

Abstract

Hematopoietic progenitor kinase 1 (HPK1) is implicated as a negative regulator of T-cell receptor-induced T-cell activation. Studies using HPK1 kinase-dead knock-in animals have demonstrated the loss of HPK1 kinase activity resulted in an increase in T-cell function and tumor growth inhibition in glioma models. Herein, we describe the discovery of a series of small molecule inhibitors of HPK1. Using a structure-based drug design approach, the kinase selectivity of the molecules was significantly improved by inducing and stabilizing an unusual P-loop folded binding mode. The metabolic liabilities of the initial 7-azaindole high-throughput screening hit were mitigated by addressing a key metabolic soft spot along with physicochemical property-based optimization. The resulting spiro-azaindoline HPK1 inhibitors demonstrated improved in vitro ADME properties and the ability to induce cytokine production in primary human T-cells.
Keywords: HPK1, immuno-oncology, structure-based design, kinase inhibitor, CYP inhibition
In contrast to traditional anticancer therapies where the goal is to target pathways that drive tumor proliferation or survival, cancer immunotherapies aim to leverage the patient’s own immune system to combat tumors.1 Over the past several years, a number of antibody inhibitors of immune checkpoints have found considerable clinical success by enhancing cancer patients’ immune T-cell activity. The survival benefits engendered by those approved antibody inhibitors have been impressive; however, the response rate to these agents remains modest. Hence, there is tremendous interest in the identification of additional agents to complement the approved immunomodulators.2
Hematopoietic progenitor kinase 1 (HPK1) is a member of the Ste20 serine/threonine family of kinases that serves as a negative regulator of T-cell receptor (TCR) induced T-cell activation.3 Studies using HPK1 kinase-dead knock-in mouse models have demonstrated that HPK1 kinase activity limits TCR signaling and cytokine production.4 In preclinical syngeneic models, loss of HPK1 kinase function was found to suppress tumor growth. Given the compelling evidence, HPK1 has been proposed to be a promising cancer immunotherapy target.5 As such, the goal of our program was to identify a selective small molecule inhibitor of HPK1.
A high-throughput screen using the kinase domain of HPK1 yielded compound 1, a literature Abl inhibitor6 (Abl IC50 < 0.51 nM), as a promising hit with respect to potency for HPK1 (Ki = 0.4 nM) and lipophilic ligand efficiency7 (LLE = 6.3). In addition to activity against Abl, 1 also showed inhibitory activity against LCK (IC50 = 24 nM, 55-fold selectivity over HPK1). Since Abl8 and LCK9 are essential components of cell survival and TCR signaling, respectively, improving selectivity against these two off-targets was a major focus of our optimization effort.
To improve selectivity against Abl, we first explored the difference in the ATP binding site gatekeeper residue between HPK1 (Met) and Abl (Thr).10 Due to the shorter Thr side chain in Abl, modeling suggested that Abl would suffer from a greater loss of favorable van der Waals interactions if the size of the C3 substituent of the azaindole motif were reduced (R1 in Table 1). Gratifyingly, conversion of the anisole substituent (1, Abl IC50 < 0.51 nM) to the smaller ethyl group (2, Abl IC50 = 22 nM) resulted in considerable improvement in selectivity against Abl while improving the LLE against HPK1. The increase in LLE suggested that the small alkyl substituent was better suited for HPK1 binding. While compound 2 exhibited only modest Abl selectivity, this promising result suggested that optimizing other vectors of the ligand using a structure-based approach may further enhance the selectivity against Abl.
Table 1. Optimization of 7-Azaindoles.
| compd | R1 | R2 | cLogP | HPK1 Ki (nM)a | LLEb | Abl selectivityc | LCK selectivityd |
|---|---|---|---|---|---|---|---|
| 1 | 2-MeOPh | H | 3.1 | 0.4 | 6.3 | <1× | 55× |
| 2 | Et | H | 2.1 | 2.1 | 6.6 | 10× | 237× |
| 3 | H | H | 1.5 | 130 | 5.4 | 66× | |
| 4 | Et | Me | 2.4 | 0.4 | 7.0 | 58× | 808× |
| 5 | Et | Et | 2.8 | 0.2 | 6.9 | 2556× | |
| 6 | Et | Cl | 2.6 | 0.5 | 6.7 | 501× |
All apparent Ki values represent arithmetic means of at least two determinations using the HPK1 Lantha binding biochemical assay.
LLE, lipophilic ligand efficiency based on HPK1 Ki.
Selectivity expressed as the ratio of Abl Ki over HPK1 Ki.
Selectivity expressed as the ratio of LCK Ki over HPK1 Ki.
