

Summary
Noradrenergic, serotonergic, and histaminergic neurons are continuously active during waking, reduce discharge during NREM sleep, and cease discharge during REM sleep. Cataplexy, a symptom associated with narcolepsy, is a waking state in which muscle tone is lost, as it is in REM sleep, while environmental awareness continues, as in alert waking. In prior work, we reported that, during cataplexy, noradrenergic neurons cease discharge, and serotonergic neurons greatly reduce activity. We now report that, in contrast to these other monoaminergic “REM-off” cell groups, histamine neurons are active in cataplexy at a level similar to or greater than that in quiet waking. We hypothesize that the activity of histamine cells is linked to the maintenance of waking, in contrast to activity in noradrenergic and serotonergic neurons, which is more tightly coupled to the maintenance of muscle tone in waking and its loss in REM sleep and cataplexy.
Introduction
REM sleep and waking share certain physiological traits, including a low-voltage electroencephalogram (EEG), similar evoked responses to sensory stimuli, and activation of brain motor and autonomic control systems (Baust et al., 1972; Salin-Pascual et al., 2001). However, there are also striking differences between the two states. By definition, an animal is aware of its environment during waking, whereas in REM sleep it is not. In waking, muscle tone is continuously present, but, in REM sleep, tone in the antigravity muscles is absent. The physiological and neurochemical mechanisms mediating these differences between waking and REM sleep are unclear.
One possible substrate for the differences between the behavioral and physiological conditions in waking and REM sleep is the activity of monoaminergic neurons. Although most brain neurons show high levels of activity in both waking and REM sleep (Siegel, 2000; Steriade et al., 1993), serotonergic (Jacobs and Azmitia, 1992), noradrenergic (Aston-Jones and Bloom, 1981), and histaminergic (Sakai et al., 1990; Strecker et al., 2002) cells are continuously and maximally active in waking but are minimally active or silent in REM sleep. The cessation of activity of these monoaminergic neurons might underlie the loss of muscle tone and loss of environmental awareness characteristic of REM sleep. If this were the case, it would remain to be determined whether all these monoaminergic cells contribute equally to these changes in muscle tone and environmental awareness.
Cataplexy is a sudden loss of muscle tone in waking, usually triggered by laughter or the strong, sudden onset of emotions in humans with narcolepsy (Guilleminault, 1976). In canine narcoleptics, cataplexy is triggered by emotional excitement, play, or the consumption of favored foods (Siegel, 1999; Wu et al., 1999). Waking and a low-voltage EEG are maintained during cataplectic attacks (Guilleminault, 1976). Humans report being alert, with clear perception and accurate memory of events occurring during cataplexy, and both human and canine narcoleptics can visually track objects during this state. However, skeletal muscle tone is absent or greatly decreased. Thus, cataplexy represents a unique natural experiment in which muscle atonia is dissociated from sleep. If the activity of one or more of the monoaminergic cell groups is correlated with muscle tone, one would expect that these cells would cease discharge in cataplexy. If their activity is linked to waking, one would predict that they would maintain their discharge during cataplexy. We have previously reported that noradrenergic cells of the locus coeruleus (LC) cease discharge during cataplexy (Wu et al., 1999) and that serotonergic cells reduce discharge to NREM sleep levels during cataplexy (Wu et al., 2004). We now report that histaminergic cells maintain waking levels of activity during cataplexy and, unlike noradrenergic and serotonergic cells, do not alter their discharge rate after the administration of drugs that increase the occurrence of cataplexy. These data suggest separate roles for different monoaminergic cell groups in the regulation of muscle tone and environmental awareness and in the pathophysiology of narcolepsy.
Results
We recorded a total of 156 neurons in the ventral posterior lateral hypothalamus (PLH) across the sleep-wake (S-W) cycle, of which 88 cells were further studied during cataplexy episodes (Table 1). The recorded cells were categorized into seven types according to their S-W discharge pattern: REM-off, NREM-off, wake/REM-active, wake-active, REM-active, state independent, and other (Table 1). The REM-off cells that we recorded from the tuberomammillary area of the posterior hypothalamus fulfilled the anatomical and physiological criteria for histamine-containing cells, as described below.
Table 1.
Average Discharge Rates of Posterior Lateral Hypothalamus Neurons during Waking, Sleep and Cataplexy
| Cell Type | N | AW | QW | NREM | REM | CAT |
|---|---|---|---|---|---|---|
| Wake/REM-active (all) | 77 (49.4%) | 3.42 ± 0.45 | 2.46 ± 0.37 | 1.44 ± 0.21 | 3.07 ± 0.41 | |
| Wake/REM-active (CAT-tested) | 45 | 4.44 ± 0.68 | 3.30 ± 0.57 | 1.74 ± 0.31 | 3.82 ± 0.63 | 4.00 ± 0.77 |
| Wake-active (all) | 19 (12.2%) | 4.10 ± 1.14 | 3.39 ± 1.06 | 2.49 ± 0.79 | 1.85 ± 0.60 | |
| Wake-active (CAT-tested) | 9 | 2.71 ± 1.73 | 2.00 ± 1.45 | 1.56 ± 1.16 | 1.40 ± 1.12 | 2.19 ± 1.51 |
| REM-off (all) | 14 (9.0%) | 4.76 ± 0.86 | 1.93 ± 0.65 | 0.61 ± 0.29 | 0.30 ± 0.07 | |
| REM-off (CAT-tested) | 9 | 5.32 ± 1.23 | 2.06 ± 1.00 | 0.87 ± 0.44 | 0.38 ± 0.08 | 3.36 ± 0.94 |
| NREM-off (all) | 12 (7.7%) | 2.56 ± 0.85 | 0.74 ± 0.36 | 0.21 ± 0.07 | 0.68 ± 0.17 | |
| NREM-off (CAT-tested) | 9 | 2.75 ± 0.97 | 0.78 ± 0.28 | 0.24 ± 0.08 | 0.64 ± 0.22 | 1.43 ± 0.51 |
| REM-active (all) | 7 (4.5%) | 0.45 ± 0.17 | 0.70 ± 0.40 | 1.17 ± 0.50 | 2.15 ± 0.76 | |
| REM-active (CAT-tested) | 5 | 0.53 ± 0.24 | 0.86 ± 0.57 | 1.36 ± 0.72 | 2.61 ± 1.03 | 1.33 ± 0.88 |
| State independent (all) | 11 (7.0%) | 7.93 ± 2.34 | 7.68 ± 2.24 | 7.42 ± 2.16 | 7.66 ± 2.25 | |
| State independent (CAT-tested) | 5 | 4.06 ± 1.66 | 3.87 ± 1.88 | 4.03 ± 1.73 | 4.12 ± 1.83 | 4.25 ± 1.63 |
| Other (all) | 16 (10.2%) | 4.93 ± 1.76 | 3.43 ± 0.80 | 3.60 ± 1.06 | 3.39 ± 1.12 | |
| Other (CAT-tested) | 6 | 9.99 ± 4.21 | 4.84 ± 1.87 | 6.83 ± 2.40 | 6.01 ± 2.84 | 9.68 ± 3.83 |
Discharge rates (Hz) are mean ± SEM. Percentages of cells recorded (total n = 156) are given in parentheses. AW, active waking; QW, quiet waking; NREM, non-REM sleep; REM, REM sleep; CAT, cataplexy.
Characterization of Histaminergic REM-Off Cells
The tuberomammillary REM-off cells that we report on here have the following properties. (1) They are located within the histamine cell region, as identified by immuno-staining. (2) They have long-duration action potentials, in contrast to adjacent cells (Sakai et al., 1990; Vanni-Mercier et al., 2003). Acute and in vitro studies have demonstrated that posterior hypothalamic cells with long-duration action potentials are histaminergic (Reiner and McGeer, 1987; Uteshev et al., 1995). (3) They have a wake-active-REM-off pattern of activity, in contrast to adjacent cells, most of which are active in both waking and REM sleep (Sakai et al., 1990; Vanni-Mercier et al., 2003). This REM-off pattern was observed in dialysis studies that measured histamine release from the posterior hypothalamus, with high and uniform levels seen during waking, reduced levels in NREM sleep, and minimal levels in REM sleep, exactly tracking the unit activity profile that we report (Strecker et al., 2002).
REM-Off Cells Were Recorded from the Region Containing Histamine-IR Cells
Histamine-immunoreactive (histamine-IR) cells (20–50 μm diameter) were largely concentrated in the tuberomammillary nucleus in the ventral PLH of the dog, as described in other species (Figure 1A; Panula et al., 1984, 1990; Watanabe et al., 1984; Watanabe and Yanai, 2001). In addition, some large, bipolar histamine-IR neurons (30 μm diameter) were seen at the level of the fornix and mammillothalamic tract in the posterior hypothalamus (Figure 1A1). REM-off cells were recorded from microelectrodes that were located within the region that contained histamine-IR cells (Figure 1B). Reconstruction of the recording sites and mapping of the recorded neurons were done with the help of Nissl- and histamine-stained sections (Figure 1B). The location of the cells was within a 1 mm anterior-posterior range of the section illustrated in Figure 1B.
Figure 1. Identification and Characterization of Neurons Recorded from the Tuberomammillary Area of the Ventral Posterior Lateral Hypothalamic Region.

(A) Photomicrograph showing histamine-immunostained cells located above the level of the fornix (A1), at the level of the fornix (A2), and adjacent to the base of the brain (A3). The location of the electrode track within the histamine cell group is shown (A4). Arrows indicate the histamine-immunoreactive cells along the electrode track. Scale bar for (A1) and (A2), 100 μm; scale bar for (A3) and (A4), 100 μm.
(B) Locations of histamine-immunostained neurons and recorded cell types. Histamine-immunostained cells (black dots) were concentrated in the ventral posterior lateral hypothalamic region (ventral PLH). Scale bar, 1 mm. OT, optic tract; Fx, fornix; Mt, mammillothalamic tract.
