

Abstract
We profiled the liver transcriptome, proteome, and metabolome in 347 individuals from 58 isogenic strains of the BXD mouse population across age (7 to 24 months) and diet (low or high fat) to link molecular variations to metabolic traits. Several hundred genes are affected by diet and/or age at the transcript and protein levels. Orthologs of two aging-associated genes, St7 and Ctsd, were knocked down in C. elegans, reducing longevity in wildtype and mutant long-lived strains. The multiomics data were analyzed as segregating gene networks according to each independent variable, providing causal insight into dietary and aging effects. Candidates were cross-examined in an independent Diversity Outbred mouse liver dataset segregating for similar diets, with ~80–90% of diet-related candidate genes found in common across datasets. Together, we have developed a large multiomics resource for multivariate analysis of complex traits and demonstrate a methodology for moving from observational associations to causal connections.
Keywords: Aging, multiomics, proteomics, genetic reference population, liver, time course, gene-by-environment interaction (GxE), causal inference, network biology, multivariate analysis
Graphical Abstract
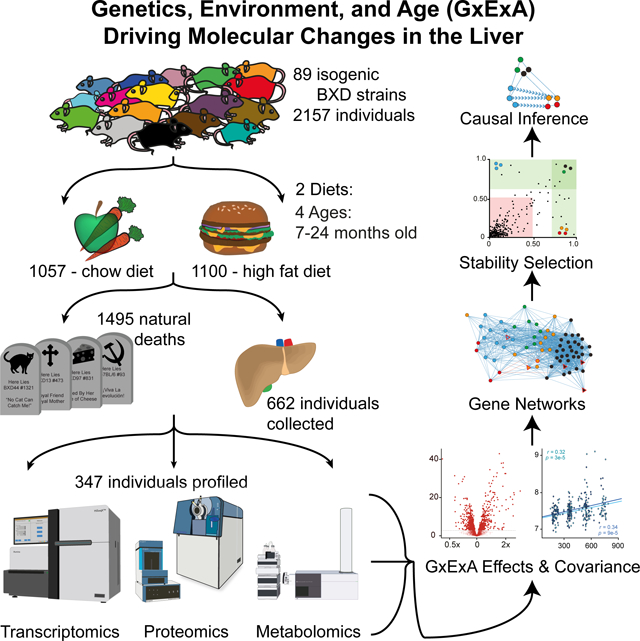
eTOC
The interaction between genetic and environmental (GxE) factors interact and over a lifetime lead to diverging metabolic phenotypes and lifespans across diverse populations. We have collected livers from 662 individuals from the BXD isogenic strain family across two diets over their natural lifespan and performed multiomic profiling of their transcriptome, proteome, and metabolome. These data allow us to examine the molecular basis of GxE and aging and to define genes and pathways which diverge in tandem with metabolic variation.
INTRODUCTION
Aging is a dynamic and multi-faceted process driven over a lifetime of interactions among genetic variants, environmental factors, and stochastic processes. Despite its complexity, longevity is a heritable trait, with genotype explaining 30–50% of its variation across laboratory mice (Belknap, 1998; Hook et al., 2018) and ~25% in humans (Hook et al., 2018). Age is a prominent “risk factor” for a wide range of diseases, such as metabolic syndrome, diabetes, heart disease, neurodegeneration, and most cancers (Kraja et al., 2006; White et al., 2014). Cells and tissues display common perturbations with increasing age such as a diminished capacity for proteostasis (Labbadia and Morimoto, 2015; Santra et al., 2019) and the accumulation of mitochondrial defects (Srivastava, 2017). These and other common endpoints are recognized, but there is substantial diversity in the mechanisms and timelines connecting individuals’ chronological age (“calendar age”), their biological age (“healthspan”), and their expected lifespan (“longevity”) across different environmental conditions (Horvath, 2013), let alone across organisms. It is now possible to measure biomarkers of both biological and chronological age, such as by DNA methylation signatures (Bell et al., 2019). However, it is unclear whether interventions that directly affect the dynamics of aging biomarkers, such as methylation sites, would causally improve either longevity or healthspan (Jazwinski and Kim, 2019; Levine et al., 2018).
Given the inherent challenges of longitudinally obtaining tissue biopsies in human clinical cohorts, populations of model organisms provide a reproducible system to study the varying biomolecular processes of aging. Additionally, isogenic cohorts permit “paired” tissue biopsies to be collected across multiple times and environments. This allows the creation of resources, such as the Tabula Muris Senis project (Tabula Muris, 2020), which establish baseline resources for molecular changes over time, such as how transcript expression changes across cell types and time in C57BL/6 mice. Further research is necessary to test how genetic variation and environmental interactions (GxE) influence molecular clocks, the extent to which relations are congruent between cognate mRNA and protein, and how changes in molecular levels link with aging and age-associated phenotypes. Getting at the causality of these relations is critical in developing more sophisticated interventions to reduce disease burden and enhance health and longevity. However, causal inference requires multiple simultaneous axes of variation and/or longitudinal sample acquisition (Kesmodel, 2018; Lancaster et al., 2020), a relative rarity for population-scale studies of gene expression which tend to focus on cross-sectional analysis for single independent variables.
In this study, we have generated transcriptome, proteome, and metabolome profiles in liver samples from 58 strains of the large and genetically diverse set of isogenic BXD mice, across their natural lifespans and in two different diets. These data were generated in 347 individuals belonging to 300 distinct genotype, age, diet, and sex-matched cohorts and combined with phenotypes collected across the entire family, including blood biomarkers, organ weights, longitudinal body weight, and longevity. We developed this dataset to examine relations between genetics, dietary environments, and age (GxExA) on gene expression. We show that the experimental design and data provide a platform for detecting, evaluating, and testing how and to what extent biomolecular processes and phenotypes vary as a function of GxExA.
Moreover, we use the dataset to highlight an approach to identify causal relationships in gene expression networks. The multiple independent variables segregating in this study (diet, age, genotype) allowed us to apply a causal inference method we recently developed (Meinshausen et al., 2016; Pfister et al., 2021) called stabilized regression. This method starts with supervised learning, using as input a target of interest (e.g. gene expression or a phenotype), next searches for any measurements which covary with the target, then evaluates how these associations change according to at least two independent variables. Regression coefficients for each independent variable are combined with a stability score that estimates whether the selected target is more likely to be upstream of a canonical pathway (i.e. causal), downstream (i.e. a biomarker), or ambiguous (i.e. a connection not affected by the secondary independent variable). We perform causal inference analysis for 23 core metabolic gene sets which are known to vary as a function of diet or age, and search for modifier genes outside the canonical gene sets which explain differences in gene expression networks as a function of genotype, age, and diet. Roughly 20% of the detected gene–pathway associations were specific to an age or dietary environment, indicating a causal relationship between these genes, the target pathway, and the independent variables (i.e. genotype and either age or diet).
Here we have generated one of the largest coherent, replicable, and extensible sets of aging multiomics gene expression data in a model population. This provides two key resources for the study of aging, metabolism, and complex trait analysis. First, these data were generated in the isogenic BXD population, thus providing a reproducible platform and extensible reference for further examining the mechanisms by which GxExA affect gene expression, metabolites, and core physiological phenotypes. Second, this multivariate study design demonstrates the capacity for new advances in statistics for the study of complex networks: stabilized regression can calculate the causality for associations which are impacted by two or more independent causal variables.
RESULTS
Clinical Analysis of Lifespan as a Function of Genotype and Diet
In this study, we initially followed 2157 mice from 89 strains of the BXD family across their natural range of lifespan, of which a subset of 662 individuals from 60 strains were sacrificed for tissue collection. Individuals were placed in the colony around 5 months of age, after which cohorts were evenly segregated into two dietary cohorts, one fed a low fat “chow” diet (CD; Harlan Teklad 2018, 6% calories from fat) and the other a high fat diet (HFD; Harlan 06414, 60% calories from fat). Pairs of individuals from each cohort (i.e. strain and diet matched) were sacrificed at 7, 12, 18, and if possible 24 months of age to collect the tissue biobank from 662 individuals, belonging to 309 distinct cohorts balanced according to age, diet, and genotype (Figure 1A, Table S1, Figure S1A), of which omics data were eventually generated for 300. The liver was selected as the primary organ of interest due to its central role in metabolism and the wide range of liver-related clinical and molecular phenotypes known to vary across the BXDs as a function of diet, sex, and genotype (Andreux et al., 2012; Williams et al., 2016). The liver was pulverized in liquid nitrogen and aliquoted in parallel for transcriptomics, proteomics, and metabolomics (Figure 1B).
Figure 1. Overview of Aging Colony.
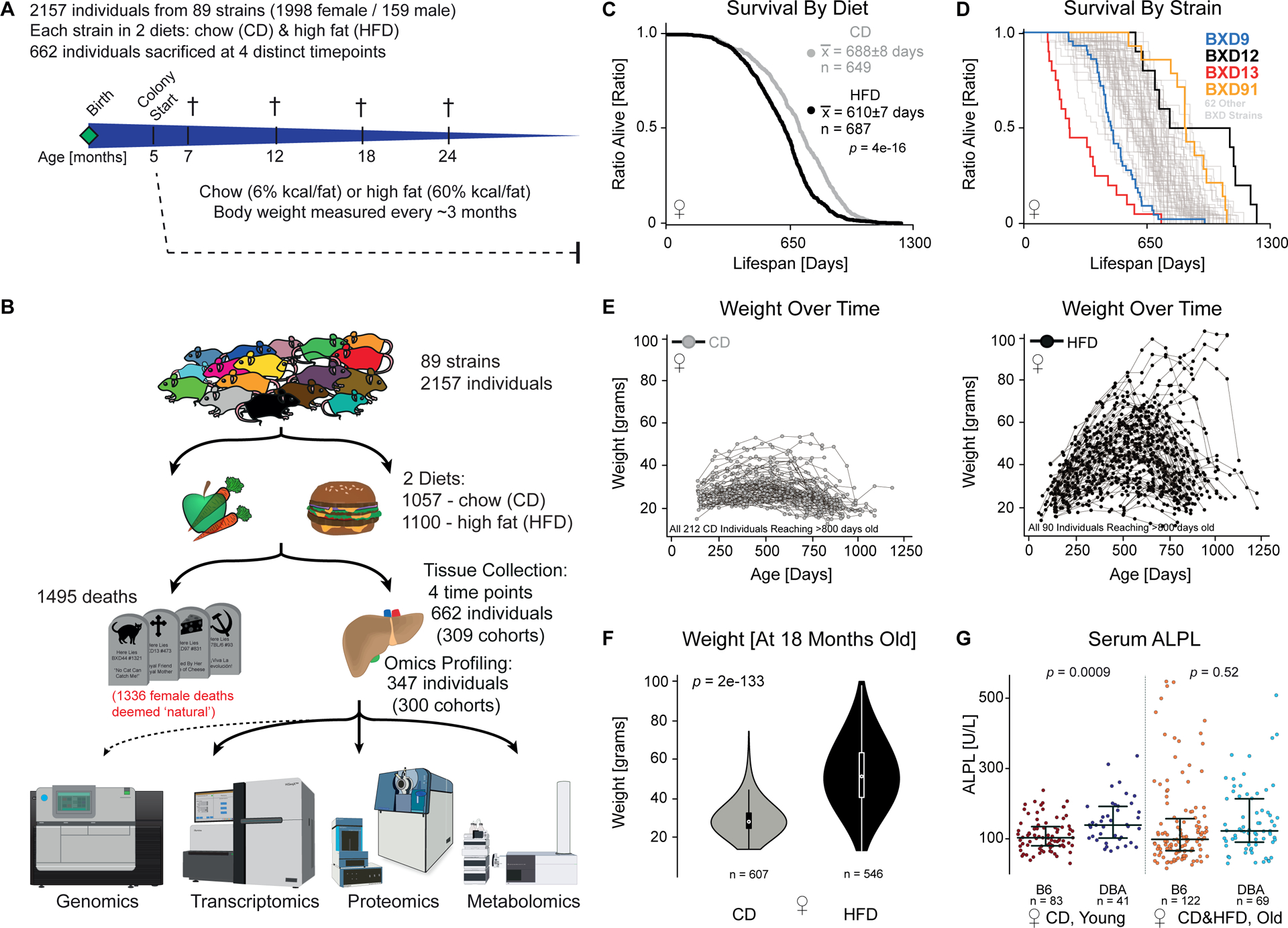
(A) Study overview. Animals entered the aging program at around 150 days of age and were set into dietary cohorts. 662 individuals were selected for sacrifice at 7, 12, 18, and/or 24 months of age for sacrifice. (B) Study design and workflow. The 662 individuals are from 309 cohorts according to diet, age, sex, and strain. 347 individuals were selected for omics profiling, corresponding to 300 distinct cohorts out of the 309 originally acquired. Of the 1495 natural deaths, 1336 were used for lifespan calculations (see Methods). (C) Kaplan–Meier survival curves for CD and HFD females, irrespective of strain. Significance indicated by a Fleming-Harrington weighted log-rank test. (D) Kaplan–Meier survival curves for all 66 BXD strains with at least 8 natural deaths in the female cohorts, irrespective of diet. (E) Weight-overtime for 212 CD (left) and 90 HFD (right) individuals which reached ≥ 800 days of age. All animals were weighed bimonthly. (F) Violin plots of body weight at 18±1.8 months of age in each diet, significance indicated by t-test. (G) ALPL serum metabolite levels across BXD strains as a function of several cofactors, significance indicated by t-test.
Earlier studies have demonstrated that expected lifespan across the BXD family of mice varies by ~3-fold—from 11 to 32 months (De Haan and Van Zant, 1999; Gelman et al., 1988; Lang et al., 2010). These differences among strains are consistent across studies, even twenty years apart (r = 0.77, Figure S1B (Gelman et al., 1988; Hook et al., 2018; Lang et al., 2010)). Correspondingly, we find significant correlations between our CD lifespans (Roy et al., 2021) and those of the previous studies (r = 0.53 and r = 0.69, for the 1988 and 2010 studies, respectively, Figure S1B). In this study, we calculated longevity using 1336 individual female mice which lived out their natural lifespans, permitting comparisons across diets (n_diet = 2, Figure 1C) and strain (n_strain = 66, Figure 1D and Table S1), with 48 strains having sufficient data in both diets for dietary comparisons (≥ 6 natural deaths in each dietary cohort; Table S1). Genetic variation across the population explained 71% of variation in expected cohort median lifespan, versus 14% by diet and 15% by gene–diet interactions (Figure S1C). Conversely, body weight at sacrifice was explained largely by diet (41%), with a significant contribution from genotype (21%) and interactions between the two (7%) (Figure S1C).
Overall mean strain lifespans (merging dietary cohorts) vary from 314±37 days (mean ± SEM for BXD13, n = 20) to 870±39 days (BXD91, n = 14) (Figure 1D, Figure S1D). While HFD feeding causes a mean 10% decrease in longevity, the magnitude of decrease varies by strain: BXD9’s lifespan is unaffected, while BXD65s tend to live nearly an additional year longer on CD than HFD (log-rank test, p = 3e-6, Figure S1E). HFD leads to a significant decrease in lifespan in 40% of the strains (p ≤ 0.05), and 64% have at least a tendency to live shorter on HFD (p ≤ 0.10). These differences notwithstanding, the HFD effect is generally consistent across strains, with mean strain lifespan correlating between diets at r = 0.68 (Figure S1F). Although diet has a relatively modest effect on longevity, it has a substantial impact on weight (Figure 1E 1F); strains had an average 78% increase in body mass, and 89% of strains gained weight significantly upon HFD feeding by 18 months of age (p < 0.05, comparing areas under the curve). As with lifespans, the effect of HFD on body weight varies depending on genetic background: BXD16s gained the least with an average increase of 11%, while BXD100s gained an average of 133% (Figure S1G, S1H).