An X-ray structure of 3 bound to HPK1 revealed a binding mode that was atypical among kinase structures (Figure 1A). While the 7-azaindole motif engaged the hinge region of the kinase in a canonical manner, the P-loop of the kinase adopted an unusual “folded” conformation. In the “folded P-loop” conformation, the side chain of Tyr28 was positioned to engage the ligand through hydrophobic interactions.11 Furthermore, the main-chain peptide bond N–H of Gly24 in the P-loop is directed toward the ligand and engaged the amide carbonyl of the ligand via a hydrogen bond.
Figure 1.
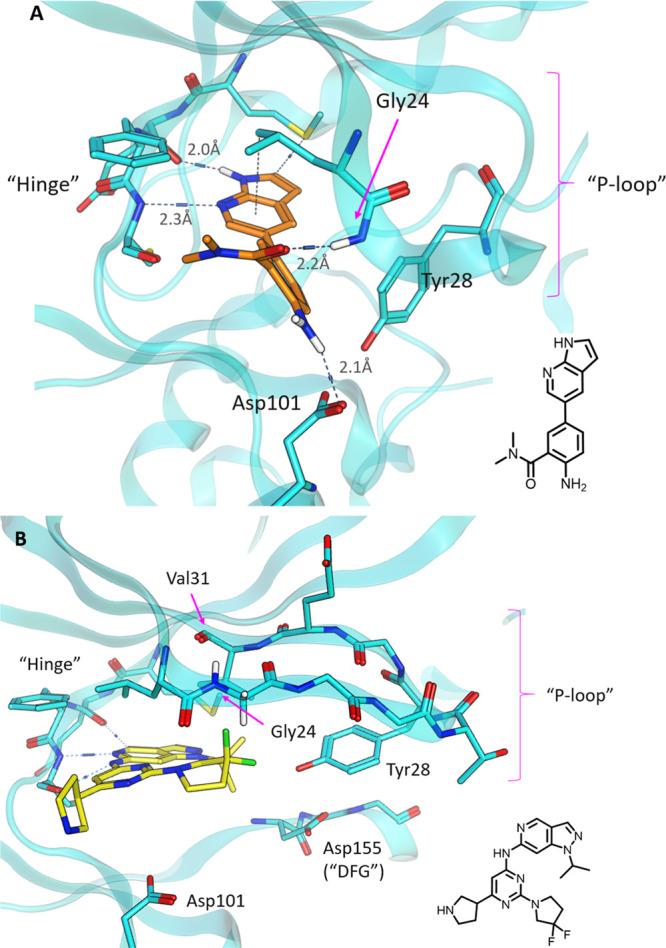
(A) X-ray structure of 3 bound in HPK1 (PDB: 7R9L) and (B) X-ray structure of G185812 bound in HPK1 (PDB: 7R9N). Predicted hydrogen bond interactions are shown as dashed lines. Distances from N/O to hydrogen are shown.
In contrast, in the more usual “extended P-loop” conformation (as exemplified by G1858;12Figure 1B), both structural features (Gly24 N–H and Tyr28 side chain) would not be available to interact with the azaindole ligands. In the “extended” conformation, the N–H of Gly24 forms a hydrogen bond with Val31 within the P-loop β-sheet, while the side chain of Tyr28 is situated in the DFG region of the binding pocket, away from the hinge region.12
The “folded” P-loop conformation is not commonly observed among kinases that have been studied.11 The ability of HPK1 to adopt the folded conformation was attributed to the unusually high number of glycine residues within its P-loop resulting in a high degree of flexibility, a feature that is absent in the LCK sequence. Literature examples suggested that stabilizing or engaging the “folded” P-loop conformation may have the potential to improve selectivity.11,13 Hence, we hypothesized that further engagement of Gly24 and Tyr28 in the folded P-loop conformation of HPK1 may result in improvement in both potency and selectivity against our key off-target LCK and the wider kinome.
To further optimize the interactions between the 7-azaindole ligands and Tyr28 in the folded P-loop, small lipophilic substituents at the C4 position (R2 in Table 1) were evaluated. Compared to the parent molecule 2 (R2 = H), installation of a methyl (4), ethyl (5), or chloro (6) group afforded gains in potency for HPK1 and improved selectivity against LCK and Abl (compound 4). It is also noteworthy that the alkyl substitutions at C4 also improved the LLE of the molecules, suggesting that productive interactions were established. Encouraged by the potency and selectivity of the 3-ethyl-7-azaindole series, our attention turned to improvement of the ADME-PK properties.