(C) Action potentials of representative cells with different sleep-wake discharge profiles. Spike durations of the cells: wake-active, 1.8 ms; wake/REM-active, 2.0 ms; NREM-off, 1.7 ms; REM-active, 1.3 ms; REM-off, 2.9 ms. Action potentials of state-independent and other cell types are not shown. Calibration for unit action potentials: 1 ms and 50 μV.
(D) The mean spike widths (ms) ± SEM of the cell groups recorded from the ventral PLH region. SI, state independent. *p < 0.05, **p < 0.01.
REM-Off Cells Had Long Action Potential Duration
The spike widths (action potential durations) differed between cell groups defined on the basis of their discharge rate profile (F = 3.7; df = 6,149; p < 0.002; ANOVA). The spike width of the REM-off cells (2.3 ± 0.2 ms) was significantly greater than those of wake/REM-active (1.8 ± 0.04 ms) and REM-active (1.3 ± 0.08 ms) cells (p < 0.01 for both; Dunn’s [Bonferroni] test; Figures 1C and 1D). The spike width of the REM-off cells was also significantly greater than those of wake-active (1.7 ± 0.1 ms), NREM-off (1.7 ± 0.08 ms), and state-independent (1.7 ± 0.1 ms) cells (p < 0.05 for all; Dunn’s [Bonferroni] test; Figures 1C and 1D). No significant differences were seen among the spike widths of other cell types.
REM-Off Cells Had Similar Discharge Rates in Quiet Waking and Cataplexy
REM-off cells had their highest firing rates in active waking (AW) and reduced their discharge rates in QW and NREM sleep; they exhibited very low discharge rates or complete cessation of activity throughout REM sleep (Figure 2A; Table 1). REM sleep firing rates were significantly lower than AW firing rates (t = 5.7; df = 13; p < 0.01; Bonferroni t test). The average activity rates in both REM sleep (t = 2.7; df = 13; p < 0.05; Bonferroni t test) and NREM sleep (t = 2.9; df = 13; p < 0.05; Bonferroni t test) were also significantly lower than that in QW.
Figure 2. Activity of a PLH REM-Off Cell during Sleep-Wake States and Cataplexy.
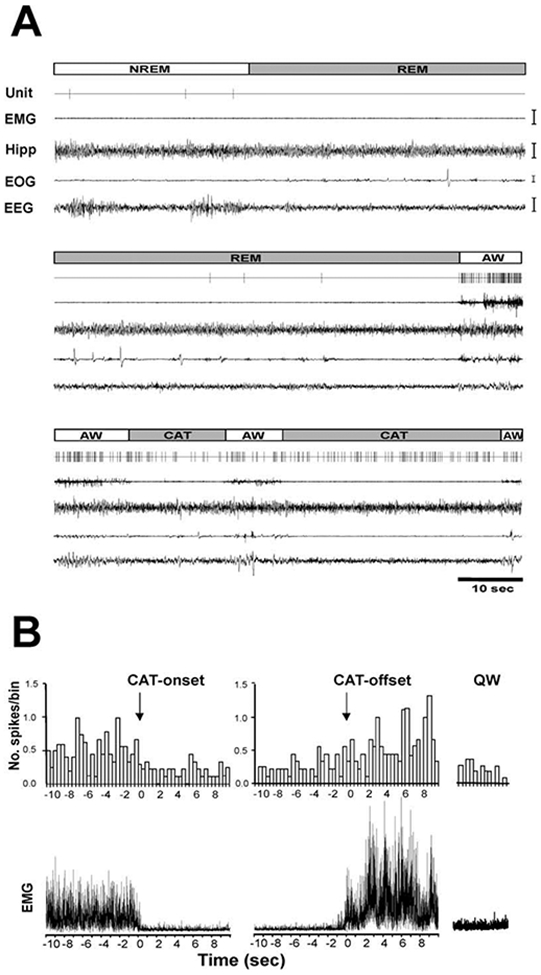
(A) Unit activity in NREM sleep, REM sleep, active waking, and cataplexy. Unit, single-cell activity; EMG, electromyogram; Hipp, hippocampal electroencephalogram; EOG, electrooculogram; EEG, electroencephalogram; AW, active waking; NREM, non-rapid eye movement sleep; REM, rapid eye movement sleep; CAT, cataplexy. Calibration bars: EMG, 100 μV; Hipp, 50 μV; EOG, 50 μV; EEG, 50 μmV.
(B) Spike histogram based on an average of nine cataplexy episodes (duration 50.6 ± 17.9 s) shows the discharge profile of a REM-off neuron in AW prior to, during, and after cataplexy. (Upper panel) Histograms of average discharge rates (400 ms bins) show change in discharge rate from wake to onset of cataplexy and from offset of cataplexy to wake. (Lower panel) The corresponding averaged electromyogram (EMG) shows atonia with the onset of cataplexy (CAT-onset) and increased activity with cataplexy offset (CAT-offset) and in quiet waking (QW).
The average discharge rate of these cells in cataplexy was nonsignificantly higher than that in QW, nonsignificantly lower than that in AW, and significantly higher than those in NREM sleep (t = 2.8; df = 8; p < 0.05; Bonferroni t test) and REM sleep (t = 3.4; df = 8; p < 0.05; Bonferroni t test; Figures 2B and 3A-3C; Table 1). This is in contrast to REM-off cells recorded from the LC (Wu et al., 1999) and dorsal raphe (Wu et al., 2004), which significantly reduce discharge in cataplexy relative to QW (Figure 4). The average firing rate of tuberomammillary hypothalamic REM-off cells during cataplexy was 10.6 times their average firing rate in REM sleep.
Figure 3. Discharge Patterns of PLH REM-Off and NREM-Off Neurons.
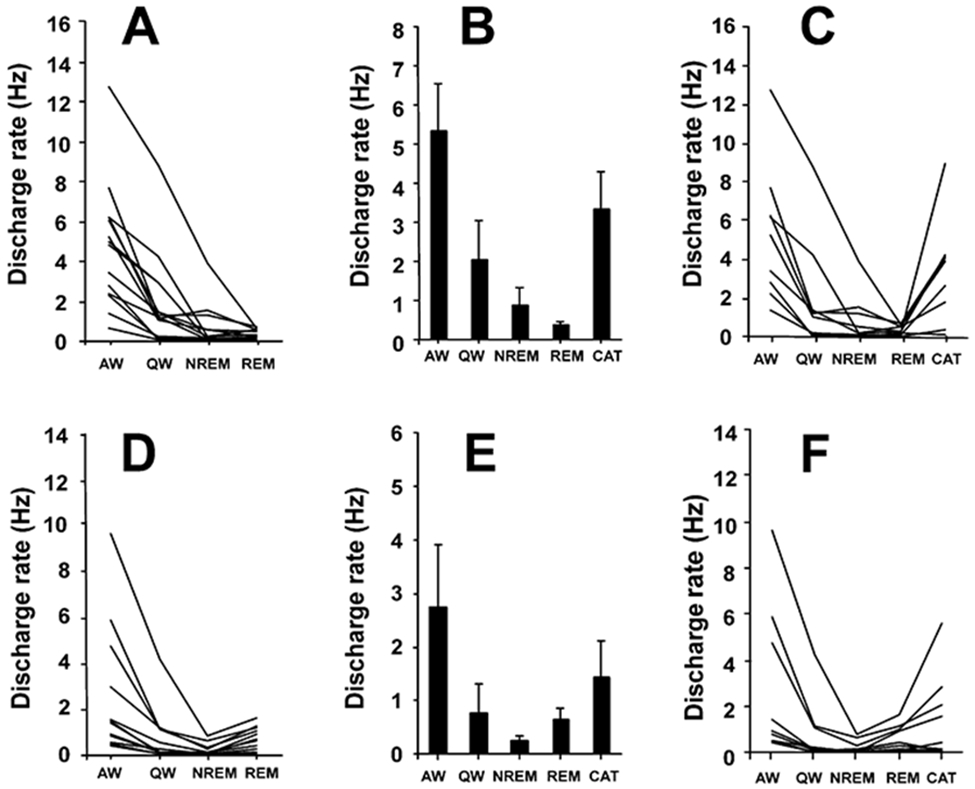
(A) Firing patterns of REM-off neurons across sleep-wake states (n = 14).
(B) Mean discharge rates ± SEM of REM-off neurons that were also recorded during cataplexy (n = 9).
(C) Activity of REM-off neurons that were recorded during cataplexy and sleep-wake states.
(D) Firing patterns of NREM-off neurons that were recorded across sleep-wake states (n = 12).
(E) Mean discharge rates ± SEM of NREM-off neurons also recorded during cataplexy (n = 9).
(F) Activity of NREM-off neurons across sleep-wake states and in cataplexy. AW, active waking; QW, quiet waking; NREM, non-rapid eye movement sleep; REM, rapid eye movement sleep; CAT, cataplexy.
Figure 4. Comparison of Mean Discharge Rates and Cataplexy-Related Activity of REM-Off Cells Recorded from Three Brain Regions.
