

Abstract
Myelodysplastic syndromes (MDS) are heterogeneous hematopoietic disorders that are incurable with conventional therapy. The incidence is increasing with global population ageing. Although many genetic, epigenetic, splicing, and metabolic aberrations have been identified in MDS patients, their clinical features are quite similar. Here we show that hypoxia-independent activation of hypoxia-inducible factor 1α (HIF1A) signaling is both necessary and sufficient to induce dysplastic and cytopenic MDS phenotypes. The HIF1A transcriptional signature is generally activated in MDS-patient bone-marrow stem/progenitors. Major MDS-associated mutations (Dnmt3a, Tet2, Asxl1, Runx1, and Mll1) activate the HIF1A signature. While inducible activation of HIF1A signaling in hematopoietic cells is sufficient to induce MDS phenotypes, both genetic and chemical inhibition of HIF1A signaling rescues MDS phenotypes in a mouse model of MDS. These findings reveal HIF1A as a central pathobiologic mediator of MDS, and as an effective therapeutic target for a broad spectrum of MDS patients.
SIGNIFICANCE:
We showed that dysregulation of HIF1A signaling could generate the clinically-relevant diversity of MDS phenotypes by functioning as a signaling funnel for MDS driver-mutations. This could resolve the disconnection between genotypes and phenotypes, and provide a new clue as to how a variety of driver-mutations cause common MDS phenotypes.
INTRODUCTION:
MDS are a group of heterogeneous clonal disorders which are characterized by ineffective hematopoiesis and uni- or multi-lineage dysplasia (1,2). Because of its diversity and complexity, the pathogenesis of MDS remains to be elucidated. Limited preclinical models are available for dissecting the pathogenesis and testing new drugs, and each model has its limitation (3,4). Stem cell transplantation is a curative strategy for MDS, however, few patients are eligible for transplantation. Further elucidation of the pathogenesis of MDS and development of novel therapeutic strategies are needed. MDS are associated with mutations in chromatin-modifying enzymes, splicing factors, transcription factors, cohesin complex, and metabolic enzymes that regulate hematopoietic stem cell (HSC) self-renewal, survival, and differentiation. Cooperating genetic lesions occur, involving signaling molecules that regulate cell growth and proliferation (1,2,5). Although a number of heterogeneous genomic aberrations have been identified in MDS patients (6,7), their key clinical phenotypes are similar. Therefore, we hypothesized that driver mutations activate common underlying mechanisms involved in MDS phenotypes. Identification of these key mediators would reveal fundamental insights into MDS pathogenesis, and present novel opportunities for therapeutic intervention beyond specific mutations or aberrance for MDS.
Hypoxia inducible factor-1α (HIF1A) is a critical transcription factor for the hypoxic response, angiogenesis, normal HSC regulation, and cancer development (8,9). Importantly, HIF1A is also essential for the activation of innate and adaptive immunity (10). HIF1A is regulated by both oxygen-dependent and oxygen-independent ways (11). HSCs and progenitor cells (HSPCs) isolated from MDS patients display abnormal self-renewal and differentiation, and accumulating clinical and research evidence suggests an important role for systemic inflammation and immune activation in MDS pathogenesis (12). Thus, we tested the impact of HIF1A signaling in MDS.
RESULTS:
Activated HIF1A Pathway in the Broad Spectrum of MDS Patients
HIF1A is mainly regulated at translational and protein levels. Thus, analyzing downstream HIF1A-signature-gene expression is a reliable approach to measure HIF1A activation. To determine whether MDS patients have an activated HIF1A gene signature, we analyzed a published cohort of CD34+ BM cells isolated from healthy donors (n = 17) or from MDS patients (n = 183) (13). This MDS cohort contains patients with refractory anemia (RA) (n = 55), refractory anemia with ring sideroblasts (RARS) (n = 48), refractory anemia with excess blast type 1 (RAEB1) (n = 37), or type 2 (RAEB2) (n = 43). HIF1A regulates many genes, which could either be HIF1A direct targets or indirect targets (such as those regulated by HIF1A-regulated miRNAs). The number of HIF1A regulated genes exceeds 1,000, and continues to increase (11). Notably, there are unique sets of target genes activated by HIF1A signaling in individual cell types (different lineage, at different maturation stage) (11). Recently, HIF1A induced genes (which include both direct and indirect targets) in human cord blood (CB) CD34+ cells have been defined (14). To limit the number of HIF1A induced genes and generate the gene set for analysis, we selected three-fold upregulated genes in the constitutively active HIF1A transduced CD34+ CB cells compared to control cells. Gene set enrichment analysis (GSEA) revealed that expression of these genes is significantly enriched in the MDS cohort (Fig. 1A and S1A). Some well-known classical HIF1A target genes were also significantly upregulated in the MDS patient dataset (Fig. S1B). Using immunohistochemical staining, we then determined HIF1A activation (protein accumulation) in the BM biopsies from MDS patients (n = 38), versus de-identified adult BM samples (diagnosed as apparently normal) (n = 6). Consistent with transcriptional analysis of CD34+ cells (Fig. 1A), we found a high frequency of HIF1A expressing cells in both low-risk and high-risk revised-international prognostic scoring system (IPSS-R) (15) groups (Fig. 1B, 1C, and S2). The expression levels of HIF1A mRNA in these MDS patients were not increased (Fig. 1D). These results suggest that the HIF1A pathway (but not Hif1a mRNA) is widely activated in a broad spectrum of MDS patients in both low-risk and high-risk groups.
Figure 1. MDS Patients Display Activated HIF1A Signaling.
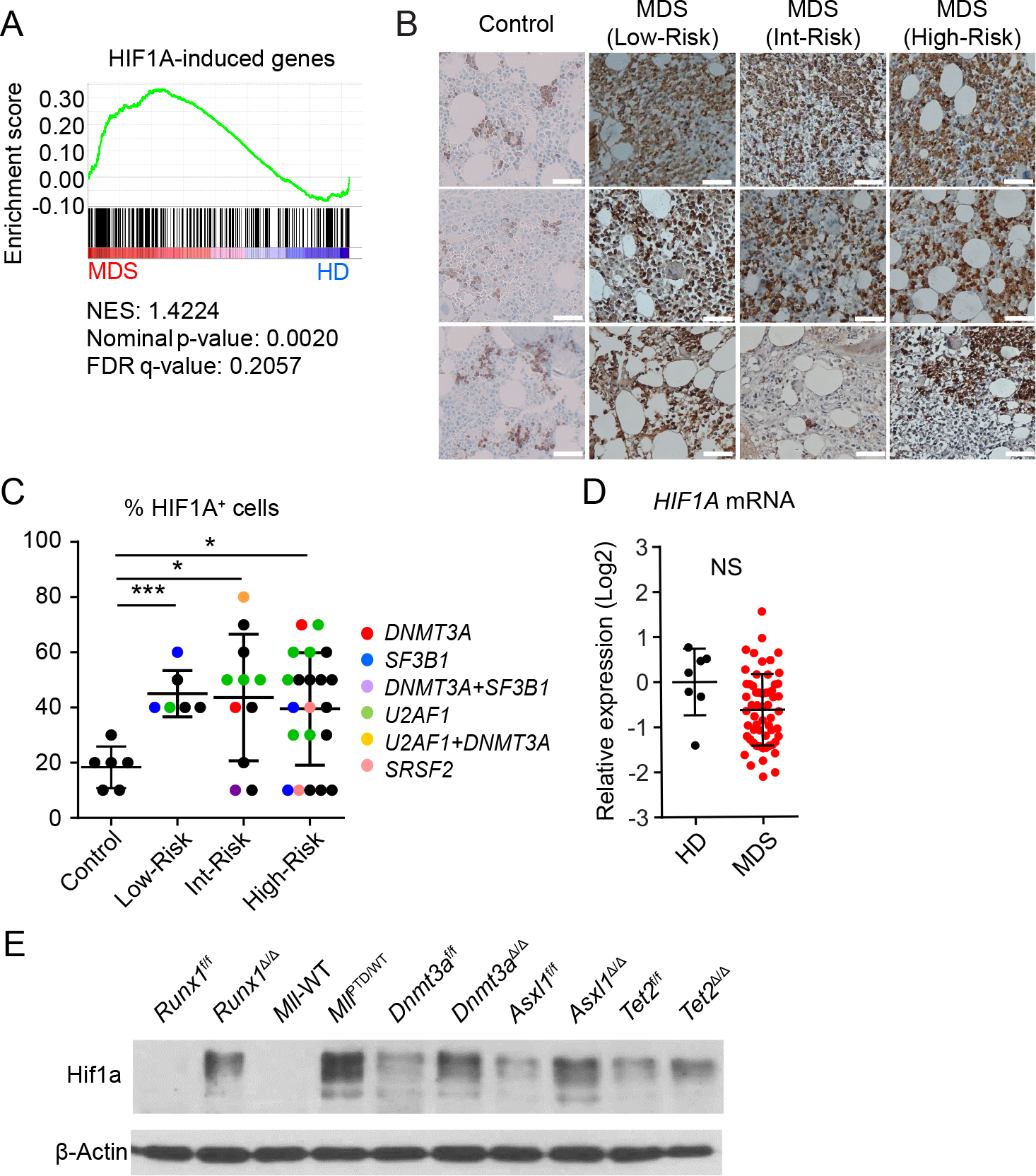
A, Gene set enrichment analysis (GSEA) plot showing increased gene expression of HIF1A induced genes in CD34+ BM cells from MDS patients (n = 183) with RA (n = 55), RARS (n = 48), RAEB1 (n = 37) and RAEB2 (n = 43) relative to healthy donor (n = 17). The normalized enrichment score (NES), P value, and false discovery rate (FDR) are shown. B-C, HIF1A immunohistochemistry staining of BM biopsy samples. Representative case (B) and frequency of HIF1A positive cells in each sample (C). Color of dots indicate mutation profile; red, DNMT3A mutation; blue, SF3B1 mutation; purple, DNMT3A/SF3B1 mutations; green, U2AF1 mutation; yellow, U2AF1/DNMT3A mutations; pink, SRSF2 mutation; black, no known mutation was detected. Data are mean ± s.d. D, HIF1A mRNA expression in BM mononuclear cells from MDS patients (n = 59) and healthy donors (n = 7). E, Hif1a protein expression in the c-Kit+ cells from indicated mice. Runx1f/f and Vav1-Cre/Runx1f/f (Runx1Δ/Δ), Mll-wild-type (WT), and MllPTD/WT mice, and poly(I:C) injected Dnmt3af/f, Mx1-Cre/Dnmt3af/f, Asxl1f/f, Mx1-Cre/Asxl1f/f, Tet2f/f, Mx1-Cre/Tet2f/f mice were used. Floxed allele without Cre recombination (control) are shown as f/f. Conditional knockout are shown as Δ/Δ. F, Quantification of Hif1a protein expression in (E). Results were normalized to the expression level in the Runx1Δ/Δ cells. *P < 0.05, ***P < 0.001; NS, not significant.
The frequency of HIF1A expressing cells did not associate with the mutational profile (Fig. 1C). Mutations in DNA methyltransferase 3A (DNMT3A), tet methylcytosine dioxygenase 2 (TET2), additional sex combs-like 1 (ASXL1), RUNX1, and Mixed lineage leukemia (MLL)-partial tandem duplication (PTD) MLL-PTD, have been reported in patients with MDS (5). To determine whether MDS-associated mutations induce HIF1A signaling, we performed immunoblot analysis of c-Kit+ BM cells from Dnmt3aΔ/Δ mice, Tet2Δ/Δ mice, Runx1Δ/Δ mice, Asxl1Δ/Δ mice, and MLL-PTD-knock-in heterozygous mice (MllPTD/WT mice). The expression of Hif1a protein in c-Kit+ cells isolated from these mice was significantly increased compared to control cells (Fig. 1E and S3 A–J). These results suggest that MDS-associated mutations induce HIF1A signaling in mouse models without obvious anemia or other MDS phenotypes.
