according to the ACMG guideline (evidences: PVS1, PM2).
Neither mother consented to their own testing for segregation analysis of the mutations nor did the mother of the adult individual consent to her child's testing. We therefore only knew the two mutations of the proband and that both were absent from the father's DNA derived from peripheral blood. TMLHE is an X-linked gene, so inheritance from the father was unlikely. Seeing the similarity between the two affected, related individuals and theoretically ruling out the rare coincidence that two unrelated healthy females previously married to the healthy father would transmit mutations in the same gene or in a different gene with very similar phenotypic consequence causing X-linked intellectual disability, we informed the father and his new spouse about the limitations of our genetic counseling and informed them about options regarding insemination with donor sperm. But before undergoing assisted reproduction, we offered sperm analysis for the MED13L mutation c.5695G > A, which the father accepted. He produced a sample with high spermium concentration (50 million/mL), from which DNA was isolated, and targeted mutation testing was performed. The MED13L variant could be identified unequivocally in the sperm cells, confirming germinal mosaicism of the father for this mutation (Fig. 1). We identified the presence of the MED13L mutation in ∼30%–50% of the sperm cells (predicted on the basis of the peak heights on the sequencing electropherograms) accountable for a high recurrence risk in the couple's future pregnancies and advised on either preimplantation genetic diagnosis or insemination with donor sperm cells. Although we could not confirm that the older child's condition is caused by the MED13L c.5695G > A mutation, we strongly believe he has the same MED13L genotype responsible for the similar phenotype.
Figure 1.
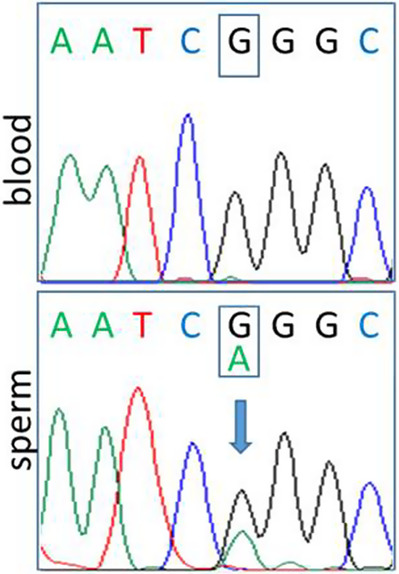
Sanger sequencing chromatograms of the father showing the presence of the c.5695G > A, p.(Gly1899Arg) variant of the MED13L gene only in the sperm cells.
DISCUSSION
MRFACD, MED13L-related intellectual disability syndrome, a recently recognized but well-described congenital malformation, is an intellectual disability syndrome with detailed clinical descriptions available for only 75 reported individuals (Muncke et al. 2003; Asadollahi et al. 2013, 2017; Redin et al. 2014; Utami et al. 2014; Adegbola et al. 2015; Cafiero et al. 2015; van Haelst et al. 2015; Caro-Llopis et al. 2016; Yamamoto et al. 2017; Gordon et al. 2018; Jiménez-Romero et al. 2018; Smol et al. 2018; Tørring et al. 2019; Yi et al. 2020; Dawidziuk et al. 2021). A further 14 cases have been identified in the frames of large high-throughput sequencing studies investigating the genetic background of intellectual disability or congenital heart defect (Musante et al. 2004; Najmabadi et al. 2011; Iossifov et al. 2012; Hamdan et al. 2014; Iglesias et al. 2014; Codina-Solà et al. 2015; Martínez et al. 2017; Mullegama et al. 2017; Aoi et al. 2019; Ji et al. 2020; Sabo et al. 2020; Tian et al. 2021). In 2015, Adegbola et al. (2015) redefined the MED13L syndrome as a novel, recognizable neurobehavioral syndrome and concluded that cardiac defect is not an invariable feature. Since then, several literature reviews and case reports have been published, and the most prominent features were found to be intellectual disability, hypotonia, motor delay, often profound language impairment, and facial dysmorphism consisting of an open-mouth appearance, macrostomia, macroglossia, large or low-set ears, and anomalies of the palpebral fissures (Adegbola et al. 2015; Tørring et al. 2019). Friendly behavior was described by Cafiero et al. (2015); other authors, however, report on challenging behavior including hyperactivity, aggression, and autistic traits (Adegbola et al. 2015; Smol et al. 2018). Deep language impairment has been reported in affected individuals with or without a heart defect and was explained with MED13L being part of a functional network in the brain important for language development (Jiménez-Romero et al. 2018). As for the molecular background, almost all cases arose de novo; deletions, duplications, and sequence variants of the MED13L gene have been reported to be deleterious (Tørring et al. 2019). Missense mutations tend to cause a more severe phenotype than other types of variants (Asadollahi et al. 2017; Smol et al. 2018), whereas copy-number gains result in a milder phenotype (Asadollahi et al. 2013). Because of the MED proteins’ role in transcriptional regulation, the “MEDs syndromes” have been acknowledged as transcriptomopathies (Yuan et al. 2015; Caro-Llopis et al. 2016).