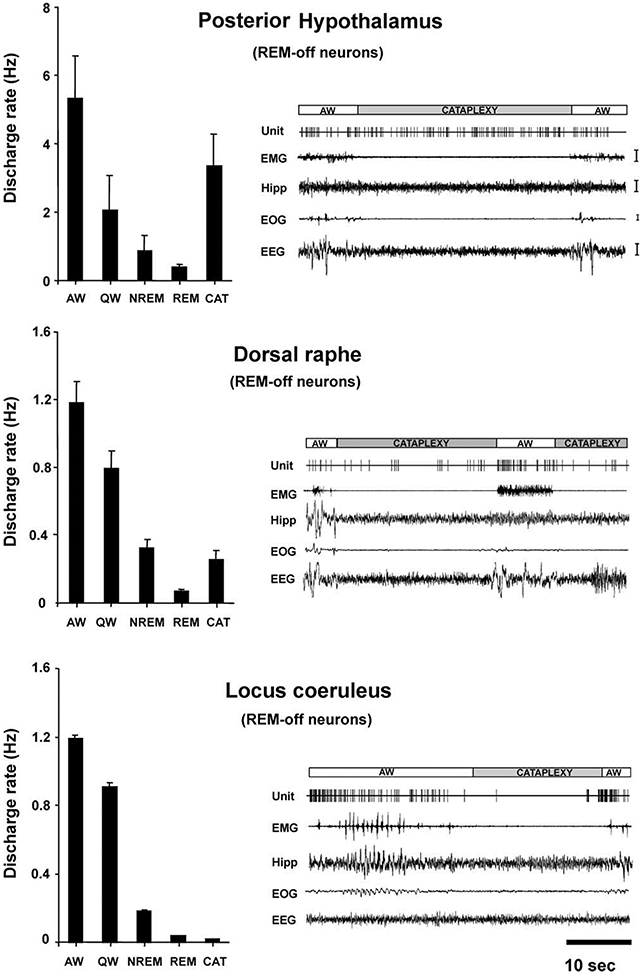
Posterior hypothalamic (histaminergic) neurons remained active, whereas dorsal raphe (serotonergic) neurons reduced discharge (Wu et al., 2004), and locus coeruleus (noradrenergic) neurons ceased discharge (Wu et al., 1999) during cataplexy. All of these cell types were active in waking, reduced discharge in NREM sleep, and were silent or nearly silent in REM sleep. Calibration: EMG, 100 μV; Hipp, 50 μV; EOG, 50 μV; EEG, 50 μV. Abbreviations as in Figure 2 and 3 legends.
Effect of Cataplexy-Enhancing Drugs on the Activity of REM-Off Cells
All three cataplexy-enhancing drugs that were tested produced no significant change in the firing rate of posterior hypothalamic REM-off cells, as compared to the pretreatment quiet waking state. After the administration of the anticholinesterase physostigmine salicylate (n = 6), the α1 adrenergic antagonist prazosin (n = 2), and the α2 adrenergic agonist BHT-920 (n = 3), these cells remained active or increased their firing rate during the period before cataplexy onset, as compared to pretreatment levels (Figure 5A). The behavior of these cells contrasts with that of LC and dorsal raphe cells, which reduce their discharge rate after administration of these drugs and further reduce their rate immediately prior to and during spontaneous cataplexy attacks (Figure 5B; Wu et al., 1999, 2004).
Figure 5. Effect of Drugs Affecting Cataplexy or Alertness on the Activity of REM-Off Cells, GABA Release, and Severity of Cataplexy.
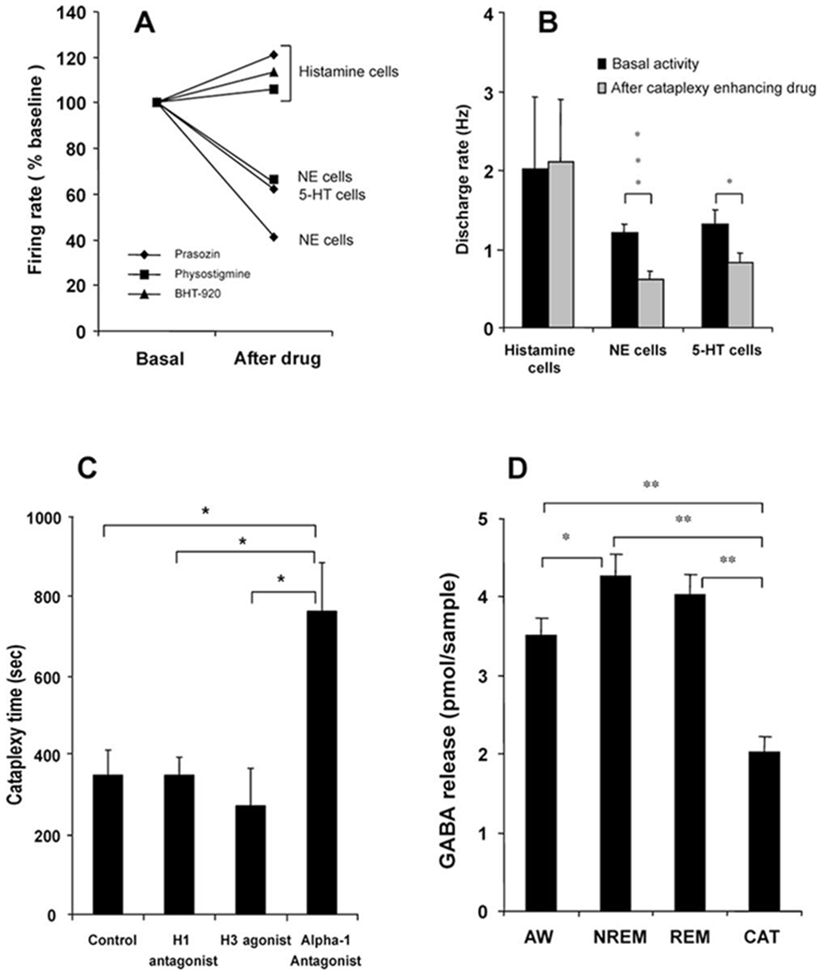
(A) Histaminergic REM-off cells remained active or increased activity in waking after the administration of the anticholinesterase physostigmine salicylate (n = 6), the α1 antagonist prazosin (n = 2), and the α2 agonist BHT-920 (n = 3), as compared to pretreatment (basal activity taken as 100%). Noradrenergic (NE) REM-off cells reduced their discharge rate after the administration of physostigmine (n = 3) and prazosin (n = 6; Wu et al., 1999). Serotonergic (5-HT) REM-off neurons also reduced their discharge rate after prazosin (n = 15; Wu et al., 2004). The rate changes that are shown preceded cataplexy attacks, which were accompanied by further rate decreases in serotonergic and noradrenergic cells, but not in histaminergic cells.
(B) The mean discharge rate in waking shows that the activity of histaminergic cells did not change (t = −0.075; df = 16; p < 0.94), and noradrenergic (t = 4.2; df = 16; p < 0.001) and serotonergic (t = 2.2; df = 28; p < 0.05) cell activity was significantly reduced after the administration of cataplexy-inducing drugs.
(C) Effect of histaminergic and noradrenergic antagonists on severity of cataplexy. The modified food elicited cataplexy test was done after intravenous administration (n = 5) of the α1 antagonist prazosin, the H1 antagonist mepyramine, the H3 (autoreceptor) agonist α-methylhistamine, and normal saline (control). The H1 antagonist and the H3 agonist did not alter cataplexy time, whereas the α1 antagonist greatly increased cataplexy. **p < 0.05.
(D) GABA release in the ventral PLH was significantly higher in NREM sleep compared to waking (*p < 0.05). In cataplexy (n = 16), GABA release was significantly reduced when compared with REM sleep (n = 9), NREM sleep (n = 23), and waking (n = 15) states. **p < 0.01. Abbreviations as in Figure 2 and 3 legends.
Effect of Histaminergic and Noradrenergic Antagonists on Cataplexy
The modified food elicited cataplexy test (Boehmer et al., 2004) showed that intravenous administration of the α1 antagonist prazosin significantly (p < 0.05; Bonferroni t test) aggravated cataplexy (762.0 ± 122.3 s), as compared to saline controls (349.4 ± 61.6 s). On the other hand, the H1 antagonist mepyramine and the H3 (autoreceptor) agonist α-methylhistamine did not significantly change the severity of cataplexy (Figure 5C).
Reduction in GABA Release in Cataplexy
High-pressure liquid chromatography (HPLC) analysis of microdialysis samples showed that γ-aminobutyric acid (GABA) release in the tuberomammillary region significantly differed across S-W states and cataplexy [F(3,66) = 34.5; p < 0.0001; ANOVA; Figure 5D]. GABA release was significantly higher in NREM sleep as compared to waking (p < 0.05; Dunn’s [Bonferroni] test). Cataplexy (n = 16) was accompanied by significant reduction in GABA release, as compared to REM sleep (n = 9), NREM sleep (n = 23), and waking (n = 15) (p < 0.01 for all; Dunn’s [Bonferroni] test), indicating that histamine neurons are disinhibited during cataplexy.
NREM-Off Cells Have Elevated Discharge Rates in REM, QW, and Cataplexy
NREM-off cells and the others listed below are presumed to be nonhistaminergic ventral PLH cells on the basis of their spike widths and their discharge rate profiles across the S-W cycle. The transmitter(s) they release is unknown.
A complete cessation of firing or a very slow firing rate was seen in NREM-off cells during NREM sleep (Figure 3D; Table 1). The firing rate was significantly lower in NREM sleep than in AW (t = 3.2; df = 11; p < 0.05; Bonferroni t test). These cells also showed significantly lower firing rates in QW (t = 3.6; df = 11; p < 0.01; Bonferroni t test) and REM sleep (t = 2.7; df = 11; p < 0.05; Bonferroni t test) than in AW, although firing rates in REM sleep were significantly higher than those in NREM sleep (Figure 3D; t = 4.8; df = 11; p < 0.01; Bonferroni t test). Their mean firing rate in cataplexy was nonsignificantly higher than those in QW, NREM sleep, and REM sleep but lower than that in AW (Figures 3E and 3F; Table 1).