As shown in Table 2, compound 2 showed high turnover in human liver microsomes and time-dependent inhibitory activity against CYP3A4. Attempts to attenuate oxidative metabolism, such as deletion of the amino group (7) or installation of an electron-withdrawing group (8 and 9), did not improve in vitro metabolic stability (as measured by lowering of human liver microsome clearance) or address time-dependent inhibition of CYP3A4. Interestingly, introduction of a fluoro substituent improved both the potency for HPK1 and selectivity against LCK. Based on modeling, we hypothesized that the fluorine atom occupies a lipophilic pocket and stabilizes the nonplanar conformation of the ligand such that its aniline N–H could effectively hydrogen-bond with Asp101 (see Figure 1A). It is also worthy to note that, compared to analogues with primary or secondary amides, analogues with a tertiary amide (such as 2) demonstrated superior potency and selectivity against LCK (data not shown). We hypothesized that the tertiary amide preferred to reside in an out-of-plane conformation relative to the aniline such that the carbonyl group was well-positioned to hydrogen bond with the backbone N–H of Gly24. Given the lack of impact on metabolism with modifications within the aniline portion of the molecule, the focus of our optimization efforts then shifted to the 7-azaindole motif.
Table 2. Optimization of Solvent-Exposed Region.

All apparent Ki values represent arithmetic means of at least two determinations using the HPK1 Lantha binding biochemical assay.
Selectivity expressed as the ratio of LCK Ki over HPK1 Ki.
LM = Liver microsomes clearance measured in mL/min/kg, H = human, R = rat, M = mouse.
CYP3A4 time-dependent inhibition (TDI) assay performed using testosterone as a probe substrate.
Despite the presence of the 7-aza substitution, the electron-rich 5-membered ring within the 7-azaindole motif was implicated as the primary cause of oxidative metabolism and time-dependent inhibitory activity against CYP3A4.14 Examination of the X-ray cocrystal structure of 3 bound to HPK1 suggested that further substitutions at the 2- and 3-positions of the 7-azaindole should be tolerated and could provide opportunities to address the metabolic issues. Installation of a chlorine atom (10) at the 2-position to sterically and inductively deactivate the azaindole toward oxidation failed to improve human liver microsome (HLM) stability and time-dependent inhibition of CYP3A4 (Table 3).
Table 3. Optimization of Hinge-Binding Azaindole Group.

All apparent Ki values represent arithmetic means of at least two determinations using the HPK1 Lantha binding biochemical assay.
LLE, lipophilic ligand efficiency based on HPK1 Ki.
Selectivity expressed as the ratio of LCK Ki over HPK1 Ki.
HLM = Human liver microsomes predicted clearance (mL/min/kg).
CYP3A4 time-dependent inhibition (TDI) assay performed using testosterone as a probe substrate.
Further modeling suggested saturation of the azaindole to the corresponding azaindoline system should be tolerated and would also provide alternate exit vectors for structure-based design. 3-Spiro-cyclopropyl-indoline 11 demonstrated encouraging potency against HPK1 while reducing human liver microsome turnover and CYP inhibition. Expansion of the spiro-cyclopropyl ring to the cyclobutyl (12) or cyclopentyl groups (13) were also well-tolerated and offered alternative design exit vectors. In general, the spiro-indoline analogues showed a promising combination of good metabolic stability (as measured by HLM), minimal time-dependent inhibition of CYP3A4, acceptable potency, and selectivity that warranted further optimization.
As predicted by the SAR from the 7-azaindole series, installation of a small lipophilic group at the 4-position of the spiro-azaindoline (14–16) to enhance the van der Waals interactions with Tyr28 led to improved selectivity against LCK (14–16) and potency for HPK1 (15, 16). However, possibly due to the increase in lipophilicity, the C4 substitution resulted in the erosion of metabolic stability and undesired CYP3A4 inhibition. This observation suggested that a reduction in general lipophilicity would be required to maintain a balance between potency and metabolic stability.
An X-ray structure of 14 bound to HPK1 (Figure 2) revealed a binding mode that was similar to that of the 7-azaindole HPK1 inhibitors, where the indoline motif formed two hydrogen bonding interactions with the hinge residues of HPK1. The carboxyl-aniline group extended toward solvent where the aniline N–H formed a hydrogen bonding interaction with Asp101. The phenolic side chain of Tyr28 engaged the inhibitor via hydrophobic interactions. The spirocyclic cyclopropyl ring occupied a pocket flanked by the Met90 gatekeeper residue, Leu144, Val31, and a water network in the direction of the catalytic Lys46. A close examination of this region of the binding pocket suggested that installation of polar groups on the spirocyclic system should not only reduce the overall lipophilicity of the molecules, but also engage polar functionalities in the vicinity of Lys46.
Figure 2.
