

Abstract
Eleven new sphingosine 1-phosphate receptor 2 (S1PR2) ligands were synthesized by modifying lead compound N-(2,6-dichloropyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl) hydrazine-1-carboxamide (JTE-013) and their binding affinities toward S1PRs were determined in vitro using [32P]S1P and cell membranes expressing recombinant human S1PRs. Among these ligands, 35a (IC50 = 29.1 ± 2.6 nM) and 35b (IC50 = 56.5 ± 4.0 nM) exhibit binding potency toward S1PR2 comparable to JTE-013 (IC50 = 58.4 ± 7.4 nM) with good selectivity for S1PR2 over the other S1PRs (IC50 > 1000 nM). Further optimization of these analogues may identify additional and more potent and selective compounds targeting S1PR2.
Keywords: Sphingosine 1-phosphate receptor 2, Multiple sclerosis, Binding affinities, Selectivity
Introduction
Sphingosine 1-phosphate (S1P) is a bioactive lysophospholipid that transmits signals through the family of G-protein coupled receptors S1PR1, 2, 3, 4, and 5, which were originally known as EDG-1, 3, 5, 6, and 8. Upon binding S1P, these five sphingosine 1-phosphate receptors (S1PRs) regulate diverse biological functions, from proliferation and survival to migration and secretion, across many cell types.1-3 S1PRs are expressed in a wide variety of tissues, with each subtype exhibiting a different cell specificity. S1PR1, 2, and 3 are expressed ubiquitously, whereas S1PR4 is confined to lymphoid and hematopoietic tissues, and S1PR5 is primarily located in the white matter of the central nervous system (CNS) and spleen.4,5
Multiple sclerosis (MS) is an inflammatory disorder of the brain and spinal cord in which focal lymphocytic infiltration leads to damage of myelin and axons.6 According to World Health Organization reports, the estimated number of people with MS increased from 2.1 million in 2008 to 2.3 million in 2013. MS significantly decreases life expectancy of patients by an average of 5–10 years.7 The mechanisms that cause progression in MS are not well understood, thus major treatment strategies are aimed at improving neuronal function and limiting progression of disease after initial diagnosis.8 The sphingolipid-like immunomodulator fingolimod (FTY720) was the first oral therapeutic agent for relapsing-remitting MS to be approved by the US Food and Drug Administration (FDA).9 [γ-35S]GTPγS binding assay showed that FTY720 is a potent agonist at four S1PRs (S1PR1, 3, 4, and 5), but inactive at S1PR2.10 Several other S1PR1 agonists have also performed well in clinical trials with MS patients.11 Robyn Klein’s group recently demonstrated that S1PR2 plays an important role in regulating blood–brain barrier (BBB) function during inflammatory demyelination. An increased expression of S1PR2 is observed in disease-susceptible regions of both female SJL mice with experimental autoimmune encephalomyelitis (EAE) and female patients with MS compared with S1PR2 expression seen in their male counterparts.12 Although therapeutics have focused on S1PR1 agonists, S1PR2 could be a potential biomarker for the prognosis and treatment of MS.
There is a need to discover potent and receptor sub-type selective ligands to better understand the biological role of these different receptors and identify therapeutic approaches for different pathological conditions.11 Although many S1P receptor ligands have been reported, most are S1PR1 specific, and very few are specific for S1PR2 (Fig. 1). JTE-013, which was first reported in 2001, is one of the most widely-used potent and selective S1PR2 antagonist; it inhibits specific binding of radiolabeled S1P to cell membranes of Chinese hamster ovary (CHO) cells stably transfected with human or rat S1PR2.13 Although CYM-5520 was reported as the first allosteric S1PR2 selective agonist, with an EC50 value of 0.48 μM, a [33P]S1P competitive binding assay showed CYM-5520 was not competitive with S1P.14 Recently, Takuya Seko’s group identified several potent and selective S1PR2 antagonists in a novel series of 1,3-bis(aryloxy)benzene derivatives.15-17 We reported a radiolabeled JTE-013 analogue, [11C]TZ34125 and its in vivo studies, suggesting the sexual dimorphism of S1PR2 expression in the cerebellum of cyclosporin pretreated SJL mice.18 Here, we have continued our medicinal chemistry exploration of JTE-013 structural analogues. We divided the structure of JTE-013 into three fragments (A, B, and C) which were subsequently modified as shown in Fig. 2. Firstly, on one side fragment A was modified by reversing the pyrazole ring and adding one more carbonyl group to check the possibility of improving binding potency and stability, as reported;19,20 on the other side, fragment A was modified by replacing the isopropyl group with a steric bioisotere group, trifluoromethyl to check its impact on the binding potency toward S1PR2.21 Secondly, fragment B was modified by replacing the urea linker with heterocycle linker; this modification will reduce hydrogen bond donor (HBD) and cause reduction of molecular total polar surface area (tPSA) to improve the physicochemical property.22 Thirdly, fragment C was modified by replacing the 2,6-dichloropyridine ring in JTE-013 with different fused heterocycles or substituted pyridines. Together, eleven new compounds were synthesized and their in vitro binding affinities were determined using [32P]S1P and cell membranes expressing recombinant human S1PRs.
Fig. 1.
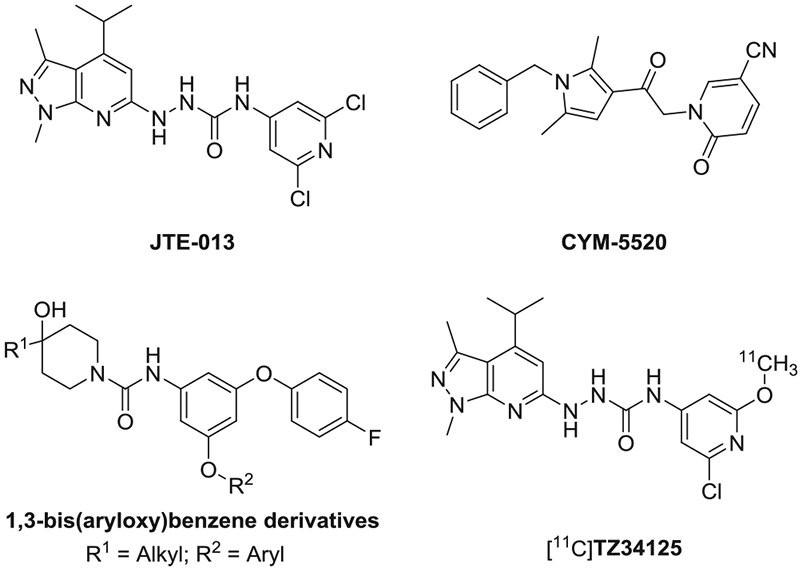
Structures of S1PR2 ligands.
Fig. 2.

Design strategy.
As shown in Scheme 1, the synthesis of compound 7 was achieved by starting with compound 1, which was synthesized as reported.19 The pyridine N-oxide 2 was obtained using meta-chloroperoxybenzoic acid (mCPBA) to perform an N-oxidation reaction. The cyano group was introduced on the pyridine ring under trimethylsilyl cyanide (TMS-CN) to give cyanide intermediate 3, which was then converted to amide 4 by hydration in the presence of 1 M NaOH and H2O2. The intermediate compound 6 was generated by reacting commercially available 2-chloro-6-methoxyisonicotinic acid 5 with diphenylphosphoryl azide. The final compound 7 was accomplished by a two-step one-pot reaction: the azide 6 was refluxed in toluene and converted to an isocyanate, which was followed by treating with 7-isopropyl-1,3-dimethyl-1H-pyrazolo-[4,3-b]pyridine-5 carbetamide 4 to afford compound 7.
Scheme 1.
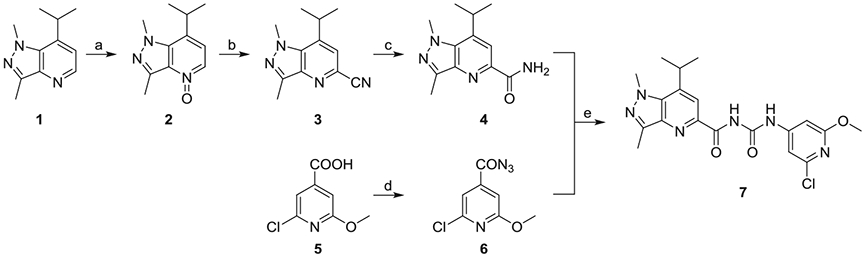
Synthesis of analogue 7. Reagents and conditions: (a) mCPBA, CHCl3, 70 °C, 79%; (b)TMS-CN, triethylamine, CH3CN, 110 °C, 75%; (c) 1 M NaOH, H2O2, methanol, rt, 90%; (d) diphenylphosphoryl azide, triethylamine, 1,4-dioxane, 0 °C-rt, 55%; (e) toluene, reflux, THF, 50 °C, 65%.