Wake/REM-Active Cells Do Not Alter Their Firing Rate in Cataplexy Compared with QW and REM Sleep
The largest group of cells recorded (49%) from the ventral PLH was wake/REM-active neurons. These cells showed high discharge rates in both AW and REM sleep (Figure 6). Firing rates during NREM sleep were significantly lower than those in AW (Figures 7A and 7B; t = 6.8; df = 76; p < 0.01; Bonferroni t test), QW (t = 4.4; df = 76; p < 0.01; Bonferroni t test), and REM sleep (t = 6.0; df = 76; p < 0.01; Bonferroni t test). The QW discharge rate was also lower than those in REM sleep (t = 4.8; df = 76; p < 0.01; Bonferroni t test) and AW (t = 8.2; df = 76; p < 0.01; Bonferroni t test). These cells were as active in cataplexy as in waking (Figure 7B). The average firing rate during cataplexy was significantly higher than that in NREM sleep (t = 4.3; df = 44; p < 0.01; Bonferroni t test) but was not significantly different from those in QW, AW, or REM sleep (Figure 7B; Table 1).
Figure 6. Activity of Ventral PLH Wake/REM-Active Neurons across the Sleep-Wake Cycle and in Cataplexy.
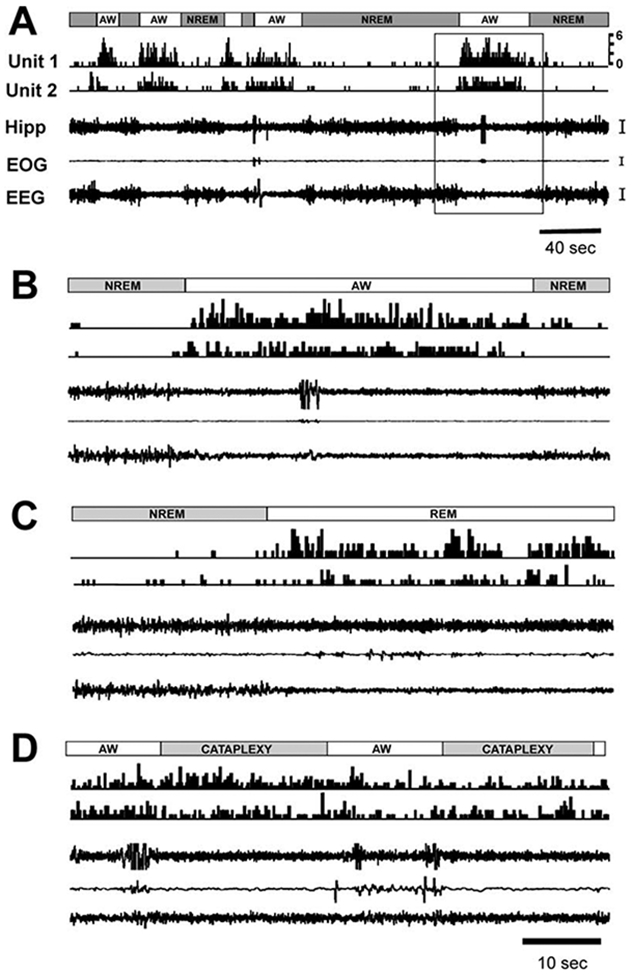
(A) Single-cell discharge (unit 1 and unit 2) patterns (0.2 s bin) across sleep-wake episodes. Gray bars, NREM sleep; unfilled bars, active waking (AW).
(B) Enlarged view of the area indicated by a box in (A).
(C) Episodes of NREM and REM sleep.
(D) Episodes of AW and cataplexy (last AW episode unlabeled). Calibration: Hipp, 50 μV; EOG, 50 μV; EEG, 50 μV. Abbreviations as in Figure 2 and 3 legends.
Figure 7. Discharge Patterns of PLH Wake/REM-Active, Wake-Active, REM-Active, and State-Independent Neurons.
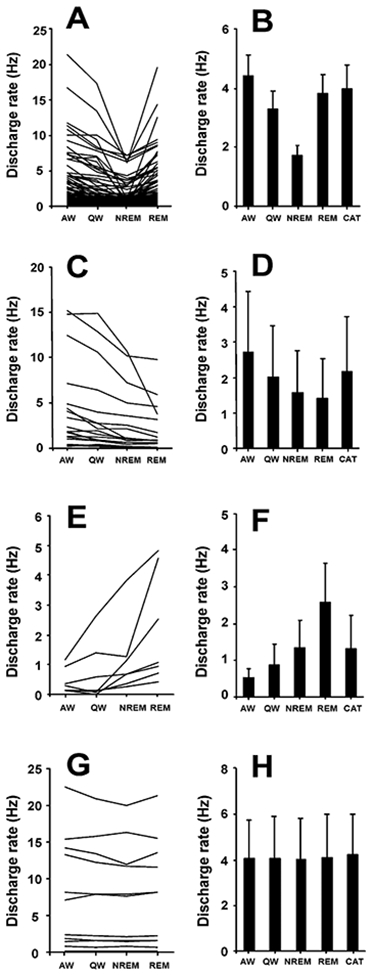
(A) Firing patterns of wake/REM-active neurons across sleep-wake states (n = 77).
(B) Mean discharge rates ± SEM of wake/REM-active neurons that were also recorded during cataplexy (n = 45).
(C) Discharge patterns of wake-active neurons across sleep-wake states (n = 19).
(D) Mean discharge rates ± SEM of wake-active neurons during cataplexy and sleep-wake states (n = 9).
(E) Firing patterns of REM-active neurons across sleep-wake states (n = 7).
(F) Mean discharge rates ± SEM of REM-active neurons that were also recorded during cataplexy (n = 5).
(G) Firing patterns of state-independent neurons across sleep-wake states (n = 11).
(H) Mean discharge rates ± SEM of state-independent neurons that were recorded during cataplexy (n = 5). Abbreviations as in Figure 3.
Wake-Active Cells Have Similar Discharge Rates in QW and Cataplexy
The second largest group of cells (12%) in the ventral PLH region, wake-active neurons, showed maximal discharge rate during AW and significantly reduced discharge rates during NREM sleep (t = 4.4; df = 18; p < 0.01; Bonferroni t test) and REM sleep (t = 3.6; df = 18; p < 0.01; Bonferroni t test) compared to AW (Figure 7C; Table 1). Mean firing rates during NREM sleep (t = 3.2; df = 18; p < 0.05; Bonferroni t test) and REM sleep (t = 2.6; df = 18; p < 0.05; Bonferroni t test) were significantly lower than that in QW (Figure 7C). The mean discharge rate during cataplexy was nonsignificantly higher than those in NREM sleep and REM sleep but was lower than that in AW (Figure 7D; Table 1). The average activity of this group of cells during cataplexy was comparable to that seen during QW (Figure 7D; Table 1).
REM-Active Cells Do Not Alter Firing in Cataplexy Compared with QW but Fire Less in Cataplexy Than in REM Sleep
REM-active cells showed maximal tonic discharge rate selectively during REM sleep (Figure 7E; Table 1). These cells had an average firing rate in REM sleep that was five times higher than that in AW (t = 3.1; df = 6; p < 0.05; Bonferroni t test; Table 1). Their firing rate during REM sleep was also significantly higher than their firing rate in QW (t = 3.4; df = 6; p < 0.05; Bonferroni t test; Table 1). REM-active cells showed a nonsignificant increase in discharge rate during cataplexy, as compared to those in AW, QW, and NREM sleep, but this rate was lower than the REM sleep discharge rate (Figure 7F; Table 1).
State-Independent and Other Cells Fire during Cataplexy
Cells in the state-independent category did not show substantial changes in their discharge rates across S-W states or in cataplexy (Figures 7G and 7H; Table 1).
In the category of “other,” three neurons in NREM sleep and two in QW showed maximal discharge, and the remaining cells showed a high discharge rate in more than one S-W state (Table 1). On average, these cells were somewhat more active during cataplexy than in QW (Table 1).
Hippocampal Theta Activity Is Present during Cataplexy
Hippocampal theta activity was pronounced during cataplexy episodes, along with muscle tone suppression (Figure 8). Theta (3.5–6 Hz) power varied significantly among REM sleep, cataplexy, NREM sleep, QW, and AW states (F = 41.4; df = 4,3; p < 0.0001; ANOVA). Theta power was significantly higher in cataplexy than in both AW and QW (p < 0.05 and p < 0.01, respectively; Dunn’s [Bonferroni] test), as was also true for REM sleep with respect to AW and QW (p < 0.01 for both; Dunn’s [Bonferroni] test). However, theta power was lower in cataplexy than in REM sleep (p < 0.01; Dunn’s [Bonferroni] test).
Figure 8. Hippocampal EEG Power Spectrum across Sleep-Wake States and in Cataplexy.