HIF1A Activation is Sufficient for the Development of MDS
To determine whether HIF1A activation is sufficient to induce MDS phenotypes, we generated transgenic mice with blood-cell-specific and inducible HIF1A expression. Using a Vav1-Cre/Rosa26-loxP-Stop-loxP (LSL) reverse-tetracycline-controlled transactivator (rtTA) driver (Rosa26-LSL-rtTA), we achieved doxycycline-inducible expression of both a stable and constitutively-active human HIF1A triple-point-mutant (TPM) (16), and wild-type ARNT (also known as HIF1B, a subunit for dimerization with HIF1A) (tet-on-TPM/ARNT) (B6J/129X1 SvJ) (Fig. 2A). After doxycycline administration, we found increased HIF1A protein expression in the c-Kit+ BM cells from Vav1-Cre/TPM mice (Fig. 2B). The resulting HIF1A protein expression level was 1.5-fold higher than in MllPTD/WT cells (Fig. 2C). To identify gene expression changes, we performed RNA-Seq analysis of Vav1-Cre/TPM and wild type c-Kit+ BM cells. Pathway enrichment analysis revealed that a number of HIF1A-related gene sets were significantly upregulated in the c-Kit+ BM cells from Vav1-Cre/TPM mice (Fig. 2D). Notably, ARNT dimerizes not only with HIF1A but also with aryl hydrocarbon receptor (AHR). Genes downregulated by the small molecule Stemregenin-1 (SR1), a known AHR antagonist, were not upregulated in the c-Kit+ BM cells from Vav1-Cre/TPM mice. This clearly suggests that co-expression of TPM protein with wild-type ARNT protein selectively activates HIF1A signaling (but not AHR signaling) in c-Kit+ BM cells (Fig. S4A).
Figure 2. Chronic HIF1A Signaling is Sufficient to Induce MDS Phenotypes.
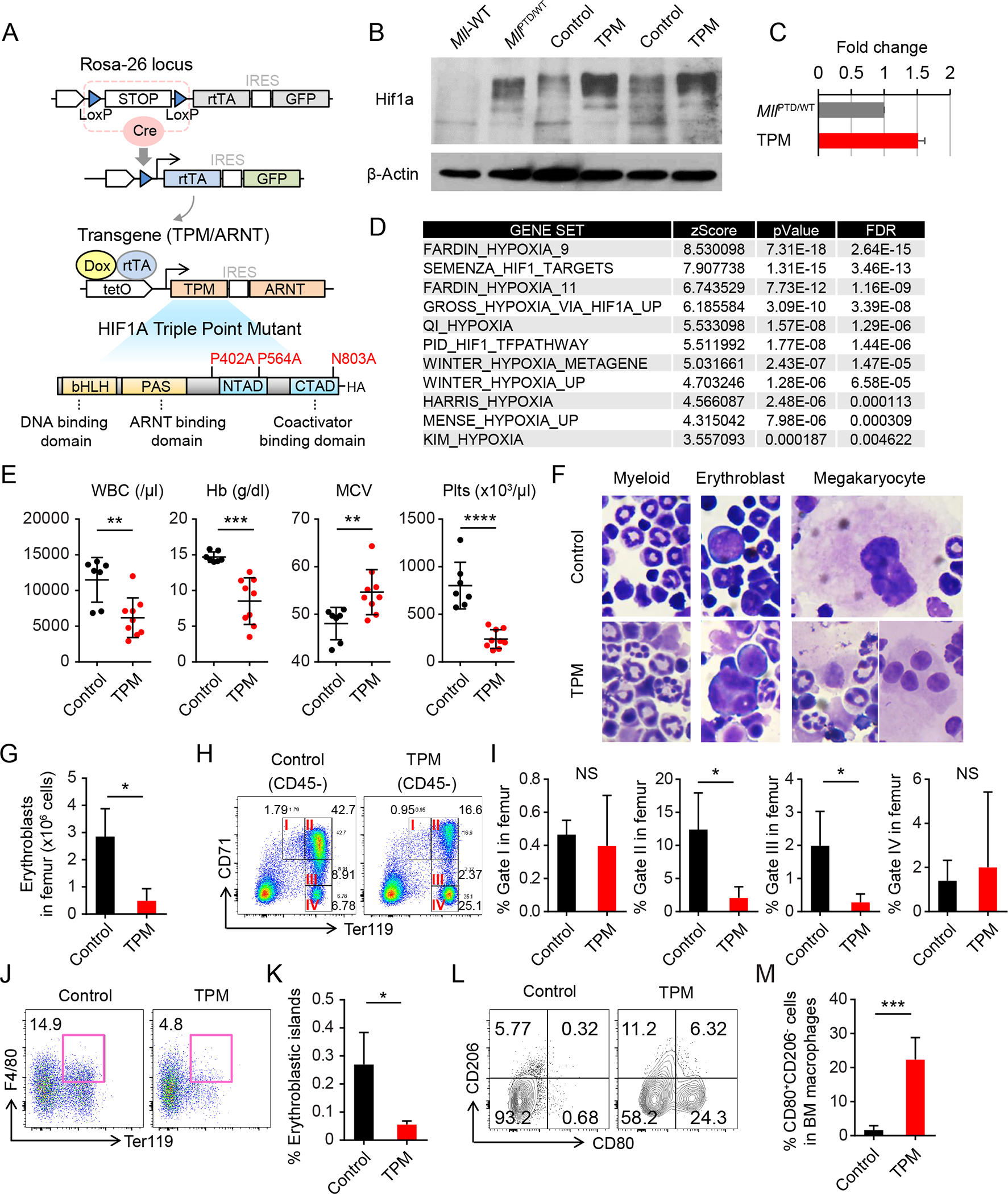
A, Schematic of the inducible HIF1A transgenic mice and HIF1A triple point mutant (TPM). B, Hif1a protein expression in the c-Kit+ cells from indicated mice. The sample from 293T cells with transient overexpression of full length TPM was used as a control (OE-TPM). C, Quantification of Hif1a protein expression in (B). Results were normalized to the expression level in the MllPTD/WT cells. Data are mean ± s.d. D, Significantly enriched gene sets in c-Kit+ BM cells from Vav1-Cre/LSL-rtTA/HIF1a TPM mice (Vav1-Cre/TPM). E, White blood cell (WBC) counts, hemoglobin (HB), mean corpuscular volume (MCV), and Platelet (Plts) counts in PB from Vav1-Cre/Control mice (n = 7) and Vav1-Cre/TPM mice (n = 9) at 12 to 20 weeks after HIF1A induction. Data are mean ± s.d. F, Wright Giemsa staining of BM cells from Vav1-Cre/Control and Vav1-Cre/TPM mice. G, Absolute number of erythroblasts (CD45−CD71high and CD45−Ter119+ cells) in femur from Vav1-Cre/Control (n = 4) and Vav1-Cre/TPM mice (n = 3). H-I, Flow cytometric analysis of 7-amino-actinomycin D (7AAD)− CD45− single BM cells from Vav1-Cre/Control (n = 4) and Vav1-Cre/TPM mice (n = 3) (H). Gated fractions indicate proerythroblasts (I), basophilic erythroblasts (II), polychromatic erythroblast (III), and orthochromatic erythroblasts (IV), respectively. Percentages of cells of each fraction in femur are shown in (I). J-K, Flow cytometric analysis of 7AAD− Gr-1− 115− multiplets from Vav1-Cre/Control (n = 5) and Vav1-Cre/TPM mice (n = 3) (J). Percentage of cells in Ter119+F4/80+ fraction in femur is shown in (K). L-M, Flow cytometric analysis of Gr-1− CD115− F4/80+ SSClow-dim BM macrophages from Vav1-Cre/Control and Vav1-Cre/TPM mice (L). Percentage of cells in CD80+ CD206− fraction in BM macrophages is shown in (M). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant.
While doxycycline administration had no observable effect on control mice, Vav1-Cre/TPM mice quickly developed thrombocytopenia, then within 2 to 8 months leukocytopenia, macrocytic anemia, and splenomegaly gradually developed (Fig. 2E and S4B). Induced Vav1-Cre/TPM BM consistently showed multi-lineage dysplasia, with micro-megakaryocytes and megakaryocytes with multiple separate nuclei, which are major diagnostic criteria for MDS (Fig. 2F). Most of the mice also developed BM reticulin (but not collagen) fibrosis (Fig. S4C) which is associated with a worse outcome in MDS patients (17). The frequency of HSPCs in BM from Vav1-Cre/TPM mice was increased (Fig. S4D and S4E). Among the myeloid progenitors, increase in the megakaryocyte-erythrocyte progenitors (MEPs) was prominent (Fig. S4E). To evaluate the clonal advantage of the Vav1-Cre/TPM cells, we performed in vitro serial CFU replating assay and in vivo competitive bone marrow transplantation (CBMT) assay (1:1 ratio) (Fig. S4F). The number of colonies derived from Vav1-Cre/TPM BM cells was significantly higher even after serial replating, than that of control cells (Fig. S4G and S4H). We also found long term repopulating advantage of Vav1-Cre/TPM cells in the competitive BMT assay (Fig. S4I). We note that the phenotypes of the primary Vav1-Cre/TPM mice were transplantable when we transplanted whole BM cells into lethally irradiated wild-type mice (Fig. S4J).
Concordant with anemia, the late stage erythroblasts were decreased, accompanied by a significant reduction of basophilic and polychromatic erythroblasts (Fig. 2G–2I). The impairment of late stage erythropoiesis causes ineffective erythropoiesis, which is one of the key features of MDS. It has been proposed for several decades that macrophages play a critical role in the late stage of erythropoiesis (18). We therefore examined the potential impact of activated HIF1A signaling on macrophage subsets, which could indirectly influence erythropoiesis. We found significant decreases in both the frequency and absolute number of F4/80+ Ter119+ BM erythroblastic islands (Fig. 2J and 2K), along with phenotypic inflammatory macrophages in the Vav1-Cre/TPM BM (Fig. 2L and 2M).
To dissect the cell intrinsic/extrinsic effect of HIF signaling activation, we utilized tissue-specific Cre transgenes to restrict HIF1A signaling within the hematopoietic system. Specifically, we combined Rosa26-LSL-rtTA and tet-on-TPM/ARNT alleles with an erythroid-lineage specific EpoR-Cre allele (EpoR-Cre/TPM), a megakaryocyte-lineage specific PF4-Cre allele (PF4-Cre/TPM), and a myelomonocytic-lineage LysM-Cre allele (LysM-Cre/TPM). Mice with the corresponding tissue-specific Cre expression served as controls. HIF1A induction in the erythrocyte lineage did not cause anemia (Fig. S5A), and the differentiation pattern of erythroblasts (Fig. S5B–S5D) and the number of BM erythroblastic islands (Fig. S5E) were not altered in the EpoR-Cre/TPM mice. When we induced HIF1A expression in the megakaryocyte lineage, the PF4-Cre/TPM mice quickly developed thrombocytopenia (Fig. S5F), and micro-megakaryocyte formation (Fig. S5G), but not anemia. On the other hand, the LysM-Cre/TPM mice showed a significant reduction of BM erythroblastic islands, and eventually developed anemia (Fig. S5H and S5I). Notably, LysM-Cre/TPM mice developed anemia more slowly compared to Vav1-Cre/TPM mice, suggesting that other additional cell types/factors participate in the development of anemia. Taken together, these data suggest that HIF1A is sufficient to trigger a variety of key MDS features via both cell-intrinsic and extrinsic mechanisms. Considering that HIF1A is broadly activated in MDS from low risk to high risk, HIF1A may be the underlying mechanism that dictates MDS phenotypes; unifying genetically-heterogeneous MDS under one umbrella.