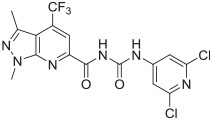
To generate trifluoromethyl substituted compounds 13a–d, Scheme 2 was followed. Starting with 3-methyl-1H-pyrazol-5-amine 8a or 1,3-dimethyl-1H-pyrazol-5-amine 8b, cyclization was accomplished by reacting with ethyl 4,4,4-trifluoro-acetoacetate in propionic acid at 140 °C to give hydroxyheter-oarenes 9a or 9b, respectively. Bromination of compounds 9a or 9b using POBr3 afforded bromoheteroarenes 10a or 10b in high yields, which were subjected to react with CuCN to afford cyanide intermediates 11a or 11b. After hydration, the amide intermediates 12a and 12b were obtained. Compounds 13a–d were prepared using a two-step one-pot reaction procedure similar to the procedure of making compound 7.
Scheme 2.
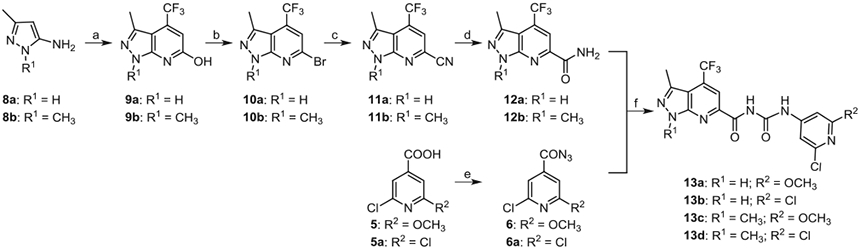
Syntheses of analogues 13a–d. Reagents and conditions: (a) ethyl 4,4,4-trifluoro-acetoacetate, propionic acid, 140 °C, 61% for 9a and 75% for 9b; (b) POBr3, anisole, 140 °C, 78% for 10a and 70% for 10b; (c) CuCN, DMF, 110 °C, 60% for 11a and 75% for 11b; (d) 1 M NaOH, H2O2, methanol, rt, 60% for 12a and 82% for 12b; (e) diphenylphosphoryl azide, triethylamine, 1,4-dioxane, 0 °C-rt, 55% for 6 and 60% for 6a; (f) toluene, reflux, THF, rt, 53% for 13a, 48% for 13b, 64% for 13c, and 67% for 13d.
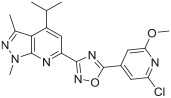
To investigate the impact of replacing the urea linker (fragment B) with a heterocycle linker, compounds 22 and 23 were synthesized by following Scheme 3. The commercially available 1,3-dimethyl-1H-pyrazol-5-amine 8b was cyclized with ethyl isobutyrylacetate to afford 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-ol 14. Under similar conditions as Scheme 2, the key cyanide intermediate 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-e-6-carbonitrile 16 was obtained. Next, the cyanide 16 was treated with hydroxylamine hydrochloride to afford N-hydroxy-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carboximidamide 17. The cyanide 16 was also converted to 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carboximidamide 18 by reacting with NH4Cl in the presence of sodium methoxide. The oxadiazole compound 22 was obtained by a two-step reaction. First, intermediate 17 and 2-chloro-6-methoxyisonicotinic acid 19 were condensed in the presence of PyBOP, DIPEA, and DMF, then the coupled product was cyclized in 1,4-dioxane at 90 °C to give compound 22. The triazole compound 23 was obtained by refluxing the intermediate 18 with 2-chloro-6-methoxy-isonicotinohydrazide 21 in ethanol.
Scheme 3.
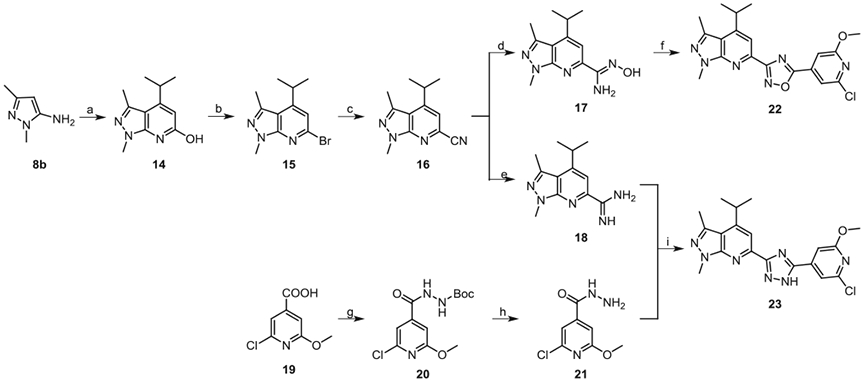
Syntheses of analogues 22 and 23. Reagents and conditions: (a) ethyl isobutyrylacetate, acetic acid, reflux, 10%; (b) POBr3, anisole, reflux, 98%; (c) CuCN, DMF, 110 °C, 60%; (d) hydroxylamine hydrochloride, NaHCO3, methanol, reflux, 81%; (e) NaOCH3, methanol, rt, NH4Cl, 70 °C, 50%; (f) (i) 2-chloro-6-methoxyisonicotinic acid 19, DIPEA, PyBOP, DMF (ii) dioxane, 90 °C, 40%; (g) (i) oxalyl chloride, DMF, CH2Cl2, rt (ii) tert-butyl carbazate, triethylamine, CH2Cl2, 0 °C-rt, 99%; (h) CF3COOH, CH2Cl2, 0 °C-rt, 25%; (i) ethanol, reflux, 63%.
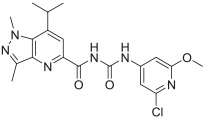
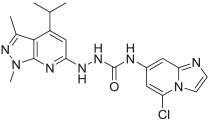
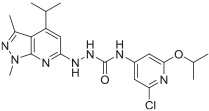
Finally, fragment C was modified as shown in Scheme 4. Key intermediate 32 was first prepared by treating intermediate 15 with hydrazine in ethanol. Cyanogen bromide treatment of 2,6-dichloropyridine-3,4-diamine 24, synthesized as reported,20 gave 4,6-dichloro-1H-imidazo[4,5-c]pyridin-2-amine 25. Compound 25 was reacted with triphosgene under triethylamine to afford the isocyanate 26, which was coupled with intermediate 32 to give compound 33. Treatment of commercially available 2-amino-6-chloroisonicotinic acid 27 using 2-bromo-1,1-dimethoxyethane in the presence of 48% HBr in ethanol afforded 5-chloroimidazo[1,2-a]pyridine-7-carboxylic acid 28. Compound 28 was reacted with dipenylphosphorylazide under triethylamine in DMF to afford 5-chloroimidazo [1,2-a]pyridine-7-carbonyl azide 29, followed by coupling with intermediate 32 afforded compound 34. Compounds 35a and 35b were obtained using a procedure similar to that used to synthesize 34, starting with the corresponding substituted isonicotinic acid.
Scheme 4.