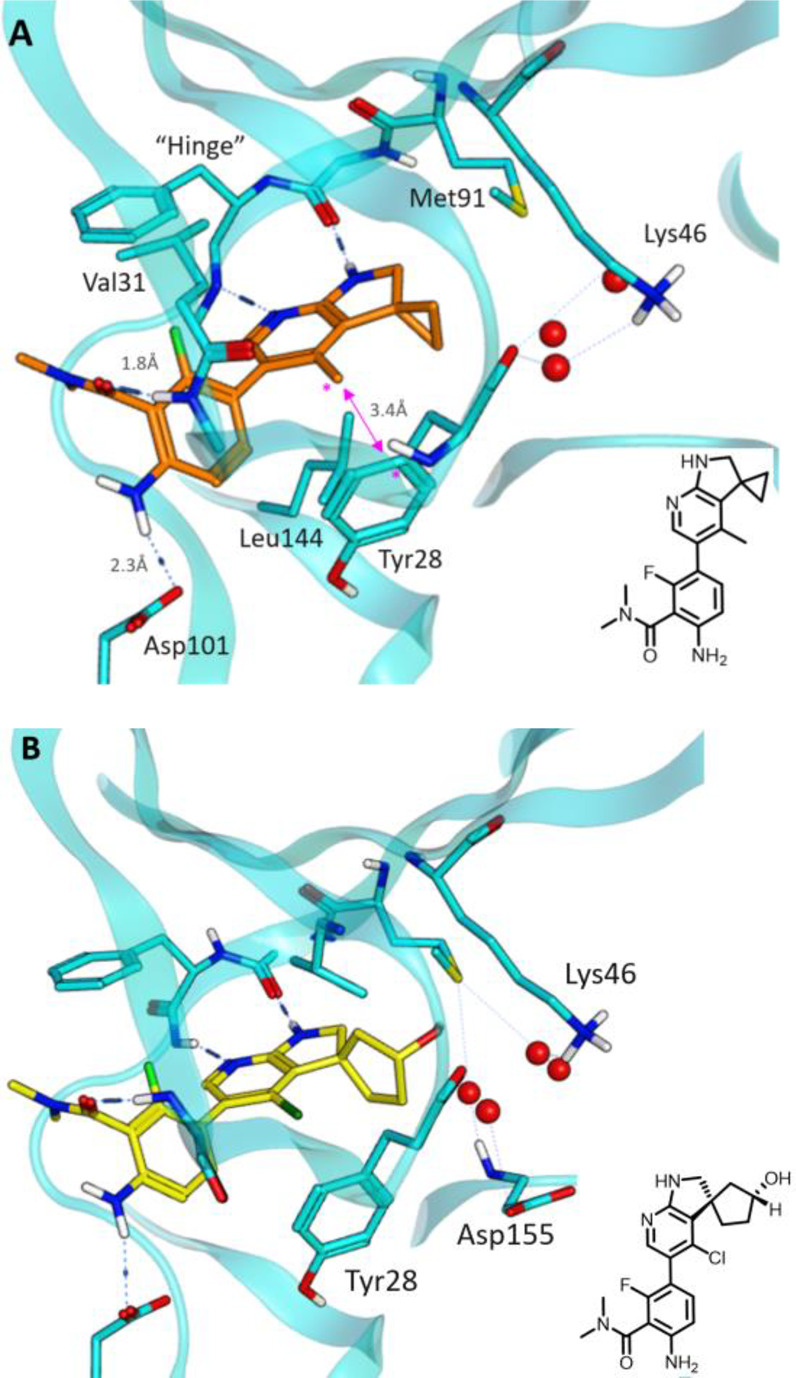
X-ray structures of (A) 14 (PDB: 7R9P) and (B) 17 (PDB: 7R9T) bound to HPK1. Red circles represent water molecules. Predicted hydrogen bonds: Interactions are shown as dashed lines; distances from N/O to hydrogen are shown. Pink arrow represents the distance between the carbons labeled with asterisks.