Activation of HIF1A signaling is Critical for MLL-PTD-mediated Clonal Advantage
The MLL-PTD is found in patients with MDS and AML, but not in other hematological malignancies (19,20). Previously, we showed that MllPTD/WT mice present with MDS-associated features, such as increased self-renewal and apoptosis in HSPCs, expansion of the myeloid progenitor population, and ineffective hematopoiesis; however, these HSPCs phenotypes are insufficient to progress to bona fide MDS or AML (19,21). Given that MDS-associated mutations induce HIF1A signaling (Fig. 1E and S3), we focused on MllPTD/WT mice (as an example) to dissect the significance of HIF1A in the context of MDS pathogenesis.
MLL is an epigenetic regulator: its C-terminal Su[var]3–9, enhancer of zeste, trithorax (SET) domain has methyltransferase activity on lysine 4 on histone 3 (H3K4) (22), which is retained in the MLL-PTD mutant. Tri-methylation of H3K4 (H3K4me3) is associated with transcription activation. To clarify the effects of MLL-PTD on target gene regulation, we performed H3K4me3 ChIP-Seq analysis of lineage marker− Sca1+ c-Kit+ cells (LSKs) isolated from wild-type (WT) and MllPTD/WT mice. Interestingly, the DNA sequences underlying MllPTD/WT-uniquely increased H3K4me3 peaks were significantly enriched for the Hif1a/Arnt hetero-dimer DNA binding motif (Fig. 3A and 3B). KEGG pathway analysis revealed enrichment in several inflammatory/immune-response-related pathways, such as systemic lupus erythematosus (P = 1.61e-48), chemokine signaling (P = 9.06e-37), cytokine-cytokine receptor (P = 1.09e-36), and toll-like receptor signaling pathways (P = 5.78e-29) (Fig. 3C; Table S1), as well as glycolysis/gluconeogenesis pathways (P = 2.84e-14). We note that HIF1A is a well-known regulator of glycolysis/gluconeogenesis (8,23). We also compared the H3K4me3 ChIP-Seq data of LSKs from MllPTD/WT mice with the ENCODE binding data of human CD34+ myeloid progenitors. We then performed KEGG pathway analysis using genes on the overlapped H3K4me3 peaks. Similar to what were enriched in the initial analysis (Fig. 3C), we found upregulation of several inflammation and HIF-signaling related pathways (Fig. S6A).
Figure 3. Downregulation of mitochondrial complex II in MLL-PTD.
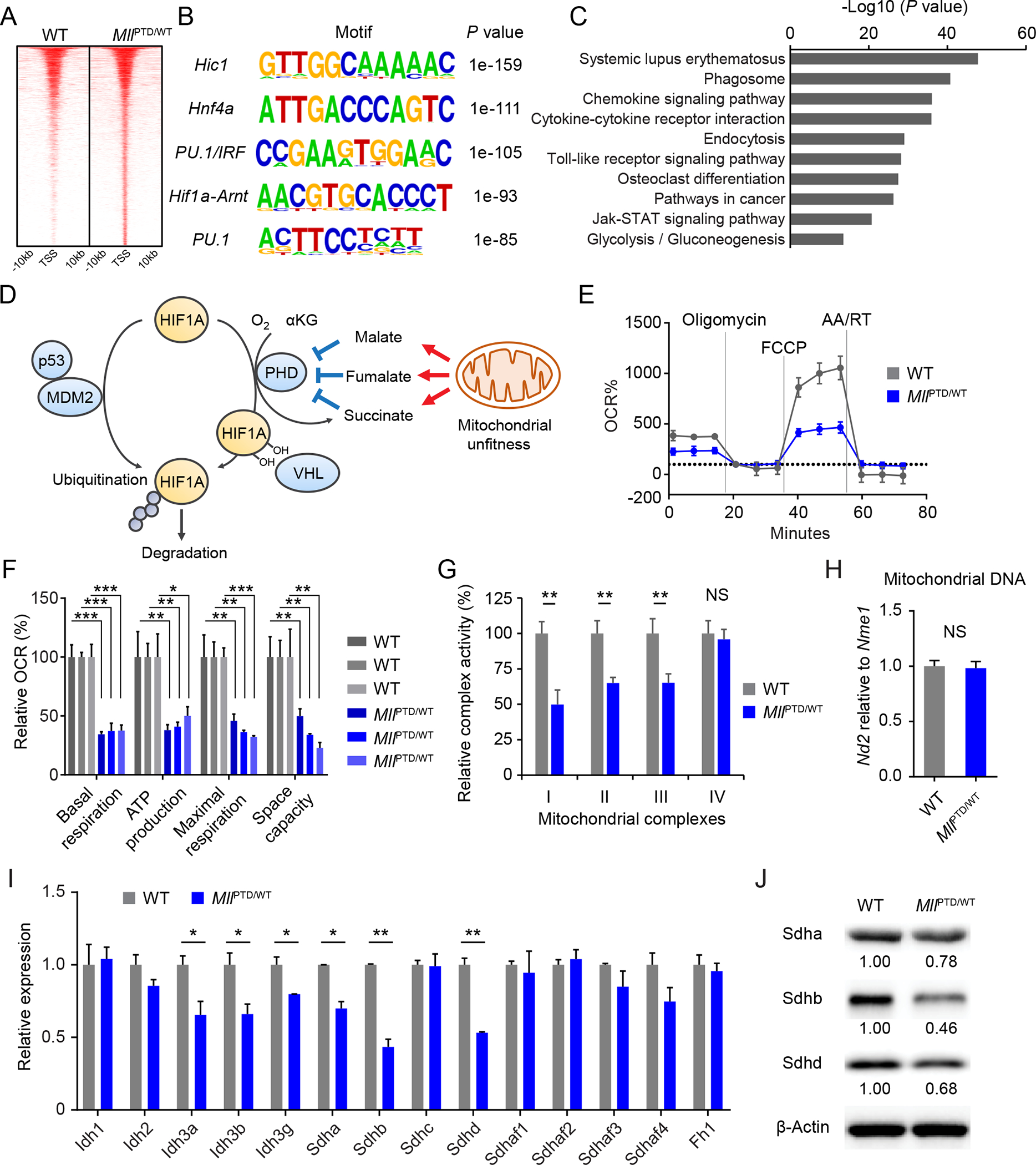
A, Heatmap showing differentially H3K4me3-marked regions (±10 kb regions from TSS). B, Enriched motifs found in the differentially H3K4me3-marked regions from indicated samples. C, KEGG pathway enrichment analysis on the gene set from the differentially H3K4me3-marked regions in MLL-PTD. False discovery rate (FDR) < 0.001 was used. D, Schematic of HIF1A protein regulation. E-F, Measurements of mitochondrial respiration in c-Kit+ BM cells from indicated mice. Average results are shown (n = 3 per group) in (E). Relative basal respiration, ATP production, maximal respiration, and space capacity are shown in (F). Data are mean ± s.d. G, Mitochondrial complex activity in the c-Kit+ cells from indicated mice. Relative activity is shown (n = 3 per group). Data are mean ± s.d. H, Mitochondrial DNA amount in the c-Kit+ cells from indicated mice. Data are mean ± s.d. I, Expression levels of genes related to TCA cycle enzymes in the c-Kit+ cells from indicated mice. Data are mean ± s.d. Representative plots from more than three independent studies are shown. J, Sdha, Sdhb, and Sdhd protein expression in the c-Kit+ cells from indicated mice. *P < 0.05, **P < 0.01, ***P < 0.001; NS, not significant.
MLL-PTD confers a clonal advantage in stress hematopoiesis (19). To evaluate the biological impact of HIF1A upregulation on MLL-PTD-induced phenotypes, we generated MllPTD/WT/Hif1aFlox/Flox/Mx1-Cre mice. It has been reported that assays of BM CFU-C and CFU-S did not show a significant difference between Hif1aFlox/Flox cells and Hif1aΔ/Δ cells (9). We have shown that MllPTD/WT HSPCs can be replated more than three times, and MllPTD/WT HSCs show significantly enhanced fitness in a competitive BMT assay (1:1 ratio), even after the secondary BMT (19). However, MllPTD/WT/Hif1aΔ/Δ cells stopped growing after the second replating (Fig. S6B and S6C) similar to the WT cells. In a competitive BMT assay (1:1 ratio), Hif1a abrogation attenuated the repopulating advantage in MllPTD/WT cells to the level of WT cells (Hif1af/f cells) (Fig. S6B, S6D and S6E). Taken together, these results suggest that, similar to Vav1-Cre/TPM mice, MLL-PTD-mediated HIF1A signaling controls HSC clonal expansion and fitness.
MLL-PTD Stabilizes HIF1A through Pseudohypoxia
Since ubiquitination is critical for HIF1A protein degradation in normoxia (or even in hypoxia), we tested whether downregulation of E3-ligases contributes to MLL-PTD-mediated HIF1A protein stabilization in normoxia. Von Hippel-Lindau tumor suppressor (VHL), which recognizes prolyl hydroxylase (PHD) mediated hydroxylation of proline residues, is a well-known E3 ubiquitin ligase for oxygen-dependent HIF1A-protein degradation (24). Murine double minute-2 (MDM2) E3-ubiquitin ligase is also involved in HIF1A protein degradation independent of oxygen condition (25) (Fig. 3D). MDM2 expression is positively regulated by TP53 while HIF1A expression is negatively regulated by TP53. However, mRNA expression levels of Vhl and Mdm2 were increased in c-Kit+ cells isolated from MllPTD/WT mice compared to control cells. Tp53 mRNA expression was not altered in the c-Kit+ BM cells from MllPTD/WT mice (Fig. S7A and S7B). This suggests that MLL-PTD stabilizes HIF1A through other mechanisms.
PHDs are α-ketoglutarate (α-KG)-dependent dioxygenase, catalyzing the hydroxylation of the proline residue using O2, Fe2+, and α-KG as substrates. Notably, α-KG is an intermediate metabolite of the tricarboxylic acid (TCA) cycle in mitochondria. Thus, accumulation of subsequent metabolites of α-KG, such as succinate, fumarate, and malate, inhibits PHD activity even in normoxia, leading to pseudohypoxia. Indeed, mitochondrial dysfunction or the Warburg effect is known to result in accumulation of TCA intermediate metabolites, leading to activation of HIF signaling (Fig. 3D). Thus, in order to determine whether MLL-PTD affects mitochondrial function, we first measured cellular respiration using c-Kit+ BM cells from MllPTD/WT mice (Fig. 3E and 3F). We found significantly decreased basal respiration, ATP production, and maximal respiration of MllPTD/WT HSPCs compared to WT cells (Fig. 3F). The activity of mitochondrial complexes I, II, and III was significantly decreased in the c-Kit+ BM cells from MllPTD/WT mice (Fig. 3G). To assess the mechanism of mitochondrial dysfunction, we first measured the mitochondrial DNA copy number. However, the copy number of mitochondrial DNA in the c-Kit+ BM cells was comparable between the WT and MllPTD/WT mice (Fig. 3H), suggesting similar mitochondrial mass in MllPTD/WT HSPCs. Among the mitochondrial electron transport chain complexes, complex II (succinate dehydrogenase, SDH) also functions as a critical enzyme in the mitochondrial TCA cycle. Inactivation of SDHs has been reported in several cancers (26–28), resulting in a blockade of the TCA cycle, impairing cellular respiration (29). It has been well-documented that the SDH inactivation causes HIF stabilization (29,30). Thus, we first determined the gene expression level of SDH subunits as well as genes that code for other TCA cycle key enzymes which may result in a similar pseudohypoxia as SDH inactivation. We found that the expression level of Sdha, Sdhb, and Sdhd mRNA was significantly decreased in MllPTD/WT HSPCs (Fig. 3I). Consistent with the reduction of the transcripts, the expression levels of their proteins were also decreased (Fig. 3J). These results indicate that a defective complex II may lead to mitochondrial unfitness and pseudohypoxia in MLL-PTD.