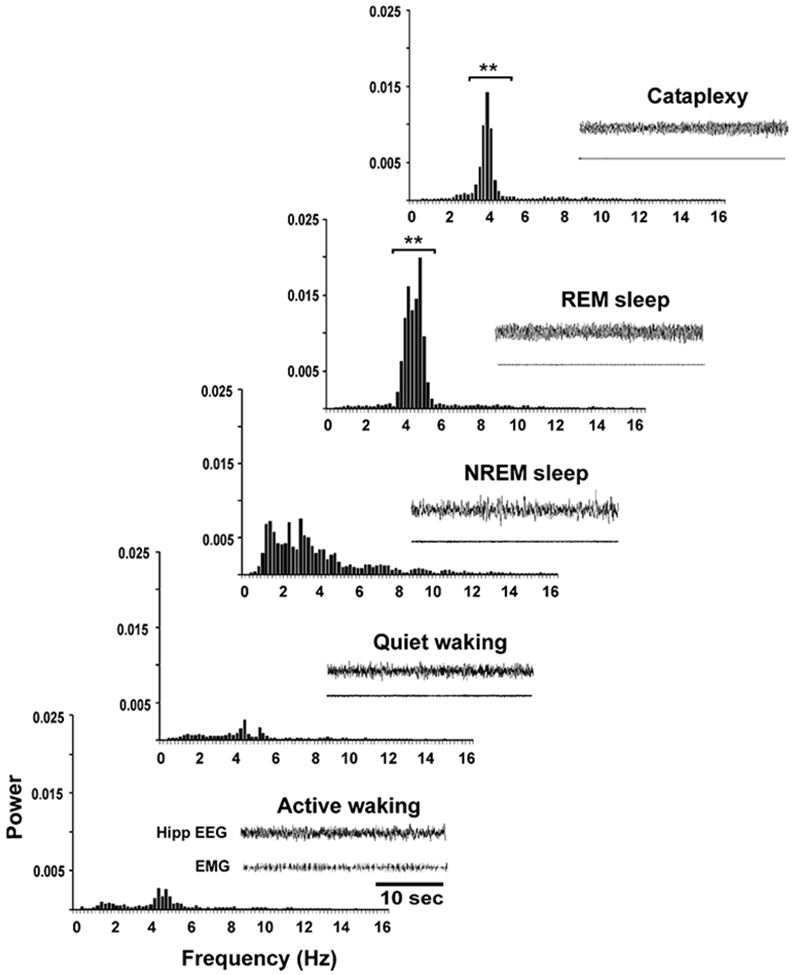
Hippocampal theta (3.5–6 Hz) power is significantly higher in REM sleep than in other sleep-waking states. Theta power is significantly higher during cataplexy than in AW and QW but lower than in REM sleep. Hippocampal EEG and EMG are also shown for each state. Power on y axis is presented in arbitrary units. **p < 0.01, compared to AW and QW.
Discussion
Identification of Histamine Cells
Histamine-containing cells are localized to the tuberomammillary region of the posterior hypothalamus in the rat, cat, and human (Lin et al., 1988; Panula et al., 1984, 1990; Reiner and McGeer, 1987; Sakai et al., 1990; Steininger et al., 1999; Vanni-Mercier et al., 2003; Watanabe and Yanai, 2001; Watanabe et al., 1984). We find a similar localization in the narcoleptic dog, with histamine-IR cells concentrated in the ventral posterior hypothalamus and adjacent mammillothalamic and perifornical regions. Histamine cells are concentrated in a region caudal to but having some overlap with the region containing hypocretin/orexin (Hcrt) cells (Thannickal et al., 2000a, 2000b).
Cells that cease discharge in REM sleep (REM-off) have been recorded in posterior hypothalamic regions that contain histamine-IR cells (Sakai et al., 1990; Vanni-Mercier et al., 2003). In rats and cats, it has been shown that these posterior hypothalamic REM-off cells have long-duration action potentials, a property shown to be unique to histamine-containing cells in this region (Reiner and McGeer, 1987; Sakai et al., 1990; Uteshev et al., 1995; Vanni-Mercier, 2003). The REM-off cells that we have observed in the canine posterior hypothalamus have significantly longer action potentials than other cell types that we have recorded from within this area.
Activity of Histamine, Norepinephrine, and Serotonin Cells in Cataplexy
We have found that, during cataplexy attacks, presumed histamine-containing cells fire at levels similar to or higher than those seen in quiet waking (QW). Our micro-dialysis-HPLC measurements showed that GABA release into the tuberomammillary nucleus was significantly reduced in cataplexy relative to sleep and waking levels. This reduction in GABA release is likely to disinhibit histamine cells (Nitz and Siegel, 1996). Along with increased forebrain excitation, this may explain the maintained discharge of histamine cells at a time when noradrenergic and serotonergic neurons are decreasing activity. Sherin et al. (1998) have demonstrated that histamine neurons receive a major GABAergic projection from the ventrolateral preoptic area (VLPO). In earlier work (Sherin et al., 1996), they demonstrated that neurons in the VLPO were selectively active during sleep. In the current work, this linkage between sleep and GABA release is shown to be maintained, with reduced GABA release under conditions of cataplexy, a state of alert waking. The presumed reduction of activity in VLPO sleep-active neurons during cataplexy allows continued activity in histamine neurons, which in turn supports other physiological changes that are required for waking.
The maintained discharge of histamine cells during cataplexy is consistent with what we know about the state of cataplexy and the role of histamine cells in maintaining wakefulness. The histamine neurons of the hypothalamus have strong projections to EEG arousal-related areas in the forebrain and brainstem (Lin et al., 1988). Injection of histamine agonists produces extended arousal (Lin et al., 1994), and antihistamines impair alertness (Bender et al., 2003).
We find that the noradrenergic, serotonergic, and histaminergic REM-off cells differ dramatically in their activity profiles during cataplexy. We have reported that the presumed noradrenergic cells of the LC decrease firing prior to cataplexy, cease firing during cataplexy, and resume firing immediately prior to the offset of cataplexy (Wu et al., 1999). This discharge profile is consistent with other work showing that muscle tone suppression that is triggered by brainstem activation is linked to cessation of norepinephrine release onto motoneurons (Lai et al., 2001). We have further shown that LC neurons are silenced when pontine or medullary stimulation is used to trigger muscle tone suppression (Mileykovskiy et al., 2000). When the pontine pathway to the LC is blocked, stimulation of motor inhibitory sites in the pons and medulla produces motor excitation rather than inhibition (Kohyama et al., 1998; Siegel et al., 1983; Siegel and Tomaszewski, 1983). Therefore, activation of the brainstem muscle tone suppression system that causes the release of glycine and GABA onto motoneurons (Chase and Morales, 2000; Kodama et al., 2003) also causes a cessation of LC discharge and consequent reduction of norepinephrine release onto motoneurons. This cessation of LC discharge is linked to the suppression of muscle tone in the decerebrate animal, to the normal atonia of REM sleep, and to the pathological loss of muscle tone in cataplexy (Siegel et al., 1991, 1992). Inactivation of the LC causes a reduction of ipsilateral muscle tone (Andre et al., 1992; D’Ascanio et al., 1988). In contrast, lesion of the histamine cells or complete disconnection of histamine cells from motor systems by transection of the neuraxis does not diminish muscle tone (T.C. Chou et al., 2001, Soc. Neurosci., abstract; Lai and Siegel, 1991).
Locus coeruleus cells, which have extensive rostral projections, are active in relation to alert waking behaviors (Aston-Jones et al., 2001). However, the alert waking state of cataplexy arises despite a complete cessation of LC discharge. Thus, although activity in LC cells contributes to cortical arousal, it is not required for maintenance of the alert waking state during cataplexy episodes.
We report that, during cataplexy, dorsal raphe neurons have a discharge pattern that is intermediate between that of cells in the LC (Wu et al., 1999, 2004) and that of histamine-containing cells. In addition, we have found that brainstem stimulation that suppresses muscle tone also reduces serotonin release onto motoneuron pools (Lai et al., 2001). The reduction in discharge that we have seen in the dorsal raphe cells of narcoleptic dogs during cataplexy might be sufficient to produce the reduction in serotonin release seen in brainstem stimulation-induced muscle tone suppression. However, since the dorsal raphe nucleus has largely ascending projections, it is unlikely that this nucleus is responsible for a significant portion of the serotonin release seen in motoneuron pools. Rather, the main sources of serotonin in spinal and brainstem motoneuronal pools are the pontine and medullary raphe groups. We speculate that, during cataplexy, these cell groups may behave more like LC neurons than dorsal raphe cells. The ascending projections of dorsal raphe cells may make some contribution to cortical arousal in cataplexy as well as in normal waking (Jacobs and Azmitia, 1992; Wu et al., 2004), along with the activity of histamine cells.
The administration of prazosin or physostigmine, both of which precipitate cataplexy, significantly decreases the activity of noradrenergic and serotonergic cells prior to the onset of cataplexy episodes, whereas these same drugs either do not change or slightly increase the activity of histaminergic cells prior to cataplexy episodes (Figure 5). The most effective drugs for treating cataplexy in both narcoleptic dogs and humans are those that manipulate the monoamine systems, with noradrenergic agonists being particularly important in treating cataplexy (Nishino et al., 1993). Antihistamines impair alertness in no6rmals and narcoleptics but do not increase cataplexy in human narcoleptics (Aldrich, 1998; Guilleminault and Anognos, 2000) or in the canine Hcrtr2 mutant narcoleptic, as shown in the current study.
Other Cell Types
We identified six cell types with different S-W discharge profiles, in addition to those of the presumed histaminergic cells of the ventral PLH. Neurons in this area exhibit a number of different neurotransmitter phenotypes and projection patterns (Lantos et al., 1995). It remains to be determined how the sleep cycle and cataplexy discharge patterns that we see relate to the other characteristics of these cell groups.
Hippocampal Theta
In this paper, we have replicated and extended prior indications that hippocampal theta is elevated during cataplexy (Kushida et al., 1985) as it is during REM sleep (Prospero-Garcia et al., 1993). Figure 8 shows that the amplitude of theta in cataplexy approaches that in REM sleep and is significantly larger than that seen in active waking.
The elevated theta, reduced muscle tone, and reduced activity of noradrenergic LC cells and serotonergic dorsal raphe cells are among the most striking-similarities between cataplexy and REM sleep. The persistent activity of histamine cells and the maintenance of environmental awareness in cataplexy, however, contrast with the cessation of histamine activity and loss of environmental awareness in REM sleep.
Histamine and Hypocretin
Most human narcolepsy is caused by a loss of Hcrt neurons (Thannickal et al., 2000a) and a consequent reduction in Hcrt levels (Peyron et al., 2000). Gliosis accompanying the loss of Hcrt cells is consistent with the hypothesis that this loss occurs postnatally and that it is accompanied by inflammation (Thannickal et al., 2000a). Hcrt cells densely innervate histamine cells (Eriksson et al., 2001). We recently found that gliosis is most intense in the posterior hypothalamus, suggesting that the loss of Hcrt terminals in this region and the consequent loss of Hcrt innervation of histamine cells may be important components of the pathology of narcolepsy (Thannickal et al., 2003).
