
Fig. 2 |. MetaRibo-Seq bioinformatic workflow.
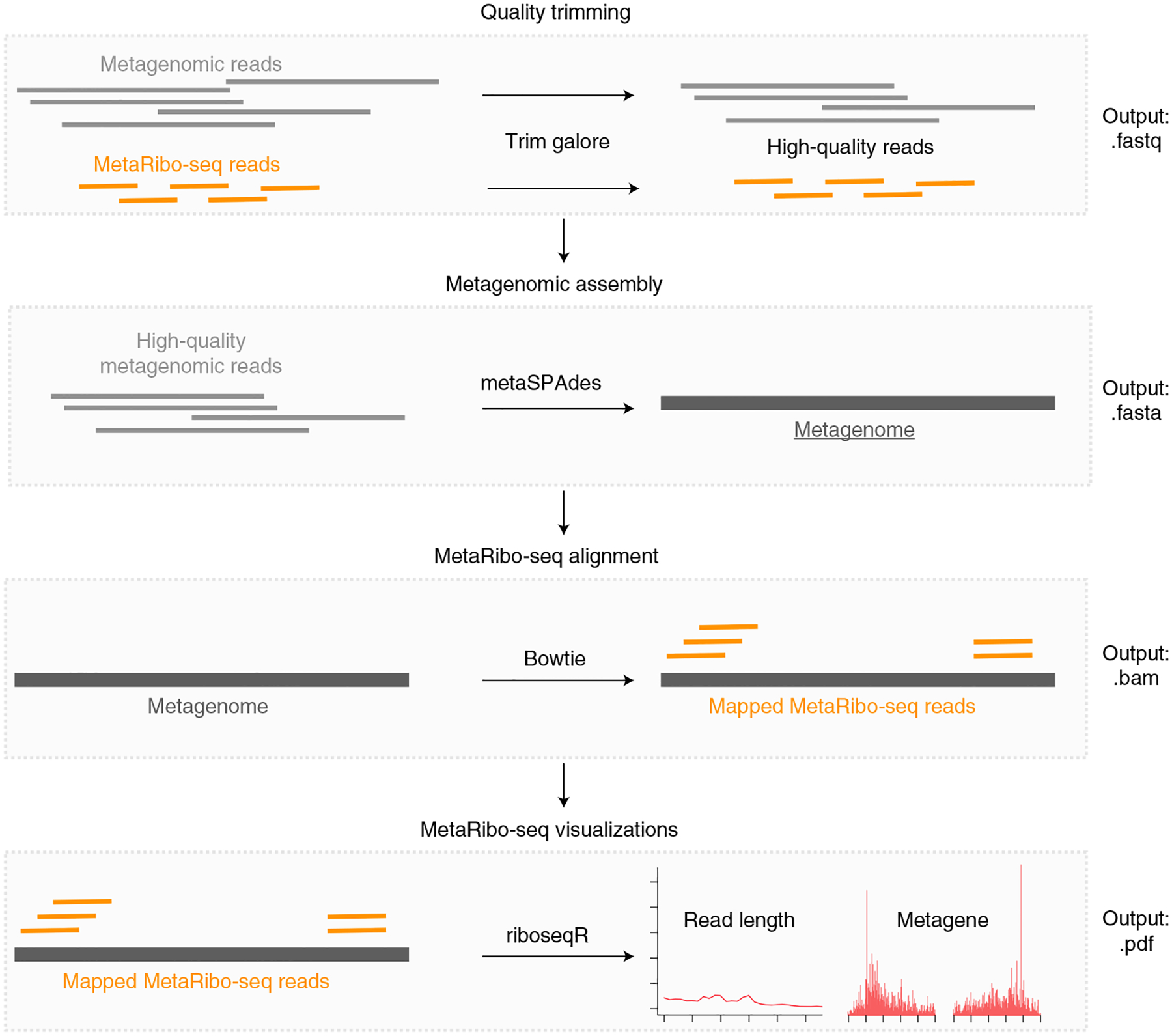
After sequencing, this computational workflow assesses MetaRibo-Seq library quality and serves as a starting point for downstream analyses. After quality trimming all relevant reads, this pipeline uses metagenomic reads for a given sample to create a de novo reference and then aligns MetaRibo-Seq reads corresponding to the sample to the assembly. These alignment files are used to create visualizations, including aligned footprint length distributions, footprint length specific triplet periodicity, and metagene plots for various footprint sizes.