In addition to body weight and longevity, we also measured 18 plasma metabolites commonly used in clinical settings such as cholesterol, iron, glucose, and alkaline phosphatase (ALPL) levels (Table S2). We observed that HFD reduces the circulating serum level of ALPL, as reported previously in the BXDs (Williams et al., 2016), along with an increase in circulating ALPL in old mice (Figure S1I), as has been observed in humans (Fenuku and Foli, 1975). Strains with the B6 allele of Alpl are known to have lower ALPL levels than those with the D2 allele (Andreux et al., 2012), which we again observed in typical control conditions (i.e. CD, young) (p = 0.0009, Figure 1G). However, the genotype effect is dependent on environment: for old females, the effect caused by Alpl sequence variants is masked by environmental interactions between age and diet (p = 0.52, Figure 1G). These interactions between age, genotype, and diet on ALPL are known (Andreux et al., 2012; Williams et al., 2016), but this illustrates the challenge of GxE and causal discovery for complex traits: a single circulating metabolite is affected by genotype (Alpl allelic variants), diet, and age.
Multiomic Molecular Analysis of the Aging Liver
We hypothesized that transcriptome, proteome, and metabolome data could indicate molecular networks involved in the etiology of hepatic aging, dietary response, their interactions with genetic variants across the population, and resulting differences in metabolic phenotypes. To examine this, we selected livers from 347 individuals for multiomic gene expression analysis out of the total collection of 662 individuals, representing 300 of the 309 collected cohorts for a total of 58 strains, 2 sexes, 4 ages, and 2 diets (Table S1, sheet “Cohorts_Harvested”). After sample quality control (QC; see Methods), RNA-seq data were retained from 291 individuals (255 cohorts) and proteomics data from 315 individuals (278 cohorts), with 275 individuals overlapping in both datasets (240 cohorts). Untargeted metabolomics data were generated by flow injection analysis TOF-MS from 624 individuals (298 cohorts), resulting in a total of 274 individuals (239 cohorts) with full data in all three layers. RNA-seq data were generated with 20 million reads per sample on a HiSeq PE150, with 25394 distinct transcripts quantified, of which 20827 are annotated as protein-coding. Proteomics data were generated using SWATH-MS on an SCIEX 6600 instrument, with 3940 proteins quantified after QC. The metabolomics data were generated on an Agilent 6550 instrument, with 464 uniquely-detected metabolites remaining after QC. The processed and normalized set of all omics data are available in Data S1 (for raw data, see Data Availability). We focused on gene expression in this study, primarily for the 3772 genes which were measured at both the mRNA and protein level to get a comparable multiomic overview of the gene expression across GxExA. These 3772 genes belong to some overrepresented ontologies (e.g. mitochondria, cytoplasm, and ribosomal proteins) while others are depleted (e.g. membrane proteins and secreted proteins) (Table S2, sheet 1). Note that some functional categories are fundamentally absent due to tissue type (e.g. olfactory receptors) or selection time (e.g. developmental proteins), while other depletions are due to technical reasons, e.g. membrane-bound proteins are difficult to extract, separate, and digest in proteomics (Whitelegge, 2013; Williams et al., 2018).
Diet has a significant impact on the expression of 893 transcripts and 1352 proteins (Figure 2A, adjusted t-test between discrete groups) while 1562 transcripts and 998 proteins significantly covary with age (Figure S2A, correlation coefficient, adjusted p-value). ANOVA determines an average 64% of observed variation in transcripts to be explained by genotype (“strain”), diet, age, and their interactions (Figure 2B). While genotype has the largest individual effect (~30%), the data come from 58 genotypes, compared to 2 diets and a range of ages in adulthood. To standardize this difference in degrees of freedom for each independent variable, we calculated the F statistic, which finds diet and age to have stronger median effects than genotype (2.87, 2.38, and 1.78 respectively). However, genotype has the most transcripts with significant F statistics; 1361 out of the 3772 overlapped transcripts are significantly impacted by genotype, versus 625 by diet and 680 by age (p < 0.001). Similar trends are observed for both protein and metabolite data (Figure S2B). Diet or age have relatively consistent effects on a gene’s mRNA and protein expression (r = 0.42 and r = 0.50 respectively; Figure 2C), while the effects of diet and age on gene expression are themselves independent (Figure S2C). We next examined the relationships between mRNA and protein levels. Across all samples, mRNA are moderately predictive for the relative abundances of their proteins (r = 0.44, Figure 2D)—that is, more abundant mRNAs tend to be the more abundant proteins and vice-versa. However, we are generally interested in how genes and pathways respond to perturbations (i.e. genotype, diet, or age). In this case, the average correlation of all 3772 mRNA with their protein as a function of GxExA is rho = 0.14, with 33% of mRNA–protein pairs covarying significantly across all measurements (adj.p < 0.05; Figure 2E). That is, knowing the variation in mRNA expression across genotype, diet, and age provides only a weak predictor for variance in its corresponding protein.
Figure 2. Multiomics Overview of mRNA, Protein, and Metabolite Liver Expression.
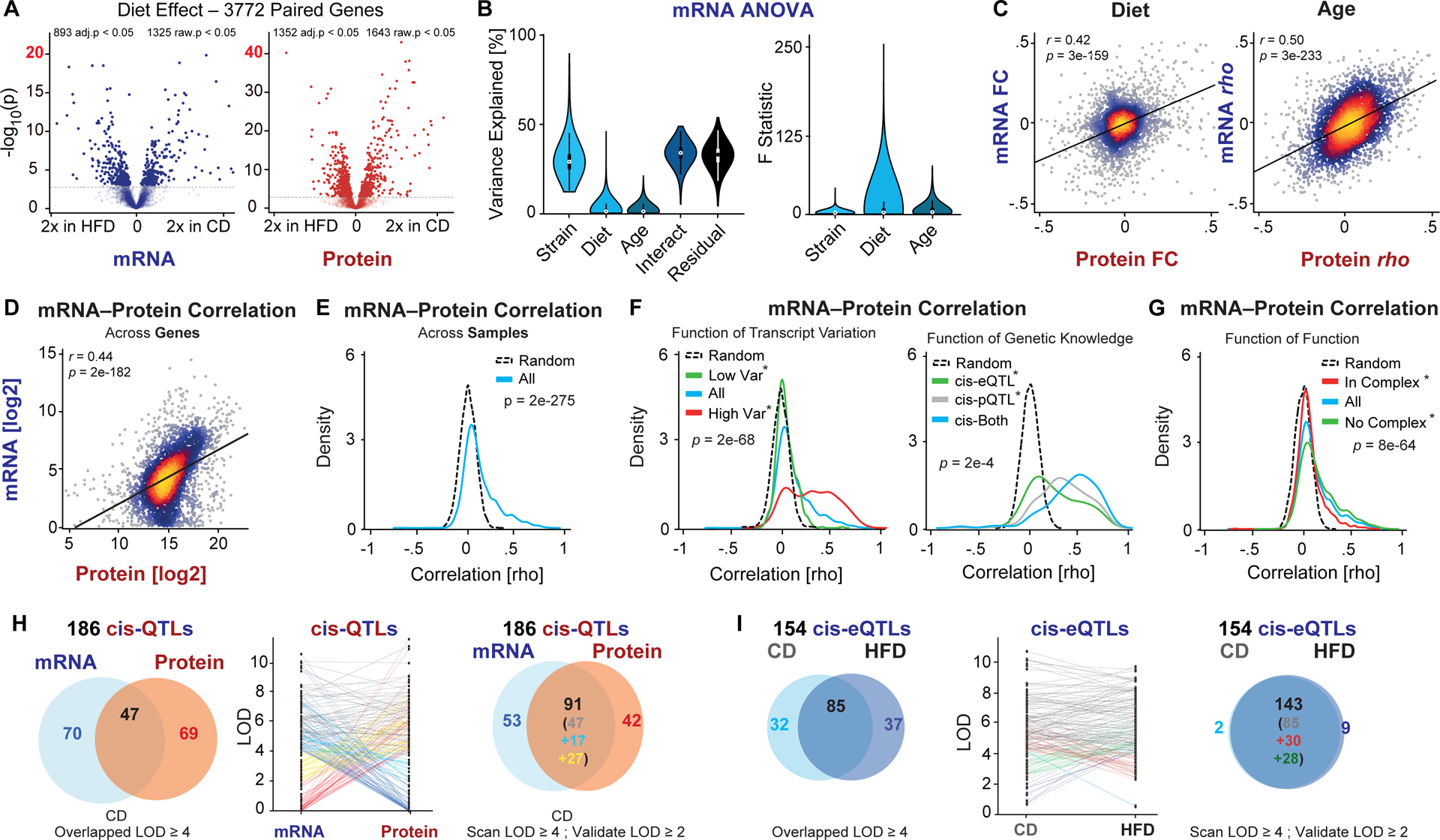
(A) Volcano plot for all 3772 transcripts and proteins measured in both expression types, and the number affected by diet, calculated by t-tests, either below a nominal p-value of 0.05 or a Benjamini-Hochberg adjusted p-value of 0.05. (B) ANOVA analysis showing variation explained and F statistic as a function of the independent variables. For variance explained, “interact” is the sum of all interactions: diet*age, diet*strain, age*strain, age*diet, and age*diet*strain. (C) Pearson correlation density plot of the relationship between the effects of dietary fold changes (left) and age correlations (right) on all 3772 paired gene products. Brighter colors represent higher density data. (D) Pearson correlation of average mRNA and protein levels across all samples and all genes. (E) Density plot showing ~33% of transcripts covary with their protein (i.e. area under the blue curve but above the black curve of randomized data). Significance is determined by a paired t-test of real correlations compared to randomized labels. (F) (Left) Density plot of mRNA–protein correlations as a function of the mRNA expression variance; (right) density plot showing correlation as a function of cis-QTL presence; (center-right) as a function of diet. Significance is determined by t-test between the groups denoted by the asterisk. (G) Density plot of correlation as a function of the gene existing in a complex. Significance is determined by t-test between the groups denoted by the asterisk. (H) (Left) Genes with highly significant cis-QTLs (LOD ≥ 4) in CD cohorts at the mRNA and protein level. (Middle) Slopegraph showing the change in LOD score between mRNA and protein; (Right) Venn diagram showing ~50% cis-QTL overlap at more permissive cutoffs (LOD ≥ 4 in discovery cohort, ≥ 2 in validation cohort). (I) (Left) Across diet at the mRNA level, roughly half of cis-eQTLs are found congruently. (Middle) Slopegraph showing the change in LOD score across diet. (Right) Venn diagram accounting for less strict alignment cutoffs, showing ≥ 90% of cis-eQTLs align across diet.
Despite this low average correlation, additional data can be used to improve the predictive capacity of mRNA for its protein product in some cases. Independent variables with a large effect size on a transcript’s expression are far more likely to have a corresponding effect on the protein’s expression. More highly variable transcripts tended to correlate better with their proteins—71% of the most abundant decile of transcripts covary with their protein, versus only 6% of the least-abundant decile (Figure 2F, Figure S2D). More abundant transcripts also tend to correlate better: only 12% of the least-abundant decile of transcripts covary with their protein, compared to 63% of the most-abundant. This could indicate higher levels of noise in low-abundance transcripts (and proteins, given abundance correlates at r = 0.44), but it is worth noting that abundance and variability are only weakly correlated (rho = 0.07, Figure S2F). Thus, measurements of variation within an omics layer indicates some cases where protein and transcript measurements can be used as reasonable proxies. For the 60 transcripts that are in the top decile of abundance and variability, 87% correlate with their protein significantly and with an average rho = 0.51—versus an average rho = 0.14 for the average correlation of the 3772 paired gene products.
Other factors stemming from prior knowledge can also be used to predict mRNA–protein covariation. For instance, genes with significant quantitative trait loci (QTLs) mapping near their own location—i.e. cis-QTLs—tend to have more significant transcript–protein relationships (Figure 2F). Such QTLs indicate nearby sequence variants causing varying transcript expression (cis-eQTL) or protein expression (cis-pQTL), and these tend to be highly robust and reproducible (Keele et al., 2020). The knowledge of which genes have cis-QTLs can also provide predictive information across expression type: transcripts that have strong cis-eQTLs (logarithm of the odds (LOD) ≥ 4) correlate substantially better with their protein (rho ~ 0.27). Other predictive patterns can be observed using knowledge about a gene’s function. For instance, genes that are involved in protein complexes (annotated by CORUM (Giurgiu et al., 2019)) have less significant mRNA–protein covariance (average rho ~ 0.06, Figure 2G). This is despite that complex-member mRNAs tend to be somewhat more abundant than average (p = 2e-6) and have no difference in their variation (p = 0.08). The size of the complex also impacts the expected correlation: at an adjusted p < 0.05, 34% of the 360 quantified genes in dimers have significant mRNA–protein correlations, against only 4% of the 431 genes in complexes of ≥ 20 subunits (i.e. not more significant than expected by chance) (Figure S2G).
Finally, we examined the relationships between gene expression and the varying genetic backgrounds of the BXD population via QTL mapping on all 3772 transcript–protein pairs. 216 genes mapped to a significant cis-eQTL or cis-pQTL at LOD ≥ 4 (Figure 2H; >99.9% true positive rate using discovery cutoffs, Figure S2H). While only 25% of cis-QTLs were observed at this threshold for both mRNA and protein levels concurrently (i.e. 53 out of 216), an additional 24% were observed at a secondary threshold when followed-up with a specific QTL hypothesis (LOD ≥ 2, corresponding to a 99.7% true positive rate when used as a validation cutoff, Figure S2H). Nearly half of cis-QTLs (49%, i.e. rightmost panel of Figure 2H) are unique to transcript or protein levels, in line with previous estimates (e.g. (Chick et al., 2016)). We next examined the reproducibility of cis-QTLs as a consequence of diet. At discovery cutoffs (i.e. LOD ≥ 4), just over half of cis-eQTLs (Figure 2I) and cis-pQTLs (Figure S2I) were observed in common across diets, while at validation cutoffs, more than 90% of cis-QTLs—for both transcripts and proteins, separately—were observed in both dietary conditions. However, it is worth noting that some genes only yield cis-QTLs under certain environmental states, such as Cyp3a11 and Cyp3a16, which map to robust cis-pQTLs, but exclusively in HFD, or Akt2 which maps to a robust cis-pQTL but only in aged animals (Table S2, sheet 2). Similar general trends are observed when comparing cis-QTLs across age instead of diet; 45% of cis-QTLs are concordantly affecting both transcript and protein within age group, while 92% of cis-pQTLs are in common across age groups (Figure S2J). Thus, while there is significant correspondence between transcript and protein variation, certain molecular changes may only be evident at the transcriptome or the proteome level (e.g. (Liu et al., 2019; Williams et al., 2016)), and genes’ mRNA and protein products cannot be ad hoc assumed to be proxies for one-another.