Canine genetic narcolepsy is caused by a mutation in the Hcrtr2 gene rather than a loss of Hcrt neurons (Lin et al., 1999; Thannickal et al., 2000b). However, canine and human narcolepsy are similar in their symptoms, physiology, behavioral triggers, and response to pharmacological agents, suggesting that most of the symptoms are triggered downstream of the Hcrt cell loss or receptor malfunction that causes the disorder, particularly at the level of monoamine neurons (Siegel, 1999).
It is important to note that the direct effects of the complete loss of Hcrtr2 function in Hcrtr2 mutant dogs cannot explain the clear-cut differences in the activity of norepinephrine, serotonin, and histamine cells during cataplexy. In the normal animal, all three cell groups are excited by Hcrt (Hagan et al., 1999; Horvath et al., 1999; Ivanov and Aston-Jones, 2000; van den Pol et al., 2002; Bourgin et al., 2000; Li et al., 2002; Bayer et al., 2001). LC cells have predominantly Hcrtr1, and histamine cells have predominantly Hcrtr2. Therefore, one might expect that histamine cells would have abnormal slowing in the Hcrtr2 mutant dogs and that LC cells would have normal discharge (and serotonergic cells would have an intermediate pattern). Instead, we see just the reverse during cataplexy. LC cells have abnormal slowing, whereas histamine cells maintain waking discharge levels during cataplexy.
It is also clear that the overall REM-off profile is present in norepinephrine, serotonin, and histamine cells even in the absence of functional Hcrtr2, indicating that this receptor does not mediate the sleep cycle modulation of discharge in the histamine cells or in LC or raphe cells. The discharge rate of histamine REM-off cells in waking has been reported to be 2.4 Hz in cats and 3.9 Hz in rats (Sakai et al., 1990; Steininger et al., 1999; Vanni-Mercier et al., 2003). We find it to be 4.8 Hz in our Hcrtr2 mutant dogs. Since Hcrt is excitatory to histamine cells, this difference in rate could not be a direct effect of the loss of Hcrtr2, but might be a result of species differences.
We present a model (Figure 9) that can explain some of our observations by incorporating known Hcrt and non-Hcrt effects, known afferents, projections and actions of LC and histamine cells, and hypothesized actions of Hcrt receptors on the self-excitation of Hcrt neurons. This model considers and incorporates recent findings by Willie et al. (2003) and Kisanuki et al. (2001) that indicate that cataplexy is more severe in ligand knockout (KO) and Hcrtr1 KO animals than in Hcrtr2 KO animals and that sleepiness is worse in Hcrtr2 KO animals.
Figure 9. Model for the Mechanism of Cataplexy and Sleepiness in Narcolepsy.

Locus coeruleus noradrenergic neurons (LC) may be involved in the maintenance of muscle tone, and posterior hypothalamus histamine neurons (HA) may be involved in the maintenance of alertness in cataplexy. In quiet waking, non-Hcrt excitatory inputs are reduced. Thicker input symbolizes more release of neurotransmitters. Net level of neuronal activity is given schematically for each state. Hcrtr, Hypocretin receptor; KO, knockout.
Model of the Role of Hcrt in the Control of Norepinephrine, Histamine, and Hcrt Neurons in Relation to Cataplexy
Type 1 Hcrt receptors, the dominant Hcrt receptor type on LC noradrenergic neurons, have an important role in facilitating muscle tone during certain types of emotionally excited waking, thereby preventing cataplexy (Figure 9). In the normal animal, during nonexcited waking, Hcrt tonically facilitates the LC via Hcrtr1. The net effect of other inputs is also facilitatory. In moderately and highly excited waking states, non-Hcrt inputs decrease, as evidenced by the transient muscle weakness seen in normal individuals during certain emotionally charged behaviors, such as laughter or surprise. Hcrt release increases to compensate for this, allowing muscle tone to be maintained.
In Hcrt receptor-1 KO animals, there is no substantial Hcrt input to the LC under any condition because of the absence of functional Hcrtr1. As non-Hcrt input decreases, cataplexy rapidly occurs.
In contrast, there is tonic Hcrt input to LC neurons in Hcrtr2 mutant dogs and mice through the intact Hcrtr1. However, we hypothesize that the lack of functional Hcrtr2 disrupts the hypothalamic positive feedback loop (Li et al., 2002) responsible for elevated levels of Hcrt release with excitement in normal animals. This prevents or diminishes excitation of Hcrt neurons, resulting in cataplexy of lesser intensity than that seen in Hcrtr1 or ligand KOs.
In Hcrtr2 mutant narcoleptic dogs, little Hcrt effect would be expected on histamine cells, which contain primarily Hcrtr2. We hypothesize that the histamine cells receive excitatory input from limbic regions activated during the emotional stimulation responsible for triggering cataplexy (Gulyani et al., 2002). GABA release, originating from hypnogenic neurons of the VLPO, is greatly reduced during cataplexy, because VLPO GABAergic neurons are inhibited by forebrain limbic regions (Chou et al., 2002) that are activated during emotional stimulation. This accounts for the maintained activity of histamine neurons during cataplexy when compared with quiet waking states.
This model explains the more intense cataplexy reported in ligand KO and Hcrtr1 KO animals as compared to Hcrtr2 KO animals and predicts that Hcrt supplementation will be more effective in blocking cataplexy in Hcrtr2 KO/mutant animals (John et al., 2000) than in Hcrtr1 KO animals (Figure 9). Humans with loss of Hcrt neurons will have symptoms of both Hcrtr1 and Hcrtr2 deficiency, attenuated by residual Hcrt from surviving Hcrt neurons (Thannickal et al., 2000a).
Model of the Role of Hcrt in the Control of Histamine and Hcrt Neurons in Relation to Sleepiness
Type 2 Hcrt receptors on histamine neurons have a major role in preventing sleepiness during waking. A reduction of histamine levels has been seen in the cerebrospinal fluid of Hcrtr2 mutant narcoleptic dogs (Nishino et al., 2001) and narcoleptic humans (Nishino et al., 2002). It is well known that antihistamines cause sleepiness and that stimulation of histamine cells produces arousal (Bender et al., 2003; Guilleminault and Anognos, 2000; Lin et al., 1994). The arousing effect of Hcrt has been shown to be heavily dependent on activation of histamine-containing cells (Huang et al., 2001; Ishizuka et al., 2002). In normals, as non-Hcrt input diminishes in extended quiet waking periods, Hcrt neurons, which are tonically active in the absence of input (Eggermann et al., 2001), continue to release the peptide at a basal level (Bayer et al., 2001; Eriksson et al., 2001; Yamanaka et al., 2002; Figure 9). In Hcrtr1 KO animals, effective Hcrt stimulation of histamine cells is possible; however, we hypothesize that the positive feedback loop underlying recruitment of Hcrt cells is impaired by the mutation. This results in an intermediate level of excitation of histamine cells and consequent sleepiness. In Hcrtr2 KO/mutant animals, no effective Hcrt excitation of histamine cells occurs, resulting in reduced histamine cell activity and severe sleepiness whenever non-Hcrt inputs to these cells are reduced (e.g., in quiet waking). In human narcoleptics, the loss of Hcrt cells will produce Hcrtr1 and Hcrtr2 KO effects. This model predicts that Hcrtr2 KOs/mutants will be sleepier than Hcrtr1 KOs and supplementation will be more effective in reversing sleepiness in Hcrtr1 KOs. Hcrt supplementation should be effective in human narcoleptics, who are likely to have intact Hcrtr1 and Hcrtr2. The selectively increased loss of Hcrt cells and axons and the increased gliosis seen in the posterior hypothalamus of human narcoleptics suggest that this region is a focus of inflammation and damage in human narcolepsy.
Conclusion
We find that cataplexy is the only condition in which the levels of activity of different monoaminergic cell groups diverge over sustained time periods. This provides a unique insight into their functional roles. Histamine cells remain active during cataplexy. The maintenance of histamine cell discharge, together with the concurrent loss of discharge in LC and activation of medullary motor inhibitory neurons that we have reported previously in canine narcoleptics (Siegel et al., 1991), can explain the unique characteristics of cataplexy, which combines loss of muscle tone with maintenance of environmental awareness. The divergent patterns of discharge of the REM-off cell groups in cataplexy support the concept that activity in histamine cell groups is strongly linked to forebrain arousal, whereas serotonergic and noradrenergic cells have important roles in the regulation of muscle tone and perhaps the coordination of motor activity with forebrain arousal.
Experimental Procedures
Animals
Four genetically narcoleptic (Hcrtr2 mutant; Lin et al., 1999) Doberman pinschers (3 males, 1 female; 4.5 ± 1.3 years) were used. All the procedures were in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals and were approved by the Animal Research Committees of the University of California at Los Angeles and of the Veterans Administration Greater Los Angeles Healthcare System.
Surgery
Under isoflurane anesthesia, electrodes for the assessment of different S-W stages (EEG, EMG, EOG, and hippocampal EEG) were chronically implanted as described earlier (Siegel et al., 1991; Wu et al., 1999). To record unit activity, microdrive assemblies with 25 and 64 μm stainless steel microwires (California Fine Wire Co., Grover Beach, CA) were stereotaxically implanted into the tuberomammillary area of the ventral PLH (A 21, L 3, H 12; Lim et al., 1960). In two dogs, cannulae were implanted into the ventral PLH for the microdialysis experiments.
Recording of Unit Activity during S-W Stages and Cataplexy
The dogs were free to move around the recording chamber (1.2 m × 12 m × 1.2 m). Signals for the assessment of different S-W stages were amplified with a Grass model 78 E Polygraph (Grass Instruments, Quincy, MA). All the recordings were carried out between 09:00 and 18:00 hr. Single-cell activity was recorded during S-W stages and food elicited cataplexy tests (FECTs).