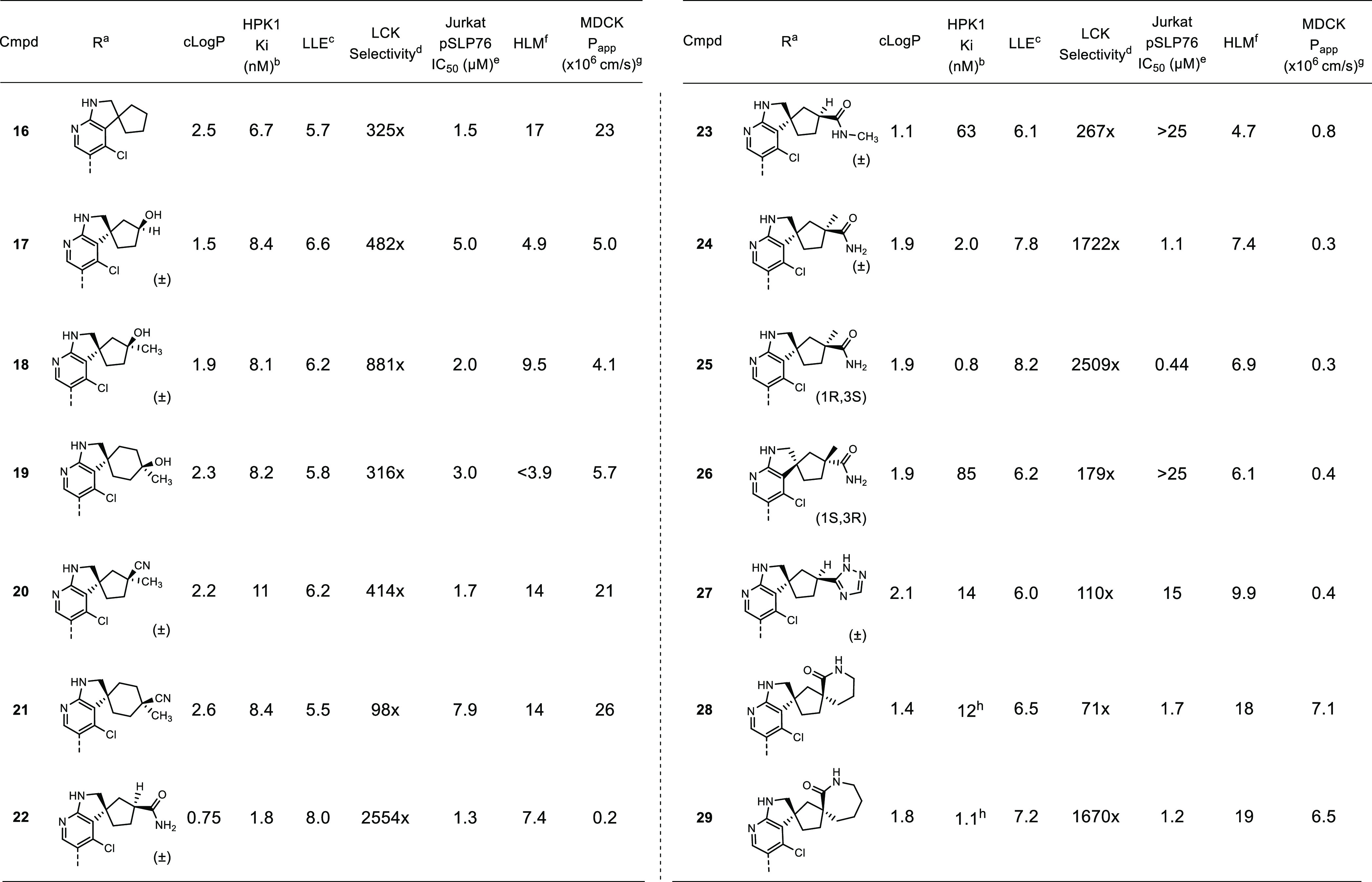
While the cyclobutyl analogue 15 was superior with respect to LLE, modeling suggested that the larger cyclopentyl (16) or cyclohexyl templates should provide vectors and distances that are more suited to target Lys46. To test this hypothesis, a series of alcohol- and nitrile-functionalized analogues of 16 were designed to indirectly interact with Lys46 via the water network. Hydroxylated analogues 17–19 demonstrated HPK1 activity comparable to the parent 16 while improving in vitro metabolic stability significantly (Table 4). The nitrile analogues 20 and 21 maintained potency without improving HLM stability. An X-ray structure of 17 bound to HPK1 (Figure 2B) showed that the binding pose was identical to that of 14 with the hydroxyl group forming several hydrogen bonding interactions with the water network adjacent to Lys46. This observation was consistent with the increase in LLE and suggested that productive interactions were formed between the polar functionalities and the binding site.
Table 4. Optimization of Spiro-indolines.
Relative stereochemistry as drawn. Absolute stereochemistry as noted.
All apparent Ki values represent arithmetic means of at least two determinations using the HPK1 Lantha binding biochemical assay.
LLE, lipophilic ligand efficiency based on HPK1 Ki.
Selectivity expressed as the ratio of LCK Ki over HPK1 Ki.
Cellular assay that measures the direct phosphorylation of SLP76 by HPK1 in Jurkat cells.
HLM = Human liver microsome predicted clearance (mL/min/kg).
MDCK permeability assay (apical-to-basolateral).
Apparent HPK1 Ki values for compounds 28 and 29 represent arithmetic means of two determinations using the HPK1 HTRF enzymatic assay.
To further improve HPK1 potency, we opted to design extended substituents that may directly engage Lys46 or its neighboring polar functionalities. Introduction of a primary amide (22) resulted in a gain in LLE, which suggested productive hydrogen bonding interactions were made. While demonstrating good HLM stability (HLM = 7.4 mL/min/kg), 22 only showed modest permeability (MDCK apical-to-basolateral Papp = 0.2 × 10–6 cm s–1), possibly due to the high number of hydrogen bond donors (five) and high topological polar surface area (114 Å2). We anticipated that this might limit oral bioavailability; thus, we turned our attention to improving the permeability of 22.