Metabolic Characteristics of Age
We next correlated all gene expression data with the measured age of the animals to look for molecular signatures of aging, both within and across dietary cohort (Table S3, sheet 6). We selected the 100 proteins and transcripts from each diet which correlated the strongest with age and analyzed their gene ontologies (GO) with DAVID (Huang da et al., 2009) (corresponding to p < 5e-4 in both CD and HFD, equivalent to rho > ~|0.34|; Table S3, sheets 2–4). This combined list of 158 mRNAs and 176 proteins was scanned for enrichment in KEGG pathways and in GO cellular compartments. The extracellular exosome is the most enriched cellular compartment with age in both mRNA (45% of genes, p = 2e-20) and protein (60% of genes, p = 8e-44), related to aging-related patterns in the extracellular matrix (ECM, or “matrisome”) recently observed in aging literature (e.g. (Ewald, 2020)). The mitochondria are the second most-enriched functional category with age (mRNA: 29% of genes, p = 7e-12, protein: 23% of genes, p = 7e-7) (Figure 3A) and the decline of mitochondrial function with age is well-known in literature (e.g. (Srivastava, 2017)). Only a few functional pathways were enriched, such as the steroid hormone biosynthesis highlighted with mRNA (p = 1e-8) and the lysosome pathway for protein (p = 1e-5). Other notable enriched pathways included the lysosome, as loss of proteostasis and declining lysosome function has been recognized as one of the hallmarks of aging (Stoka et al., 2016). We then examined these relationships in more detail using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). For the 127 genes in the “NABA Matrisome” gene set measured at the mRNA and protein level, we observed ECM genes are disproportionately and directionally associated with age: increasing at both the transcript and protein level (Figure 3B). Previous research in collagens (a key ECM component) has shown that COL1A1 mutant mice have decreased lifespans—showing that a disbalance in the ECM can in fact drive aging (Vafaie et al., 2014), rather than being a simple bystander.
Figure 3. Aging Candidate Discovery & C. elegans.
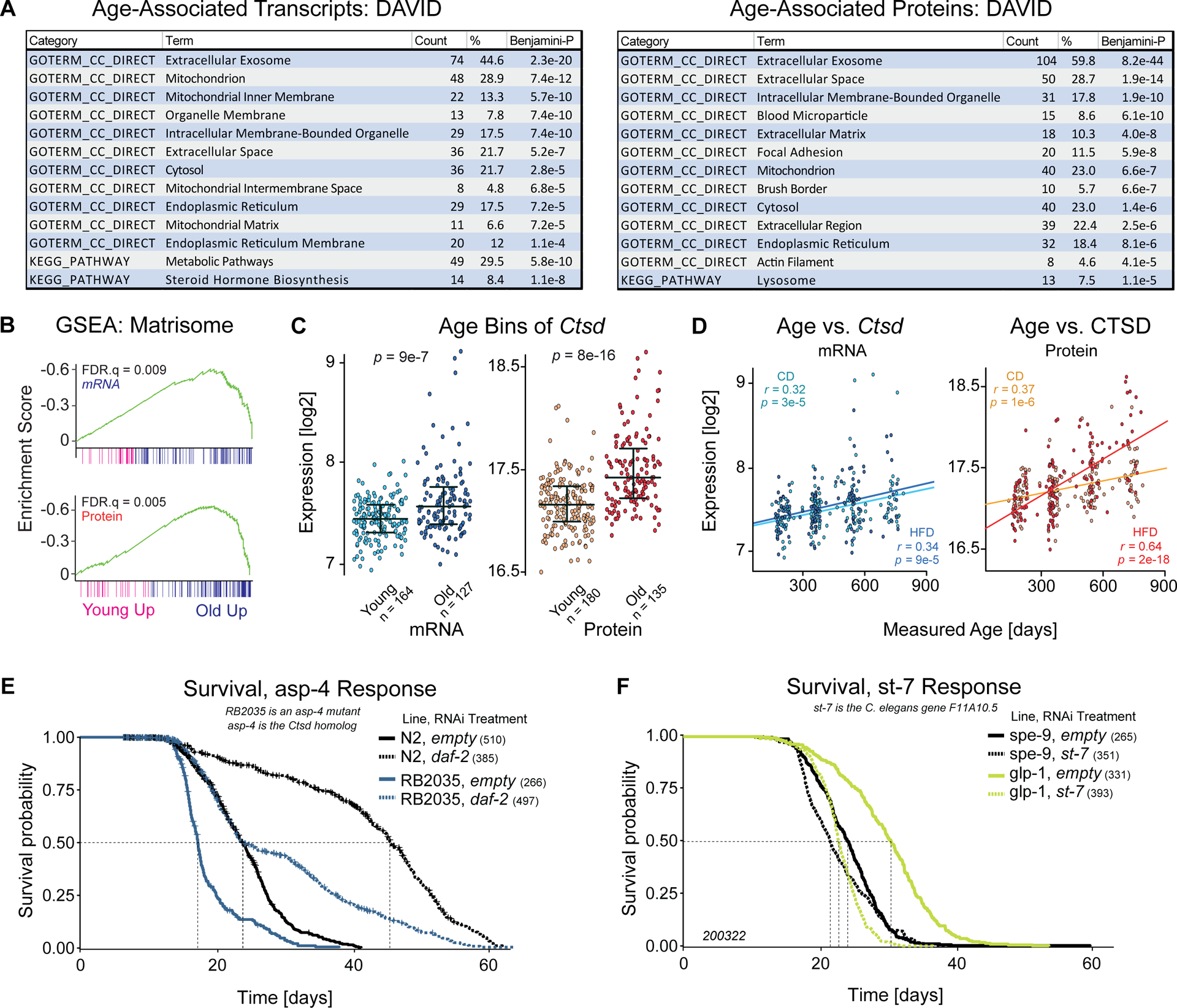
(A) DAVID analysis of the top transcripts and proteins which correlate with the measured age of the mouse when the tissue was taken. (B) GSEA for the 127 genes measured at the mRNA and protein level in the “NABA” matrisome gene set (a superset of ECM genes) showing an enrichment with age for both mRNA and protein. (C) Ctsd mRNA (left) and protein (right) expression as a function of age, using a bimodal cutoff, significance determined by t-test. (D) Pearson correlation plots of age versus Ctsd mRNA and CTSD protein. (E) Longevity analysis of RB2035, a mutant C. elegans with the removal of the Ctsd homolog asp-4, compared to wildtype N2 C. elegans with or without the inhibition of daf-2 for lifespan extension. The sample size is included in parentheses after each group. (F) Longevity analysis of st-7 RNAi knockdown on two C. elegans backgrounds; the “normal” lifespan of spe-9 mutants compared to the long-lived glp-1 mutant line. Significances between groups in panels E and F are indicated in Table S4 according to Fleming-Harrington weighted log-rank tests.
With these gene candidates and pathways in mind, we looked for candidate longevity genes which could be interventionally tested in C. elegans. 52 of the top aging-associated genes (~17%) had a single clear C. elegans ortholog according to WormBase (Harris et al., 2020) (Table S3, sheet 5). Among these, Ctsd was highlighted as a gene of interest due to its dual involvement in both the lysosome process (i.e. protein degradation) and ECM (it specifically targets ECM proteins (Benes et al., 2008)). The single Ctsd ortholog, asp-4 (BLAST p = 1e-107, score=386), has not been examined for longevity in the C. elegans literature. The Ctsb ortholog W07B8.4 has been shown to increase in expression with age and to affect reproductive aging, but not lifespan, in wildtype worms and in long-lived daf-2 mutants (Templeman et al., 2018; Wiederanders and Oelke, 1984). Literature in mammals also shows a general increase in lysosome proteins with age (Cellerino and Ori, 2017), including Ctsd (Sato et al., 2006), despite that lysosome activity tends to decrease with age. Inhibition of genes involved in autophagy have also been shown to inhibit lifespan extension interventions (Sun et al., 2020). Ctsd thus ties into an aging hypothesis regarding proteostasis which states that misfolded and malfunctional proteins increase in relative abundance with age, triggering an increase in lysosome expression, compounded by the lysosome itself becoming less able to maintain proteostasis. In our study, we observed significant increases in Ctsd as a function of age in mRNA and protein data both categorically (Figure 3C) and in a quantitative correlation with age (Figure 3D).
Given this functional knowledge of the lysosome, we hypothesized asp-4 may affect longevity, and that like the Ctsb ortholog W07B8.4, it could interact with daf-2, a gene whose knockdown leads to decreased protein turnover (around 30%) and large increases in lifespan (more than 50%) (Visscher et al., 2016). We found that RB2035 (the asp-4 mutant C. elegans line) has a significant decrease in lifespan compared to wildtype when both were on empty vector treatment (L4440), with a median lifespan of 17.2 days, versus 23.7 days for control (p = 7e-27, Figure 3E). Adulthood-specific daf-2 treatment resulted in the expected doubling of the lifespan (45.1 days, p = 2e-95) in wildtype animals, while in the RB2035 background, this effect was much reduced, having the same median lifespan as controls (23.7 days) although with a far longer lifespan tail, and an overall lifespan extension (p = 1e-15, Figure 3E). These patterns were confirmed in a complete experimental replicate (Figure S3A; data in Table S4).
We hypothesized that genes which negatively correlate with expected lifespan (rather than calendar age) in the BXDs might also provide candidates for lifespan modification in C. elegans. We examined correlates for all data with expected lifespan (Table S3, sheet “AllCorrs_ExpectedLifespan”), which yielded ~10% as many candidates as compared to the analysis with calendar age. Given the paucity of candidates, we chose to look across all transcripts (not only the 3772 overlapping) and only a single gene—suppressor of tumorigenicity 7 (St7)—correlates below the false discovery cutoff in both CD and HFD cohorts (p < 0.0005; Figure S3B and Table S3). Moreover, St7 does not correlate with measured age (Figure S3B), nor are there categorical differences between young and old mice (Figure S3C). That is, St7 expression does not change with lifespan, but strains with higher levels of it tend to live less long. St7 has a single, strong ortholog in C. elegans, called F11A10.5 (BLAST p = 1e-112, score 403; referred to here as st-7). No published longevity data nor relationships with lifespan are available for st-7, but we hypothesized that its suppression may increase expected lifespan. For the sterile control C. elegans spe-9, st-7 inhibition with RNAi caused a minor reduction in lifespan of around 11% (p = 0.001, Figure 3F and S3D, Table S3 sheet “overview”). When combined with the long-lived glp-1 mutant model, which loses germline stem cells, st-7 inhibition halved lifespan (p = 5e-48, Figure 3F and S3D, Table S3). st-7 inhibition therefore did not increase lifespan, but as with Ctsd, the direction of a causal effect can be different from that expected by correlation analysis. With these findings in mind, we set out to develop targeted hypotheses about how GxExA drives divergences in gene expression, metabolic pathways, and phenotypes.
Using gene-environment-age interactions to understand liver physiology
In addition to the aging-associated pathways detected by DAVID, we hypothesized that other core metabolic pathways may have modifier genes which are GxExA-dependent and which can be used to understand the molecular basis behind metabolic shifts in the BXD population. In order to reduce multiple testing, we pre-selected 23 gene sets from GSEA (Subramanian et al., 2005) which are associated in literature to at least one of our independent study variables (diet, age, or BXD genotype; Table S5). A further 2 “false” gene sets were also selected: one of entirely random genes, and one of random metabolic genes. Prior hypotheses are detailed in Table S5, including e.g. that CYP450 gene family is downregulated in HFD-fed individuals due to a reduction in plant-based xenobiotics (Sadler et al., 2018), oxidative phosphorylation (OXPHOS) subunits are downregulated in aged individuals (Kruse et al., 2016), and DBA/2J genetic variants upregulate supercomplex assembly in the electron transport chain (Houtkooper et al., 2013). To identify modifier genes, we first focused on two types of association: (1) molecular coexpression networks which are significant for our data types (i.e. mRNA and/or protein) across genotypes, and (2) to examine molecular signatures that as a function of age, diet, or data type. 22 gene sets formed significant protein coexpression networks, and 17 pathways formed significant mRNA coexpression networks (Table S5).
Given the discrepancies between mRNA and protein behavior for complexes (i.e. Figure 2G), we first examined the OXPHOS pathway (“REACTOME Respiratory Electron Transport”) as it is composed of large protein complexes, is known to decrease as a function of age (Houtkooper et al., 2011), and has variant supercomplex assembly across the BXDs due to genetic variants in Cox7a2l (Williams et al., 2016). Gene expression of both OXPHOS mRNA and protein corresponds to strong correlation networks (p < 1e-4, Figure 4A), but no correlation is observed between the mRNA and protein expression networks (p = 0.54, Figure 4B and Figure S4A). Furthermore, two key substructural elements of OXPHOS are evident. Uniquely at the mRNA level, the mitochondrially-encoded OXPHOS subunits (red-highlighted triangles, Figure 4A) are distinct from the nuclear-encoded primary cluster. Uniquely at the protein level, each complex of OXPHOS forms a distinct subnetwork within the overall structure, with no difference observed between nuclear and mitochondrially-encoded OXPHOS proteins (Figure 4A).
Figure 4. Functional Gene Networks of Transcripts and Their Proteins.
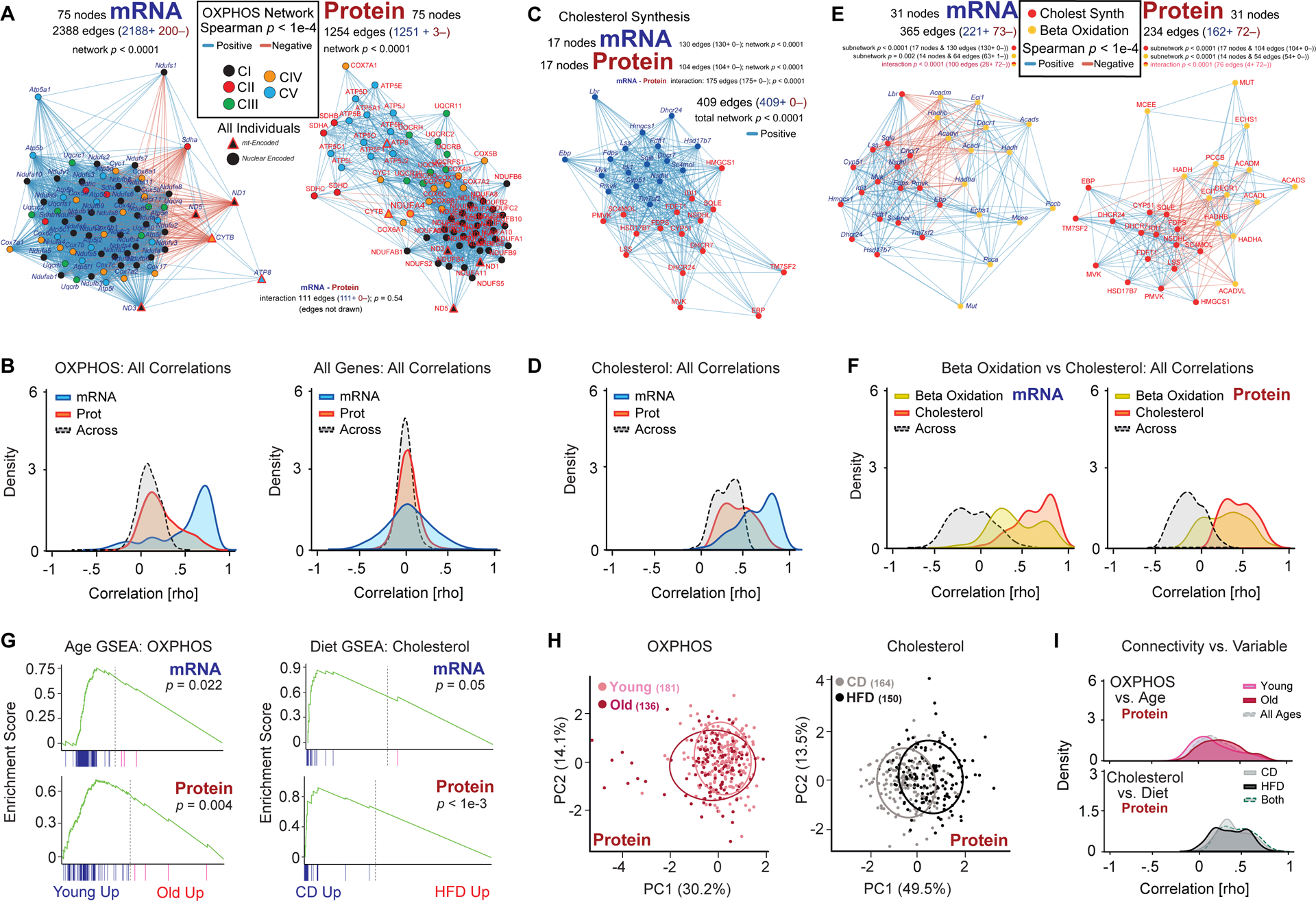
(A) OXPHOS Spearman correlation networks for network connectivity between 75 genes with both mRNA (left) and protein (right) measurements. Node color represents to which component of OXPHOS the gene belongs. Red-highlighted nodes are mitochondrially-encoded. NDUFA4 is a Complex IV member, despite its gene symbol (Balsa et al., 2012). In panels A, C, and E, edges with Spearman correlation p-values < 1e-4 are displayed and counted. Overall network significance in A, C, and E is determined by comparing the number of edges at p < 1e-4 to 10,000 randomly-generated gene networks of the same node number from the same source data. (B) Spearman correlation density plot corresponding to panel A, now showing all 5550 correlations for mRNA and protein networks (i.e. 75^2 minus identity) and “Across” for the 11000 correlations in the mRNA–protein correlation network (i.e. all possible connections between nodes where one node is an mRNA and the other is a protein). (C) Spearman correlation network for both mRNA (blue) and protein (red) in the cholesterol biosynthesis network. (D) Density plot corresponding to panel C. Of all 1088 possible edges (272 within mRNA and within protein, and 544 across), 409 have Spearman correlations with p < 0.0001 (i.e. the edges drawn in C). (E) Spearman correlation network for the cholesterol biosynthesis and beta oxidation gene networks for mRNA (left) and protein (right) drawn together. (F) Spearman correlation density plot for the above graphs. “Across” means within mRNA and protein, but across beta oxidation to cholesterol nodes. (G) GSEA enrichment for the OXPHOS and cholesterol gene sets as a function of age for mRNA and protein levels. (H) PCA biplot of the first two principal components of the OXPHOS and cholesterol protein pathways as a function of age and diet, respectively, visualizing the moderate, significant, separation by these two variables. (I) Correlation density plot of OXPHOS versus age and cholesterol versus diet.