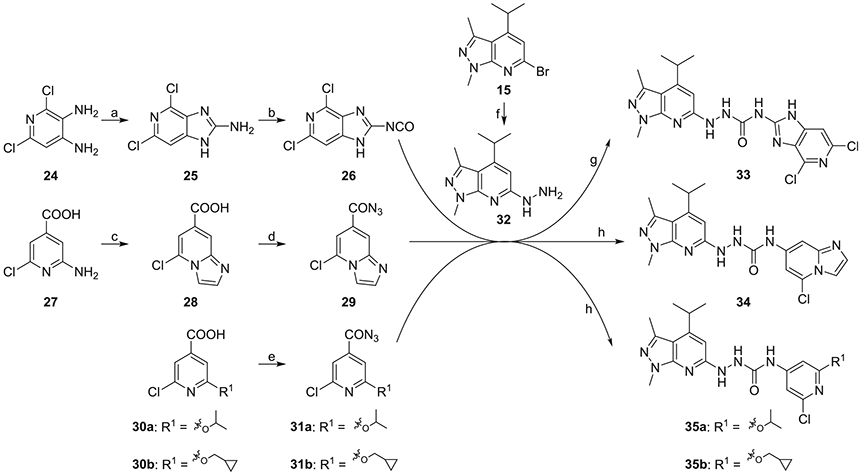
Syntheses of analogues 33, 34, 35a, and 35b. Reagents and conditions: (a) cyanogen bromide, ethanol, reflux, 64%; (b) triphosgene, triethylamine, toluene, rt, 63%; (c) 2-bromo-1,1-dimethoxyethane, 48% HBr, ethanol, reflux, 79%; (d) diphenylphosphoryl azide, triethylamine, DMF, 55%; (e) diphenylphosphoryl azide, triethylamine, 1,4-dioxane, 0 °C-rt, 64% for 31a and 69% for 31b; (f) hydrazine, ethanol, reflux, 65%; (g) THF, rt, 20%; (h) toluene, reflux, THF, rt, 57% for 34, 67% for 35a, and 83% for 35b.
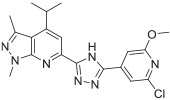
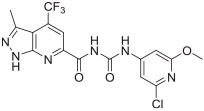
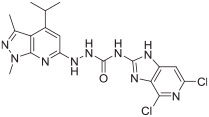
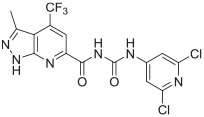
The in vitro binding potency of newly synthesized analogues and reference compound JTE-013 was evaluated using competitive radioligand binding assay with cell membranes following published protocol.23 As shown in Table 1, compound 7, having a reversed pyrazole ring and one more carbonyl group showed decreased binding potency compared to JTE-013; compounds 13a–d, containing trifluoromethyl group used as bioisotere of isopropyl also exhibited poor binding potency (IC50 > 1000 nM). Modification of fragment A did not improve binding potency. Modification of fragment B gave compounds 22 and 23, with oxadiazole and triazole rings instead of the urea linker. The IC50 values of 22 and 23 are >1000 nM, indicating that the heterocyclic linker reduced binding potency. Modification of fragment C gave compounds 33, 34, and 35a–b. Compounds 33 and 34, possessing imidazo[4,5-c]pyridine and imidazo[1,2-a]-pyridine, respectively, were also not potent (IC50 > 1000 nM); Compounds 35a and 35b, possessing 2-chloro-6-isopropoxypyridine and 2-chloro-6-cyclopropoxypyridine, respectively, had modestly improved binding potency (35a, IC50 = 29.1 ± 2.6 nM; 35b, IC50 = 56.5 ± 4.0 nM) compared to JTE-013 (IC50 = 58.4 ± 7.4 nM). The competitive binding curves of compounds 35a, 35b, and JTE-013 for S1PR2 were presented in Fig. 3. These results indicated that replacement of 2,6-dichloropyridine moiety with imidazo[4,5-c]pyridine and imidazo [1,2-a]pyridine had a negative impact on S1PR2 binding potency, and the replacement of a chloro on 2,6-dichloropyridine ring with isopropoxyl and cyclopropoxyl resulted in a modest improvement in the binding potency. Although most of compounds exhibited poor binding potency with IC50 > 1000 nM, useful structure-activity relationship (SAR) information of these new analogues was generated. The 1H-pyrazolo[3,4-b]pyridine in fragment A and urea linker in fragment B are important pharmacophores, modifications of either fragment resulted in loss of binding activity. The modification of 2,6-dichloropyridine in the fragment C is able to remain or improve the binding potency toward S1PR2 when the different substituted groups were introduced. Because compounds 35a and 35b showed good binding affinity for S1PR2, their binding potency toward other S1P receptor subtypes were determined to evaluate their selectivity for S1PR2. As shown in Table 2, compounds 35a and 35b exhibited high selectivity toward S1PR2 over S1PR1, 3, 4, and 5, indicating these two new ligands are selective for S1PR2. Our future work will focus on the continued exploration of structure modifications of fragment C building on this SAR information.
Table 1.
The IC50 values (mean ± SD)a of new compounds toward S1PR2.
| Compd. | Structure | S1PR2 IC50 (nM) | Compd. | Structure | S1PR2 IC50 (nM) |
|---|---|---|---|---|---|
| JTE-013 |
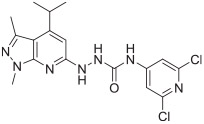
|
58.4 ± 7.4 | 22 |
|
>1000 |
| 7 |
|
>1000 | 23 |
|
>1000 |
| 13a |
|
>1000 | 33 |
|
>1000 |
| 13b |
|
>1000 | 34 |
|
>1000 |
| 13c |

|
>1000 | 35a |
|
29.1 ± 2.6 |
| 13d |
|
>1000 | 35b |

|
56.5 ± 4.0 |
IC50 values were determined at least two independent experiments, each run was performed in duplicate; for compounds with IC50 < 100 nM, at least three independent experiments were performed, each run was performed in duplicate.
Fig. 3.

Competitive binding curves of compounds 35a, 35b, and JTE-013 for S1PR2. A CHO cell membrane containing recombinant human S1PR2 was used in a [32P]S1P competitive binding assay to measure the binding affinities.
Table 2.
The IC50 values (mean ± SD)a of 35a and 35b binding toward other S1PRs.
| Compd. | IC50 (nM) |
||||
|---|---|---|---|---|---|
| S1PR1 | S1PR2 | S1PR3 | S1PR4 | S1PR5 | |
| JTE-013 | >1000 | 58.4 ± 7.4 | >1000 | ND | ND |
| S1P | 1.4 ± 0.3 | 3.6 ± 0.5 | 0.4 ± 0.2 | 151 ± 82 | 3.1 ± 1.1 |
| 35a | >1000 | 29.1 ± 2.6 | >1000 | >1000 | >1000 |
| 35b | >1000 | 56.5 ± 4.0 | >1000 | >1000 | >1000 |
IC50 values were determined at least two independent experiments, each run was performed in duplicate; for compounds with IC50 < 100 nM, at least three independent experiments were performed, each run was performed in duplicate, ND-not determined.
Experimental methods and characterization data for intermediates and target compounds are provided in the Notes along with the binding assay methods.24
In summary, a series of pyrazolopyridine derivatives were synthesized and evaluated for their S1PR2 binding potency and selectivity. The in vitro data indicated compounds 35a and 35b have comparable binding affinities for S1PR2 with IC50 values of 29.1 ± 2.6, 56.5 ± 4.0 nM, respectively compared to compound JTE-013 (IC50 = 58.4 ± 7.4 nM). Further evaluation of the selectivity of S1PR2 over other S1PRs (1, 3, 4, and 5) indicated that both 35a and 35b are selective for S1PR2. The initial SAR analysis suggest 1H-pyrazolo[3,4-b]pyridine in fragment A and urea linker in fragment B are essential pharmacophores, but the 2,6-dichloropyridine in fragment C can be modified. Further optimization of these analogues is needed to identify additional and more potent and selective compounds targeting S1PR2.
Acknowledgments
This study was supported by the National Multiple Sclerosis Society [RG150705331] and USA Department of Energy (DOE) Training Grants [DESC0008432 and DESC0012737]. It was partially supported by USA National Institutes of Health (NIH) through the National Institute of Neurological Disorders and Stroke [NS075527] and the National Institute of Mental Health [MH092797]. Mass spectrometry was generated from the Washington University Mass Spectrometry facility that is supported by NIH.