Our strategy to improve permeability involved (a) reducing the number of hydrogen bond donors and (b) sterically masking the polar functionalities. Conversion of the primary amide to a secondary amide (compound 23) failed to maintain potency. Installing a methyl group adjacent to the amide (24) maintained potency and metabolic stability but did not improve permeability. Replacing the amide with other bioisosteres such as a triazole (27) resulted in loss of potency. Interestingly, expansion to the cyclic lactams (28 and 29) improved permeability while maintaining potency. When compared to 23 (where the N-methyl-amide likely existed preferentially in the s-trans conformation), the amide moiety in 28 and 29 was restricted to the s-cis conformation that may have maintained productive H-bonding interactions with the protein. We hypothesized that the increase in lipophilicity and steric shielding of the polar amide drove the improvement in permeability. However, the increase in lipophilicity also eroded metabolic stability.
Despite its low permeability as measured by the MDCK assay, we were encouraged by the potency and metabolic stability in HLMs of 24. Hence, its single enantiomers were isolated to give compounds 25 and 26. The two single enantiomers were tested in a cellular assay that measures the direct phosphorylation of SLP76 by HPK1 in Jurkat cells. Gratifyingly, compound 25 showed strong inhibitory effects in the cellular assay despite its modest MDCK permeability. Compound 25 also showed excellent selectivity against key off-targets such as LCK as well as in a broader kinase panel (5/47 kinases inhibited >70% at 0.1 μM; Abl inhibited at <35% at 0.1 μM). To evaluate its stimulatory effect on immune response, human pan T-cells were treated with 25 for 4 h followed by stimulation with αCD3 and αCD28. The resulting increase in cytokine production (as measured by IL2 concentration) was then determined. As shown in Figure 3, treatment with 25 resulted in dose-dependent augmentation of IL2 production as one would expect from inhibition of HPK1 kinase activity. Further ADME-PK profiling showed that 25 addressed the two major liabilities of the original 7-azaindole HPK1 inhibitors: 25 demonstrated good metabolic stability in human hepatocytes (human hepatocyte clearance for 25 = 9.2 mL/min/kg; human hepatocyte clearance for 2 = 15 mL/min/kg) and minimal inhibitory activity against CYP3A4 (TDI IC50 > 10 μM).
Figure 3.

Dose–response curve measuring IL2 levels in human pan T-cell cultures after treatment with 25 followed by stimulation with αCD3 and αCD28 (calculated EC50 = 1.56 ± 0.10 μM). Activity displayed as percent increase in IL2 concentration relative to stimulated/untreated controls. Data points represent results from two replicates.
After an iv bolus dose in mouse, 25 showed a blood clearance of 57 mL/min/kg (Table 5). The good in vitro–in vivo correlation for mouse suggested that human liver microsomes and hepatocytes could be a reasonable predictor of metabolic stability in human. Despite its modest MDCK permeability, 25 showed 13% oral bioavailability in mouse after oral dosing using a suspension formulation (25 mg/kg; MCT suspension).
Table 5. Pharmacokinetic Profile of 25.
LM = Liver microsome predicted clearance (mL/min/kg), H = human, R = rat, M = mouse.
Hep = Hepatocyte clearance measured in mL/min/kg, H = human, R = rat, M = mouse.
Mouse PK: C57BL/6, 1 mg/kg iv dose or 25 mg/kg po dose, blood clearance measured in mL/min/kg, Vss = volume of distribution (L/kg).
HLM and Hep clearance values represent arithmetic means of two determinations.
Herein, we described the lead optimization of a series of HPK1 inhibitors where improvement in selectivity was achieved by optimizing interactions with the induced, noncanonical P-loop conformation in HPK1. The key transformation in the lead optimization process to address metabolic liabilities included the conversion of the 7-azaindole to a series of spiro-indoline motifs as the hinge-binding element. Results from optimization of 25 to further improve its ADME properties will be disclosed in due course.