We next examined the cholesterol biosynthesis process, a comparatively linear molecular pathway of enzymatic reactions driven by individual genes, rather than protein complexes like OXPHOS. For cholesterol biosynthesis, the two layers of gene expression strongly correlate within expression type, and across from mRNA to protein (Figure 4C, 4D). The beta oxidation pathway is also found to have a strong negative correlation with cholesterol biosynthesis genes for both mRNA and protein (Figure 4E), indicative of their complementary underlying functions (Fungwe et al., 1994). As for cholesterol, beta oxidation genes yield a significant network for both their mRNA and protein (Figure 4F). Gene sets of metabolic pathways that are predominantly made of protein complexes tend to have weaker across-layer correlation than within-layer correlation as compared to pathways which are predominantly non-protein complexes (e.g. beta oxidation, TCA cycle, Figure S4B). However, transcripts in all pathways covary more closely with other transcripts in the same pathway than they do to their protein equivalents (Figure S4B). We next examined the impact of diet and age on each of the selected functional gene networks. All pathways except the proteasome were affected by diet or age at either the mRNA and/or protein level (p < 0.01), a predictably high overall enrichment given that the sets were selected with diet and age hypotheses from literature in mind (Figure 4G, Table S5). While age and diet had significant impacts on these pathways, genetic variation across strain still played the largest role, precluding a reliable categorization of any given animal into an age or diet cohort purely based on PCA of a single gene set (Figure 4H). Furthermore, even for gene sets impacted by diet or age with strong effect sizes, e.g. cholesterol biosynthesis induced by HFD, the overall network connectivity across diet remained similar (Figure 4I). This suggests that genetic mechanisms driving the networks’ responses to the causal study variable may lie outside of the canonical gene sets. We thus set out to identify genes interacting with these canonical metabolic pathways as a function of GxExA.
Data-Driven Approaches to Non-Consensus Networks & Causal Inference
Functional gene ontologies provide a crucial platform for moving from data-driven hypothesis generation to molecular mechanisms. However, gene set annotations necessitate cutoffs for categorization which can be arbitrary as metabolic pathways are subsets of larger sets of interconnected genetic mechanisms. Furthermore, the majority of the genome still remains relatively unexplored in the literature (Stoeger et al., 2018). Data-driven approaches can identify relationships between gene expression and other pathways or diseases, including by building off reference gene sets (Lee et al., 2011). Furthermore, associations between genes and a target pathway may only be significant in a subset of the data, such as in aged or HFD-fed individuals. When covariation between two dependent variables diverges in response to an independent variable, this can be used to more precisely determine the causal relationship between the two dependent variables. To identify such associations between gene expression and phenotypes and functional pathways, we developed and applied a machine learning technique which compares the effects of multiple independent variables on a target trait or network (Pfister et al., 2021). This method allows for a linear regression-based variable selection (similar to lasso regression (Meinshausen and Buhlmann, 2006)) and is combined with stability selection (Meinshausen and Buhlmann, 2010) which uses resampling to control for false discovery. This significantly reduces false discovery compared to correlation networks or hierarchical clustering. Furthermore, the method performs a causal analysis (Pfister et al., 2021) to assess if strongly-associated candidate genes mediate the effect of a secondary independent variable (i.e. strain is the primary variable, then diet or age is the secondary). A conceptual figure of how stability analysis can be thought of roughly in terms correlation analysis and differential expression analysis is available (Figure S5A).
We looked across all 3772 gene products with mRNA and protein measurements which associate with traits and pathways that were strongly driven by genotype and either diet or age. We first checked which gene products associate with body weight as a factor of diet and genotype (Figure 5A). The resulting stability plot indicates on the y axis the permuted probability that the gene is part a model which is the most stable across body weight in both diets, while the x axis indicates the probability that the gene is among the most predictive factors of body weight in at least one diet. Thus, genes with high y axis values will tend to be strongly affected by body weight (e.g. Cd81 transcript, FABP2 protein, Figure 5B, 5C). Genes with high x axis values (e.g. Cd81, CES2C) will tend to have strong correlation coefficients with body weight in at least one diet (Figure 5B, 5D). This indicates three broad “categories” of hits. First, those in the upper-right (Figure 5A) are strongly affected by diet and are robustly predictive of body weight, whether diets are separated or combined (e.g. Cd81). These gene associations are highly robust, but their causal relationship with body weight cannot be determined (i.e. diet → Cd81 variation → body weight gain, diet → body weight gain → Cd81 variation, or Cd81 variation ← diet → body weight gain). Next, we have genes in the upper-left such as FABP2. Such genes are also affected by diet, but they are not among the most predictive genes for body weight in dietary groups when considered separately, indicating that the causal effect of diet on their expression happens independently of body weight gain. Thus, these genes are statistically upstream of body weight in a causal pathway. Lastly, we have genes in the bottom-right such as CES2C. These genes are not part of the most stable models associated with body weight as a function of diet, yet they are part of the strongest predictive model in at least one diet. Diet causally affects body weight but not CES2C – yet CES2C correlates strongly with body weight – which indicates the gene is distal from the effects of diet on the target trait, and thus it is associated with body weight regardless of diet.
Figure 5. Network Expansion and Functional Gene Discovery.
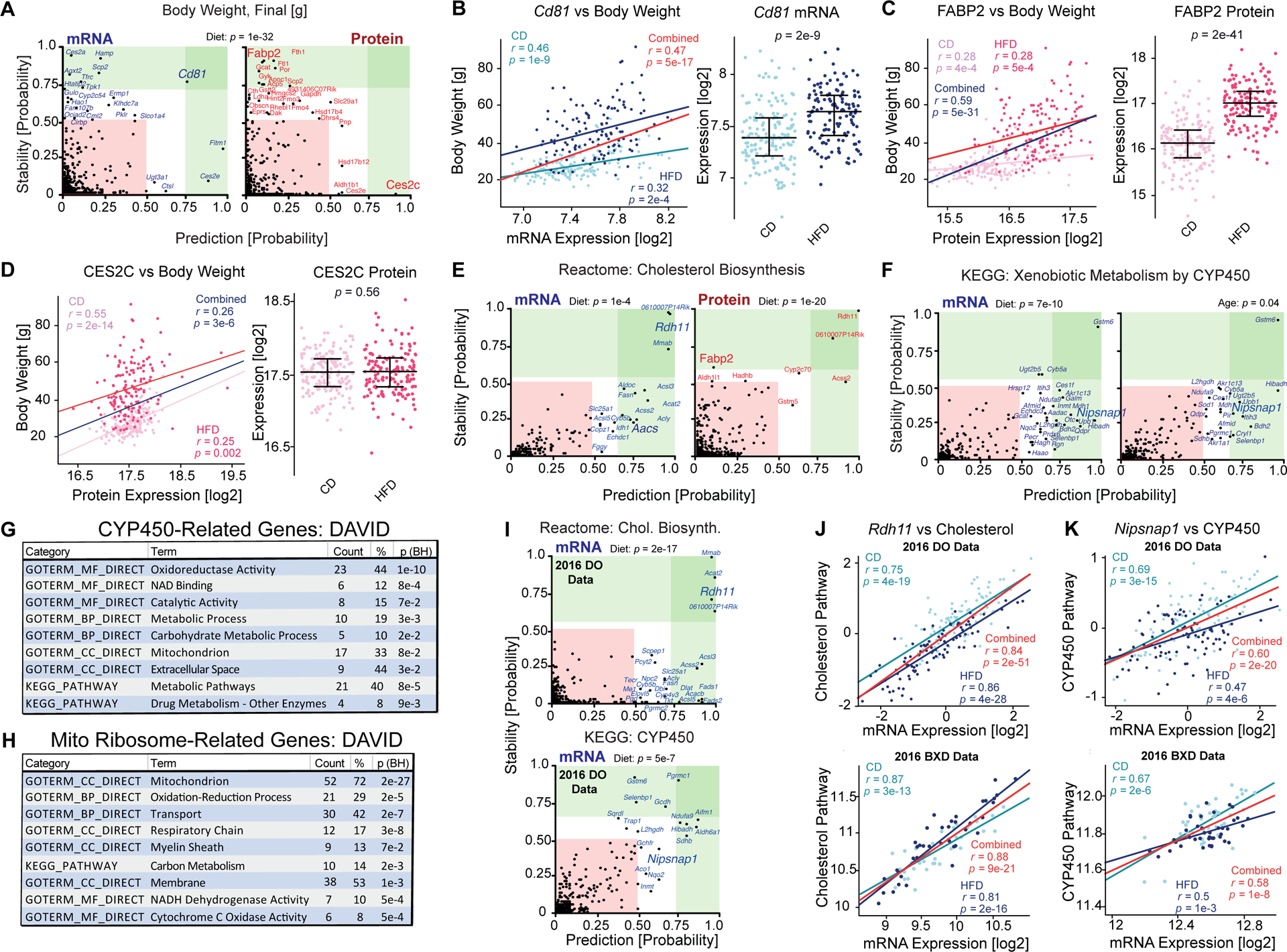
(A) Prediction–stability plot to identify which of the 3772 target gene products (mRNA: left; protein: right) associate with body weight as a function of diet. The p-value at the top of each stability plot refers to the t-test result of comparing the target trait (here, body weight) as a function of the independent variable (here, diet). (B) Pearson correlation plot of the Cd81 transcript versus body weight in CD, HFD, and across diets, with the dietary effect and t-test on Cd81 shown at right. (C) Pearson correlation plot of the FABP2 protein versus body weight in CD, HFD, and across diets, with the dietary effect and t-test on FABP2 shown at right. (D) Pearson correlation plot of the CES2C protein versus body weight in CD, HFD, and across diets, with the dietary effect and t-test on CES2C shown at right. (E) Prediction–stability plot for the cholesterol biosynthesis pathway in mRNA and protein as a function of diet. The cholesterol biosynthesis pathway has a t-test significance of 1e-4 across diet in mRNA and 1e-20 in protein. (F) Prediction–stability plot for the CYP450 pathway showing transcript hits as a function of diet or age, with the t-test result as a function of diet or age given at top. (G) DAVID enrichment analysis of the top candidate genes found through stability analysis of the CYP450 pathway. (H) DAVID enrichment analysis of the candidate genes found through stability analysis of the mitochondrial ribosome pathway. (I) Prediction–stability plots using data from a 2016 study of DO mice segregating across diet and genotype. The same target gene sets for cholesterol biosynthesis (top) and CYP450 metabolism (bottom) were examined against the transcriptome data from that study, showing general alignment with candidates identified in the aging BXD study. The t-test significance of the effect of diet on cholesterol biosynthesis gene expression is 2e-17, and 5e-7 for the CYP450 pathway. (J) Pearson correlation analyses for Rdh11 and the cholesterol pathway in the DO study with 96 CD and 94 HFD individuals (top) and a previous, independent BXD liver study with 41 CD and 40 HFD individuals (bottom). (K) Equivalent plots for Nipsnap1 and CYP450 pathway in the DO (top) and BXD (bottom) studies.
We next looked for genes associated with the same functional pathways that we examined for covariation across gene expression type and the effects of diet and age (i.e. Figure 4). For such pathways, covarying genes outside the canonical pathway can inform gene functions (e.g. the cholesterol synthesis and beta oxidation networks, Figure 4E). Stability analysis allowed us to scan all pathways across transcripts and proteins as a function of age and or diet and identify gene products which are related to the canonical pathways, and whether the relationship is consistent across conditions or conditional, such as a relationship only apparent under HFD. All significant and suggestive results for each pathway are in Table S6 (cutoffs selected empirically from estimated false discovery rates, Figure S5B). As a function of diet and age, we detected an average of 23 gene candidate hits per mRNA network and 19 per protein network, for a total of 2101 associations belonging to 748 distinct genes. Candidate genes appear multiple times, as they can be associated with multiple independent gene sets and as a function diet, age, and mRNA/protein measurement type. 450 of the 2101 total associations—around 21%—had putative directional relationships to their target pathway (prediction or stability score of ≥ 0.50 and at least 0.30 units away from the X=Y linear axis) (Table S6).
For example, cholesterol biosynthesis genes form a strong coexpression network in both CD and HFD conditions (Figure S5C), despite HFD significantly decreasing the general pathway expression (Figure 4G). Consequently, we expected to find genes which strongly covary with cholesterol biosynthesis in both dietary conditions as well as genes which have diet-dependent associations. Stability analysis highlighted 17 transcripts and 8 proteins outside of the canonical pathway, but which related closely to it across genotypes as a function of diet (Figure 5E). For instance, Rdh11 was highly stable and highly predictive at both the mRNA and protein level, indicating the expression of this gene will change as a function of diet and that it will similarly correlate with the cholesterol biosynthesis pathway in both dietary conditions. Conversely Aacs expression is not affected by diet, but the transcript nevertheless correlates similarly with the cholesterol biosynthesis pathway within and across dietary states, while FABP2 is affected by diet, but its correlation substantially strengthens when using all data. This can be interpreted directly in the stability plots, but as with body weight, the finding can be approximated in terms of correlations and groupwise-comparisons (Figure S5D).