Paternal age has been shown to be the primary factor influencing the number of de novo mutations in a child and is therefore expected to be a risk factor for pathogenic de novo mutations (Kong et al. 2012; Rahbari et al. 2016). In our patient, the father was in his late teens when his first child was born, in his 30s when his second child was born, and in his 40s at the time of seeking medical help prior to having more children because of the apparent recurrence of intellectual disability in his offspring. We could only test his second child, in whom whole-exome sequencing revealed two variants in two genes associated with intellectual disability: a heterozygous mutation c.5695G > A, p.(Gly1899Arg) in the MED13L gene and a c.638 + 2del (r.spl) hemizygous variant in the TMLHE gene not yet described in the literature but predicted to be damaging by in silico tools. The latter mutation may have influenced the clinical picture in our patient by shifting his mental status toward autism spectrum disorder, but the MED13L mutation dominated his cognitive, behavioral, and morphological status so that we could not separate the possible effect of the TMLHE variant or identify key features that would not fit in the symptoms of MED13L-related intellectual disability. The missense mutation in MED13L in our patient had been described in only two patients before, published by Caro-Llopis et al. (2016) (Patient 2) and McRae et al. (2017), but from these, detailed clinical data are accessible for the one reported by Caro-Llopis (Caro-Llopis et al. 2016; McRae et al. 2017). This patient had low body weight, psychomotor delay, behavioral problems including aggression, autism spectrum disorder, brain MRI abnormalities such as ventriculomegaly and megacysterna magna, normal myelination, cardiac anomaly (patent ductus arteriosus), hearing impairment, and dysmorphic features consisting of low-set ears, hypertelorism, antimongoloid palpebral fissures, strabismus, ptosis, epicanthal folds, and wide and depressed nasal root. Our patient shared overlapping features, but instead of aggressive behavior, he had a friendly, sociable personality, a complex cardiac defect with transposition of the pulmonary veins and atrial septal defect, and normal brain MRI, but epilepsy (Table 2).
Table 2.
Comparison of the clinical features of our patient and the patient described by Caro-Llopis et al. having MED13L c.5695G > A, p.(Gly1899Arg) mutation based on the clinical table created by Tørring et al. (2019)
| Clinical feature | Caro-Llopis et al. (2016) | Our patient |
|---|---|---|
| Intellectual disability | Yes | Yes |
| Speech delay | Yes | Yes |
| Anomalies of the hands and feet | NA | Yes; fattened eminences, overriding toes, pedes plani, halluces valgi |
| Hypotonia | Yes | Yes |
| Ophthalmological anomalies | Yes; strabismus, ptosis | No |
| Anomalies on cerebral MRI | Yes; ventriculomegaly, mega cisterna magna | No |
| Congenital heart defect Complex Simple (PFO/PDA) |
Yes; PDA | Yes; ASD, transposition of the pulmonary veins |
| Autistic features | Yes | NA |
| Bulbous nasal tip | NA | No; pointed |
| Open mouth appearance | Yes | Yes |
| Depressed/broad nasal root | Yes; both | No; rather high nasal bridge |
| Ear anomalies | Yes; low-set | Yes; prominent |
| Macrostomia | NA | Yes |
| Upslanting palpebral fissures | No; downslanting | No; downslanting |
| Macroglossia | NA | Yes |
| Bitemporal narrowing | NA | No; short forehead |
| Brachycephaly | NA | NA |
| Horizontal eyebrows | NA | NA |
| Others | Unilateral hearing loss, atopic dermatitis | Epilepsy, broad-based gait, crowded teeth, maxillary hypoplasia, large frontal incisors, hypotrophic muscles |
(NA) Not assessed, (MRI) magnetic resonance imaging, (PFO) patent foramen ovale, (PDA) patent ductus arteriosus, (ASD) atrial septal defect.