Chemistry
As outlined in Scheme 1, the synthesis of 25 began with SEM protection of the commercially available 5-bromo-4-chloro-7-azaindole 30. The azaindole motif was then converted to the 3,3-dibromoxindole 31 via oxidative bromination with NBS. Zinc reduction then yielded oxindole 32. The spirocyclic cyclopentenyl ring was then installed via bis-alkylation using allyl bromide, followed by a ring-closing metathesis to give cyclopentene 34. The oxindole was then reduced and N-protected to give indoline 36. Hydroboration/oxidation then yielded a diastereomeric mixture of 37 and 38. The alcohol 37 was then advanced to the corresponding nitrile 39 by activating the alcohol via mesylation and cyanide displacement. α-Alkylation of the nitrile using methyl iodide then afforded a diastereomeric mixture of 40a and 40b (dr 4:1). Separation of the diastereomers was achieved via silica gel flash chromatography to yield 40a (racemic). Deprotection of the PMB group with concomitant hydrolysis using TFA yielded both the nitrile 41 (result of incomplete hydrolysis) and primary amide 42. Compound 42 was then coupled with the boronate 43(15) to give (±)-25. The single enantiomers were separated using chiral supercritical fluid chromatography (SFC) to give 25 and 26. Other analogues described herein were prepared using a similar synthetic scheme. (Experimental procedures can be found in the Supporting Information.)
Scheme 1. Synthesis of Compound 25.

Acknowledgments
We thank the Genentech Analytical Research, DMPK, in vivo Studies Group and Compound Management colleagues for their contributions. We also thank the Chemistry groups at Pharmaron, WuXi AppTec, and ChemPartner for their technical contributions. We thank the beamline staff at Advance Photon Source and Stanford Synchrotron Radiation Light-Source for their assistance in data collection.
Glossary
Abbreviations
- A
apical
- Abl
Abl kinase
- ADME
absorption, distribution, metabolism, elimination
- B
basolateral
- CL
clearance
- CYP
cytochrome P450
- F
oral bioavailability
- Hep
hepatocytes
- HLM
human liver microsomes
- HPK1
Hematopoietic progenitor kinase 1
- HTS
high-throughput screening
- IL2
interleukin-2
- iv
intravenous
- LCK
lymphocyte-specific protein tyrosine kinase
- LLE
ligand lipophilic efficiency
- Liver Mic
liver microsomes assay
- MCT
0.5% methylcellulose with 0.2% Tween in water
- MH
mouse hepatocytes
- MLM
mouse liver microsomes
- NBS
N-bromosuccinimide
- Papp
apparent permeability
- PK
pharmacokinetics
- PMB
para-methoxybenzyl
- po
oral administration
- SEM
trimethylsilylethoxymethyl
- SFC
supercritical fluid chromatography
- SLP76
SH2-domain-containing leukocyte protein of 76 kDa
- T
testosterone, TCR, T-cell receptor
- TDI
time-dependent inhibition
- TFA
trifluoroacetic acid
- Vss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00473.
Experimental procedures, compound characterization, detailed kinase selectivity panel data for compound 25, crystallographic statistics for protein cocrystal structures of 3, G1858, 14 and 17 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Chen D. S.; Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013, 39, 1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]; b Barbee M. S.; Ogunniyi A.; Horvat T. Z.; Dang T.-O. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann. Pharmacother. 2015, 49, 907–937. 10.1177/1060028015586218. [DOI] [PubMed] [Google Scholar]; c Sharma P.; Allison J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015, 161, 205–214. 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mahoney K. M.; Rennert P. D.; Freeman G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discovery 2015, 14, 561–584. 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]; b Heffron T. P.; Chan B. K. Small molecules for cancer immunotherapy. Med. Chem. Rev. 2017, 52, 219–241. 10.29200/acsmedchemrev-v52.ch12. [DOI] [Google Scholar]
- Sawasdikosol S.; Zha R.; Yang B.; Burakoff S. HPK1 as a novel target for cancer immunotherapy. Immunol. Res. 2012, 54, 262–265. 10.1007/s12026-012-8319-1. [DOI] [PubMed] [Google Scholar]
- Hernandez S.; Qing J.; Thibodeau R. H.; Du X.; Park S.; Lee H.-M.; Xu M.; Oh S.; Navarro A.; Roose-Girma M.; Newman R. J.; Warming S.; Nannini M.; Sampath D.; Kim J. M.; Grogan J. L.; Mellman I. The kinase activity of Hematopoietic Progenitor Kinase 1 is essential for the regulation of T cell function. Cell Rep. 2018, 25, 80–94. 10.1016/j.celrep.2018.09.012. [DOI] [PubMed] [Google Scholar]