To broadly determine the relevance of hits from the stability analysis of these pathways, we used DAVID (Huang da et al., 2009) to determine GO categories for the hits and their potential functional relationships. For the CYP450 target gene set, we identified 35 related candidate transcripts and 22 candidate proteins as a function of diet and age (mRNA shown in Figure 5F). This set of 52 genes (5 were found by both mRNA and protein analysis) included 23 were associated with the “oxidoreductase activity” (p = 1e-10, Figure 5G). This candidate list included those with clear functional interactions to CYP450 activity, such as two genes in glutathione metabolism (Gstm6 and Gstm7) and four carboxylesterase genes (e.g. Ces1e). However, at least two dozen candidate genes have no clear known connection with CYP450 or any proximal pathway, such as Nipsnap1, Echdc3, or Rgn (Figure 5F, Table S6). Similar patterns were seen with other gene sets. For instance, the mitochondrial translation gene set (Reactome, M27446) associates with 72 candidate genes, of which 52 which are known to be mitochondria-associated, including 12 in the respiratory transport chain (Figure 5H). The remaining candidates had no established functional or positional connection to the mitochondria, such as Mien1, Nedd8, and Tmed1 (Table S6).
Lastly, we considered that some of the robust hits with no known literature relationship should be observable in two previous population studies of the mouse liver which had been done CD and HFD conditions (Chick et al., 2016; Williams et al., 2016). One such study, on 192 mice from the Diversity Outbred (DO) cross, allowed sufficient sample sizes for applying stability analysis on the cholesterol biosynthesis and the CYP450 gene sets as a function of diets. 10 of the top 17 hits for cholesterol biosynthesis in the BXD liver dataset were also among the most significant candidates in the DO dataset (Figure 5I top and Table S6, sheet “Meta-Analysis”), and 15 of the 17 candidates (all except Aacs and Fggy) significantly covaried with the cholesterol biosynthesis pathway. For the 28 candidate transcripts related to CYP450, 8 were also top candidates in the DO dataset, and 25 significantly covaried with the pathway (Figure 5I bottom and Table S6). We also examined data from a 2016 study of ours on the effects of CD and HFD on liver gene expression in 81 cohorts of young BXD males. While this sample size was not sufficient for significant discoveries with stability analysis, we could examine how target genes covaried with the target pathway and the effect of diet. Again, 15 of the 17 cholesterol candidates covaried significantly (all except Aacs and Fggy), while among the 28 CYP450-related candidates, 23 covaried significantly (Table S6). This substantial enrichment highlighted several genes with very strong associations across independent studies and variables, but of unknown functional connections such as Rdh11 and cholesterol (Figure 5J) or Nipsnap1 and CYP450 (Figure 5K). Finally, as a general control, we examined how the 28 CYP450 candidate transcripts covaried with an unrelated gene set, that of mitochondrial translation. Only 2 candidates covaried in the DO data (Ndufa9 and Hagh) and 8 candidates in the male BXD data, of which only Ndufa9 was in common across all datasets—which as part of OXPHOS has a clear mechanistic connection to mitochondrial translation. Consequently, we can observe that stability analysis can identify robust and functionally-relevant candidate genes to target traits and pathways.
DISCUSSION
Aging is a dynamic process driven by a complex longitudinal mixture of genetic predestination, environmental effects, stochastic processes, and their interactions. Despite the relatively high heritability of longevity and wealth of knowledge about aging, much remains unknown about molecular causality even for well-studied aging processes such as mitochondrial stress, telomere shortening, and DNA methylation. For instance, greying hair and shortened telomeres have strong, clear associations with age, but it remains a challenge to causally determine whether a hypothetical telomere-lengthening treatment would improve lifespan any more than does black hair dye. Even when causal interventional effects on lifespan have been shown, such as the effect of caloric restriction (CR) on lifespan, it is essential to deconvolute the effects of genetic background. In mammals, CR has been causally shown to both shorten and extend lifespan, depending on genetic background (Mattison et al., 2012; Rikke et al., 2010). These phenotypic effects are highly reproducible, indicating that variant molecular mechanisms may only activate and be evident under certain genotypes and environments. Here, we provide a large, multiomics aging dataset and demonstrate how multivariate experimental designs can be combined with causal data analysis strategies to examine longstanding questions in how molecular factors vary and cause complex traits across GxExA.
We measured the transcriptional, proteomic, and metabolomic landscapes of livers from 300 cohorts of the BXD mouse population as a function of age, sex, strain, or diet. Genetic differences alone explained ~30% of variation for all molecular measurement layers, versus only ~5% for age and diet. However, the genotype axis contains more degrees of freedom: dozens of different BXD strains were measured, compared to only two diets. Additionally, while there is a spectrum of age, all timepoints are in adulthood; variance explained by aging that included developmental timepoints would likely be significantly higher. Thus, diet had the strongest adjusted impact in this study (i.e. the F statistic), but the influence of independent variables on gene expression will change according to the precise parameters selected for a study. While a gene’s transcript and protein respond broadly similarly to the impact of genotype, age, or diet, that does not mean transcripts are fundamentally a reliable proxy measurement for proteins. Further information about the dependent variable (e.g. high variation) or its function (e.g. not in a protein complex) can affect the predicted reliability of the mRNA–protein correlation. However, the majority of variance in protein levels caused by an independent variable is not predictable by mRNA measurements.
We first examined our multiomic dataset to uncover candidate genes related to age and lifespan and uncovered a few dozen candidates (Table S3). We examined two candidate genes which had not been previously studied in C. elegans: the Ctsd ortholog (asp-4) and the St7 ortholog (dubbed st-7). On the surface, the sign of correlation in the BXD data would lead one to expect their inhibition to extend lifespan (Ctsd is positively correlated with measured age, St7 negatively correlated with expected lifespan), yet the reduction of both genes inhibited lifespan. The decrease in lifespan for asp-4 knockdown was expected due to a wealth of prior literature on its mechanism in the lysosome, while little was known for st-7. This highlights two challenges when moving forward with aging research. First, a molecular component which robustly and strongly correlates with age may itself have no causal impact on age. Second, even when a causal association is expected, the directionality may be more unpredictable. For example, a gene pathway which causally affects lifespan may increase in individuals with shorter lifespans, but this could be a protective pathway whose diminution may further shorten lifespan, or it could be a maladaptive pathway whose diminution would lengthen lifespan.
Numerous studies on aging gene expression have shown that few genes have large expression differences (e.g. >2-fold) as a consequence of age (Edwards et al., 2007; Walther and Mann, 2011). However, one should consider gene expression changes in the context of entire pathways: a 2-fold change in expression of the entire OXPHOS pathway is a huge impact, as is a 2-fold change in a phenotype such as exercise capacity, insulin response, or lifespan. It is now possible to quantify the transcriptome and proteome across hundreds of samples with sufficient precision to significantly detect candidate genes with ever-smaller fold changes (Poulos et al., 2020). By aggregating such effects together (such as by GSEA), many small effects can highlight an aggregate shift in an entire pathway. While an n of 10,000 transcriptomes or proteomics would allow tiny effect sizes to be determined “significant” (and at enormous project costs), causally validating such an effect in a traditional mechanistic genetics experiment targeting a single gene remains a challenge. Causal statistics provide an avenue to determine which parts of a large network are most proximal to the independent variable and which have the largest effect the target trait. Rather than looking for consensus across input datasets to find the most stable associations (Marbach et al., 2012), we have looked for which elements of a network diverge as a consequence of independent study variables. For instance, the genes which drive body weight variation in response to HFD may not be apparent by looking at weight variance in CD cohorts. While fully unsupervised machine learning algorithms require extremely large sample sizes (e.g. >20,000 transcriptomes), false discovery can be reduced by limiting the possible search space using prior knowledge, e.g. from literature or exploratory analyses.
Here, we have applied a stability inference algorithm that we recently developed (Pfister et al., 2021) which takes advantages of two aspects of this study design. First, the study’s three independent variables (i.e. genetics, diet, age) permit stability analysis. That is, correlation networks and regression analyses can be first performed across the large primary axis of genotypes. Discrepancies in network connectivity as a function of diet or age can then be quantified by changes in edge strength and centrality. Stability analysis allows inference for when this difference is due to the intervention or when it is a response. Second, the study’s acquisition of both mRNA and protein data provides for a second “type” of consensus: results that are consistent across mRNA and protein gain improved confidence, while results that are inconsistent can be flagged according to certain criteria (e.g. presence of target gene in a protein complex). We analyzed these results across two independent mouse population studies diverging for similar diets (Chick et al., 2016; Williams et al., 2016) and found overlap of ~80–90% of associations between the three studies for candidate genes related to gene sets which were highly modulated by HFD. This combined analysis uncovered some robust connections for genes with known functional relationships to the target pathway (e.g. Gstm6 and CYP450), as well those with no clear connection in literature (e.g. Rdh11 and cholesterol).
Altogether, this dataset and method demonstrate how the simultaneous study of how multiple independent variables impact gene expression can be used for the study of complex traits. Candidate genes related to longevity can still be identified through gene coexpression analysis, such as Ctsd and St7. However, numerous hurdles are between the selection of candidate genes related to complex pathways and traits and subsequent mechanistic validation. Study designs which incorporate multiple simultaneous independent variables in a full (or nearly-full) fractional design (Bate and Clark, 2014) can be used to identify stable factors related to a target trait. This allows the implementation of causal inference to gene expression studies, providing information not on only a positive or negative correlation, but also whether it is statistically upstream, downstream, or confounded. With such developments in biostatistics and study designs in systems biology, we can move hypothesis discovery in data-driven studies from correlation networks to include causal knowledge.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Evan Williams (evan.williams@uni.lu).
MATERIALS AVAILABILITY
This study did not generate new materials.
DATA AND CODE AVAILABILITY
The full transcriptomics data are available on GeneNetwork.org under Species: mouse, Group: BXD NIA Longevity Study, Type: Liver mRNA, and Dataset: UTHSC BXD Harvested Liver RNA-Seq (Aug18) RPKM Log2. Raw mass spectrometry data for proteomics are available on ProteomeXchange (Vizcaino et al., 2016) under accession ID PXD009160. Raw mass spectrometry data for metabolomics are available on the MassIVE resource, ID MSV000081441 (ftp://massive.ucsd.edu/MSV000081441 login is “guest”, no password). All processed data, for the omics layers and the phenotypes, are available in a “ready to use” Data S1, which is the version of the data that was used to generate the figures. All code required for generating the figures, along with some helper files, are included in Data S2.
METHOD DETAILS
Mouse Care and Handling
All animal care was handled according to the NIH’s Guidelines for the Care and Use of Laboratory Animals and was also approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center (UTHSC). 2157 mice from 89 strains of the BXD family (including parents and both F1s) were followed in the colony. 159 animals were males and 1998 animals were females. Animals were maintained in the UTHSC vivarium in Specific Pathogen-Free (SPF) housing throughout the longevity experiment. The housing environment was a 12-hour day/night cycle in 20–24°C temperature with housing cages of 145 in2 with up to 10 animals per cage. Diets were either Harlan Teklad 2018 (CD; 24% calories from protein, 18% from fat, 58% from carbohydrates) or Harlan Teklad 06414 (HFD; 18.3% calories from protein, 60.3% from fat, 21.4% from carbohydrates). Water was Memphis city municipal tap water. Food and water were ad libitum. All animals were followed from their point of entry into the colony (typically around 5 months of age) until death. Animals were checked daily for morbidity and were weighed approximately every 2–3 months throughout their lives. 662 animals were sacrificed at specific ages for tissue collection across cohort (i.e. diet, strain, sex, and age) while all other animals lived out their natural lifespans. For animals living out their natural lifespan, ~90% died naturally while the remaining ~10% were euthanized according to AAALAC guidelines and made by an independent veterinarian at the UTHSC facility. Euthanized animals were retained for lifespan calculations, with the expectation that they would have otherwise died shortly thereafter.
Of note: on April 28, 2016 all mice were moved from the study’s major housing facility (“Nash”, which was slated for demolition) to a new building (“TSRB”). By this point, 94% of sacrificed individuals had been born, raised, and sacrificed in the Nash facility, so only 6% of individuals processed for omics analysis were moved, all of which were sacrificed between 1 September and 26 October 2016, i.e. after 4 to 5 months of acclimatization.
Aging Calculations
Lifespan calculations were made using the “survival” package on R with the Surv and survfit functions. Significance tests were calculated using survdiff in the same package, which uses a weighted-log rank test. 1495 deaths were recorded, of which 1386 were female. 50 of these female deaths were suppressed prior to lifespan calculations for various reasons, e.g. 7 mice died due to flooded cages, 2 animals were accidentally entered at far too old an age (>1.5 years), 2 mice were found with broken limbs, 6 were sacrificed for an urgent revision for an unrelated paper, 3 mice died before the average age of entry into the colony (7 months), and the rest were removed by the veterinarian for non-definitively-aging related reasons (e.g. seizures noted during body weighings). The 662 animals which were sacrificed for this study’s tissue collection aim were not used for lifespan calculations.
Cohort Sacrifice Selection
Animals were selected for tissue harvest with the following aims: 2 animals per strain, diet, and age, for a target of 4 age points, i.e. up to a target maximum of 16 sacrificed animals per strain (2 replicates * 2 diets * 4 ages). In the final sample collection database, an average of 11 animals were available per strain (60 strains, 662 animals), with molecular profile data acquired in the end for 58 strains. The target ages were 7, 12, 18, and 24 months of age. Roughly every 3 months for the duration of the experiment, ~40 animals were selected for sacrifice, with approximately 15 animals sacrificed per day over the course of 3 or 4 continuous days. Animals were removed from the aging colony the night prior to sacrifice, but they retained access to food and water. Sacrifices started at approximately 9am with the anesthetic Avertin used via intraperitoneal injection of 0.2 mL per 10 g of body weight. Animals were perfused with ice cold phosphate-buffered saline. The liver was the first organ harvested. The gall bladder was removed, the liver weighed, and then immediately frozen in liquid nitrogen in 20 mL scintillator vials. When reporting the number of strains analyzed for each part of this study, we count the two F1s—B6D2 and D2B6—and the parental strains. Although F1 hybrids are not isogenic (or “inbred”) strains, they can be reliably and reproducibly generated to provide biological replicates, and thus can be used as reliably as inbred strains for studies on gene-by-environment interactions. C57BL/6J and DBA/2J are counted as “BXD strains” for simplicity, although like the F1s, they do not help with QTL in the context of this study. A more detailed breakdown of BXD genetics has been published recently (Ashbrook et al., 2021).
Time Points for Aging Calculations
While CD and HFD comparisons were binary, comparisons across age were somewhat more challenging. For some analyses, linear regression was used for time at sacrifice (or measurement) against the target variable. However, for other analyses, particularly GSEA, age-QTLs, and causal inference, discrete bins were used. For binning animals based on age group, animals were binned by measured age—i.e. not by expected cohort lifespan—with young mice considered those sacrificed before 419 days of age, and old mice considered those beyond 431 days of age, with a mean±σ of 293±80 vs 615±97, respectively. Note that the bins are more distinct than the standard deviations suggest as the age distribution for sacrificed individuals is not normal; e.g. only 12 animals were sacrificed between the ages of 400 and 500 days; see Table S1 or Figure S1D for more details.