Indeed, concordant with decreased complex II activity, NMR assay showed the metabolic shift in the MllPTD/WT mice (Fig. 4A). Several TCA cycle metabolites, such as fumarate and malate, were significantly accumulated in the plasma of MllPTD/WT mice, while the concentration of pyruvate was increased in MllPTD/WT mice as a result of activation of glycolysis (Fig. 4B and 4C). Notably, the ratio of fumarate/α-KG and malate/α-KG was significantly increased (Fig. 4D). This concentration gradient could inhibit the α-KG-dependent dioxygenases including PHDs (Fig. 4D and 4E). Using the Vhlf/f Vav1-Cre allele, we confirmed that HIF1A protein is constantly degraded through PHD-VHL axis in c-Kit+ HSPCs (Fig. S8). However, adding succinate, fumarate, and malate into the culture medium stabilized HIF1A protein even in normoxia (Fig. 4F). Besides PHDs, Ten-eleven translocation methylcytosine dioxygenases (TETs) and histone demethylases are also α-KG-dependent dioxygenases (Fig. 4E) (31–34). Both TETs and histone demethylase are critical epigenetic modifiers that are involved in DNA demethylation through oxidizing 5-methylcytosine (5mC) on DNA, or regulation methylation status on histone. Inactivation of TETs and histone demethylase function results in hypermethylation of DNA or histone. Notably, aberrant methylation status of DNA and histone are well known features of MDS. To assess the levels of 5mC and 5-hydroxymethylcytosine (5hmC), we performed dot blot analysis using genomic DNA in c-Kit+ cells from MllPTD/WT mice. We found increased 5mC level and decreased 5hmC level in DNA from MllPTD/WT cells compared to WT cells, indicating DNA hypermethylation in MllPTD/WT mice (Fig. 4G). We also found increased levels of histone methylation in MllPTD/WT HSPCs (Fig. 4H) Taken together, these results suggest that Warburg effect and metabolic rewiring lead to accumulating of TCA-cycle intermediate metabolites, which results in pseudohypoxia-mediated HIF1A signaling activation in addition to the hypermethylation features in HSPCs of MllPTD/WT mice.
Figure 4. MLL-PTD induces pseudohypoxia to stabilize HIF1A.
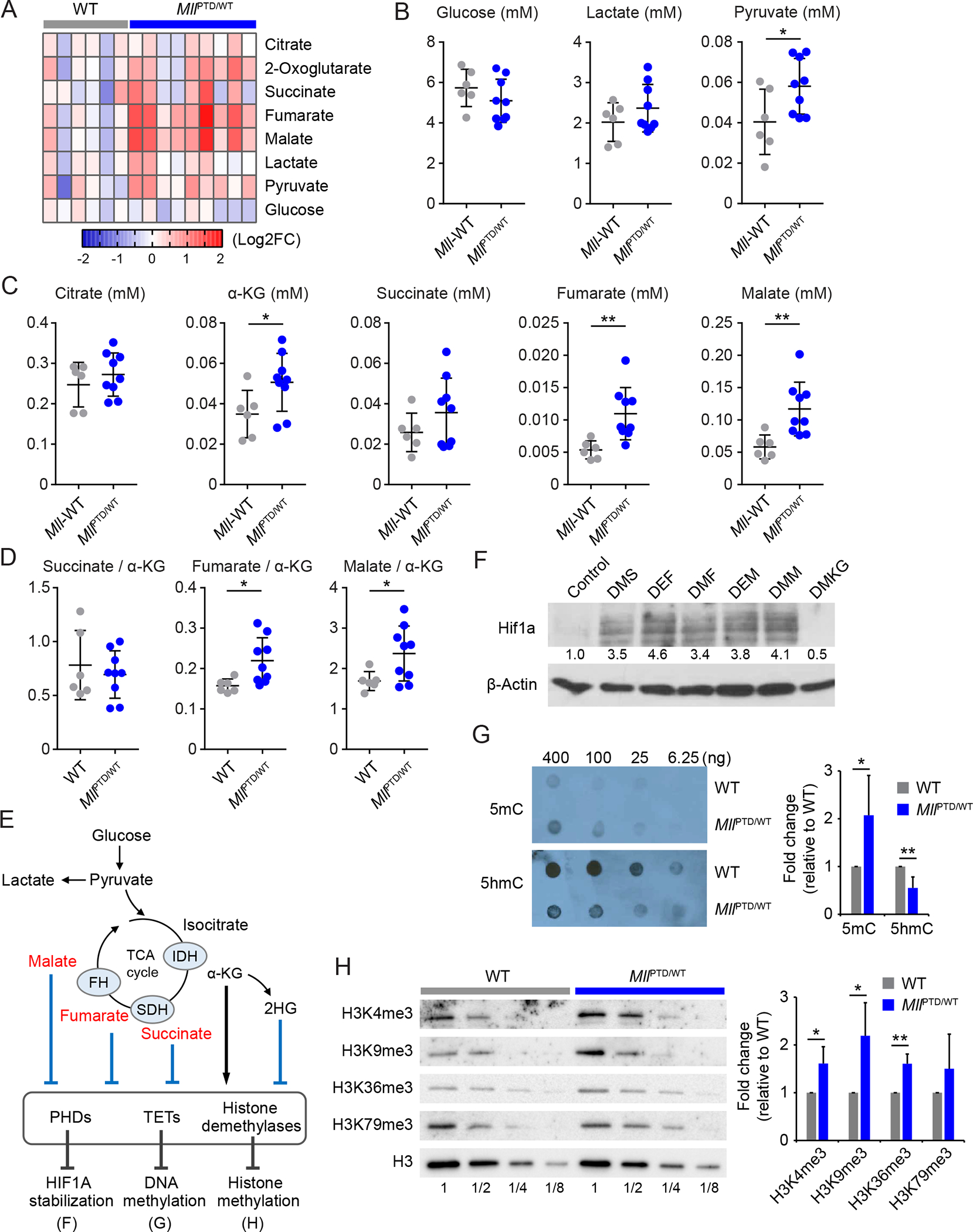
A, Heatmap showing relative concentration of TCA cycle and glycolysis metabolites in the plasma from indicated mice (WT, n = 5; MllPTD/WT, n = 9). B-C, Concentration of the TCA cycle metabolites in plasma from indicated mice. Data are mean ± s.d. D, Ratio of succinate/α-KG, fumarate/α-KG, and malate/α-KG in the plasma from indicated mice. E, Schematic of TCA cycle, metabolites and α-KG dependent dioxygenases. F, HIF1A protein stabilization by TCA cycle metabolites in CD34+ cord blood cells. Cells were incubated for 24 hours in normoxia. DMS, dimethyl succinate (5 mM); DEF, diethyl fumarate (2.5 mM); DMF, dimethyl fumarate (120 μM); DEM, diethyl malate (2.5 mM); DMM, dimethyl malate (2.5 mM); DMKG, dimethyl-α-ketoglutarate (5 mM). G, Dot blot assay and quantifications for 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) using genomic DNA of c-Kit+ BM cells from indicated mice. H, The tri-methylation levels and their quantifications of H3K4, H3K9, H3K36, and H3K79 in c-Kit+ BM cells from indicated mice. *P < 0.05, **P < 0.01; NS, not significant.
MLL-PTD and Mutant RUNX1 Cooperate to Induce MDS Phenotypes
Although MllPTD/WT mice present several MDS related phenotypes, MllPTD/WT mice do not generate MDS or AML in primary or transplant recipient mice (19,35). This suggests that additional genetic and/or epigenetic defects are necessary for transformation to MDS or AML. Importantly, almost all MDS patients have more than one mutation (36), and several mouse models of MDS have been established by combining multiple MDS-associated mutations (37–39). In secondary AML and de novo AML, MLL-PTD mutation is significantly associated with mutations in RUNX1 gene. Thus, we combined an MLL-PTD allele with an MDS-patient-derived RUNX1 mutant, RUNX1-S291fsX300 (40). We retrovirally introduced the RUNX1-S291fsX300 mutant into MllPTD/WT BM cells, and examined their functional properties in vitro and in vivo. In CFU assays, introducing RUNX1 mutation into the MllPTD/WT BM cells allowed the cells to undergo indefinite serial replating, while MllPTD/WT/Empty cells replated 3–4 times (Fig. 5A). The MllPTD/WT/RX1-S291fs BMT mice developed leukocytopenia, macrocytic anemia, thrombocytopenia (Fig. 5B and 5C), and multi-lineage dysplasia in the BM (Fig. 5D). Total BM erythroblasts were significantly decreased in MllPTD/WT/RX1-S291fs mice (Fig. 5E). We found a significant reduction of basophilic and polychromatic erythroblasts (Fig. 5F and 5G), and BM erythroblastic islands in MllPTD/WT/RX1-S291fs mice, compared to the control mice (Fig. 5H and 5I). MllPTD/WT/RX1-S291fs mice also developed BM fibrosis (Fig. S9A). Eventually all MllPTD/WT/RX1-S291fs mice died of MDS complications, mainly severe anemia or infection (Fig. S9B).
Figure 5. MLL-PTD and RUNX1 Mutations Cooperate to Induce MDS Phenotypes.