Stable single units with a signal-to-noise ratio of at least 3:1 were studied for at least three episodes of various S-W stages (Wu et al., 2004). Many of these cells were also recorded during cataplexy attacks in waking. All slow wave and unit activity signals were digitized using a CED 1401 Plus interface and analyzed using Spike2 software (Cambridge Electronic Design Ltd., Cambridge, UK). Six samples from each episode were used for power spectral analysis. Spike duration was determined from the onset of the negative component to the offset of the positive component using averaged wave-forms taken from 200 action potentials.
Cells were held for recording as long as needed to complete the experimental protocol, up to 3 days in some cases. The spike waveform shape, absence of abrupt change in amplitude or optimal microwire derivations, maintenance of discharge profile over the sleep cycle, and the waking behavioral correlates of discharge (Siegel and Tomaszewski, 1983; Siegel et al., 1983) allowed us to confirm that the same cells were recorded across multiple recording sessions.
Classification of S-W Stages and Cataplexy
Sleep-wake stages and cataplexy were classified based on electrophysiological parameters and visual observation as previously described (Siegel et al., 1991; Wu et al., 1999). In this study, cataplexy was induced by the introduction of food or play objects or occurred spontaneously as previously described (John et al., 2000; Wu et al., 1999). The discharge profile of each neuron was recorded over a minimum of five complete cataplexy episodes.
Classification of Neurons Based on Their Discharge Profile
The wake/REM-active type was defined by high firing rates in both AW and REM sleep and lowest firing rates in NREM sleep (<80% of mean AW firing rate). Wake-active cells were defined to be those cells with highest activity in AW, with firing rates during REM and NREM sleep that were 10%–80% of the mean firing rates during AW. NREM-off cells were defined as those cells whose firing rates were <10% in NREM sleep and 10%–50% in REM sleep of the mean firing rates in AW. REM-off cells were defined to be those cells whose firing rates in REM sleep were <10% of the mean firing rates in AW. REM-active cells were defined to be those cells whose highest discharge rates occurred during REM sleep. State-independent (SI) cells were defined to be those whose firing rates in NREM and REM sleep were within 20% of their mean AW firing rate. Cells that did not fit into any of the above categories were classified as “other.”
Effects of Cataplexy-Inducing Drugs on REM-Off Cell Activity
The following cataplexy-enhancing drugs were administered to study their effects on the discharge rates of REM-off cells: the anticholinesterase physostigmine salicylate (50 μg/kg, i.v.; Forest Pharmaceuticals, Inc., St. Louis, MO), the α1 antagonist prazosin hydrochloride (0.5 mg/kg, p.o.; UDL Laboratories Inc., Rockford, IL), and the α2 agonist BHT-920 (2 μg/kg, i.v.). The effect of prazosin on cell activity was monitored between 2 and 2.5 hr after drug administration, and the effect of physostigmine was assessed between 5 and 10 min after injection, the time periods during which they have their most marked effects on cataplexy probability (Wu et al., 1999). The effect of BHT-920 was assessed between 5 and 30 min after injection.
Microdialysis and High-Pressure Liquid Chromatography
Microdialysis probes (A-I-Model; Eicom Corp., Kyoto, Japan) with 1 mm long membranes were inserted through the guide cannulae 12 hr prior to the study. Artificial cerebrospinal fluid (Harvard Apparatus, MA) was continuously perfused at 2 μl/min through the ventral PLH probes for 2 hr prior to the experiment. Dialysate was collected every 5 min during waking, NREM sleep, REM sleep, and cataplexy. For the induction of prolonged cataplexy, apomorphine (D-2 agonist; 4 mg/kg, i.v.; Sigma) was given 90 min after the administration of the dopamine receptor antagonist domperidone (1 mg/kg, i.v.; Sigma), which does not cross the blood-brain barrier (M.-F. Wu et al., 2003, Soc. Neurosci., abstract). Apomorphine produces complete cataplexy within 2 min of injection, and this lasted more than 40 min.
Samples were collected prior to, during, and after cataplexy episodes. The concentration of GABA in the dialysate was detected by HPLC (EP-300; Eicom Corp., Kyoto, Japan) with fluorescent detection (excitation/emission = 340/440 nm), as described in our prior studies (John et al., 2003; Kodama et al., 2003).
Effect of Histaminergic and Noradrenergic Antagonists on Cataplexy
The modified food elicited cataplexy test (Boehmer et al., 2004) was done 10 min after intravenous administration (n = 5) of the α1 antagonist prazosin (80 μg/kg; Sigma), the H1 antagonist mepyramine (500 μg/kg; Tocris, UK), the H3 (autoreceptor) agonist α-methylhistamine (500 μg/kg; Tocris, UK), and 0.9% saline (as control).
Immunohistochemical Localization of Histaminergic Neurons
At the end of the study, a 15 μA anodal current was passed through the microelectrodes for 20 s to make an iron deposit at the tip of the electrode (Wu et al., 1999). Dogs were sacrificed by an overdose of sodium pentobarbital (50 mg/kg, i.v.) and transcardially perfused with buffered saline and paraformaldehyde. The brains were equilibrated in 30% sucrose, and 40 μm coronal sections of the hypothalamus were cut on a cryostat.
Histamine staining was done on a one-in-four series of hypothalamic sections (120 μm interval). Free-floating sections were incubated for 72 hr at 4°C on an agitator with rabbit antihistamine antiserum (DiaSorin, Stillwater, MN) diluted 1:7500 in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 and 4% normal goat serum. They were rinsed and incubated with biotinylated anti-rabbit IgG (1:200 in PBS) and 4% normal goat serum for 2.5 hr. The sections were then rinsed and incubated in 1:100 ABC in PBS for 2 hr (Vector Laboratories, Inc., Burlingame, CA). After being rinsed with Tris-buffered saline, sections were incubated for 5–10 min in 0.05% diaminobenzidine and 0.001% H2O2, yielding a brown precipitate in histamine-IR neurons. Control sections processed without the antihistamine antiserum showed no specific staining or labeled neurons.
One series of hypothalamic sections was rinsed in 5% potassium ferrocyanide for the Prussian blue reaction to locate the iron deposit at the tip of the electrode and then stained with cresyl violet. Mapping of histamine-IR neurons and reconstruction of the electrode tract were done with the help of a Neurolucida Imaging System (Microbrightfield, Colchester, VT).
Data Analysis
All mean discharge rate values were expressed as action potentials per second ± standard error. Spike widths, hippocampal theta power, and microdialysis data were compared by one-way analysis of variance (ANOVA), followed by post hoc comparisons using Dunn’s (Bonferroni) test.
Acknowledgments
This work was supported by the Medical Research Service of the US Department of Veterans Affairs and USPHS grants NS14610, MH64109, and HL41370. The authors thank Tohru Kodama for his help with preliminary experiments and Bao Phan, David Yau, and Giselle Nguyen for their technical assistance.