RNA and Transcriptomics
Roughly 20 mg of pre-pulverized liver tissue was mixed with 1 mL of TRIzol reagent at 4°C for RNA extraction. The sample was then further homogenized in TRIzol with a metallic bead for 2×30s at 25 Hz. The homogenate was transferred to a new tube (without the bead) and 200 μL of chloroform was added and mixed. Samples were centrifuged at 12,000g for 15 minutes at 4°C. ~400 μL of the clear top phase was taken and added to a tube with 400 μL isopropanol, followed by vortexing and the same centrifugation. The RNA pellet was observed and the liquid discarded. The RNA pellet was resuspended in ethanol by pipetting, then the samples were centrifuged again. The ethanol was removed and the sample was air dried and quantified by Nanodrop. Next, samples were cleaned up using the RNEasy MinElute kits (Qiagen) as per manufacturing instructions. RNA-seq and RNA integrity (RIN) checks were performed by an Agilent 2100 Bioanalyzer, and samples with RIN ≥ 6 were retained for RNA-seq, which was run on a NovoGene HiSeq 3000 with 150 bp end paired reads after polyA+ enrichment at the University of Tennessee Health Science Center transcriptomics platform. Samples were measured with an average of 24±2.7 million reads (mean±standard deviation). Raw fastq files were aligned to the reference mouse genome using STAR version 2.6.0c, using the UCSC genome assembly version GRCm38 (mm10). An average of 86.4±2.6% of reads were mapped per sample. Reads were counted using RSeQC version 2.6.4. Read counts were normalized to RPKM values using gene lengths from ENSEMBL82 v2015-10-02. All RNA-seq data were then scaled by adding 1 to the normalized counts and then taking the log2. 127 genes were removed from paired analysis due to the measurements being > 50% “0” at the mRNA level. Transcripts with more than half zeroes were not considered for mRNA–protein correlation due to this high number of matching values which throw off Spearman correlations. A further 38 genes had up to 100 counts of 0 which have lower than average (rho = 0.05) correlation with their transcript. Of these 163 genes, twelve of these genes were significantly affected by diet, and one by age, while all genes had significantly lower than average correlation with their protein level (rho = 0.05). Good data could potentially be contained for these transcripts, but given the preponderance of noise, we have discounted all mRNAs with more than half read counts of zero. All measurements, including those with high “0” counts, are included in Data S1. Note that for multiomics analyses we only used the 3772 genes with overlapping mRNA and protein data, but all RNA-seq data for all transcripts is included in Data S1.
Protein and Proteomics
We have previously published a detailed step-by-step protocol has been published for the protein extraction and peptide digestion (Wu et al., 2017). In brief: liver samples were first entirely pulverized by mortar and pestle in liquid nitrogen, proteins were then extracted from ~20–50 mg of powered liver in 750 μL of RIPA-M buffer. The remaining cell pellet was then lysed fully in 8M of urea. The fractions were combined and 100 μg of each sample precipitated with acetone overnight at −20°C. The precipitated sample was resuspended in urea, reduced with dithiothreitol, and alkylated with iodoacetamide. Samples were diluted to 1.5M urea and then digested overnight (22 hours) using modified porcine trypsin. Peptides were cleaned C18 MACROSpin plates (Nest Group). Roughly 1.5 μg of each peptide sample was loaded onto a PicoFrit emitter on an Eksigent LC system coupled to an AB Sciex 6600 TripleTOF mass spectrometer and acquired in SWATH data-independent acquisition mode (DIA) (Gillet et al., 2012) with 100 variable windows in a 60 minute gradient. A recent review also provides more detail on the full DIA pipeline (Ludwig et al., 2018). In brief, the resulting .wiff files were converted to mzXML using Proteowizard 3.0.5533 before being run through the OpenSWATH pipeline v2.4.0 (Rost et al., 2014). The library used was merged from our prior mouse library (Williams et al., 2018) together with part of the PanHuman library (Rosenberger et al., 2014). All peptides in the PanHuman library were BLASTed against the canonical mouse proteome from UniProt, version downloaded July 2017. Peptides which were found to be proteotypic in mouse, and not already extant in the mouse library, were then merged with the mouse library. Note that all runs for both human and mouse library generation were acquired with on the same machine (a TripleTOF 5600+) with the same settings (detailed in (Rosenberger et al., 2014)). This merged “PanMouse” library contains 103,644 proteotypic peptides corresponding to 8219 unique proteins. This library was used to search the mzXML files for the OpenSWATH pipeline using the msproteomicstools package available on GitHub. Scoring and filtering were done by PyProphet at 1% peptide FDR, followed by cross-run alignment with TRIC using a max retention time difference of 60 seconds and a target 1% FDR, 29935 proteotypic peptides were identified which corresponding to 3694 unique proteins, were quantified across the 375 retained MS injections. Proteome data were segregated into batches based on noted changes during mass spectrometry (LC column change, MS tuning and cleaning). Due to the unexpected complexity of the batch effect correction and normalization, we turned this approach and technical sample QC into a separate publication (Cuklina et al., 2021). In brief, all data were log2 transformed and then quantile normalized. Data were the analyzed for batch effects, including continuous effects (e.g. signal drift over time as the LC column gets dirtier) and discrete batch effects (e.g. when the LC column is changed by the operator after too much signal attenuation, also caused by preparation batches as the peptide samples were processed and cleaned in 96 well plates). We observed 7 distinct batches, all of which corresponded to expected experimental factors (i.e. sample preparation or LC column changes with MS machine cleanings). We then performed batch effect correction with two steps for the continuous and discrete batch effects, with the continuous correction done on a batch-by-batch process followed by a discrete correction across all corrected batches. The continuous correction was done by LOESS curve fitting, followed by across-batch normalization with ComBat, a synthetic approach based off of similar concerns with other large omics datasets.
Metabolomics
Pre-homogenized liver samples of were precisely weighed (to a target of ~20 mg) and then extracted in ~7 mL of a solution of 40% acetonitrile, 40% methanol, and 20% water, incubated for 24 hours at −20°C. The suspension was then centrifuged, the supernatant transferred to a new tube, and then lyophilized. Dried samples were kept at −80°C until ready to be injected on the mass spectrometer, when they were resuspended in water according to the weight of the input tissue sample to a target of 5 mg/mL. The same extraction of all 621 samples was done in duplicate, starting from the same pre-homogenized liver sample, approximately one month apart, which is detailed by the “Run1” and “Run2” suffix in the data (Data S1). Untargeted metabolomics analysis was performed by flow injection analysis with negative ionization on an Agilent 6550 QTOF instrument scanning between 50–1000 Da in 4 GHz HighResolution mode (Fuhrer et al., 2011). All samples were injected in technical duplicates in both experiments, and nearly all samples were injected in biological duplicates, i.e. each liver sample was injected 4 times to allow measurement stability to be calculated, so most cohorts had 8 measurements (2 full-process replicates * 2 technical replicates * 2 biological replicates (usually) per age-strain-diet cohort). Flow injection is special because it omits a chromatographic column. It was preferred over canonical LC-MS because it is much superior in speed, allowing to analyze all samples in a single day and thereby minimizing drifts and batch effects common to untargeted mass spectrometry. A shortcoming of the method is that isomers cannot be distinguished. An average of 19,000 features were detected in the runs, of which about 400 could be tentatively annotated as deprotonated metabolites listed in the Human Metabolome Database by matching accurate mass (tolerance 0.001 Da) and isotopic patterns. According to Metabolomics Reporting Standards, this corresponds to Level 4. In case of ambiguous assignment, we enumerate all putative identities. Both experimental runs were run approximately one month apart, with all samples in each run being extracted and run with back-to-back technical injection duplicates both times. Technical injection replicates correlated at an average rho = 0.99, significantly better than back-to-back injections that were not technical replicates (p = 1e-59, Figure S6A). The average metabolite correlated at rho = 0.90 across all technical injection replicates, with around 5% of metabolites having rho < 0.70 across injection replicates (Figure S6A). We previously identified allelic variants between the C57BL/6 and DBA/2 copy of the gene D2hgdh modulate the liver levels of this metabolite (Williams et al., 2016). We observe in both runs a significant decrease in the metabolite levels for individuals with the DBA/2 allele (p = 1e-19 and p = 3e-12 for run 1 and 2, respectively, Figure S6B), confirming the ability of the metabolomics to detect genetic signatures. Lastly, we examined metabolites measured in this study and in a previous study of ours on the effects of HFD on young BXD males (Williams et al., 2016), finding general congruence in metabolites most affected by diet between the two studies (e.g. pyruvate is elevated in HFD cohorts by ~1.3-fold, p = 1e-14 and p = 6e-11 in the previous male study and current female study, respectively; Figure S6C). The metabolomics data were quality controlled and are included in the manuscript for data completion, as this manuscript focuses on genes’ paired mRNA and protein expression, in which metabolites do not neatly fit.
C. elegans Testing
For selecting orthologs, each of the top 100 genes from CD and HFD for mRNA and protein—a total of 300 unique genes as roughly 25% of the top 100 were picked up in at least two conditions—was checked in WormBase for orthologs. Genes with a single annotated ortholog were considered for further analysis. Genes such as Cyp4f18 matched to dozens of orthologs—essentially the entire family of cytochrome P450 genes—and were thus considered too non-specific for reasonable cross-species analysis, but could potentially be of further use for checking the associations between pathways and aging. To test Ctsd, C. elegans populations were maintained on NGM plates seeded with Escherichia coli bacteria at room temperature, with the exception that temperature-sensitive mutants were incubated at 15°C. For this work the C. elegans strains wildtype N2, and mutants TJ1060 spe-9(hc88) I; rrf-3(b26) II, CB4037 glp-1 (e2141), and RB2035 asp-4 (ok2693) from the Caenhorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). The following RNAi clones were used as well: L4440 (empty vector control), F11A10.5 (st-7), and daf-2 (Vidal library (Rual et al., 2004)). Animals were age-synchronized using population lysis (Teuscher et al., 2019) and then transferred at the late L4 stage to plates seeded with the selected RNAi clone and containing 50 μM 5-Fluoro-2’deoxyuridine (FUDR). Depending on the genotype, animals were placed at 15°C (N2 and RB2035) to complete their development. After the L4 stage all animals were shifted permanently to 20°C and their survival was quantified. Manual survival scoring (by hand) was conducted as described previously (Ewald et al., 2017). Briefly, individuals which did not move in response to being prodded were classified as dead. Automated lifespan measurements were conducted using air-cooled Epson V800 flatbed scanners at scanning intervals of 30 minutes as described (Stroustrup et al., 2013). For the automated measurements the animals were transferred to fresh plates (BD Falcon Petri Dishes, 50×9mm) at day four of adulthood to facilitate the image detection process by removing as many eggs as possible. The survival data was analyzed using R in combination with the survival (v3.1–12) and survminer (v0.3.1) libraries. In the analysis, all animals which were observed to burrow, undergo bagging, explode or have escaped the agar surface were censored, and the L4 stage was set as the timepoint zero. The survival function was estimated utilizing the product-limit (Kaplan-Meier) approach and the null hypothesis was tested using the log-rank (Mantel-Cox) method.
Stability Analysis, Hypothesis Discovery, and Machine Learning Methods
The stability selection is described in mathematical detail in a separate methods paper (Pfister et al., 2021), but we provide here a brief additional summary of how to interpret the graphs and data. Our goal of this analysis was to determine which genes are functionally related to a target pathway, followed by determining if the gene was varying as a function of a secondary independent variable (e.g. diet, age), and if it was, then the causal directionality of this association with the target pathway and its sign (positive or negative, i.e. promotive or inhibitory). To this end, we compute the average expression level of the given pathway and use it as a response variable. Then, we randomly sample subsets of the pre-selected predictors and regress each subset onto the response resulting in a single regression coefficient. The individual regression coefficients are finally combined by a weighted average, where the weights are selected to ensure both good predictive performance and stability across the independent variable (diet, age, or a Mendelian QTL). By assessing the predictive performance of these regressions, we then rank the genes by their functional relation to the response (large values indicate a strong functional relation, small values a weak functional relation).
Additional context for reading stability–prediction plots is as follows. Figure S5A shows a target trait that is significantly and causally impacted by the independent variable of diet (p = 1e-20). This target trait is associated with a number of gene products (dots on the graph) which also covary as a function of genotype and diet. The stability analysis looks for the mediating factors transferring the variation from Diet → [Gene Products] → [Target Trait]. In each stability plot, we have up to 3772 points (each representing a gene product) arrayed on two axes. The y axis of “Stability” means that the functional relationship of the target trait with respect to these gene products does not change according to the selected variable (e.g. diet). For instance, if diet has a huge aggregate effect on the target trait, then genes with large y axis values will also be affected by diet. The y axis is a permuted probability of the predicted model and not t-test results, but it will generally correspond to the effect of the independent variable on the associated gene’s expression. The x axis corresponds to the probability that a given gene product is part of a model that – in at least one of the diets – is among the most predictive (i.e., a linear regression model based on a subset of the predictors with a small mean squared error on at least one of the diets). Simplifying slightly, large x axis values indicate that a gene generally “correlates” well in at least one diet. It does not need to be that they correlate similarly in both diets, for example if the sign of the correlation would flip but be strongly significant in at least one environment, this would result in a large x axis value. Gene products with high x but low y axis values will correlate significantly different between the diets, with the combined analysis providing a less significant correlation coefficient. Such factors are likely either downstream (Diet → [Target Trait] → [Gene Product]) or confounded ([Target Trait] ← Diet → [Gene Product]). Genes with high x and high y axis values will correlate in both cases, with the combined analysis providing a more significant correlation coefficient. Such factors are robust but it is undetermined whether they are statistically upstream or downstream. Conversely, genes with low x but high y axis values will correlate more significantly in the combined test of CD and HFD compared to the two when considered separately. These genes are expected to be upstream of the target trait (i.e. Diet → [Gene Product] → [Target Trait]). Note that additional unknown factors may lie between the arrows. Genes with a low x and low y axis values will not correlate in any case and likely have no relationship to the target trait.
For Mendelian separation using COX7A2L and HMGCS2, the 20 heterozygous F1 animals were removed. The selection procedure also uses stability selection (Meinshausen and Buhlmann, 2010) in order to control false discoveries and improve reliability. To empirically benchmark the false discovery rate, we perform a permutation based analysis as follows (Figure S5B): We apply the entire selection procedure 100 times by permuting the observations of the response variable in each iteration and keeping everything else fixed (and hence preserving the correlation structure between the predictors). This analysis was performed for all the pathways we considered. Note that the gene set “Random75” finds significant correlations—this is expected as there is 75 random genes are using their true expression data, thus true correlations are expected, just they are not expected to be related to any specific pathway.
Meta-Analysis
Two recent mouse population studies of the effect of CD and HFD on liver gene expression were selected for cross-validation of gene products related to target pathways affected by HFD. The supplemental data tables were downloaded from these two datasets (BXD: (Williams et al., 2016); DO: (Chick et al., 2016)) and the file structures were reformatted to fit the same format as the datasets for this study. These adapted tables are included in the Data S2 section, with the Figure 5 plotting code; two files including the full datasets for each of those prior studies, and one additional file for the DO mRNA data which was used for stability analysis.