References
- 1.Hla T. Pharmacol Res. 2003;47:401. [DOI] [PubMed] [Google Scholar]
- 2.Rosen H, Goetzl EJ. Nat Rev Immunol. 2005;5:560. [DOI] [PubMed] [Google Scholar]
- 3.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Annu Rev Biochem. 2009;78:743. [DOI] [PubMed] [Google Scholar]
- 4.Chun J, Goetzl EJ, Hla T, et al. Pharmacol Rev. 2002;54:265. [DOI] [PubMed] [Google Scholar]
- 5.Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. Pharmacol Rev. 2010;62:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Compston A, Coles A. Lancet. 2008;372:1502. [DOI] [PubMed] [Google Scholar]
- 7.Browne P, Chandraratna D, Angood C, et al. Neurology. 2014;83:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore P, Methley A, Pollard C, et al. J Neurol Sci. 2016;360:4. [DOI] [PubMed] [Google Scholar]
- 9.Kappos L, Radue E-W, O’Connor P, et al. N Engl J Med. 2010;362:387. [DOI] [PubMed] [Google Scholar]
- 10.Brinkmann V, Davis MD, Heise CE, et al. J Biol Chem. 2002;277:21453. [DOI] [PubMed] [Google Scholar]
- 11.Abbasi T, Garcia JG. Handb Exp Pharmacol. 2013;216:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz-Orengo L, Daniels BP, Dorsey D, et al. J Clin Invest. 2014;124:2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osada M, Yatomi Y, Ohmori T, Ikeda H, Ozaki Y. Biochem Biophys Res Commun. 2002;299:483. [DOI] [PubMed] [Google Scholar]
- 14.Satsu H, Schaeffer M-T, Guerrero M, et al. Bioorg Med Chem. 2013;21:5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusumi K, Shinozaki K, Kanaji T, et al. Bioorg Med Chem Lett. 2015;25:1479. [DOI] [PubMed] [Google Scholar]
- 16.Kusumi K, Shinozaki K, Yamaura Y, et al. Bioorg Med Chem Lett. 2015;25:4387. [DOI] [PubMed] [Google Scholar]
- 17.Kusumi K, Shinozaki K, Yamaura Y, et al. Bioorg Med Chem Lett. 2016;26:1209. [DOI] [PubMed] [Google Scholar]
- 18.Yue X, Jin H, Liu H, Rosenberg AJ, Klein RS, Tu Z. Org Biomol Chem. 2015;13:7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang WK, Wang L, Corpuz EG, Chow K, Im WB. PCT Inl. Appl WO2011/041287A1; 2011. [Google Scholar]
- 20.Swenson RE, Raju N, Estrella-Jimenez ME, Ramalingam K. PCT Inl. Appl WO2011/159864A1; 2011. [Google Scholar]
- 21.Jagodzinska M, Huguenot F, Candiani G, Zanda M. ChemMedChem. 2009;4:49. [DOI] [PubMed] [Google Scholar]
- 22.Rankovic Z J Med Chem. 2017;60:5943. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg AJ, Liu H, Tu Z. Appl Radiat Isot. 2015;102:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(A) General: Commercially available starting materials, reagents, and solvents were used as received. Unless otherwise indicated, all reactions were conducted in oven dried glassware. Reactions were monitored by thin-layer chromatography (TLC) carried out on pre-coated glass plates of silica gel 60 F254 from EMD Chemicals Inc. Visualization was accomplished under ultraviolet light (UV 254 nm). Flash column chromatography was performed using 230–400 mesh silica gel purchased from Silicycle. Yields refer to isolated yield by chromatography, unless otherwise stated. Melting points were determined on a MEL-TEMP 3.0 apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on Varian 400 MHz instrument. Chemical shifts are reported in parts per million (ppm) and are calibrated using residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; DMSO d6: δ 2.50 ppm; Acetone d6: δ 2.05 ppm). High resolution positive ion mass was acquired by a Bruker MaXis 4G Q-TOF mass spectrometer with electrospray ionization source. Chemical purities of finial compounds were determined by reversed phase HPLC (Agilent C-18 column; mobile phase: 80% Acetonitrile in 0.1 M Ammonium formate, pH 4.5; flow rate: 1.0 mL/min; UV: 254 nm).7-Isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine 4-oxide (2): To a solution of 7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine (1) (0.66 g, 3.5 mmol) in 50 mL of chloroform was added 77% 3-chloroperbenzoic acid (mCPBA) (1.2 g, 7.0 mmol) at 0 °C. The resulting reaction mixture was stirred at 90 °C for 3 h. After cooling to room temperature, the mixture was neutralized with aqueous NaHCO3 and extracted with dichloromethane (3 × 100 mL). The combined organic phases were washed with water, saturated brine, and concentrated under reduced pressure. The crude was purified by flash chromatography with hexane/ethyl acetate (2/1, v/v) to give 2 (0.57 g, 79%). 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 6.3 Hz, 1H, Ar─H), 6.78 (d, J = 6.3 Hz, 1H, Ar─H),. 4.21 (s, 3H, ─N─CH3), 3.60 (hept, J = 6.7 Hz, 1H, ─CH─), 2.83 (s, 3H, ─C─CH3), 1.37 (d, J = 6.7 Hz, 6H, ─CH─CH3).7-Isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine-5-carbonitrile (3): To a solution of 7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine N-oxide (2) (0.72 g, 3.5 mmol) and triethyl amine (0.71 g, 7.0 mmol) in 20 mL acetonitrile was added trimethylsilyl cyanide (TMS-CN) (0.53 g, 5.2 mmol). The resulting solution was stirred at reflux for 18 h. Then another portion of trimethylsilyl cyanide (0.26 g, 2.6 mmol) was added and the reaction was continued to stir for another 7 h. The solvent was removed under reduced pressure and the residue was purified by flash chromatography with hexane/ethyl acetate (3/1, v/v) to give 3 (562 mg, 75%). 1H NMR (400 MHz, CDCl3): δ 7.46 (s, 1H, Ar─H), 4.26 (s, 3H, ─N─CH3), 3.69 (hept, J = 6.7 Hz, 1H, ─CH─), 2.64 (s, 3H, ─C─CH3), 1.40 (d, J = 6.7 Hz, 6H, ─CH─CH3).7-Isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine-5-carbox-amide (4): To a solution of 7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine-5-carbonitrile (3) (81 mg, 0.38 mmol) in methanol (5 mL) was added 1 M NaOH (7 mL) and 35% H2O2 (1.3 mL). The reaction mixture was stirred at room temperature for 2 h. Then, the reaction mixture was diluted with dichloromethane. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product 4 was obtained as a white solid without further purification (79 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H, Ar─H), 7.84 (br, 1H, CONH2), 5.43 (br, 1H, CONH2), 4.18 (s, 3H, ─N─CH3), 3.63 (hept, J = 7.0 Hz, 1H, ─CH─), 2.58 (s, 3H, ─C─CH3), 1.38 (d, J = 7.0 Hz, 6H, ─CH─CH3).2-Chloro-6-methoxyisonicotinoyl azide (6): To a round-bottomed flask equipped with a magnetic stir bar was added 2-chloro-6-methoxyisonicotinic acid (5) (1.88 g, 10.0 mmol), diphenylphosphorylazide (3.03 g, 11.0 mmol), and 1,4-dioxane (20 mL). Triethylamine (2.53 g, 25.0 mmol) was added slowly through syringe under nitrogen at 0 °C. The reaction was warmed and stirred at room temperature for 5 h. Then, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (30/1, v/v) to afford 6 (1.12 g, 55%). 