- a Sawasdikosol S.; Burakoff S. A perspective on HPK1 as a novel immuno-oncology drug target. eLife 2020, 9, e55122 10.7554/eLife.55122. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Degnan A. P.; Kumi G. K.; Allard C. W.; Araujo E. V.; Johnson W. L.; Zimmermann K.; Pearce B. C.; Sheriff S.; Futran A.; Li X.; Locke G. A.; You D.; Morrison J.; Parrish K. E.; Stromko C.; Murtaza A.; Liu J.; Johnson B. M.; Vite G. D.; Wittman M. D. Discovery of Orally Active Isofuranones as Potent, Selective Inhibitors of Hematopoetic Progenitor Kinase 1. ACS Med. Chem. Lett. 2021, 12, 443–450. 10.1021/acsmedchemlett.0c00660. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yu E. C.; Methot J. L.; Fradera X.; Lesburg C. A.; Lacey B. M.; Siliphaivanh P.; Liu P.; Smith D. M.; Xu Z.; Piesvaux J. A.; Kawamura S.; Xu H.; Miller J. R.; Bittinger M.; Pasternak A. Identification of Potent Reverse Indazole Inhibitors for HPK1. ACS Med. Chem. Lett. 2021, 12, 459–466. 10.1021/acsmedchemlett.0c00672. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vara B. A.; Levi S. M.; Achab A.; Candito D. A.; Fradera X.; Lesburg C. A.; Kawamura S.; Lacey B. M.; Lim J.; Methot J. L.; Xu Z.; Xu H.; Smith D. M.; Piesvaux J. A.; Miller J. R.; Bittinger M.; Ranganath S. H.; Bennett D. J.; DiMauro E. F.; Pasternak A. Discovery of Diaminopyrimidine Carboxamide HPK1 inhibitors as Preclinical Immunotherapy Tool Compounds. ACS Med. Chem. Lett. 2021, 12, 653–661. 10.1021/acsmedchemlett.1c00096. [DOI] [PMC free article] [PubMed] [Google Scholar]; e You D.; Hillerman S.; Locke G.; Chaudhry C.; Stromko C.; Murtaza A.; Fan Y.; Koenitzer J.; Chen Y.; Briceno S.; Bhadra R.; Duperret E.; Gullo-Brown J.; Gao C.; Zhao D.; Feder J.; Curtin J.; Degnan A. P.; Kumi G.; Wittman M.; Johnson B. M.; Parrish K. E.; Gokulrangan G.; Morrison J.; Quigley M.; Hunt J. T.; Salter-Cid L.; Lees E.; Sanjuan M. A.; Liu J. Enhanced antitumor immunity by a novel small molecule HPK1 inhibitor. J. Immunother. Cancer 2021, 9, e001402 10.1136/jitc-2020-001402. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Linney I. D.; Kaila N. Inhibitors of immuno-oncology target HPK1 – a patent review (2016–2020). Expert Opin. Ther. Pat. 2021, 31, 893–910. 10.1080/13543776.2021.1924671. [DOI] [PubMed] [Google Scholar]
- Arnold W. D.; Bounaud P.; Chen C.; Eastman B.; Gosberg A.; Gradl S. N.; Hopkins S.; Li Z.; McDonald I.; Sprengeler P. A.; Steensma R. W.; Wilson M. E.. Preparation of pyrrolo[2,3-b]pyridine derivatives as kinase modulators. US patent 20090143352.
- a Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]; b Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]; c Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- Wang J. Y. J. The capable ABL: what is its biological function?. Mol. Cell. Biol. 2014, 34, 1188–1197. 10.1128/MCB.01454-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios E. H.; Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Hantschel O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer 2012, 3, 436–446. 10.1177/1947601912458584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães C. R.; Rai B. K.; Munchhof M. J.; Liu S.; Wang J.; Bhattacharya S. K.; Buckbinder L. Understanding the impact of the P-loop conformation on kinase selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204. 10.1021/ci200153c. [DOI] [PubMed] [Google Scholar]
- Wu P.; Sneeringer C. J.; Pitts K. E.; Day E. S.; Chan B. K.; Wei B.; Lehoux I.; Mortara K.; Li H.; Wu J.; Franke Y.; Moffat J. G.; Grogan J. L.; Heffron T. P.; Wang W. Hematopoietic Progenitor Kinase-1 structure in a domain-swapped dimer. Structure 2019, 27, 125–133. 10.1016/j.str.2018.10.025. [DOI] [PubMed] [Google Scholar]
- Collie G. W.; Michaelides I. N.; Embrey K.; Stubbs C. J.; Börjesson U.; Dale I. L.; Snijder A.; Barlind L.; Song K.; Khurana P.; Phillips C.; Storer R. I. Structural Basis for Targeting the Folded P-Loop Conformation of c-MET. ACS Med. Chem. Lett. 2021, 12, 162–167. 10.1021/acsmedchemlett.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. G.; Liu J. J.; Li A.-R.; van Lengerich B.; Wang S.; Medina J. C.; Collins T. L.; Danao J.; Seitz L.; Willee A.; D’Souza W.; Budelsky A. L.; Fan P. W.; Wong S. G. W. Solving time-dependent CYP3A4 inhibition for a series of indole-phenylacetic acid dual antagonists of the PGD2 receptors CRTH2 and DP. Bioorg. Med. Chem. Lett. 2014, 24, 2877–2880. 10.1016/j.bmcl.2014.04.092. [DOI] [PubMed] [Google Scholar]
- Synthesis of compound 43 is described in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.