Germinal mosaicism resulting in the recurrence of MRFACD syndrome in a carrier mother's children have been reported only once in the literature. In the affected family there was a large intragenic deletion of 12 exons responsible for the disruption of the MED13L gene (Yamamoto et al. 2017). We presented a case of paternal germinal mosaicism causing MRFACD syndrome in one of the father's children and being the likely cause of intellectual disability and facial dysmorphism in the father's child from a previous marriage. We also presented our diagnostic approach that circumvented the difficulties of inexecutable segregation analysis and provided a basis for the couple's knowledgeable reproductive decision.
METHODS
Genetic tests were done for a diagnostic purpose after obtaining written informed consent.
G-banding chromosome analysis was performed according to standard procedures. FMR1 CGG repeat expansion was tested using triplet repeat primed polymerase chain reaction (PCR) (AmplideX FMR1 PCR kit, Asuragen). Array CGH was performed using Affymetrix CytoScan 750K and Affymetrix Chromosome Analysis Suite (ChAS) v2.0 Software (Affymetrix, Thermo Fisher Scientific).
Exome enrichment (Agilent SureSelectXT Human All Exon 50Mb) and exome sequencing (Illumina HiSeq) were done at BGI Europe. After read alignment (BWA), variant calling (GATK), and copy-number variant calling (by CoNIFER), variant annotation and prioritizing was done by the Department of Human Genetics, Radboud UMC, using a strategy developed in-house. This pipeline also filters the data for genes defined in the intellectual disability gene panel (DG-2.13). The panel contains 1116 genes that were sequenced with an average target coverage of 100.3242×. The whole-exome sequencing metrics are provided in Table 3. Confirmation of reported variants by Sanger sequencing was only performed for low-quality variant calls (GATK score < 500). Variants were checked in the databases gnomAD (http://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and HGMD (www.hgmd.cf.ac.uk). In silico tools (MutationTaster, SIFT, DANN, Revel, and SpliceAl) were used to predict the effects of the variants. The variants were classified according to the ACMG 2015 guideline (Richards et al. 2015).
Table 3.
Whole-exome sequencing metrics
| Total number of reads | Mapped reads | On-target reads | Mean coverage at target regions |
|---|---|---|---|
| 64,892,925 | 64,888,420 | 51,399,652 | 100.3242 |
To investigate the presence of the MED13L variant in the father, targeted bidirectional Sanger sequencing of exon 25 was performed on the paternal DNA samples extracted from whole blood and sperm cells.
A semen sample was obtained from the father to study germinal mosaicism. After 30 min of liquefaction at 37°C, routine semen analysis was performed. The main parameters were the following: volume 1.9 mL, pH 7.8, sperm concentration 51 million/mL, and total motility 85%. Sperm cells were isolated from the semen sample using the swim-up technique. The sample was diluted with preheated (37°C) Sperm Preparation Medium (Origio), and centrifugated for 10 min at 600g. The pellet was layered with Sequential Fert solution (Origio). The motile sperm cells were allowed to swim up for 1.5 h at 37°C in a 5% CO2 thermostat. The final sperm concentration was 40 million/mL in the supernatant.
DNA was extracted from sperm cells using a spin column–based method (QIAamp DNA Mini kit, QIAGEN) with a slight modification: dithiothreitol was added to ATL buffer in the first step of the extraction to enhance the breakdown of the nuclear membrane.
ADDITIONAL INFORMATION
Data Deposition and Access
Sequencing data are not publicly available, because parental consent was obtained for diagnostic purposes only. The variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers VCV001240004.1 and SCV001976436.1.
Ethics Statement
Informed consent from the father for genetic analyses was obtained for genetic testing and for publication; the father was allowed to read the manuscript and provide his feedback on it. Because maternal consents could not be obtained, some data regarding phenotypic descriptions appear restricted; these are available after direct contact with the corresponding author. All tests were done for a diagnostic purpose. At all times, testing and examination procedures fulfilled the criteria of the 2008 Hungarian Law on Human Genetics and the World Medical Association (WMA) Declaration of Helsinki.
Author Contributions
K.S. performed the clinical genetic examination and genetic counseling of the family, reviewed the literature, and prepared the manuscript; R.P. analyzed and interpreted the exome sequencing data and revised the manuscript; A.U. performed the molecular cytogenetic testing and evaluated it; B.B. analyzed and interpreted the sperm analyses data and drafted and revised the manuscript; A.M. had a role in sperm analyses; and I.B. analyzed data and revised the manuscript.