Miscellaneous Bioinformatics and Statistical Tests
All functions were tested to work in R 4.0.4. QTL calculations were performed with r/QTL2 v0.22–11 (2020-07-09) (Broman et al., 2019) using a linear mixed model and a kinship matrix generated by the “leave one chromosome out” (loco) method. Genotypes used were from the 2019 build of the BXD genotypes from GeneNetwork (Mulligan et al., 2017). Transcript QTLs are referred to as eQTLs (“expression” QTLs). Protein QTLs are referred to as pQTLs, and metabolite QTLs as mQTLs. The blood serum measurements are considered as clinical phenotypes rather than as a part of metabolomics or proteomics due partly for historical categorization standards, and also due to differences in the measurement technologies and (in this particular study) source tissue. QTLs were declared as “cis” if the peak LOD region was within 10 MB of the gene location. While the BXDs generally have a resolution of under 4 MB (Ashbrook et al., 2021), the empirically calculated false positive rate at LOD > 4 (i.e. Figure S2H) using a 10 MB window was under 0.1%. All cis-QTLs are included in a supplemental data file as their generation is computationally intensive (Table S2, sheet 2). The code for generating all QTLs (cis or otherwise) is also included (attached code files, filename b_Figure2_HelperFile_QTLs.r).
Distributions (e.g. Figure 4B) were compared using a chi-squared test or Kalmogorov–Smirnov, as indicated. The R package “corrgram” v1.13 was used for generating correlation matrices. Two group comparisons were made by Welch’s t-test. The contribution of each independent variable to trait expression was calculated using the ANOVA (aov) function in R using categorical age (i.e. young and old, as used for the volcano plots). Correlations were performed using Pearson (r) or Spearman (rho), as indicated. To determine community structure in correlation networks, the package ggbiplot v0.55 was used. Adjusted p-values were calculated using the Benjamini-Hochberg method of the p.adjust function in the R library stats (v4.0.4). Spearman correlation networks were plotted using the imsbInfer v0.2.4 library in R (itself based off of the R library called iGraph). Lifespan calculations for mice and significance tests were made using the “survival” v3.2–7 package on R with the survfit and survdiff functions. Lifespan calculations for C. elegans are detailed in the C. elegans section. The output longevity data were retained for strains with ≥ 6 recorded natural deaths within a cohort. To compare lifespan across diet, a minimum of 12 natural deaths were thus necessary. Outliers were removed with the R library “outliers” (v0.14) using the “rm.outlier” function.
Reference gene sets for the functional analyses in Figures 3–5 were taken from the “C2” curated gene set lists on the GSEA website using version 4.0.0. The exact reference names of all 25 gene sets are in Table S5. Note that this analysis only considers the 3772 genes with both mRNA and protein data. Canonical functional gene assignments were curated either from GSEA (for pathway membership) or for CORUM (for complex membership). To determine the significance of correlation networks, random networks were permuted using the same number of nodes as in the target network, but randomly selected from amongst the 3772 other proteins and mRNA. For networks with multiple categories of gene sets, the target and random networks were computed both on a per-set basis as well as the total set and the interaction between the sets. 10,000 random networks were permuted for each comparison, and networks were assigned p-values based on this permutation, according to how many random gene sets had at least as many edges as the target set at the given cutoff, or assigned p < 0.0001 if no random network had as many edges as the input network at the cutoff. All figures were generated either in R and refined in Adobe Illustrator, or were hand-drawn in Adobe Illustrator (e.g. Figure 1B, Figure 3A).
The code necessary for generate all R-output figure panels is included in Data S2, including stand-alone files for each figure (with the related supplemental figure). The input file format for stability inference is different than for the other analyses. Separate input files are included for using as input for stability analysis (e.g. b_Figure5_HelperFile_mRNAData.csv). These use the exact same data provided in Data S1, but they are separated by data type (e.g. mRNA) and use the subset of genes with measurements at both mRNA and protein.
Supplementary Material
Figure S1. Supplemental Overview of Phenotype Data. Related to Figure 1. (A) Histogram and dot plot of the animals with tissues collected at each time and diet, with the fraction on CD or HFD noted. Due to decreased lifespan in HFD, there is a disbalance in the CD/HFD ratio for the oldest timepoint at 24 months. (B) Significant Pearson lifespan correlations are observed between BXD strains in all three lifespan studies; 15 strains overlap in the left panel, 18 strains overlap in the studies portrayed in blue on the right panel and 16 strains for the red. (C) Proportion of variance explained by genotype, diet, age, or the interactions between these three variables, for key phenotypes in female mice, as calculated by ANOVA. (D) Lifespan calculations as a function of genotype for two strains with extreme differences in lifespan: BXD13 (very short lived) and BXD91 (very long lived). Significance in panels D and E are indicated by a Fleming-Harrington weighted log-rank test. (E) Kaplan–Meier curves for two strains with extreme effects of diet on lifespan. (F) Pearson correlation of measured lifespan by strain between CD and HFD fed cohorts in this study. 48 strains are shown, but with reciprocal B6D2F1 and D2B6F1s crosses are displayed separately although they diverge only for their mitochondrial genome (all are females, so the Y chromosome is irrelevant). (G–H) Body weight over time chart for two strains with extreme effects of diet on weight, showing the t-test result for the difference in body weight between CD and HFD using the weight for each animal taken between 470–530 days of age. (I) Serum alkaline phosphatase (ALPL) levels, segregated by different independent variables; t-test significance between groups is indicated.
Figure S2. Supplemental Patterns in Multifactorial, Multiomic Analysis. Related to Figure 2. (A) Volcano plot showing the effect of age on mRNA and protein levels between old and young individuals. The number of genes which cross the t-test significance threshold below the nominal p-value of 0.05 or a Benjamini-Hochberg adjusted p-value of 0.05 are indicated. (B) Variation explained and F statistic for proteins and metabolites a function of the independent variables and their interactions. (C) Spearman correlation density plot of the relationship between diet (fold change) and age (correlation) on mRNA levels for all genes. The equivalent correlation for protein is not significant (r = 0.01, p = 0.55; not shown). (D) Histogram of the percentage of significant Pearson correlations for mRNA–protein pairs as a function of the variance of mRNA expression across the population. The color scale indicates each decile. (E) Density plot of mRNA–protein Pearson correlations as a function of transcript abundance. (F) Spearman correlation plot between expression variance and abundance for mRNA. (G) Histogram of the percentage of significantly correlated mRNA–protein pairs as a function of the size of the protein complex to which the gene belongs, using CORUM annotation. (H) Empirical calculations of the false discovery rate of cis-eQTLs as a function of LOD score depending on if the gene is detected in only one dataset (“discovery”) or if it is the validation of expectations from independent cis-QTL data. (I) Left: Venn diagram of the overlap of cis-pQTLs for CD or HFD cohorts using a strict cutoff of LOD ≥ 4. Middle: Slopegraph of LOD scores of all 165 genes with significant cis-pQTLs. Right: The same Venn diagram, but now using more flexible cutoffs. (J) cis-QTL consistency on a gene-level basis between mRNA and protein levels for just young individuals (left) and as a function across age for protein levels (right). The same patterns are observed here as for across-diet comparisons.
Figure S3. Supplemental Analysis of Aging Candidate Discovery & C. elegans. Related to Figure 3. (A) Replicate study of the effect of Ctsd (asp-4) on lifespan in C. elegans. (B) (Left) St7 has a negative Pearson correlation with expected lifespan of the individual in both CD (n = 114) and HFD (n = 88); (right) However, St7 does not correlate with the measured age of the animal when it was harvested for expression analysis in either CD (n = 161) or HFD (n = 129). Note that not all individuals have “expected lifespan” measurements (i.e. indicating the diet and strain cohort to which the individual belonged had < 6 natural deaths). (C) The equivalent data as categorical comparisons, showing the t-test results and fold changes. (D) Repeat of St7 (st-7) lifespan in C. elegans. Full details and significance tests for panels A and D are in Table S4.
Figure S4. Supplemental Functional Network Analysis and False Discovery. Related to Figure 4. (A) Spearman correlation network for the 75 OXPHOS genes measured at both the mRNA and protein level, showing minimal connectivity between the two layers (~3% of edges are across mRNA to protein). Network p-values are compared against 10,000 randomly selected gene sets of the same size from the same data. (B) Correlation density plots of all Spearman correlations for the ribosome, mitochondrial ribosome, beta oxidation, and TCA cycle as a function of mRNA level, protein level, or across the two. The correlations of random genes within mRNA/protein or across layer is shown at the top-right. The average correlation of two mRNAs is similar to the average of two random proteins (+0.04 for mRNA, +0.03 for protein), but the standard deviation is different: ±0.252 for mRNA, ±0.12 for protein.
Figure S5. Supplemental Stability Inference and False Discovery. Related to Figure 5. (A) A conceptual schematic for how to interpret prediction–stability plots. (B) The overlap of 100 negative control permutation networks using the mitochondrial translation gene set (top) and the PPAR signalling pathway gene set (bottom). The theoretical upper-bound false discovery in this area is 1 discovery per test in the green area regardless of gene set. Here, for this randomized gene set, the sum of the permutation tests discovers only 4 nodes in the “green” sections of the mRNA and 2 in the protein network, empirically indicating a false positive of approximately 0.03 per test averaged across all runs. Permuted false discovery in the white area is approximately 1 node per test. Note that the exact cutoff values change slightly depending on input. The reported p-values are for the effect of the stated independent variable (e.g. age) on the target pathway (e.g. mitochondrial translation) by t-test. (C) Spearman correlation plot of the cholesterol biosynthesis transcripts in CD and HFD conditions. (D) Pearson correlation plots between the mean cholesterol biosynthesis pathway expression and three candidates selected from different “quadrants” of the prediction–stability plots.
Figure S6. Supplemental Quality Control for Metabolomics. Related to Figure 2. (A) Violin plot of correlations between all 629 back-to-back technical injection replicates in metabolomics Run 2 (mean rho = 0.99), showing significant differences with the correlations of back-to-back injections of 629 unrelated samples (mean rho = 0.91; p = 1e-59 t-test). (B) Comparison of the effect of D2hgdh allele on D-2-hydroxyglutarate levels. Animals with the C57BL/6 allele of D2hgdh had significantly higher D-2-hydroxyglutarate than those with the DBA/2 allele in both runs (t-test). (C) Comparison of three metabolites with large fold changes caused by HFD in our earlier 2016 BXD liver metabolome study (Williams et al., 2016), showing a general concordance in the effect of diet across study (t-test).
Table S1. Processed Phenotype, mRNA, Protein, and Metabolite Data. Related to Figure 1.
Table S2. GO Enrichment Categories of 3772 Paired mRNA & Protein Quantified & cis-QTLs. Related to Figure 2.
Table S3. Aging-Associated Genes. Related to Figure 3.
Table S4. C. elegans Longevity Data. Related to Figure 3.
Table S5. Expression Analysis of Metabolic Pathways Affected By Age, Diet, and Genotype. Related to Figure 4 and Figure 5.
Table S6. Stability Analysis: Gene Hits Related to Metabolic Pathways. Related to Figure 5.
HIGHLIGHTS.
Generation of a large multivariate, multiomic study on GxExA in liver
Isogenic cohort study, allowing separation of each variable’s contribution to variance
Ctsd and St7 are aging-associated in mice, and causally affect lifespan in C. elegans
Stability selection and regression provide causal inference to supplement covariation
ACKNOWLEDGEMENTS
Research was further supported by the EPFL, ETHZ, the NIH (R01AG043930 to RW), the European Research Council (Proteomics4D (AdvG grant 670821 and Proteomics v3.0; AdvG-233226 to RA), and SNSF (31003A-140780, 31003A-143914 and CSRII3-136201 to RA, PP00P3_163898 to CS and CYE). NP and PB were supported by ERC No 786461 (CausalStats - ERC-2017-AdvG). EGW was supported by an NIH F32 Ruth Kirchstein Fellowship (F32GM119190). Thanks to Lorne Rose at the University of Tennessee Center of Excellence Sequencing Facility for transcriptomics, and to Sebastien Lamy at the EPFL phenotyping unit (UDP) for clinical blood analysis, to Casey Chapman for BXD colony maintenance, and to Ludovic Gillet, Patrick Pedrioli, Özgen Eren, Chloe Lee, and Yansheng Liu for proteomics discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy MF, Henry H, Schoonjans K, Williams RW, and Auwerx J (2012). Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell 150, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, Lutz CM, Valenzuela A, Bohl CJ, Ingels JF, et al. (2021). A Platform for Experimental Precision Medicine: The Extended BXD Mouse Family. Cell Syst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landazuri MO, and Enriquez JA (2012). NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab 16, 378–386. [DOI] [PubMed] [Google Scholar]
- Bate ST, and Clark RA (2014). The design and statistical analysis of animal experiments. (Cambridge, United Kingdom: Cambridge University Press; ). [Google Scholar]
- Belknap JK (1998). Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav Genet 28, 29–38. [DOI] [PubMed] [Google Scholar]
- Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, Christensen BC, Gladyshev VN, Heijmans BT, Horvath S, et al. (2019). DNA methylation aging clocks: challenges and recommendations. Genome Biol 20, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes P, Vetvicka V, and Fusek M (2008). Cathepsin D--many functions of one aspartic protease. Crit Rev Oncol Hematol 68, 12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen S, Yandell BS, and Churchill GA (2019). R/qtl2: Software for Mapping Quantitative Trait Loci with High-Dimensional Data and Multiparent Populations. Genetics 211, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, and Ori A (2017). What have we learned on aging from omics studies? Semin Cell Dev Biol 70, 177–189. [DOI] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, and Gygi SP (2016). Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuklina J, Lee CH, Williams EG, Sajic T, Collins BC, Rodriguez Martinez M, Sharma VS, Wendt F, Goetze S, Keele GR, et al. (2021). Diagnostics and correction of batch effects in large-scale proteomic studies: a tutorial. Molecular systems biology, e10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan G, and Van Zant G (1999). Genetic analysis of hematopoietic cell cycling in mice suggests its involvement in organismal life span. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 13, 707–713. [DOI] [PubMed] [Google Scholar]
- Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, and Prolla TA (2007). Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC genomics 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY (2020). The Matrisome during Aging and Longevity: A Systems-Level Approach toward Defining Matreotypes Promoting Healthy Aging. Gerontology 66, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald CY, Hourihan JM, Bland MS, Obieglo C, Katic I, Moronetti Mazzeo LE, Alcedo J, Blackwell TK, and Hynes NE (2017). NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenuku RI, and Foli AK (1975). Variations in total serum alkaline phosphatase activity with age and sex in adult and adolescent Ghanaians. Clin Chim Acta 60, 303–306. [DOI] [PubMed] [Google Scholar]
- Fuhrer T, Heer D, Begemann B, and Zamboni N (2011). High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Analytical chemistry 83, 7074–7080. [DOI] [PubMed] [Google Scholar]
- Fungwe TV, Fox JE, Cagen LM, Wilcox HG, and Heimberg M (1994). Stimulation of fatty acid biosynthesis by dietary cholesterol and of cholesterol synthesis by dietary fatty acid. Journal of lipid research 35, 311–318. [PubMed] [Google Scholar]