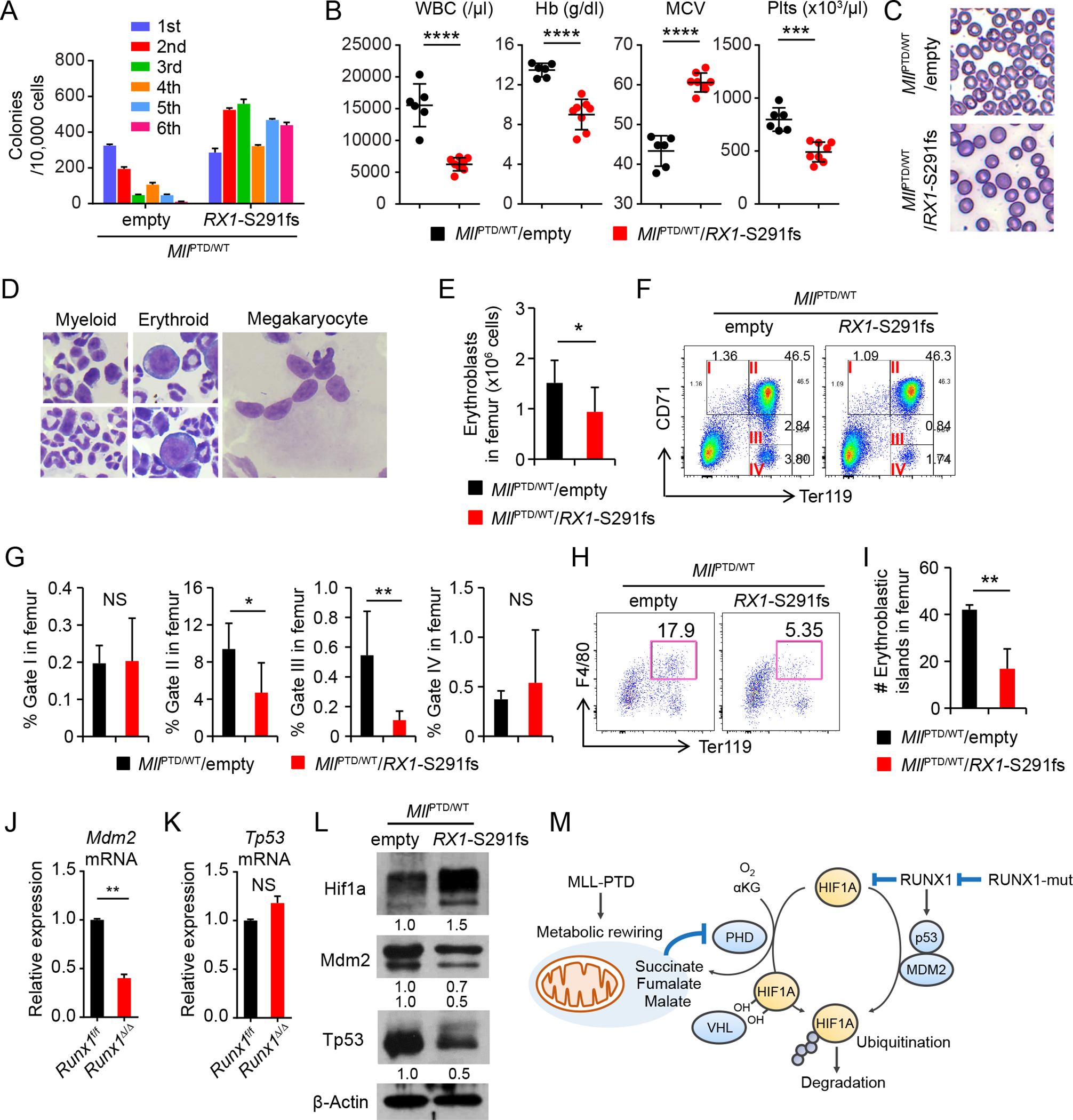
A, Serial CFU replating assay of BM cells from indicated genotypes. Data are mean ± s.d. from triplicate cultures. B, WBC counts, HB, MCV, and Plts counts in PB from the MllPTD/WT/empty (n = 6) and MllPTD/WT/Rx1-S291fs mice (n = 8) at 8 weeks after BMT. C, Morphology of RBC in PB smear after fixation. D, Multi-lineage dysplasia in the BM from MllPTD/WT/Rx1-S291fs mice. E, Absolute number of erythroblasts (CD45−CD71high and CD45−Ter119+ cells) in femur from MllPTD/WT/empty (n = 4) and MllPTD/WT/Rx1-S291fs mice (n = 6). F-G, Flow cytometric analysis of 7AAD− CD45− single BM cells from MllPTD/WT/empty (n = 4) and MllPTD/WT/Rx1-S291fs mice (n = 6) (F). Percentages of cells of each fraction in femur are shown in (G). H-I, Flow cytometric analysis of 7AAD− Gr-1− 115− multiplets from MllPTD/WT/empty (n = 3) and MllPTD/WT/Rx1-S291fs mice (n = 3) (H). Absolute number of cells in Ter119+F4/80+ fraction in femur are shown (I). J-K, Mdm2 mRNA (J) and Tp53 mRNA (K) expression in c-Kit+ BM cells from Runx1Δ/Δ mice and control mice. Data are mean ± s.d. from duplicate samples. Results are representative of three independent experiments. L, Hif1a, Mdm2, and Tp53 protein expression in the c-Kit+ cells from indicated mice. M, Model for mechanisms of HIF1A protein stabilization and activation by MLL-PTD and RUNX1 mutant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant.
Notably, RUNX-family proteins have also been shown to affect HIF1A transaction activity, cooperative target gene regulation, and protein degradation (41–43). Wild-type RUNX1 has been reported to inhibit HIF1A transcriptional activity (41). Indeed, Runx1 deleted cells display a dramatic increase of Hif1a protein compared to WT cells (Fig. 1E and S3A). Interestingly, as contrasted with the case of MllPTD/WT cells, we found that expression level of Mdm2 mRNA was significantly decreased in c-Kit+ BM cells from Runx1Δ/Δ mice compared to WT cells (Fig. 5J). Tp53 mRNA expression was not altered in the c-Kit+ BM cells from Runx1 deficient mice (Fig. 5K), which is consistent with the results reported in the recently published paper showing decreased Tp53 protein (but not mRNA) expression levels and ribosome biogenesis (RiBi) in Runx1 deficient HSPCs (44). On the other hand, the expression levels of SDH subunits, which were decreased in the MllPTD/WT cells, were not decreased in c-Kit+ HSPCs from Runx1Δ/Δ mice compared to WT cells (Fig. S9C). In CFU assays, the expression levels of Hif1a protein in RUNX1 mutant-transduced MllPTD/WT BM cells (MllPTD/WT/RX1-S291fs cells) were further increased compared to those in control vector-transduced MllPTD/WT BM cells (MllPTD/WT/Empty cells) (Fig. S9D and S9E). The expression of Hif1a protein in c-Kit+ cells isolated from MllPTD/WT/RX1-S291fs mice was increased compared to MllPTD/WT/Empty-vector cells (Fig. 5L). Importantly, this expression Hif1a protein expression level was similar to that in Vav1-Cre/TPM mice which presented a variety of clinically relevant MDS phenotypes. The expression level of known HIF1A target genes was also increased, suggesting the additive activation of transcriptional activity (Fig. S9F). In contrast, Mdm2 and Tp53 protein were decreased in MllPTD/WT/RX1-S291fs mice compared to MllPTD/WT/Empty-vector cells (Fig. 5L). The down regulation of Mdm2 and Tp53 is consistent with results obtained from MDS patients with RUNX1 mutations (Fig. S9G and S9H), and (given the ability of MDM2 to control HIF1A protein (25)) may explain the additive accumulation of HIF1A protein in MDS with RUNX1 mutations (Fig. 5M) as the expression levels of Sdh subunits remained low in MllPTD/WT/RX1-S291fs mice (Fig. S9I). Taken together, these results suggest that RUNX1 and MLL1 mutations cooperate to additively activate Hif1a expression in a hypoxia-independent manner which is sufficient to induce phenotypic MDS in a mouse model.
Essential Role of HIF1A in MDS Development
Neither MllPTD/WT mice alone nor Runx1Δ/Δ mice alone develop MDS or AML phenotypes while each of them results in increased Hif1a protein expression and clonal advantage of HSPCs. On the other hand, additively increased expression of Hif1a protein was found in the MllPTD/WT/RUNX1-mutant MDS model, which induces a variety of clinically-relevant MDS phenotypes. This suggests a threshold effect; requiring the additive effect of hypoxia-independent HIF1A signaling activation for MDS development. To test the essential role of HIF1A in MDS, we genetically eliminated Hif1a expression in our MDS model. We generated MllPTD/WT/Runx1Flox/Flox/Mx1-Cre mice (to model loss-of-function RUNX1 mutations (44,45)) and MllPTD/WT/Runx1Flox/Flox/Hif1aFlox/Flox/Mx1-Cre mice. Similar to mutant-RUNX1 overexpression, the expression of Hif1a protein in c-Kit+ cells isolated from MllPTD/WT/Runx1Δ/Δ mice was approximately 1.5-fold higher than in cells isolated from MllPTD/WT mice (Fig. 6A and S10A). In CFU assays; Runx1 deleted MllPTD/WT BM cells (MllPTD/WT/Runx1Δ/Δ cells) produced a higher number of colonies than WT or MllPTD/WT BM cells, and could be serially replated more than six times (Fig. S10B). MllPTD/WT/Runx1Δ/Δ mice also presented with multi-lineage dysplasia (Fig. S10C–G), and recipient mice transplanted with MllPTD/WT/Runx1Δ/Δ cells developed MDS (Fig. 6B). In contrast, Hif1a deletion rescued dysplasia formation (Fig. S10C–G). Hif1a deletion also abrogated disease development (Fig. 6B). This was not due to the elimination of the donor-derived cells (Fig. 6C), as most of donor-derived cells in MllPTD/WT/Runx1Δ/Δ BMT mice were CD11b+ myeloid cells (Fig. 6D), and after deletion of Hif1a these cells were still CD11b+ myeloid cells (Fig. 6D). Interestingly, the recipient mice transplanted with MllPTD/WT/Runx1Δ/Δ/Hif1aΔ/Δ cells did not develop macrocytic anemia (Fig. 6E). Hif1a deletion also partially rescued the thrombocytopenia and megakaryocyte phenotype of the MllPTD/WT/Runx1Δ/Δ BMT mice (Fig. 6E and S10F), likely because RUNX1 is a critical regulator of megakaryocytopoiesis. These results suggest that aberrantly stabilized HIF1A protein and activated HIF1A signaling play critical roles in the development of key MDS phenotypes (ineffective erythropoiesis, thrombocytopenia, and BM dysplasia).
Figure 6. Essential Role of HIF1A for MDS Development and MDS Phenotypes.