1H NMR (400 MHz, Acetone d6) d = 7.21 (s, 1H, Ar─H), 6.99 (s, 1H, Ar─H), 3.82 (s, 3H, ─CH3).N-((2-Chloro-6-methoxypyridin-4-yl)carbamoyl)-7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine-5-carboxamide (7): To a round-bottomed flask equipped with a magnetic stir bar was added 2-chloro-6-methoxyisonicotinoyl azide (6) (54 mg, 0.25 mmol) and toluene (10 mL). The mixture was refluxed for 2 h and cooled to 50 °C before 7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine-5-carboxamide (4) (50 mg, 0.21 mmol) in THF (10 mL) was added. The reaction was stirred for another 10 h. The reaction then was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (1/1, v/v) to give 7 as a white solid (57 mg, 65%). MP: 117–119 °C. 1H NMR (400 MHz, CDCl3) δ 10.90 (s, 1H, CONHCO), 10.16 (s, 1H, CONH), 8.10 (s, 1H, Ar─H), 7.17 (d, J = 1.0 Hz, 1H, Ar─H), 6.94 (d, J = 1.0 Hz, 1H, Ar─H), 4.27 (s, 3H, ─N─CH3), 3.92 (s, 3H, ─OCH3), 3.78–3.67 (m, 1H, ─CH─), 2.64 (s, 3H, ─C─CH3), 1.45 (d, J = 6.8 Hz, 6H, ─CH─CH3). 13C NMR (101 MHz, CDCl3) δ 165.75, 164.65, 149.97, 149.07, 148.24, 143.63, 142.17, 141.68, 140.32, 133.71, 115.87, 107.57, 98.22, 54.12, 39.51, 28.22, 23.11, 10.90. HRMS (ESI) calcd. for C19H21ClN6NaO3 [M+Na]+, 439.1256, found 439.1247. HPLC purity: 98%, tR = 10.1 min.3-Methyl-4-(trifluoromethyl)-1,7-dihydro-6H-pyrazolo[3,4-b]-pyridin-6-one (9a): To a round-bottomed flask equipped with a magnetic stir bar was added 3-methyl-1H-pyrazol-5-amine (8a) (1.0 g, 10 mmol), ethyl 4,4,4-trifluoroacetoacetate (2.0 mL, 13 mmol), and propionic acid (50 mL). The mixture was heated at 140 °C for 12 h. After cooling, the mixture was concentrated under reduced pressure and the residue was treated with ethyl acetate. The precipitate was filtered and dried as product 9a (1.3 g, 61%). 1H NMR (400 MHz, CD3OD) δ 6.47 (s, 1H, Ar─H), 2.42 (s, 3H, ─C─CH3).1,3-Dimethyl-4-(trifluoromethyl)-1,7-dihydro-6H-pyrazolo[3,4-b]pyridin-6-one (9b): To a round-bottomed flask equipped with a magnetic stir bar was added 1,3-dimethyl-1H-pyrazol-5-amine (8b) (10.0 g, 90 mmol), ethyl 4,4,4-trifluoroacetoacetate (17 mL, 110 mmol), and propionic acid (100 mL). The mixture was heated at 140 °C for 12 h. After cooling, the mixture was concentrated under reduced pressure and the residue was treated with ethyl acetate. The precipitate was filtered and dried as product 9b (15.5 g, 75%). 1H NMR (400 MHz, CD3OD) δ 6.60 (s, 1H, Ar─H), 3.87 (s, 3H, ─N─CH3), 2.41 (s, 3H, ─C─CH3).6-Bromo-3-methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]-pyridine (10a): A suspension of 3-methyl-4-(trifluoromethyl)-1,7-dihydro-6H-pyrazolo[3,4-b] pyridin-6-one (9a) (1.7 g, 7.7 mmol) and POBr3 (2.9 g, 10 mmol) in anisole (100 mL) was heated at 140 °C for 3 h under nitrogen. The reaction mixture was diluted with toluene (50 mL). The organic layer was then washed with water (3 × 50 mL) and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (3/1, v/v) to give 10a (1.7 g, 78%). 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H, Ar─H), 2.65 (s, 3H, ─C─CH3).6-Bromo-1,3-dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]-pyridine (10b): A suspension of 1,3-dimethyl-4-(trifluoromethyl)-1,7-dihydro-6H-pyrazolo [3,4-b]pyridin-6-one (9b) (800 mg, 2.7 mmol) and POBr3 (1.3 g, 4.5 mmol) in anisole (100 mL) was heated with stirring at 140 °C for 3 h under nitrogen. The reaction mixture was diluted with toluene (50 mL). The organic layer was then washed with water (3 × 50 mL) and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (3/1, v/v) to give 10b (556 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 7.41 (s, 1H, Ar─H), 4.03 (s, 3H, ─N─CH3), 2.54 (s, 3H, ─C─CH3).3-Methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (11a): A mixture of 6-bromo-3-methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine (10a) (1.1 g, 4.0 mmol), CuCN (0.7 g, 8.0 mmol), and anhydrous DMF (20 mL) was heated at 110 °C for 7 h. After cooling, the mixture was diluted and filtered. The filtrate was concentrated and purified by flash chromatography with hexane/ethyl acetate (3/1, v/v) to give 11a (542 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H, Ar─H), 2.66 (s, 3H, ─C─CH3).1,3-Dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (11b): A mixture of 6-bromo-1,3-dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine (10b) (1.5 g, 5.1 mmol), CuCN (1.37 g, 15.5 mmol), and anhydrous DMF (100 mL) was heated at 110 °C for 7 h. After cooling, the mixture was diluted and filtered. The filtrate was concentrated and purified by flash chromatography with hexane/ethyl acetate (3/1, v/v) to give 11b (919 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H, Ar─H), 4.10 (s, 3H, ─N─CH3), 2.60 (s, 3H, ─C─CH3).3-Methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carboxamide (12a): To a solution of 3-methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (11a) (750 mg, 3.3 mmol) in methanol (5 mL) was added 1 M NaOH (6.5 mL) and 35% H2O2 (1.2 mL). The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with dichloromethane. Then, the organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product 12a was obtained without further purification (483 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H, Ar─H), 2.58 (s, 3H, ─C─CH3).1,3-Dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carboxamide (12b): To a solution of 1,3-dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (11b) (670 mg, 2.8 mmol) in methanol (5 mL) was added 1 M NaOH (7 mL) and 35% H2O2 (1.3 mL). The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with dichloromethane. Then, the organic layer was washed with brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product 12b was obtained without further purification (593 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H, Ar─H), 4.15 (s, 3H, ─N─CH3), 2.65 (s, 3H, ─C─CH3).2,6-Dichloroisonicotinoyl azide (6a): To a round-bottomed flask equipped with a magnetic stir bar was added 2,6-dichloroisonicotinic acid (5a) (1.92 g, 10.0 mmol), diphenylphosphorylazide (3.03 g, 11.0 mmol), and 1,4-dioxane (20 mL). Triethyamine (2.53 g, 25.0 mmol) was added slowly through syringe under nitrogen at 0 °C. The reaction was warmed and stirred at room temperature for 5 h. The mixture then was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (50/1, v/v) to afford 6a (1.30 g, 60%). 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 2H, Ar─H).General procedure for the synthesis of 13a–d: To a round-bottomed flask equipped with a magnetic stir bar was added 6 or 6a (1.2 eq) and toluene (10 mL). The mixture was refluxed for 2 h and cooled to 50 °C before 12a or 12b (1.0 eq) in THF (10 mL) was added. The reaction was stirred for another 10 h. Then, the reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate to give 13a-d as a white solid. N-((2-Chloro-6-methoxypyridin-4-yl)carbamoyl)-3-methyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carboxamide (13a): Yield: 53%. MP: 91–94 °C. 1H NMR (400 MHz, DMSO d6) δ 9.65 (s, 1H, CONH), 8.08 (s, 1H, Ar─H), 6.21 (s, 1H, Ar─H), 5.78 (s, 1H, Ar─H), 3.70 (s, 3H, ─CH3), 2.58 (s, 3H, ─C─CH3). 