Funding
Part of the diagnostic procedure was supported by the Ministry of National Economy, Hungary (GINOP-2.3.2-15-2016-00039, Nr of ethical certificate: 28676-4/2017EÜIG).
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Adegbola A, Musante L, Callewaert B, Maciel P, Hu H, Isidor B, Picker-Minh S, Le Caignec C, Chiaie BD, Vanakker O, et al. 2015. Redefining the MED13L syndrome. Eur J Hum Genet 23: 1308–1317. 10.1038/ejhg.2015.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus SP, Nevins JR. 2012. A role for Mediator complex subunit MED13L in Rb/E2F-induced growth arrest. Oncogene 31: 4709–4717. 10.1038/onc.2011.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi H, Mizuguchi T, Ceroni JR, Kim VEH, Furquim I, Honjo RS, Iwaki T, Suzuki T, Sekiguchi F, Uchiyama Y, et al. 2019. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J Hum Genet 64: 967–978. 10.1038/s10038-019-0643-z [DOI] [PubMed] [Google Scholar]
- Asadollahi R, Oneda B, Sheth F, Azzarello-Burri S, Baldinger R, Joset P, Latal B, Knirsch W, Desai S, Baumer A, et al. 2013. Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability. Eur J Hum Genet 21: 1100–1104. 10.1038/ejhg.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadollahi R, Zweier M, Gogoll L, Schiffmann R, Sticht H, Steindl K, Rauch A. 2017. Genotype–phenotype evaluation of MED13L defects in the light of a novel truncating and a recurrent missense mutation. Eur J Med Genet 60: 451–464. 10.1016/j.ejmg.2017.06.004 [DOI] [PubMed] [Google Scholar]
- Cafiero C, Marangi G, Orteschi D, Ali M, Asaro A, Ponzi E, Moncada A, Ricciardi S, Murdolo M, Mancano G, et al. 2015. Novel de novo heterozygous loss-of-function variants in MED13L and further delineation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet 23: 1499–1504. 10.1038/ejhg.2015.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Llopis A, Rosello M, Orellana C, Oltra S, Monfort S, Mayo S, Martinez F. 2016. De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy? Pediatr Res 80: 809–815. 10.1038/pr.2016.162 [DOI] [PubMed] [Google Scholar]
- Codina-Solà M, Rodríguez-Santiago B, Homs A, Santoyo J, Rigau M, Aznar-Laín G, Del Campo M, Gener B, Gabau E, Botella MP, et al. 2015. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol Autism 6: 21. 10.1186/s13229-015-0017-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawidziuk M, Kutkowska-Kaźmierczak A, Gawliński P, Wiszniewski W, Gos M, Stawiński P, Rydzanicz M, Kosińska J, Własienko P, Malinowska Kordowska O, et al. 2021. The MED13L haploinsufficiency syndrome associated with de novo nonsense variant (P.GLN1981*). J Mother Child 24: 32–36. 10.34763/jmotherandchild.20202403.2021.d-20-00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CT, Chopra M, Oufadem M, Alibeu O, Bras M, Boddaert N, Bole-Feysot C, Nitschké P, Abadie V, Lyonnet S, et al. 2018. MED13L loss-of-function variants in two patients with syndromic Pierre Robin sequence. Am J Med Genet A 176: 181–186. 10.1002/ajmg.a.38536 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, Massicotte C, Ambalavanan A, Spiegelman D, Diallo O, et al. 2014. De novo mutations in moderate or severe intellectual disability. PLoS Genet 10: e1004772. 10.1371/journal.pgen.1004772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, Sisson R, Egan C, Chung WK. 2014. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med 16: 922–931. 10.1038/gim.2014.58 [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y, Narzisi G, Leotta A, et al. 2012. De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285–299. 10.1016/j.neuron.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W, Ferdman D, Copel J, Scheinost D, Shabanova V, Brueckner M, Khokha MK, Ment LR. 2020. De novo damaging variants associated with congenital heart diseases contribute to the connectome. Sci. Rep 10: 7046. 10.1038/s41598-020-63928-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Romero S, Carrasco-Salas P, Benítez-Burraco A. 2018. Language and cognitive impairment associated with a novel p.Cys63Arg change in the MED13L transcriptional regulator. Mol Syndromol 9: 83–91. 10.1159/000485638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Roeder RG. 2010. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 11: 761–772. 