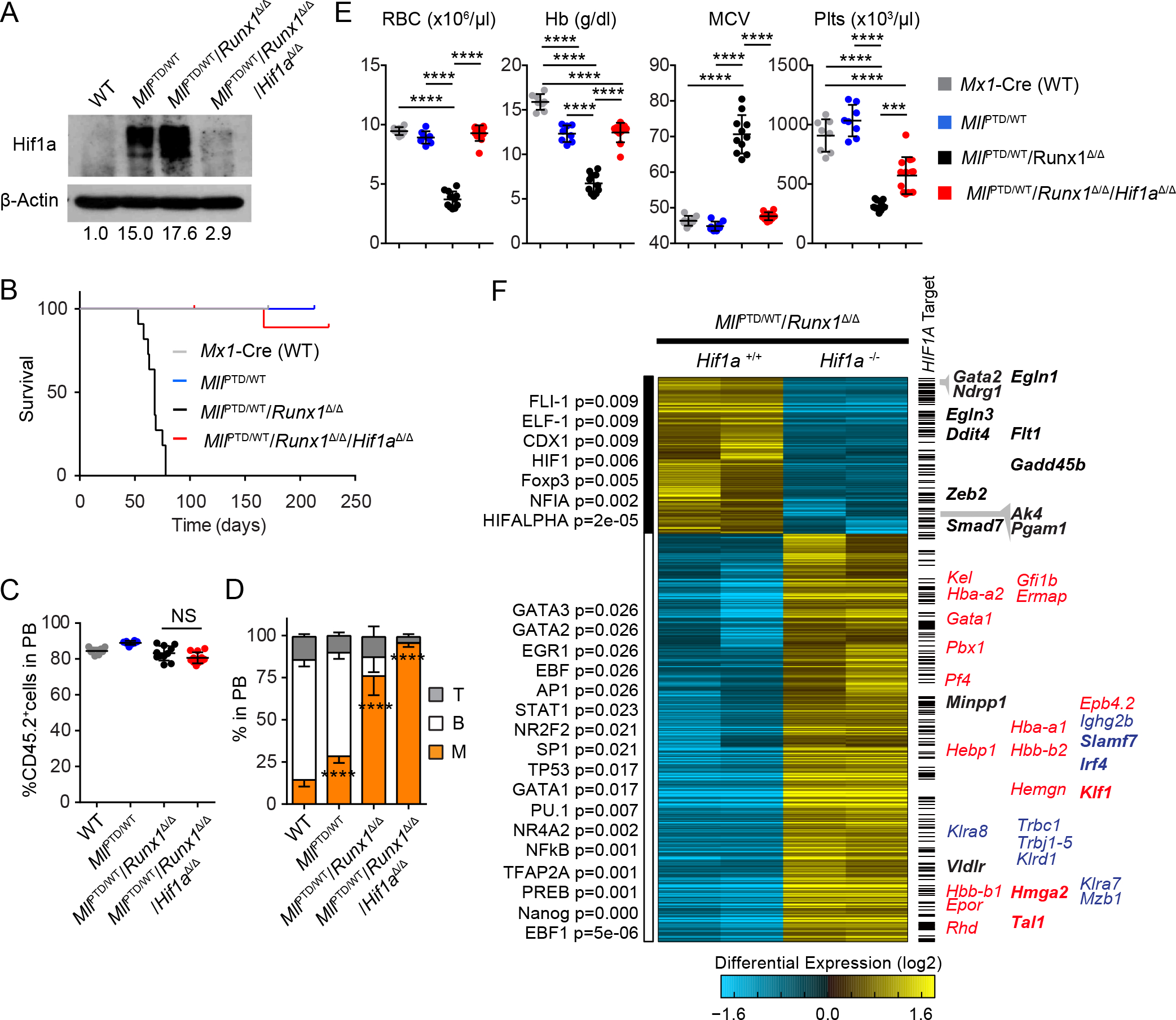
A, Hif1a protein expression in the c-Kit+ cells from indicated mice. B, Survival of Mx1-Cre/WT (n = 8), MllPTD/WT (n = 8), MllPTD/WT/Runx1Δ/Δ (n = 11), and MllPTD/WT/Runx1Δ/Δ/Hif1aΔ/Δ BMT mice (n = 11). C, PB chimerism (as measured by CD45.2 marker) of the recipients transplanted with Mx1-Cre/WT (n = 8), MllPTD/WT (n = 8), MllPTD/WT/Runx1Δ/Δ (n = 11), and MllPTD/WT/Runx1Δ/Δ/ Hif1aΔ/Δ BM cells at 7 weeks after the BMT. D, Flow cytometric analysis of CD45.2+ PB cells from the recipients transplanted with Mx1-Cre/WT (n = 8), MllPTD/WT (n = 8), MllPTD/WT/Runx1Δ/Δ (n = 11), and MllPTD/WT/Runx1Δ/Δ/ Hif1aΔ/Δ BM cells. E, Red blood cells (RBC) counts, HB, MCV, and Plts counts in PB from the Mx1-Cre/WT (n = 8), MllPTD/WT (n = 8), MllPTD/WT/Runx1Δ/Δ (n = 11), and MllPTD/WT/Runx1Δ/Δ/ Hif1aΔ/Δ BMT mice. F, Heatmap showing 927 differentially expressed genes comparing MllPTD/WT/Runx1Δ/Δ and MllPTD/WT/Runx1Δ/Δ/ Hif1aΔ/Δ (fold > 2, moderated t-test p-value < 0.05). RNA-Seq data were clustered using the hopach algorithm in AltAnalyze. To the left of the heatmap are top predicted transcription factors using TF-target enrichment analysis using the GO-Elite algorithm in AltAnalyze (48). Bars to the right of the heatmap indicate HIF1A target genes from multiple sources, including re-analyzed HIF1A ChIP-Seq analyses from hematopoietic cells (see Methods, and Supplementary Table S2). Representative HIF1A targets (black) and blood lineage markers (erythroid/megakaryocyte = red, other = blue) are shown to the right of the heatmap. All predicted HIF1A targets are bolded. ***P < 0.001, ****P < 0.0001; NS, not significant.
To better characterize the effect of HIF1A in this model, we first performed RNA-Seq analysis of c-Kit+ BM cells comparing MllPTD/WT/Runx1Δ/Δ with or without Hif1a (Fig. 6F). To define HIF1A target genes within the data set, we intersected differentially expressed genes with both published HIF1A ChIP-Seq datasets from murine hematopoietic cell populations (GSE40918, GSE36104)(46,47), as well as HIF1A binding sites based on the literature and DNA sequence conservation (GO-Elite TF-target database “PAZAR” (48), Broad Molecular Signatures Database “MSigDB” Pathway Commons (49), and Whole Genome R-Vista) (Table S2). We found that several previously evident HIF1A targets, such as Gata2, Ndrg1, Pgam1, Ak4, Zeb2, Egln3, and Ddit4 were downregulated in response to Hif1a deletion (Fig. 6F and S10H). In contrast, several key genes related to erythroid-megakaryocyte differentiation, such as Klf1, Gfi1b, Gata1, Pbx1, and Tal1, were upregulated in response to Hif1a deletion (Fig. 6F and S10H). Moreover, the loss of HIF1A also upregulated Irf4 (Fig. 6F), which is a key regulator for alternative macrophage activation (but not for inflammatory macrophages). Among these upregulated genes, Klf1, Tal1, and Irf4 were also predicted as HIF1A targets (Fig. 6F). We note that Hif1a deletion did not affect the pseudohypoxia-related glycolysis/oxidative phosphorylation gene signature in this model, suggesting that Hif1a signaling activation is downstream of pseudohypoxia conditions (Fig. S11).
Echinomycin is an inhibitor of HIF1A-mediated target gene activation (50). In CFU assays, the MllPTD/WT BM cells were more sensitive to Echinomycin than the WT BM cells (Fig. S12A), suggesting that there is a therapeutic window. MllPTD/WT/Runx1Δ/Δ cells were also sensitive to Echinomycin (Fig. 7A). In CFU replating assay using sorted cells from primary MllPTD/WT/Runx1Δ/Δ CFU cells, only the CD11b and Gr-1 double-negative population formed colonies, suggesting that this fraction contains CFU-initiating cells (Fig. 7B). Notably, Echinomycin reduced this CFU-initiating population and induced differentiation to mature granulocytic cells (Fig. 7C). To determine the therapeutic efficacy of Echinomycin in vivo, we first treated WT mice to assess its effect on normal hematopoiesis (Fig. S12B). There was no significant change over time in PB cell counts between Echinomycin and vehicle treated groups (Fig. S12C). Competitive BMT (CBMT) assay showed that the frequency of functional HSCs was also comparable between Echinomycin and vehicle treated groups (Fig. S12D). We then treated mice transplanted with BM cells from moribund MllPTD/WT/RX1-S291fs MDS mice with echinomycin and saw a significant prolongation of their survival (Fig. 7D and 7E). To determine the effect of Echinomycin on human MDS cells, we xenografted MDS patient derived MDSL cells (51) into NOD/SCID-IL2Rγ3GS (NSGS) mice (Fig. 7F). After the Echinomycin treatment, we first evaluated the chimerism of MDSL cells in BM and SP (Fig. 7F–L). MDSL cells were significantly decreased in both BM and SP from the Echinomycin treated group compared to the vehicle treatment group (Fig. 7G–L). Echinomycin treatment also significantly prolonged the survival of MDSL xenografted NSGS mice (Fig. 7M and 7N). Together, our genetic deletion and chemical inhibition data strongly suggest that the HIF1A signaling pathway is a novel therapeutic target in MDS.
Figure 7. Pharmacological inhibition of HIF1A in MDS pathogenesis.
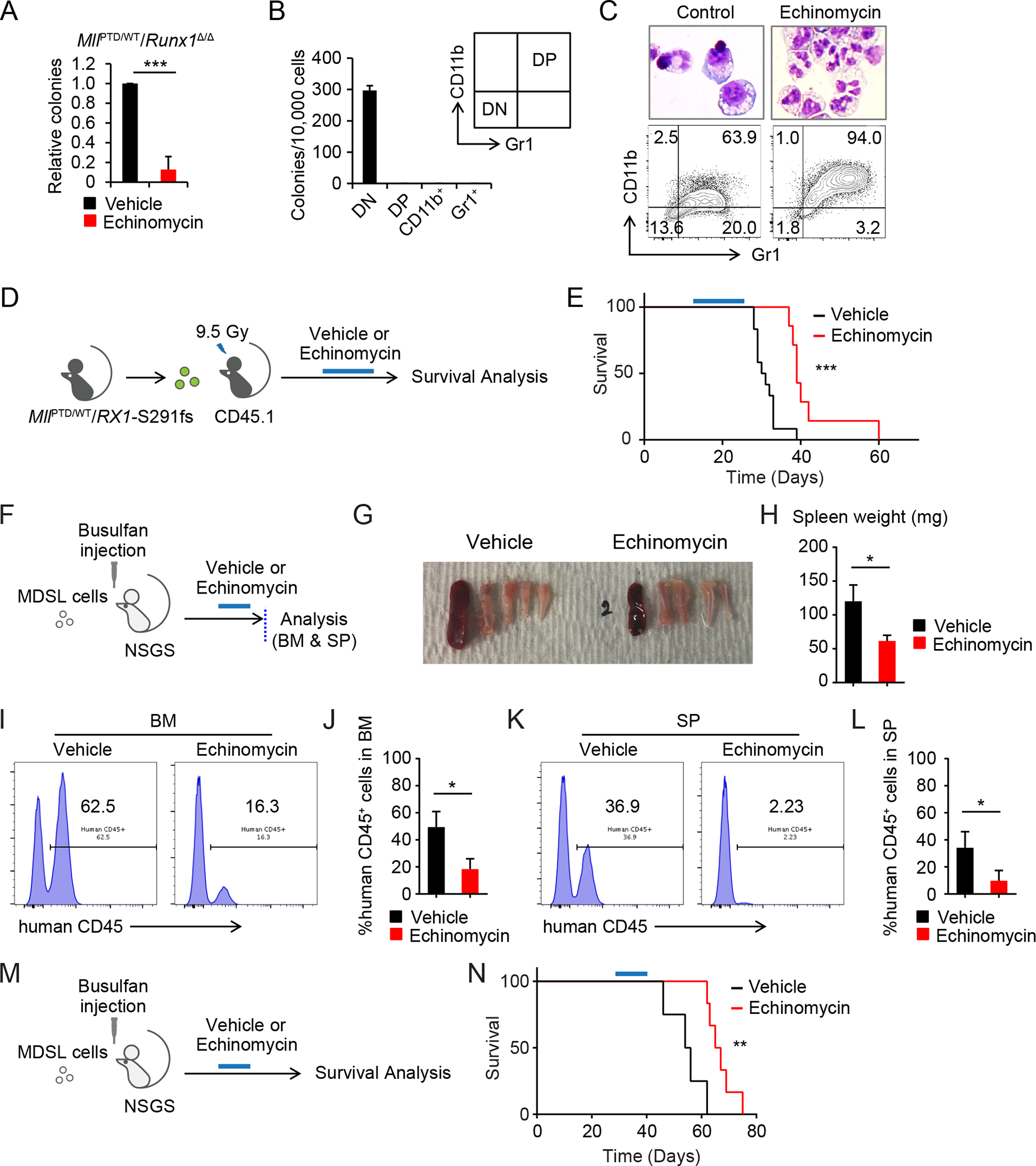
A, CFU assay of MllPTD/WT/Runx1Δ/Δ cells treated with Echinomycin (1.25 nM). Colony numbers were normalized to those in the control. Data are mean ± s.d. from independent 3 experiments. B, Secondary CFU assay using sorted 1st CFU of MllPTD/WT/Runx1Δ/Δ BM cells. CD11b− Gr1− cells (double negative, DN), CD11+ Gr1− cells (CD11b+), CD11+ Gr1+ cells (double positive, DP), and CD11− Gr1+ cells (Gr1+) were used. Data are mean ± s.d. from triplicate. C, Wright Giemsa staining and flow cytometric analysis of colony forming cells from MllPTD/WT/Runx1Δ/Δ BM cells. D, Schematic of Echinomycin treatment in the MllPTD/WT/Rx1-S291fs BMT mice model. E, Survival of MllPTD/WT/Rx1-S291fs BMT mice with or without Echinomycin treatment (control, n = 12; Echinomycin, n = 8) (E). F-L, Schematic of xenograft of NSGS with MDS-L cells is shown in (F). BM and SP were analyzed after the Echinomycin treatment (n = 4, per each group). Representative appearance of bones and SP (G), spleen weight (H), chimerism of human CD45+ cells in BM (I and J), and SP (K and L) from the indicated group are shown. Data (H, J, and L) are mean ± s.d. M, Schematic of survival analysis of xenograft model. N, Survival of MDS-L transplanted NSGS mice with or without Echinomycin treatment. Treatment was started 4 weeks after xenograft (control, n = 4; Echinomycin, n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
Complex human diseases may bear disparate mutation profiles in individual patients, but still share a common underlying mechanism. During development, different tissues re-utilize key regulators, such as HIF1A. Using genetic mouse models, human data sets, and unbiased approaches, we have identified an important role for hypoxia-independently activated HIF1A signaling in MDS development (Fig. S13). HIF1A is ubiquitously expressed and most HIF1A regulation is post-transcriptional, with HIF1A protein stability being regulated by both oxygen-dependent and oxygen-independent degradation (8,52). Furthermore, the HIF1A transactivation domain is also inactivated via hypoxia inducible factor 1 alpha subunit inhibitor (HIF1AN) (also known as FIH1) mediated hydroxylation of an asparagine residue. Both protein stability and transcriptional activity are required to activate HIF1A signaling. While mouse modeling of individual MDS-associated mutations showed the significant accumulation of HIF1A protein, single mutations are not enough to develop an MDS phenotype in mice. This might indicate a threshold effect preventing MDS development; requiring the additive effect of both HIF1A protein accumulation and activated transcriptional potential for MDS development. Individual mutations may have direct or indirect effects, leading to stabilization of HIF1A protein and activation of HIF1A signaling in both HSPCs and mature cell populations. Combination of two MDS-associated mutations induced additive accumulation of HIF1A protein (in a hypoxia-independent manner), and resulted in MDS phenotypes. Underscoring this point, a stable and constitutively active form of HIF1A induced in hematopoietic cells (Vav1-Cre/TPM mice) resulted in a variety of MDS related phenotypes (recapitulating the HIF1A accumulation, transcriptional activation, and phenotypes induced by combining multiple MDS-associated genetic events).