13C NMR (101 MHz, DMSO d6) δ 165.54, 164.66, 159.09, 152.83, 151.85, 150.74, 150.27, 148.15, (d, J = 22.2 Hz), 139.62, 123.11 (q, J = 274.7 Hz), 110.89, 106.66, 103.30, 96.73, 53.64, 14.45. HRMS (ESI) calcd. for C16H12ClF3N6NaO3 [M+Na]+, 451.0504, found 451.0536. HPLC purity: 96%, tR = 4.0 min.N-((2,6-Dichloropyridin-4-yl)carbamoyl)-3-methyl-4-(trifluoro-methyl)-1H-pyrazolo[3,4-b]pyridine-6-carboxamide (15): Yield: 48%. MP: 141–145 °C. 1H NMR (400 MHz, DMSO d6) δ 10.01 (s, 1H, CONH), 8.17 (s, 1H, Ar─H), 6.49 (s, 2H, Ar─H), 2.58 (s, 3H, ─C─CH3). 13C NMR (101 MHz, DMSO d6) δ 165.56, 158.98, 152.79, 150.28, 150.11, 149.60, 139.81, 123.14, (q, J = 279.8 Hz), 111.25, 110.83, 106.94, 14.58. HRMS (ESI) calcd. for C15H9Cl2F3N6NaO2 [M+Na]+, 455.0008, found 451.0047. HPLC purity: 97%, tR = 3.9 min.N-((2-Chloro-6-methoxypyridin-4-yl)carbamoyl)-1,3-dimethyl-4-(trifluoromethyl)-1H pyrazolo[3,4-b]pyridine-6-carboxamide (13c): Yield: 64%. MP: 103–106 °C. 1H NMR (400 MHz, CDCl3) δ 10.74 (s, 1H, CONHCO), 9.93 (s, 1H, CONH), 8.30 (s, 1H, Ar─H), 7.19 (s, 1H, Ar─H), 6.97 (s, 1H, Ar─H), 4.22 (s, 3H, ─N─CH3), 3.93 (s, 3H, ─CH3), 2.68 (s, 3H, ─C─CH3). 13C NMR (101 MHz, CDCl3) δ 164.71, 164.11, 150.44, 149.44, 149.25, 147.85, 145.30, 140.16, 133.46 (d, J = 36.4 Hz), 122.15 (q, J = 275.7 Hz), 112.61, 111.15, 107.65, 98.46, 54.20, 34.47, 14.18. HRMS (ESI) calcd. for C17H14ClF3N6NaO3 [M+Na]+, 465.0660, found 465.0674. HPLC purity: 95%, tR = 10.0 min.N-((2,6-Dichloropyridin-4-yl)carbamoyl)-1,3-dimethyl-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine-6-carboxamide (13d): Yield: 67%. MP: 133–135 °C. 1H NMR (400 MHz, CDCl3) δ 10.95 (s, 1H, CONHCO), 9.99 (s, 1H, CONH), 8.31 (s, 1H, Ar─H), 7.60 (s, 2H, Ar─H), 4.24 (s, 3H, ─N─CH3), 2.70 (s, 3H, ─C─CH3). 13C NMR (101 MHz, CDCl3) δ 164.30, 151.33, 150.45, 149.37, 147.94, 145.01, 140.25, 133.56 (d, J = 32.3 Hz), 122.11 (q, J = 274.7 Hz), 112.79, 111.26, 111.21, 34.53, 14.20. HRMS (ESI) calcd. for C16H11Cl2F3N6NaO2 [M+Na]+, 469.0165, found 469.0191. HPlC purity: 99%, tR = 9.0 min.4-Isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-ol (14): To a round-bottomed flask equipped with a condenser and magnetic stir bar was added commercial 1,3-dimethyl-1H-pyrazol-5-amine (8b) (5.56 g, 50 mmol), ethyl isobutyrylacetate (8.70 g, 55 mmol), and acetic acid (40 mL). The reaction vessel was immersed in a 120 °C preheated oil bath for 24 h. After cooling, the mixture was concentrated and the resulting oil was triturated with ethyl acetate (20 mL). The resulting solid was filtered and dried under vacuum to afford 14 (1.06 g, 10.3%). 1H NMR (400 MHz, CDCl3) δ 13.85 (s, 1H, ─H), 6.16 (d, J = 0.6 Hz, 1H, Ar─H), 3.99 (s, 3H, ─N─CH3), 3.37–3.25 (m, 1H, ─CH─), 2.51 (s, 3H, ─C─CH3), 1.31 (d, J = 6.8 Hz, 6H, ─CH─CH3).6-Bromo-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (15): To a round-bottomed flask equipped with a magnetic stir bar was added 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-ol (14) (1.03 g, 4.89 mmol), POBr3 (2.10 g, 7.33 mmol), and anisole (10 mL). The reaction vessel was immersed in a 120 °C preheated oil bath for 3 h. After cooling, saturated NaHCO3 was added to quench the mixture. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate to (10/1, v/v) to afford 15 (1.28 g, 98%). 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H, Ar─H), 4.02 (s, 3H, ─N─CH3), 3.71–3.58 (m, 1H, ─CH─), 2.66 (s, 3H, ─C─CH3), 1.35 (d, J = 6.9 Hz, 6H, ─CH─CH3).4-Isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (16): To a round-bottomed flask equipped with a magnetic stir bar were added 6-bromo-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (15) (1.34 g, 5.0 mmol), CuCN (1.80 g, 20.0 mmol), and DMF (15.0 mL). The reaction vessel was immersed in a 110 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (5/1) to afford 16 (0.64 g, 60%). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H, Ar─H), 4.07 (s, 3H, ─N─CH3), 3.72–3.60 (m, 1H, ─CH─), 2.71 (s, 3H, ─C─CH3), 1.38 (d, J = 6.9 Hz, 6H, ─CH─CH3).(Z)-N′-Hydroxy-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]-pyridine-6-carboximidamide (17): To a suspension of hydroxylamine hydrochloride (0.42 g, 6.0 mmol) in methanol (10 mL) was added NaHCO3 (0.55 g, 6.6 mmol), followed by adding 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (16) (0.64 g, 3.0 mmol). The reaction vessel was immersed in 80 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the solvent was removed under vacuum, the residue was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product 17 was obtained. (0.6 g, 81%). 1H NMR (400 MHz, DMSO d6) δ 10.04 (s, 1H, ─H), 7.56 (s, 1H, Ar─H), 5.95 (s, 2H, ─NH2), 4.00 (s, 3H, ─N─CH3), 3.65–3.50 (m, 1H, ─CH─), 2.60 (s, 3H, ─C─CH3), 1.30 (d, J = 6.8 Hz, 6H, ─CH─CH3).4-Isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carboximidamide hydrochloride (18): To a solution of NaOCH3 (0.18 g, 3.3 mmol) in methanol (5.0 mL) was added 4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carbonitrile (16) (0.41 g, 0.78 mmol). The mixture was stirred at room temperature and monitored by TLC. After the 16 disappeared, NH4Cl (0.17 g, 3.2 mmol) was added to the mixture and stirred at 70 °C until the reaction was completed as determined by TLC. After cooling, the solvent was removed under vacuum and ethyl acetate was added into the residue, the suspension was stirred at room temperature for 3 h. The mixture was filtered and the filter cake was collected as product hydrochloride salt. 1H NMR (400 MHz, CD3OD) 7.97 (s, 1H, Ar─H), 4.06 (s, 3H, ─N─CH3), 3.76–3.64 (m, 1H, ─CH─), 2.67 (s, 3H, ─C─CH3), 1.36 (d, J = 6.8 Hz, 6H, ─CH─CH3).tert-Butyl 2-(2-chloro-6-methoxyisonicotinoyl)hydrazine-1-carboxylate (20): To a solution of 2-chloro-6-methoxyisonicotinic acid (19) (188 mg, 1.0 mmol) in dichloromethane (10 mL) was added oxalyl chloride (381 mg, 3.0 mmol) and 3 drops of DMF as catalyst. The mixture was stirred at room temperature for 1 h and the solvent and excess oxalyl chloride was removed under vacuum. The residue was dissolved in the dichloromethane. To the solution was added tert-butyl hydrazinecarboxylate (132 mg, 1.0 mmol) and triethylamine (303 mg, 3.0 mmol) at 0 °C. The mixture was stirred at room temperature until the reaction was completed as determined by TLC. Then, the mixture was washed with water, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the crude product was collected. 