References
- Aldrich MS (1998). Diagnostic aspects of narcolepsy. Neurology Suppl. 50, S2–S7. [DOI] [PubMed] [Google Scholar]
- Andre P, D’Ascanio P, Ioffe M, and Pompeiano O (1992). Micro-injections of vasopressin in the locus coeruleus complex affect posture and vestibulospinal reflexes in decerebrate cats. Pflugers Arch. 420, 376–388. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G,and Bloom FE (1981). Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci 1, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Chen S, Zhu Y, and Oshinsky ML (2001). A neural circuit for circadian regulation of arousal. Nat. Neurosci 4, 732–738. [DOI] [PubMed] [Google Scholar]
- Baust W, Holzbach E, and Zechlin O (1972). Phasic changes in heart rate and respiration correlated with PGO-spike activity during REM sleep. Pflugers Arch. 331, 113–123. [DOI] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Serafin M, Saint-Mleux B, Machard D, Jones B, and Muhlethaler M (2001). Orexins (hypocretins) directly excite tuberomammillary neurons. Eur. J. Neurosci 14, 1571–1575. [DOI] [PubMed] [Google Scholar]
- Bender BG, Berning S, Dudden R, Milgrom H, and Tran ZV (2003). Sedation and performance impairment of diphenhydramine and second-generation antihistamines: a meta-analysis. J. Allergy Clin. Immunol 111, 770–776. [DOI] [PubMed] [Google Scholar]
- Boehmer LN, Wu M-F, John J, and Siegel JM (2004). Pharmacological treatment delays onset of canine genetic narcolepsy and reduces symptom severity. Experimental Neurology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgin P, Huitron-Resendiz S, Spier AD, Fabre V, Morte B, Criado JR, Sutcliffe JG, Henriksen SJ, and De Lecea L (2000). Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J. Neurosci 20, 7760–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH, and Morales FR (2000). Control of motoneurons during sleep. In Principles and Practices of Sleep Medicine, Dement WC, ed. (Philadelphia: W.B. Saunders; ), pp. 155–168. [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, and Saper CB (2002). Afferents to the ventrolateral preoptic nucleus. J. Neurosci 22, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascanio P, Pompeiano O, and Stampacchia G (1988). Noradrenergic and cholinergic mechanisms responsible for the gain regulation of vesibulospinal reflexes. Prog. Brain Res 76, 361–374. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, and Muhlethaler M (2001). Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience 108, 177–181. [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, and Haas HL (2001). Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J. Neurosci 21, 9273–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilleminault C (1976). Cataplexy. In Narcolepsy, Guilleminault C, Dement WC, and Passouant P, eds. (New York: Spectrum; ), pp. 125–143. [Google Scholar]
- Guilleminault C, and Anognos A (2000). Narcolepsy. In Principles and Practice of Sleep Medicine, Dement WC, ed. (Philadelphia: W.B. Saunders; ), pp. 676–686. [Google Scholar]
- Gulyani S, Wu M-F, Nienhuis R, John J, and Siegel JM (2002). Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience 112, 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, et al. (1999). Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. USA 96, 10911–10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, Kilduff TS, and van den Pol AN (1999). Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J. Comp. Neurol 415, 145–159. [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, Urade Y, and Hayaishi O (2001). Arousal effect of orexin A depends on activation of the histaminergic system. Proc. Natl. Acad. Sci. USA 98, 9965–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Yamamoto Y, and Yamatodani A (2002). The effect of orexin-Aand -B on the histamine release in the anterior hypothalamus in rats. Neurosci. Lett 323, 93–96. [DOI] [PubMed] [Google Scholar]
- Ivanov A, and Aston-Jones G (2000). Hypocretin/orexin depolarizes and decreases potassium conductance in locus coeruleus neurons. Neuroreport 11, 1755–1758. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, and Azmitia EC (1992). Structure and function of the brain serotonin system. Physiol. Rev 72, 165–229. [DOI] [PubMed] [Google Scholar]
- John J, Wu M-F, and Siegel JM (2000). Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res. Online 3, 23–28. [PMC free article] [PubMed] [Google Scholar]
- John J, Wu M-F, Kodama T, and Siegel JM (2003). Intravenously administered hypocretin-1 alters brain amino acid release: an in vivo microdialysis study in rats. J. Physiol 548, 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Chemelli RM, Tokita S, Willie JT, Sinton CM, and Yanagisawa M (2001). Behavioral and polysomnographic characterization of orexin-1 receptor and orexin-2 receptor double knockout mice. Sleep 24, A22. [Google Scholar]
- Kodama T, Lai YY, and Siegel JM (2003). Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J. Neurosci 23, 1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama J, Lai YY, and Siegel JM (1998). Inactivation of the pons blocks medullary-induced muscle tone suppression in the decerebrate cat. Sleep 21, 695–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushida CA, Baker TL, and Dement WC (1985). Electroenceph-alographic correlates of cataplectic attacks in narcoleptic canines. Electroencephalogr. Clin. Neurophysiol 61, 61–70. [DOI] [PubMed] [Google Scholar]
- Lai YY, and Siegel JM (1991). Ponto-medullary glutamate receptors mediating locomotion and muscle tone suppression. J. Neurosci 11, 2931–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Kodama T, and Siegel JM (2001). Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J. Neurosci 21,7384–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantos TA, Gorcs TJ, and Palkovits M (1995). Immunohistochemical mapping of neuropeptides in the premamillary region of the hypothalamus in rats. Brain Res. Brain Res. Rev 20, 209–249. [DOI] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, and van den Pol AN (2002). Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron–a potential mechanism for orchestrating the hypothalamic arousal system. Neuron 36, 1169–1181. [DOI] [PubMed] [Google Scholar]
- Lim RKS, Liu C, and Moffitt RL (1960). A Stereotaxic Atlas of the Dog’s Brain (Springfield, IL: Thomas; ). [Google Scholar]
- Lin JS, Sakai K, and Jouvet M (1988). Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology 27, 111–122. [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, and Jouvet M (1994). Hypothalamo-preoptic histaminergic projections in sleep-wake control in the cat. Eur. J. Neurosci 6, 618–625. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Kadotani H, Rogers W, Lin X, Qui X, de Jong P, Nishino S, and Mignot E (1999). The REM sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor gene. Cell 98, 365–376. [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Kodama T, Lai YY, and Siegel JM (2000).Activation of pontine and medullary motor inhibitory regions reduces discharge in neurons located in the locus coeruleus and the anatomical equivalent of the midbrain locomotor region. J. Neurosci 20, 8551–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino S, Arrigoni J, Shelton J, Dement WC, and Mignot E (1993). Desmethyl metabolites of serotonergic uptake inhibitors are more potent for suppressing canine cataplexy than their parent compounds. Sleep 16, 706–712. [DOI] [PubMed] [Google Scholar]
- Nishino S, Fujiki N, Ripley B, Sakurai E, Kato M, Watanabe T, Mignot E, and Yanai K (2001). Decreased brain histamine content in hypocretin/orexin receptor-2 mutated narcoleptic dogs. Neurosci. Lett 313, 125–128. [DOI] [PubMed] [Google Scholar]
- Nishino S, Sakurai E, Nevisimalova S, Vankova J, Yoshida Y, Watanabe T, Yanai K, and Mignot E (2002). CSF histamine content is decreased in hypocretin-deficient human narcolepsy. Sleep 25, A476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz D, and Siegel JM (1996). GABA release in the posterior hypothalamus of the cat as a function of sleep/wake state. Am. J. Physiol 40, R1707–R1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panula P, Yang HY, and Costa E (1984). Histamine-containing neurons in the rat hypothalamus. Proc. Natl. Acad. Sci. USA 81, 2572–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panula P, Airaksinen MS, Pirvola U, and Kotilainen E (1990). A histamine-containing neuronal system in human brain. Neuroscience 34, 127–132. [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, et al. (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med 6, 991–997. [DOI] [PubMed] [Google Scholar]
- Prospero-Garcia O, Miller DR, and Henriksen SJ (1993). Hippocampal interneuron activity in unanesthetized rats: relationship to the sleep-wake cycle. Neurosci. Lett 156, 158–162. [DOI] [PubMed] [Google Scholar]
- Reiner PB, and McGeer EG (1987). Electrophysiological properties of cortically projecting histamine neurons of the rat hypothalamus. Neurosci. Lett 73, 43–47. [DOI] [PubMed] [Google Scholar]
- Sakai K, El Mansari M, Lin JS, Zhang JG, and Vanni-Mercier G (1990). The posterior hypothalamus in the regulation of wakefulness and paradoxical sleep. In The Diencephalon and Sleep, Marini G, ed. (New York: Raven Press; ), pp. 171–198. [Google Scholar]
- Salin-Pascual R, Gerashchenko D, Greco M, Blanco-Centurion C, and Shiromani PJ (2001). Hypothalamic regulation of sleep. Neuropsychopharmacology Suppl. 25, S21–S27. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, and Saper CB (1996). Activation of ventrolateral preoptic neurons during sleep. Science 271, 216–219. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, and Saper CB (1998). Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J. Neurosci 18, 4705–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (1999). Narcolepsy: A key role for hypocretins (orexins). Cell 98, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2000). Brainstem mechanisms generating REM sleep. In Principles and Practice of Sleep Medicine, Kryger MH, Roth T, and Dement WC, eds. (Philadelphia: W.B. Saunders Company; ), pp. 112–133. [Google Scholar]
- Siegel JM, and Tomaszewski KS (1983). Behavioral organization of reticular formation: Studies in the unrestrained cat. I. Cells related to axial, limb, eye, and other movements. J. Neurophysiol 50, 696–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, and Tomaszewski KS (1983). Rostral brainstem contributes to medullary inhibition of muscle tone. Brain Res. 268, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer H, Paul R, Shiromani P, Dement WC, Mignot E, and Chiu C (1991). Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science 252, 1315–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer HM, Chiu C, Dement WC, Mignot E, and Lufkin R (1992). Activity of medial mesopontine units during cataplexy and sleep-waking states in the narcoleptic dog. J. Neurosci 12, 1640–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Alam MN, Gong H, Szymusiak R, and McGinty D (1999). Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 840, 138–147. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, and Sejnowski TJ (1993). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. [DOI] [PubMed] [Google Scholar]
- Strecker RE, Nalwalk J, Dauphin LJ, Thakkar MM, Chen Y, Ramesh V, Hough LB, and McCarley RW (2002). Extracellular histamine levels in the feline preoptic/anterior hypothalamic area during natural sleep-wakefulness and prolonged wakefulness: an in vivo microdialysis study. Neuroscience 113, 663–670. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, and Siegel JM (2000a). Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Nienhuis R, Ramanathan L, Gulyani S, Turner K, Chestnut B, and Siegel JM (2000b). Preservation of hypocretin neurons in genetically narcoleptic dogs. Sleep 23, A296. [Google Scholar]
- Thannickal TC, Siegel JM, Nienhuis R, and Moore RY (2003). Pattern of hypocretin (orexin) soma and axon loss, and gliosis, in human narcolepsy. Brain Pathol. 13, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev V, Stevens DR, and Haas HL (1995). A persistent sodium current in acutely isolated histaminergic neurons from rat hypothalamus. Neuroscience 66, 143–149. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Ghosh PK, Liu RJ, Li Y, Aghajanian GK, and Gao XB (2002). Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J. Physiol 541, 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni-Mercier G, Gigout S, Debilly G, and Lin JS (2003). Waking selective neurons in the posterior hypothalamus and their response to histamine H3-receptor ligands: an electrophysiological study in freely moving cats. Behav. Brain Res 144, 227–241. [DOI] [PubMed] [Google Scholar]
- Watanabe T, and Yanai K (2001). Studies on functional roles of the histaminergic neuron system by using pharmacological agents, knockout mice and positron emission tomography. Tohoku J. Exp. Med 195, 197–217. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, Tohyama M, and Wada H (1984). Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res. 295, 13–25. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, et al. (2003). Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 38, 715–730. [DOI] [PubMed] [Google Scholar]
- Wu M-F, Gulyani S, Yau E, Mignot E, Phan B, and Siegel JM (1999). Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience 91, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M-F, John J, Boehmer LN, Yau D, Nguyen GB, and Siegel JM (2004). Activity of dorsal raphe cells across the sleep-waking cycle and during cataplexy in narcoleptic dogs. J. Physiol 554, 202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, Wang QP, Tominaga M, Goto K, Shioda S, and Sakurai T (2002). Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem. Biophys. Res. Commun 290, 1237–1245. [DOI] [PubMed] [Google Scholar]