Mechanistically, we found that MLL-PTD was associated with Warburg-effect-type metabolic rewiring and hypoxia-independent activation of HIF1A signaling along with DNA hypermethylation. In contrast, HIF1A expression by mutant RUNX1 proteins was associated with downregulation of both MDM2 E3 ubiquitin ligase and TP53 protein (Fig. 5M). We note that Vhl and Mdm2 expression were upregulated in the HSPCs from MllPTD/WT mice. Thus, it is possible that RUNX1 mutant or Runx1 deficiency further upregulate Hif1a protein levels and signaling in the MllPTD/WT/RUNX1-mutant mouse model by disrupting the RUNX1-p53-MDM2 axis in an MllPTD/WT background. While DNMT3A, TET2, and ASXL1 also significantly upregulate HIF1A protein expression, further elucidation of the detailed mechanisms for these and other major MDS-associated mutations is required for a better understanding of the MDS pathobiology and target identification. Nonetheless, (given the common upregulation of HIF1A protein) HIF1A could be a promising target for a broad spectrum of MDS.
When we induced HIF1A expression in individual blood cell lineages, the megakaryocyte-lineage specific HIF1A induction caused thrombocytopenia and megakaryocyte dysplasia, indicating a sufficient, HIF1A mediated, cell-intrinsic mechanism for deregulated megakaryocytopoiesis. Erythrocyte lineage specific HIF1A induction did not cause aberrant erythropoiesis or anemia, suggesting an extrinsic mechanism for the ineffective erythropoiesis. On the other hand, HIF1A induction in the myelomonocytic lineage affected BM erythroblastic island formation, by inducing inflammatory macrophages, triggering anemia that had a slower tempo than the pan-hematopoietic cell lineage induction of HIF1A. In Vav1-Cre/TPM mice, not only innate immune cells, such as monocytes and macrophages, but also lymphoid cells were significantly activated by HIF1A. We noted that HIF1A induction in blood causes phenotypic inflammatory macrophages. Although we did not characterize the role of T cells and dendritic cells in our MDS models, activated inflammatory macrophages, T cells, and dendritic cells may cooperate to impair the late stage erythropoiesis and cause anemia. Thus, one possible explanation could be the lack of direct T cell activation in the setting of myelomonocytic lineage induction of HIF1A. Indeed, activated innate and adaptive immunity have been reported in patients with MDS (12,53). Aberrant immune activation could also interact with the BM microenvironment, and affect disease development through a blood cell-extrinsic manor. The interaction between innate and adaptive immunity might be involved in the ineffective erythropoiesis, resulting in the complex refractory anemia found in MDS patients.
Genetic deletion of Hif1a does not dramatically alter steady-state hematopoiesis (9). On the other hand, genetic deletion of Hif1a abrogated clonal advantage of MDS-related mutant cells and also reversed MDS phenotypes, including anemia and thrombocytopenia in our mutant and MDS mouse models. These results indicate that the gain-of-function effect of HIF1A signaling driven by mutations induces MDS-related phenotypes in different lineages and at various differentiation stages. Our analysis of RNA-Seq data indicates that the major impact of HIF1A abrogation in the disease context is to relieve the block in erythroid and megakaryocyte differentiation. HIF1A abrogation resulted in Gata2 downregulation and Gata1 upregulation (Gata factor switching), and upregulation of several critical genes for erythroid and megakaryocyte differentiation, such as Klf1, Gfi1b, Tal1, and Pbx1. Erythropoiesis is regulated by both intrinsic and extrinsic mechanisms. Macrophages at erythroblastic islands play a critical extrinsic role for erythropoiesis. Macrophages are regulated by other immune cells, especially T cells. HIF1A abrogation resulted in the reduction of Zeb2 expression and upregulation of Irf4 expression. ChIP-Seq data analysis indicated ZEB2 and IRF4 as HIF1A targets. ZEB2 is a key regulator for macrophage, dendritic cells, and also memory and cytotoxic T cells (54,55). IRF4 is a key regulator for alternative macrophage activation (but not for inflammatory macrophages). We noted that HIF1A induction in blood causes phenotypic inflammatory macrophages, impairment of late stage erythropoiesis, and decrease of BM erythroblastic islands. Thus, downregulation of Zeb2 and upregulation of Irf4 expression may explain the rescue of anemia phenotype through an erythroid cell-extrinsic mechanism.
A number of genetic, epigenetic, splicing, cohesin, and metabolic aberrations have been identified in MDS. Interestingly, for instance, DNMT3A and TET2 have an opposite function on DNA methylation, despite the high frequency of loss-of-function mutations in both genes. Moreover, in many cases, these aberrations are not specific for MDS, as the same mutations have also been identified in other hematologic disorders (5,56). However, there is a clear distinction in the phenotypes, clinical course, and treatment responsiveness between MDS and other hematologic malignancies, implying the complex interplay between the combination of various mutations, the genetic background, and likely the microenvironment. MDS HSPCs retain differentiation potential and keep producing aberrant mature cell populations that have the same driver mutations as their HSPCs. Once MDS HSPCs lose their differentiation ability, and acquire additional mutations, the disease can transform to acute leukemia. Current studies and recent evidence indicate that not only the HSPCs, but also cells at more mature stages, such as immune cell populations, play an important role for triggering complex MDS phenotypes (12).
Although future studies are required to elucidate the detailed mechanisms and critical downstream events in individual lineages which contribute to MDS phenotypes, our studies clearly provide new clues for dissecting and targeting MDS. Given the current focus on genomics in dissecting human disease pathobiology, we believe that our work illustrates the utility of comprehensive, unbiased genomics approaches to identify/target the critical signaling funnel for human diseases. The identification of such signaling funnels may help integrate big-data genome sequencing efforts and identify new research directions with therapeutic potential.
METHODS
Mice
The mice with the genotype of MllPTD/WT, Runx1flox/flox, Dnmt3aflox/flox, Vav1-Cre, EpoR-Cre, HIF1A-TPM, and NOD/LtSz-scid IL2RG–SGM3 (NSGS) were previously described (16,19,45,57–60). Mx1-Cre, Hif1aflox/flox, Vhlflox/flox, LysM-Cre, PF4-Cre, Rosa26-LSL-rtTA (129X1/SvJ), Asxl1flox/flox, and Tet2flox/flox mice were purchased form Jackson Laboratory. B6.SJL (CD45.1) and C57BL/6 (CD45.2) mice were obtained from the Cincinnati Children’s Hospital Medical Center (CCHMC) / Cancer and Blood Diseases Institute (CBDI) mouse core. Polyinosine-polycytosine (poly (I:C)) (12 to 18 μg/g) (GE Healthcare) was injected into mice with Mx1-Cre transgene and their control (without Mx1-Cre) intraperitoneally every other day for 3 to 5 times. All animals were housed in the animal barrier facility at CCHMC. All animal studies were conducted according to an approved Institutional Animal Care and Use Committee protocol and federal regulations.
Transient Transfection and Retrovirus Infection
Retroviruses were generated by calcium phosphate transient co-transfection of the retroviral vectors (pMY-RUNX1-S291fs-IRES-eGFP and pMY-IRES-eGFP) with the packaging plasmids Gag and Eco-env into 293T cells. 293T cells were maintained in DMEM with 10% fetal bovine serum (FBS) in a humidified incubator at 37°C and 5% CO2. BM cells were collected 3 days after a single dose of 5-fluorouracil (150 mg/kg, intraperitoneally) treatment. BM mononuclear cells were isolated using Histopaque (Sigma-Aldrich) and cultured in IMDM medium containing 10 % FBS, 100 ng/ml mouse stem cell factor (Peprotech), 100 ng/ml mouse thrombopoietin (Peprotech), and 100 ng/ml human G-CSF for overnight at 37 °C in 5 % CO2. First round retroviral infection was performed using RetroNectin (Takara Bio). Polybrene was used for the second round infection.
Western Blot Analysis
To isolate c-Kit+ BM HSPCs, freshly harvested BM cells were incubated with CD117 MicroBeads and separated on an AutoMACS Pro separator (Miltenyi) according to manufacturer specifications. Whole cell extract was prepared by sonicating cells in a sodium dodecyl sulfate (SDS) sample buffer containing 10mM NaF, 0.2mM Na3VO4, 10mM β-Glycerophosphate, 1mM phenylmethylsulfonyl fluoride (93482, Sigma-Aldrich), 1mM 4-Amidinophenylmethanesulfonyl Fluoride Hydrochloride (A6664, Sigma-Aldrich), 2.5mM dithiothreitol, 5% 2-mercaptoethanol, and proteinase inhibitors (Sigma-Aldrich, P8849; Roche Diagnosis, 11873580001). After boiling at 95 °C for 10 minutes, samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore). The following primary antibodies were used in this study: anti-HIF1A (NB100–105, Novus), anti-MDM2 (sc-965, Santa Cruz), anti-mouse p53 (1C12) (2524, Cell Signaling), anti-SDHA (ab14715, Abcam), anti-SDHB (ab14714, Abcam), anti-SDHD (ABT110, Millipore), anti-H3K4me3 (ab12209, Abcam), anti-H3K9me3 (millipore 07–442, Millipore), anti-H3K36me3 (ab9050, Abcam), anti-H3K79me3 (ab2621–100, Abcam), anti-Histone H3 (ab1791, Abcam), and anti-β-Actin (sc-1616R, Santa Cruz). Horseradish peroxidase-conjugated antibodies to rabbit (NA934V, GE Healthcare) or mouse (NA931V, GE Healthcare) were used as the secondary antibody and Super Signal West Dura Chemiluminescent Substrate (Pierce) was used for ECL detection. ImageJ software was used for quantification (61).