1H NMR (400 MHz, DMSO d6) δ 10.50 (s, 1H, ─OCONH), 9.08 (s, 1H, CONH), 7.42 (s, 1H, Ar─H), 7.19 (s, 1H, Ar─H), 3.90 (s, 3H, ─O─CH3), 1.43 (s, 9H, ─C─CH3).3-Chloro-5-methoxybenzohydrazide (21): To a solution of tert-butyl 2-(2-chloro-6-methoxyisonicotinoyl)hydrazine-1-carboxylate (20) (300 mg, 1.0 mmol) in CH2Cl2 (5 mL) was added CF3COOH (1.0 mL) at 0 °C and the mixture was stirred at room temperature until the reaction was completed as determined by TLC. The mixture was washed with saturated NaHCO3 solution, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (1/3, v/v) to afford 21 (52 mg, 25%). 1H NMR (400 MHz, DMSO d6) δ 10.10 (s, 1H, CONH), 7.40 (s, 1H, Ar─H), 7.16 (s, 1H, Ar─H), 4.65 (s, 2H, NH2), 3.89 (s, 3H, ─O─CH3).5-(2-Chloro-6-methoxypyridin-4-yl)-3-(4-isopropyl-1,3-dimethyl-1H-pyrazolo [3,4-b]pyridin-6-yl)-1,2,4-oxadiazole (22): To a solution of 2-chloro-6-methoxyisonicotinic acid (19) (132 mg, 0.70 mmol) and DIPEA (136 mg, 1.05 mmol) in DMF (5 mL) was added PyBOP (406 mg, 0.78 mmol) at 0 °C. After stirring for 15 min, (Z)-N′-hydroxy-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine-6-carboximidamide (17) (90 mg, 0.36 mmol) was added into the mixture, the reaction was stirring at room temperature monitored by TLC. Then, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the residue was dissolved with 1,4-dioxane (10 mL) and stirred at 100 °C for 6 h. After cooling, the reaction mixture was evaporated with silica gel and purified by flash chromatography with hexane/ethyl acetate (2/1, v/v) to afford 22 (56 mg, 40%). MP: 212–215 °C. 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H, Ar─H), 7.75 (s, 1H, Ar─H), 7.50 (s, 1H, Ar─H), 4.18 (s, 3H, ─N─CH3), 4.04 (s, 3H, ─O─CH3), 3.76–3.69 (m, 1H, ─CH─), 2.74 (s, 3H, ─C─CH3), 1.45 (d, J = 6.9 Hz, 6H, ─CH─CH3). HRMS (ESI) calcd. for C19H21ClN6O2 [M+H]+ 399.1332, found 399.1340. HPLC purity: 98%, tR = 13.4 min.6-(5-(2-Chloro-6-methoxypyridin-4-yl)-4H-1,2,4-triazol-3-yl)-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (23): To a round-bottomed flask equipped with a magnetic stir bar were added 4-isopropyl-1,3-dimethyl-1H-pyrazolo [3,4-b]pyridine-6-carboximidamide hydrochloride (18) (66 mg, 0.25 mmol), 3-chloro-5-methoxybenzohydrazide (21) (50 mg, 0.25 mmol), and ethanol (3.0 mL). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated NaHCO3, saturated brine, and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (3/2, v/v) to afford 23 (63 mg, 63%). MP: 267–269 °C. 1H NMR (400 MHz, DMSO d6) δ 7.82 (s, 1H, Ar─H), 7.65 (s, 1H, Ar─H), 7.38 (s, 1H, Ar─H), 4.06 (s, 3H, ─N─CH3), 3.93 (s, 3H, ─O─CH3), 3.74–3.60 (m, 1H, ─CH─), 2.65 (s, 3H, ─C─CH3), 1.39 (d, J = 6.8 Hz, 6H, ─CH─CH3). 13C NMR (101 MHz, DMSO) δ 164.51, 158.67, 155.54, 151.03, 148.57, 145.23, 144.02, 139.89, 135.87, 113.81, 113.50, 110.02, 105.78, 54.70, 33.72, 29.61, 23.26, 15.39. HRMS (ESI) calcd. for C19H21ClN7O [M+H]+, 398.1491, found 398.1487. HPLC purity: 99%, tR = 9.2 min.4,6-Dichloro-1H-imidazo[4,5-c]pyridin-2-amine (25): To a round-bottomed flask equipped with a magnetic stir bar were added 2,6-dichloropyridine-3,4-diamine (24) (0.18 g, 1.0 mmol), cyanogen bromide (0.16 g, 1.5 mmol), and ethanol (3.0 mL). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, ammonia solution was added and the precipitated was filtered as gray product 25 (0.13 g, 64%). 1H NMR (400 MHz, DMSO d6) δ 11.12 (s, 1H, ─NH─), 7.12 (s, 1H, Ar─H), 6.92 (s, 2H, ─NH2). 13C NMR (101 MHz, DMSO) δ 158.62, 146.97, 137.38, 135.12, 130.41, 105.96.4,6-Dichloro-2-isocyanato-1H-imidazo[4,5-c]pyridine (26): To a round-bottomed flask equipped with a magnetic stir bar were added 4,6-dichloro-1H-imidazo [4,5-c]pyridin-2-amine (25) (0.12 g, 0.6 mmol), triphosgene (89 mg, 0.3 mmol), and toluene (5.0 mL). After cooling to 0 °C, triethyamine was added to the mixture dropwise and the reaction mixture was stirred at room temperature for 3 h until the reaction was completed as determined by TLC. Then, the mixture was quenched with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was obtained which was used directly for the next step (87 mg, 63%).5-Chloroimidazo[1,2-a]pyridine-7-carboxylic acid (28): To a round-bottomed flask equipped with a magnetic stir bar was added 2-amino-6-chloropyridine-4-carboxylic acid (27) (0.5 g, 2.9 mmol), 2-bromo-1-dimethoxyethane (0.61 g, 3.6 mmol) and ethanol (25 mL). To the mixture was added 48% HBr (0.33 mL, 2.0 mmol) slowly through syringe under nitrogen. The reaction vessel was immersed in a 100 °C preheated oil bath for 3 h. After cooling to room temperature, the precipitate was filtered to afford 28 (0.45 g, 79%). 1H NMR (400 MHz, DMSO d6) δ 8.51 (d, J = 1.8 Hz, 1H, Ar─H), 8.34 (s, 1H, Ar─H), 8.31 (d, J = 0.7 Hz, 1H, Ar─H), 7.88 (s, 1H, Ar─H).5-Chloroimidazo[1,2-a]pyridine-7-carbonyl azide (29): To a round-bottomed flask equipped with a magnetic stir bar was added 5-chloroimidazo[1,2-a] pyridine-7-carboxylic acid (28) (0.45 g, 2.3 mmol), diphenylphosphorylazide (0.63 g, 2.3 mmol) and DMF (10 mL). After cooling to 0 °C, triethyamine (0.70 g, 6.9 mmol) was added slowly through syringe under nitrogen. The reaction was stirred at room temperature for 5 h. Then, the mixture was diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (3/2, v/v) to afford 29 (0.28 g, 55%). 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H, Ar─H), 7.92 (d, J = 1.4 Hz, 2H, Ar─H), 7.55 (d, J = 1.4 Hz, 1H, Ar─H).2-Chloro-6-isopropoxyisonicotinoyl azide (31a): To a round-bottomed flask equipped with a magnetic stir bar was added 2-chloro-6-isopropoxyisonicotinic acid (30a) (0.56 g, 2.6 mmol), diphenylphosphorylazide (1.0 g, 3.6 mmol), and 1,4-dioxane (10 mL). triethylamine (0.37 g, 3.6 mmol) was added slowly through syringe under nitrogen at 0 °C. The reaction was warmed and stirred at room temperature for 5 h. The mixture then was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (20/1, v/v) to afford 31a (0.43 g, 69%).2-Chloro-6-cyclopropoxyisonicotinoyl azide (31b): To a round-bottomed flask equipped with a magnetic stir bar was added 2-chloro-6-cyclopropoxyisonicotinic acid (30a) (0.43 g, 1.9 mmol), diphenylphosphorylazide (0.73 g, 2.6 mmol), and 1,4-dioxane (10 mL). triethylamine (0.26 g, 2.6 mmol) was added slowly through syringe under nitrogen at 0 °C. The reaction was warmed and stirred at room temperature for 5 h. Then, the mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with hexane/ethyl acetate (20/1, v/v) to afford 31b (0.31 g, 65%).6-Hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]-pyridine (32): To a round-bottomed flask equipped with a magnetic stir bar was added 6-bromo-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (15) (1.29 g, 4.8 mmol), hydrazine (4.62 g, 144 mmol) and ethanol (10 mL). The reaction vessel was immersed in a 100 °C preheated oil bath for 18 h. After cooling, the mixture was filtered, washed with cold ethanol to afford 32 (0.68 g, 65%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.23 (s, 1H, Ar─H), 5.87 (s, 1H, ─NH─), 4.00 (br, 2H, ─NH2), 3.92 (d, J = 1.5 Hz, 3H, ─N─CH3), 3.51–3.36 (m, 1H, ─CH─), 2.58 (s, 3H, ─C─CH3), 1.36 (d, J = 6.8 Hz, 6H, ─CH─CH3).N-(4,6-dichloro-1H-imidazo[4,5-c]pyridin-2-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (33): To a round-bottomed flask equipped with a magnetic stir bar was added 4,6-dichloro-2-isocyanato-1H-imidazo[4,5-c]pyridine (26) (0.12 g, 0.6 mmol), 6-hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (32) (0.11 g, 0.48 mmol) and THF (3.0 mL). The reaction mixture was stirred at room temperature for 12 h until the reaction was completed as determined by TLC. Then, the mixture was concentrated and purified by flash chromatography with dichloromethane/methanol (10/1,v/v) (54 mg, 20%). MP: 143–147 °C. 1H NMR (400 MHz, CD3OD) δ 7.38 (s, 1H, Ar─H), 6.51 (s, 1H, Ar─H), 3.81 (s, 3H, ─N─CH3), 3.54–3.46 (m, 1H, ─CH─), 2.55 (s, 3H, ─C─CH3), 1.32 (d, J = 6.8 Hz, 6H, ─CH─CH3). 