10.1038/nrg2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, et al. 2017. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542: 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama SV, Jensik P, Li C, Dorrani N, Kantarci S, Blumberg B, Grody WW, Strom SP. 2017. Coupling clinical exome sequencing with functional characterization studies to diagnose a patient with familial Mediterranean fever and MED13L haploinsufficiency syndromes. Clin Case Rep 5: 833–840. 10.1002/ccr3.904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncke N, Jung C, Rüdiger H, Ulmer H, Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schön K, et al. 2003. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (Transposition of the Great Arteries). Circulation 108: 2843–2850. 10.1161/01.CIR.0000103684.77636.CD [DOI] [PubMed] [Google Scholar]
- Musante L, Bartsch O, Ropers HH, Kalscheuer VM. 2004. cDNA cloning and characterization of the human THRAP2 gene which maps to Chromosome 12q24, and its mouse ortholog Thrap2. Gene 332: 119–127. 10.1016/j.gene.2004.02.044 [DOI] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. 2011. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478: 57–63. 10.1038/nature10423 [DOI] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. 2016. Timing, rates and spectra of human germline mutation. Nat Genet 48: 126–133. 10.1038/ng.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A, Masurel-Paulet A, Willems M, Lesca G, El-Chehadeh S, et al. 2014. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 51: 724–736. 10.1136/jmedgenet-2014-102554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo A, Murdock D, Dugan S, Meng Q, Gingras MC, Hu J, Muzny D, Gibbs R. 2020. Community-based recruitment and exome sequencing indicates high diagnostic yield in adults with intellectual disability. Mol Genet Genomic Med 8: e1439. 10.1002/mgg3.1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smol T, Petit F, Piton A, Keren B, Sanlaville D, Afenjar A, Baker S, Bedoukian EC, Bhoj EJ, Bonneau D, et al. 2018. MED13L-related intellectual disability: involvement of missense variants and delineation of the phenotype. Neurogenetics 19: 93–103. 10.1007/s10048-018-0541-0 [DOI] [PubMed] [Google Scholar]
- Tian T, Cao X, Chen Y, Jin L, Li Z, Han X, Lin Y, Wlodarczyk BJ, Finnell RH, Yuan Z, et al. 2021. Somatic and de novo germline variants of MEDs in human neural tube defects. Front Cell Dev Biol 9: 641831. 10.3389/fcell.2021.641831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tørring PM, Larsen MJ, Brasch-Andersen C, Krogh LN, Kibæk M, Laulund L, Illum N, Dunkhase-Heinl U, Wiesener A, Popp B, et al. 2019. Is MED13L-related intellectual disability a recognizable syndrome? Eur J Med Genet 62: 129–136. 10.1016/j.ejmg.2018.06.014 [DOI] [PubMed] [Google Scholar]
- Utami KH, Winata CL, Hillmer AM, Aksoy I, Long HT, Liany H, Chew EGY, Mathavan S, Tay SKH, Korzh V, et al. 2014. Impaired development of neural-crest cell-derived organs and intellectual disability caused by MED13L haploinsufficiency. Hum Mutat 35: 1311–1320. 10.1002/humu.22636 [DOI] [PubMed] [Google Scholar]
- van Haelst MM, Monroe GR, Duran K, van Binsbergen E, Breur JM, Giltay JC, van Haaften G. 2015. Further confirmation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet 23: 135–138. 10.1038/ejhg.2014.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Shimojima K, Ondo Y, Shimakawa S, Okamoto N. 2017. MED13L haploinsufficiency syndrome: a de novo frameshift and recurrent intragenic deletions due to parental mosaicism. Am J Med Genet A 173: 1264–1269. 10.1002/ajmg.a.38168 [DOI] [PubMed] [Google Scholar]
- Yi Z, Zhang Y, Song Z, Pan H, Yang C, Li F, Xue J, Qu Z. 2020. Report of a de novo c.2605C>T (p.Pro869Ser) change in the MED13L gene and review of the literature for MED13L-related intellectual disability. Ital. J. Pediatr 46: 95. 10.1186/s13052-020-00847-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, et al. 2015. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest 125: 636–651. 10.1172/JCI77435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data are not publicly available, because parental consent was obtained for diagnostic purposes only. The variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers VCV001240004.1 and SCV001976436.1.