- Gelman R, Watson A, Bronson R, and Yunis E (1988). Murine chromosomal regions correlated with longevity. Genetics 118, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. MCP 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Arnaboldi V, Cain S, Chan J, Chen WJ, Cho J, Davis P, Gao S, Grove CA, Kishore R, et al. (2020). WormBase: a modern Model Organism Information Resource. Nucleic Acids Res 48, D762–D767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook M, Roy S, Williams EG, Bou Sleiman M, Mozhui K, Nelson JF, Lu L, Auwerx J, and Williams RW (2018). Genetic cartography of longevity in humans and mice: Current landscape and horizons. Biochim Biophys Acta Mol Basis Dis 1864, 2718–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biol 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J (2011). The metabolic footprint of aging in mice. Scientific reports 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, and Auwerx J (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jazwinski SM, and Kim S (2019). Examination of the Dimensions of Biological Age. Frontiers in genetics 10, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele G, Zhang T, Pham D, Vincent M, Bell T, Hock P, Shaw G, Munger S, Pardo-Manuel de Villena F, Ferris M, et al. (2020). Regulation of protein abundance in genetically diverse mouse populations. Biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesmodel US (2018). Cross-sectional studies - what are they good for? Acta Obstet Gynecol Scand 97, 388–393. [DOI] [PubMed] [Google Scholar]
- Kraja AT, Borecki IB, North K, Tang W, Myers RH, Hopkins PN, Arnett D, Corbett J, Adelman A, and Province MA (2006). Longitudinal and age trends of metabolic syndrome and its risk factors: the Family Heart Study. Nutr Metab (Lond) 3, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse SE, Karunadharma PP, Basisty N, Johnson R, Beyer RP, MacCoss MJ, Rabinovitch PS, and Marcinek DJ (2016). Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner. Aging Cell 15, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI (2015). The biology of proteostasis in aging and disease. Annual review of biochemistry 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster SM, Sanghi A, Wu S, and Snyder MP (2020). A Customizable Analysis Flow in Integrative Multi-Omics. Biomolecules 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D, Gerhard G, Griffith J, Vogler G, Vandenbergh D, Blizard D, Stout J, Lakoski J, and McClearn G (2010). Quantitative Trait Loci (QTL) Analysis of Longevity in C57BL/6J by DBA/2J (BXD) Recombinant Inbred Mice. Aging Clinical and Experimental Research 22, 8–19. [DOI] [PubMed] [Google Scholar]
- Lee I, Blom UM, Wang PI, Shim JE, and Marcotte EM (2011). Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome research 21, 1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, et al. (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YS, Mi Y, Mueller T, Kreibich S, Williams EG, Van Drogen A, Borel C, Franks M, Germain PL, Bludau I, et al. (2019). Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nature biotechnology 37, 314–322. [DOI] [PubMed] [Google Scholar]
- Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, and Aebersold R (2018). Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Molecular systems biology 14, e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Consortium, D., Kellis M, Collins JJ, et al. (2012). Wisdom of crowds for robust gene network inference. Nature methods 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, et al. (2012). Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinshausen N, and Buhlmann P (2006). High-dimensional graphs and variable selection with the Lasso. Ann Stat 34, 1436–1462. [Google Scholar]
- Meinshausen N, and Buhlmann P (2010). Stability selection. J R Stat Soc B 72, 417–473. [Google Scholar]
- Meinshausen N, Hauser A, Mooij JM, Peters J, Versteeg P, and Buhlmann P (2016). Methods for causal inference from gene perturbation experiments and validation. PNAS 113, 7361–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Mozhui K, Prins P, and Williams RW (2017). GeneNetwork: A Toolbox for Systems Genetics. Methods Mol Biol 1488, 75–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister N, Williams E, Peters J, Aebersold R, and Bühlmann P (2021). Stabilizing Variable Selection and Regression. The Annals of Applied Statistics. [Google Scholar]
- Poulos RC, Hains PG, Shah R, Lucas N, Xavier D, Manda SS, Anees A, Koh JMS, Mahboob S, Wittman M, et al. (2020). Strategies to enable large-scale proteomics for reproducible research. Nat Commun 11, 3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikke BA, Liao CY, McQueen MB, Nelson JF, and Johnson TE (2010). Genetic dissection of dietary restriction in mice supports the metabolic efficiency model of life extension. Exp Gerontol 45, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger G, Koh CC, Guo T, Rost HL, Kouvonen P, Collins BC, Heusel M, Liu Y, Caron E, Vichalkovski A, et al. (2014). A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci Data 1, 140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, et al. (2014). OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nature biotechnology 32, 219–223. [DOI] [PubMed] [Google Scholar]
- Roy S, Sleiman MB, Jha P, Ingels JF, Chapman CJ, McCarty MS, Ziebarth JD, Hook M, Sun A, Zhao W, et al. (2021). Gene-by-environmental modulation of longevity and weight gain in the murine BXD family. Nature Metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, et al. (2004). Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome research 14, 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler NC, Webb-Robertson BM, Clauss TR, Pounds JG, Corley R, and Wright AT (2018). High-Fat Diets Alter the Modulatory Effects of Xenobiotics on Cytochrome P450 Activities. Chem Res Toxicol 31, 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M, Dill KA, and de Graff AMR (2019). Proteostasis collapse is a driver of cell aging and death. Proceedings of the National Academy of Sciences of the United States of America 116, 22173–22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Suzuki Y, Ito E, Shimazaki S, Ishida M, Yamamoto T, Yamamoto H, Toda T, Suzuki M, Suzuki A, et al. (2006). Identification and characterization of an increased glycoprotein in aging: age-associated translocation of cathepsin D. Mechanisms of ageing and development 127, 771–778. [DOI] [PubMed] [Google Scholar]
- Srivastava S (2017). The Mitochondrial Basis of Aging and Age-Related Disorders. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeger T, Gerlach M, Morimoto RI, and Nunes Amaral LA (2018). Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol 16, e2006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoka V, Turk V, and Turk B (2016). Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev 32, 22–37. [DOI] [PubMed] [Google Scholar]
- Stroustrup N, Ulmschneider BE, Nash ZM, Lopez-Moyado IF, Apfeld J, and Fontana W (2013). The Caenorhabditis elegans Lifespan Machine. Nature methods 10, 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha V, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Li M, Zhao D, Li X, Yang C, and Wang X (2020). Lysosome activity is modulated by multiple longevity pathways and is important for lifespan extension in C. elegans. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris C (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeman NM, Luo S, Kaletsky R, Shi C, Ashraf J, Keyes W, and Murphy CT (2018). Insulin Signaling Regulates Oocyte Quality Maintenance with Age via Cathepsin B Activity. Current biology : CB 28, 753–760 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuscher AC, Statzer C, Pantasis S, Bordoli MR, and Ewald CY (2019). Assessing Collagen Deposition During Aging in Mammalian Tissue and in Caenorhabditis elegans. Methods Mol Biol 1944, 169–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafaie F, Yin H, O’Neil C, Nong Z, Watson A, Arpino JM, Chu MW, Wayne Holdsworth D, Gros R, and Pickering JG (2014). Collagenase-resistant collagen promotes mouse aging and vascular cell senescence. Aging Cell 13, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher M, De Henau S, Wildschut MHE, van Es RM, Dhondt I, Michels H, Kemmeren P, Nollen EA, Braeckman BP, Burgering BMT, et al. (2016). Proteome-wide Changes in Protein Turnover Rates in C. elegans Models of Longevity and Age-Related Disease. Cell reports 16, 3041–3051. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DM, and Mann M (2011). Accurate Quantification of More Than 4000 Mouse Tissue Proteins Reveals Minimal Proteome Changes During Aging. Molecular & Cellular Proteomics 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MC, Holman DM, Boehm JE, Peipins LA, Grossman M, and Henley SJ (2014). Age and cancer risk: a potentially modifiable relationship. Am J Prev Med 46, S7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelegge JP (2013). Integral membrane proteins and bilayer proteomics. Analytical chemistry 85, 2558–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederanders B, and Oelke B (1984). Accumulation of inactive cathepsin D in old rats. Mechanisms of ageing and development 24, 265–271. [DOI] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, et al. (2016). Systems proteomics of liver mitochondria function. Science 352, aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Ryu D, Kim JY, Lan J, Hasan M, Wolski W, Jha P, Halter C, Auwerx J, et al. (2018). Quantifying and Localizing the Mitochondrial Proteome Across Five Tissues in A Mouse Population. MCP 17, 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams EG, and Aebersold R (2017). Application of SWATH Proteomics to Mouse Biology. Curr Protoc Mouse Biol 7, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Supplemental Overview of Phenotype Data. Related to Figure 1. (A) Histogram and dot plot of the animals with tissues collected at each time and diet, with the fraction on CD or HFD noted. Due to decreased lifespan in HFD, there is a disbalance in the CD/HFD ratio for the oldest timepoint at 24 months. (B) Significant Pearson lifespan correlations are observed between BXD strains in all three lifespan studies; 15 strains overlap in the left panel, 18 strains overlap in the studies portrayed in blue on the right panel and 16 strains for the red. (C) Proportion of variance explained by genotype, diet, age, or the interactions between these three variables, for key phenotypes in female mice, as calculated by ANOVA. (D) Lifespan calculations as a function of genotype for two strains with extreme differences in lifespan: BXD13 (very short lived) and BXD91 (very long lived). Significance in panels D and E are indicated by a Fleming-Harrington weighted log-rank test. (E) Kaplan–Meier curves for two strains with extreme effects of diet on lifespan. (F) Pearson correlation of measured lifespan by strain between CD and HFD fed cohorts in this study. 48 strains are shown, but with reciprocal B6D2F1 and D2B6F1s crosses are displayed separately although they diverge only for their mitochondrial genome (all are females, so the Y chromosome is irrelevant). (G–H) Body weight over time chart for two strains with extreme effects of diet on weight, showing the t-test result for the difference in body weight between CD and HFD using the weight for each animal taken between 470–530 days of age. (I) Serum alkaline phosphatase (ALPL) levels, segregated by different independent variables; t-test significance between groups is indicated.
Figure S2. Supplemental Patterns in Multifactorial, Multiomic Analysis. Related to Figure 2. (A) Volcano plot showing the effect of age on mRNA and protein levels between old and young individuals. The number of genes which cross the t-test significance threshold below the nominal p-value of 0.05 or a Benjamini-Hochberg adjusted p-value of 0.05 are indicated. (B) Variation explained and F statistic for proteins and metabolites a function of the independent variables and their interactions. (C) Spearman correlation density plot of the relationship between diet (fold change) and age (correlation) on mRNA levels for all genes. The equivalent correlation for protein is not significant (r = 0.01, p = 0.55; not shown). (D) Histogram of the percentage of significant Pearson correlations for mRNA–protein pairs as a function of the variance of mRNA expression across the population. The color scale indicates each decile. (E) Density plot of mRNA–protein Pearson correlations as a function of transcript abundance. (F) Spearman correlation plot between expression variance and abundance for mRNA. (G) Histogram of the percentage of significantly correlated mRNA–protein pairs as a function of the size of the protein complex to which the gene belongs, using CORUM annotation. (H) Empirical calculations of the false discovery rate of cis-eQTLs as a function of LOD score depending on if the gene is detected in only one dataset (“discovery”) or if it is the validation of expectations from independent cis-QTL data. (I) Left: Venn diagram of the overlap of cis-pQTLs for CD or HFD cohorts using a strict cutoff of LOD ≥ 4. Middle: Slopegraph of LOD scores of all 165 genes with significant cis-pQTLs. Right: The same Venn diagram, but now using more flexible cutoffs. (J) cis-QTL consistency on a gene-level basis between mRNA and protein levels for just young individuals (left) and as a function across age for protein levels (right). The same patterns are observed here as for across-diet comparisons.
Figure S3. Supplemental Analysis of Aging Candidate Discovery & C. elegans. Related to Figure 3. (A) Replicate study of the effect of Ctsd (asp-4) on lifespan in C. elegans. (B) (Left) St7 has a negative Pearson correlation with expected lifespan of the individual in both CD (n = 114) and HFD (n = 88); (right) However, St7 does not correlate with the measured age of the animal when it was harvested for expression analysis in either CD (n = 161) or HFD (n = 129). Note that not all individuals have “expected lifespan” measurements (i.e. indicating the diet and strain cohort to which the individual belonged had < 6 natural deaths). (C) The equivalent data as categorical comparisons, showing the t-test results and fold changes. (D) Repeat of St7 (st-7) lifespan in C. elegans. Full details and significance tests for panels A and D are in Table S4.
Figure S4. Supplemental Functional Network Analysis and False Discovery. Related to Figure 4. (A) Spearman correlation network for the 75 OXPHOS genes measured at both the mRNA and protein level, showing minimal connectivity between the two layers (~3% of edges are across mRNA to protein). Network p-values are compared against 10,000 randomly selected gene sets of the same size from the same data. (B) Correlation density plots of all Spearman correlations for the ribosome, mitochondrial ribosome, beta oxidation, and TCA cycle as a function of mRNA level, protein level, or across the two. The correlations of random genes within mRNA/protein or across layer is shown at the top-right. The average correlation of two mRNAs is similar to the average of two random proteins (+0.04 for mRNA, +0.03 for protein), but the standard deviation is different: ±0.252 for mRNA, ±0.12 for protein.
Figure S5. Supplemental Stability Inference and False Discovery. Related to Figure 5. (A) A conceptual schematic for how to interpret prediction–stability plots. (B) The overlap of 100 negative control permutation networks using the mitochondrial translation gene set (top) and the PPAR signalling pathway gene set (bottom). The theoretical upper-bound false discovery in this area is 1 discovery per test in the green area regardless of gene set. Here, for this randomized gene set, the sum of the permutation tests discovers only 4 nodes in the “green” sections of the mRNA and 2 in the protein network, empirically indicating a false positive of approximately 0.03 per test averaged across all runs. Permuted false discovery in the white area is approximately 1 node per test. Note that the exact cutoff values change slightly depending on input. The reported p-values are for the effect of the stated independent variable (e.g. age) on the target pathway (e.g. mitochondrial translation) by t-test. (C) Spearman correlation plot of the cholesterol biosynthesis transcripts in CD and HFD conditions. (D) Pearson correlation plots between the mean cholesterol biosynthesis pathway expression and three candidates selected from different “quadrants” of the prediction–stability plots.
Figure S6. Supplemental Quality Control for Metabolomics. Related to Figure 2. (A) Violin plot of correlations between all 629 back-to-back technical injection replicates in metabolomics Run 2 (mean rho = 0.99), showing significant differences with the correlations of back-to-back injections of 629 unrelated samples (mean rho = 0.91; p = 1e-59 t-test). (B) Comparison of the effect of D2hgdh allele on D-2-hydroxyglutarate levels. Animals with the C57BL/6 allele of D2hgdh had significantly higher D-2-hydroxyglutarate than those with the DBA/2 allele in both runs (t-test). (C) Comparison of three metabolites with large fold changes caused by HFD in our earlier 2016 BXD liver metabolome study (Williams et al., 2016), showing a general concordance in the effect of diet across study (t-test).
Table S1. Processed Phenotype, mRNA, Protein, and Metabolite Data. Related to Figure 1.
Table S2. GO Enrichment Categories of 3772 Paired mRNA & Protein Quantified & cis-QTLs. Related to Figure 2.
Table S3. Aging-Associated Genes. Related to Figure 3.
Table S4. C. elegans Longevity Data. Related to Figure 3.
Table S5. Expression Analysis of Metabolic Pathways Affected By Age, Diet, and Genotype. Related to Figure 4 and Figure 5.
Table S6. Stability Analysis: Gene Hits Related to Metabolic Pathways. Related to Figure 5.
Data Availability Statement
The full transcriptomics data are available on GeneNetwork.org under Species: mouse, Group: BXD NIA Longevity Study, Type: Liver mRNA, and Dataset: UTHSC BXD Harvested Liver RNA-Seq (Aug18) RPKM Log2. Raw mass spectrometry data for proteomics are available on ProteomeXchange (Vizcaino et al., 2016) under accession ID PXD009160. Raw mass spectrometry data for metabolomics are available on the MassIVE resource, ID MSV000081441 (ftp://massive.ucsd.edu/MSV000081441 login is “guest”, no password). All processed data, for the omics layers and the phenotypes, are available in a “ready to use” Data S1, which is the version of the data that was used to generate the figures. All code required for generating the figures, along with some helper files, are included in Data S2.