13C NMR (101 MHz, Acetone d6) δ 164.33, 154.42, 150.75, 146.59, 146.06, 143.38, 138.99, 120.04, 109.96, 106.29, 103.55, 103.26, 98.20, 32.22, 29.49, 22.31, 14.41. HRMS (ESI) calcd. for C19H20Cl2N9O [M+H]+, 448.1164, found 448.1175. HPLC purity: 96%, tR = 2.9 min.N-(5-Chloroimidazo[1,2-a]pyridin-7-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo [3,4-b]pyridin-6-yl)hydrazine-1-carbox-amide (34). To a round-bottomed flask equipped with a magnetic stir bar was added 5-chloroimidazo[1,2-a]pyridine-7-carbonyl azide (29) (0.28 g, 1.26 mmol) and toluene (10 mL). The mixture was refluxed for 2 h and cooled to 50 °C before 6-hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (32) (0.27 g, 1.26 mmol) in THF (10 mL) was added. The reaction was stirred for another 10 h until the reaction was completed as determined by TLC. Then, the reaction was diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with dichloromethane/methanol (15/1, v/v) to afford 34 (0.3 g, 57%). MP: 160–163 °C. 1H NMR (400 MHz, DMSO d6) δ 9.22 (s, 1H, Ar─NHCO─), 8.64 (s, 1H, Ar-NH─), 8.50 (s, 1H, ─CONH─), 7.88–7.72 (m, 2H, Ar─H), 7.59–7.40 (m, 2H, Ar─H), 6.37 (s, 1H, Ar─H), 3.71 (s, 3H, ─N─CH3), 2.58–2.36 (m, 4H, ─CH-, ─C─CH3), 1.32–1.17 (m, 6H, ─CH─CH3). 13C NMR (101 MHz, DMSO d6) δ 160.08, 154.38, 151.23, 146.42, 145.26, 139.13, 137.82, 134.08, 128.64, 125.38, 110.73, 107.82, 107.25, 98.73, 33.20, 29.36, 23.18, 15.26. HRMS (ESI) calcd. for C19H22ClN8O [M+H]+,413.1600, found 413.1598. HPLC purity: 95%, tR = 3.3 min.N-(2-Chloro-6-isopropoxypyridin-4-yl)-2-(4-isopropyl-1,3-di-methyl-1H-pyrazolo [3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (35a): To a round-bottomed flask equipped with a magnetic stir bar was added 2-chloro-6-isopropoxyisonicotinoyl azide (31a) (48 mg, 0.2 mmol) and toluene (5 mL). The mixture was refluxed for 2 h and cooled to 50 °C before 6-hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (32) (34 mg, 0.15 mmol) in THF (5 mL) was added. The reaction was stirred for another 10 h until the reaction was completed as determined by TLC. Then, the reaction was diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with dichloromethane/methanol (30/1, v/v) to afford 35a (45 mg, 67%). MP: 110–112 °C. 1H NMR (400 MHz, Acetone d6) δ 8.94 (br, 1H, Ar-NHCO─), 7.78 (br, 2H, ─NHCONH), 7.17 (s, 1H, Ar─H), 6.90 (s, 1H, Ar─H), 6.38 (s, 1H, Ar─H), 5.10–5.02 (m, 1H, O─CH─), 3.66 (s, 3H, ─N─CH3), 3.40–3.30 (m, 1H, ─CH─), 2.38 (s, 3H, ─C─CH3), 1.15 (d, J = 6.8 Hz, 6H, O─CH─CH3), 1.13 (d, J = 6.0 Hz, 6H, Ar-CH─CH3); 13C NMR (101 MHz, Acetone d6) δ 163.8, 159.5, 155.8, 155.1, 151.0, 148.2, 139.1, 108.0, 106.0, 97.9, 97.8, 97.2, 68.4, 54.1, 32.3, 22.3, 21.2, 14.4. HRMS (ESI) calcd. for C20H26ClN7NaO2 [M+Na]+, 454.1729, found 454.1726. HPLC purity: 98%, tR = 4.2 min.N-(2-Chloro-6-(cyclopropylmethoxy)pyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (35b): To a round-bottomed flask equipped with a magnetic stir bar was added 2-Chloro-6-cyclopropoxyisonicotinoyl azide (31b) (76 mg, 0.30 mmol) and toluene (5 mL). The mixture was refluxed for 2 h and cooled to 50 °C before 6-hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (32) (51 mg, 0.23 mmol) in THF (5 mL) was added. The reaction was stirred for another 10 h until the reaction was completed as determined by TLC. Then, the reaction was diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified by flash chromatography with dichloromethane/methanol (30/1, v/v) to afford 35b (86 mg, 83%). MP: 189–191 °C. 1H NMR (400 MHz, Acetone d6) δ 8.80 (br, 1H, Ar-NHCO─), 7.78 (s, 1H, ArNHCO), 7.63 (s, 1H, CONH), 7.62 (s, 1H, Ar─H), 7.03 (s, 1H, Ar─H), 6.22 (s, 1H, Ar─H), 3.74 (d, J = 7.2 Hz, 2H, ─O─CH2), 3.50 (s, 3H, ─N─CH3), 3.23–3.08 (m, 1H, Ar-CH─), 2.21 (s, 3H, ─C─CH3), 1.76 (t, J = 32.8 Hz, 1H, ─CH─), 0.98 (d, J = 7.2 Hz, 6H, Ar-CH─CH3), 0.25–0.22 (m, 2H, ─CH2CH2), 0.04–0.01 (m, 2H, ─CH2CH2); 13C NMR (101 MHz, Acetone) δ 161.7, 156.9, 153.7, 152.6, 148.5, 148.4, 145.6, 136.5, 105.8, 103.7, 95.3, 94.1, 68.2, 52.1, 29.7, 19.7, 11.8, 7.2, 0.0. HRMS (ESI) calcd. for C21H26ClN7NaO2 [M+Na]+, 466.1729, found 466.1723. HPLC purity: 98%, tR = 4.2 min.(B) Sphingosine-1-phosphate receptor binding assay: 32P Labeled S1P was prepared by incubating sphingosine and [γ-32P]ATp with sphingosine kinase 1 as previously reported. [32P]S1P was dissolved in DMSO, and then diluted in the assay buffer [50 mM HEPES-Na (pH 7.5), 5 mM MgCl2, 1 mM CaCl2, 0.5% fatty acid-free BSA]. Test compounds in DMSO were pre-incubated with commercial cell membranes expressing recombinant human S1PRs (1, 2, 3, 4, and 5) in the assay buffer for 30 min at room temperature. [32P]S1P solutions were added to give a final volume of 150 μL, 0.1 nM [32P]-S1P and 1 μg membrane protein per well. Competitive binding was performed for 60 min at room temperature and terminated by collecting the membranes onto 96-well glass fiber (GF/B) filtration plates (Millipore, Billerica, MA). Each filter was washed with 200 μL of assay buffer for a total of five times. The filter bound radionuclide was measured by a Beckman LS 3801 scintillation counter using Cherenkov counting. Specific binding was calculated by subtracting radioactivity that remained in the presence of 1000-fold excess of non-labeled S1P (1 μM) diluted with binding buffer from a 1 mM S1P stock solution in DMSO. The reported IC50 values were calculated using the 4 parameter equation, least-square non-linear regression curve-fit, with GraphPad Prism software (GraphPad Software, Inc). Each assay was repeated three times with duplicate wells for each sample; the reported values (mean ± SD, nM) are calculated from average of all assays. Assays for compounds which showed no activity (IC50 >1000 nM) were only repeated twice.