Bone Marrow Transplantation
Lethally irradiated (9.5 Gy) 2- to 4-month-old B6.SJL mice (CD45.1) were used as recipients. Total BM cells (1.0 – 2.0 ×106 cells) from primary mice (CD45.2) or retrovirus vector transduced BM cells (CD45.2) were injected into the tail vein. For radioprotection, 1×105 cells of freshly harvested whole BM cells from B6.SJL mice were co-transplanted. For MllPTD/WT/Rx1-S291fs mice in vivo experiment, GFP+ BM cells (1.0 – 2.0 ×106 cells) from primary BMT mice were used. In the case of 1:1 ratio competitive BMT assay, recipient mice received equal number of competitive BM cells from B6.SJL mice or C57BL/6 mice. For secondary transplantation of 1:1 ratio competitive BMT assay, BM cells from primary recipients at 16 weeks after transplantation purified by CD45.1 depletion were transplanted into lethally irradiated secondary mice along with B6.SJL BM cells.
Flow Cytometric Analysis
Flow cytometric analysis and cell sorting were performed with FACSCanto I, FACSCanto II, or FACSAria instrument (BD Biosciences). Colony formed cells, PB, and BM cells were immunostained using the following anti-mouse antibodies: CD45.1 (A20), CD45.2 (104), B220 (RA3–6B2), CD3 (17A2), CD45 (30-F11), CD71 (C2), c-Kit (2B8), CD34 (RAM34), Sca-1 (D7) (BD Pharmingen), CD11b (M1/70), CD16/32 (93) (eBioscience), CD115 (AFS98), Gr-1 (RB6–8C5), F4/80 (BM8), Ter119 (Ter-119), CD80 (16–10A1), and CD206 (C068C2) (Biolegend). For mature cell lineage exclusion, a lineage marker cocktail containing anti-mouse CD3e (145–2C11), B220 (RA3–6B2), Ter119 (Ter-119), CD11b (M1/70), Gr-1 (RB6–8C5), CD11c (HL3) (BD Bioscience), NK1.1 (PK136) (Biolegend) antibodies was used. Anti-human CD45 (HI-30) (BD Bioscience) or anti-human CD45 (2D1) (eBioscience) antibodies were used for detecting human cells. Data were analyzed using the FlowJo software (Tree Star).
Patient Samples
BM samples from unselected subjects with MDS were obtained from Blood Diseases Hospital at diagnosis or referral and gave informed consent compliant with the Declaration of Helsinki. Written informed consent was obtained in all cases according to the Institutional Review Board. BM mononuclear cells (BM MNCs) were isolated from BM aspirates of MDS patients and healthy donors using a Ficoll-Histopaque 1077–1 (Sigma-Aldrich) density gradient centrifugation. Genomic DNA was extracted using the AxyPrep blood genomic DNA miniprep kit (Axygen Biosciences). Mutation sequencing PCR primers were designed to amplify the coding sequences and splice sites of DNMT3A, SF3B1, U2AF1 and SRSF2. All products were confirmed by 2% agarose gel, purified using QIAquick Spin Kit (Qiagen) and sequenced using ABI PRISM 3730xl DNA Analyzer (Applied Biosystems). The sequence data files were analyzed using the Mutation surveyor3.25 software.
Immunohistochemistry Staining
The 2-μm-thick formalin-fixed, paraffin-embedded BM biopsy sections were used. The BM biopsy sections were submitted to xylene and alcohol for deparaffinization, and then blockades of endogenous peroxidase were performed. Samples were incubated in HIF1A antibody (mgc3, Abcam) for 4 hours at room temperature. A Biotin Link was used as a secondary antibody for 40 min followed by streptavidin-peroxidase method, visualization with the DAB chromogen and counter-staining using hematoxylin. De-identified and discarded adult BM samples with a diagnosis of “no abnormalities” (n = 6) were obtained from the Pathology Research Core at Cincinnati Children’s Hospital Medical Center. An exemption from the Institutional Review Board was obtained for these samples. Expression was reported as the percentage of positive cells per sample.
Quantitative RT-PCR
For human BM MNC samples, the Trizol reagent (Invitrogen) was used to isolate total RNA. reverse transcription was carried out with 1ug total RNA from each specimen using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher). The cDNA was amplified with an Applied Biosystems 7500 Real-Time PCR System Instrument (Applied Biosystems). For murine c-Kit+ BM cell and kidney samples, total RNA was isolated using RNeasy Micro Kit (Qiagen) and converted to cDNA using SuperScript III First-Strand Synthesis System for RT-PCR Kit (Invitrogen). The cDNA was amplified using an Applied Biosystems Step One Plus thermal cycler (Applied Biosystems). SYBR Green Master Mix (Life Technologies) and specific primers for each gene (Supplementary Table S3) were used.
RNA-Seq
Total BM cells were incubated with CD117 MicroBeads and c-Kit+ BM cells were separated on an AutoMACS Pro separator (Miltenyi) according to manufacturer specifications. Total RNA was isolated using RNeasy Mini Kit (Qiagen). NEBNext Ultra Directional RNA Library Prep Kit (New England BioLabs) was used for library preparation, which used dUTP in cDNA synthesis to maintain strand specificity. In short, the isolated RNA was Mg2+/heat fragmented (~200 bp), reverse transcribed to 1st strand cDNA, followed by 2nd strand cDNA synthesis labelled with dUTP. The purified cDNA was end repaired and dA tailed, and then ligated to adapter with a stem-loop structure. The dUTP-labelled 2nd strand cDNA was removed by USER enzyme to maintain strand specificity. After indexing via PCR (12 cycles) enrichment, the amplified library was cleaned up by AMPure XP beads for QC analysis. To check the quality and yield of the purified library, one μl library was analyzed by Bioanalyzer (Agilent) using DNA high sensitivity chip. To accurately quantify the library concentration for the clustering, the library was 1:104 diluted in dilution buffer (10 mM Tris-HCl, pH 8.0 with 0.05% Tween 20), and qPCR measured by Kapa Library Quantification kit (Kapabiosystem) using Applied Biosystems 9700HT real-time PCR system (Applied Biosystems). To study differential gene expression, individually indexed and compatible libraries were proportionally pooled (20–50 million reads per sample in general) for clustering in cBot system (Illumina). Libraries at the final concentration of 15 pM were clustered onto a single read (SR) flow cell using Illumina TruSeq SR Cluster kit v3, and sequenced to 50 bp using TruSeq SBS kit on Illumina HiSeq system (Illumina).
RNA-Seq Data Analysis
For the analyses of RNA-Seq data of Vav1-Cre/Control mice and Vav1-Cre/TPM mice samples, sequence reads were aligned to the reference genome using BowTie2/TopHat2(62), and reads aligning to each known transcript were quantified using Bioconductor (63). The differential expression analysis between Vav1-Cre/Control mice and Vav1-Cre/TPM mice samples was performed using the negative binomial statistical model of read counts as implemented in the edgeR Bioconductor package (64). The statistical significance of differential expression is established based on the FDR-adjusted p-values (65). The pathway enrichment analysis was performed separately for upregulated and downregulated genes using the LRpath methodology (66) as implemented in the CLEAN package (67), and the gene lists from the MSigDB database (68).
For the analyses of RNA-Seq data of MllPTD/WT/Runx1Δ/Δ and MllPTD/WT/Runx1Δ/Δ/Hif1aΔ/Δ samples, the FASTQ files were processed in the software AltAnalyze (version 2.1.0). Transcript per million estimates were calculated in AltAnalyze through a built-in call to Kallisto for read pseudoalignment and transcript quantification (Ensembl 72, mm10)(69). Differentially expressed genes between MllPTD/WT/Runx1Δ/Δ and MllPTD/WT/Runx1Δ/Δ/Hif1aΔ/Δ samples were selected based on a fold change > 2 and an empirical Bayes moderated t-test p < 0.05 in AltAnalyze. TF-target enrichment analysis using the GO-Elite algorithm (PAZAR and Amadeus databases) in AltAnalyze(48). HIF1A targets were obtained from multiple sources and data analyses including two re-analyzed published ChIP-Seq datasets (GSE40918, GSE36104) (46,47), HIF1A target annotations from the GO-Elite TF-target database (PAZAR)(48), prior predicted targets from the Broad Molecular Signatures Database (MSigDB), Pathway Commons(49), and Whole Genome R-Vista binding site predictions(70). The FASTQ files from the ChIP-Seq studies were mapped using bowtie2 to the Mus Musculus genome. Duplicate reads were removed using Picard [https://broadinstitute.github.io/picard/] and Multi-mapped reads were filtered with SAMtools (version 1.1.19) (71). MACS2 (72) was used for peak calling. Then followed by closest gene annotation to identify peaks with the software GREAT (http://bejerano.stanford.edu/great) using the default options. The ATAC–Seq data from LSK cells was obtained from GEO record (GSM1463179). For visualization, data were processed with the software biowardrobe (73), and tracks were displayed using the UCSC Genome Browser (74). RNA-Seq data sets are deposited at GEO under accession numbers GSE83988 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=srwtkoyctnynjot&acc=GSE83988) and GSE86953 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=qduhsmckvvwfzcn&acc=GSE86953).
H3K4me3 ChIP-Seq and Data Analysis
BM LSK cells from WT and MllPTD/WT mice were isolated as described (75). These cells were subjected to ChIP-Seq as previously described (76). Anti-histone H3K4me3 antibody (ab8580, Abcam) was used. The ChIP-enriched DNA was purified using the MinElute Reaction Cleanup Kit (Qiagen).
Differential bindings were analyzed using HOMER (v4.7) package (77) using find Peaks function (FDR < 0.001, F (fold enrichment over control tag count) > 4 fold, P (poisson p-value threshold relative to control tag count) < 0.001). To show the tag intensities around TSS (+10/−10 kbp regions), heatmaps were generated using HOMER’s annotate Peaks function and R’s heatmap package. No clustering method was used to visualize data; instead, the rows in the heatmap are arranged by the coverage from strong to weak. For each differentially bound peak, RefSeq genes whose transcription start sites (TSS) are located within +/− 20 kbp from the peak center were chosen for Gene set enrichment test using webGestalt (78) to get statistically significant (FDR < 0.05) gene sets or pathways. The TSS coordinates of each Refseq genes were extracted from UCSC table browser (mm9). Enriched motifs were detected using find Motifs Genome function with regional size (-size) 200 for TFs and 1000 for H3K4me3. The data sets are deposited at GEO under accession number GSE75608 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wbwpqagurtmdbyl&acc=GSE75608).
Measurements of mitochondrial respiration
The rates of oxygen consumption in fibroblast cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Biosciences, North Billerica, MA, USA), as detailed elsewhere (79,80). XF96 creates a transient, 7 μl-chamber in specialized microplates that allows for the determination of oxygen and proton concentrations in real time. The cultured cells were transferred to a XF96 assay plate at 100,000 cells/well and allowed to grow overnight. The parameters included basal respiration, ATP production, and maximal respiratory capacity. To allow comparison between different experiments, data are expressed as the rate of oxygen consumption in pmol/min or the rate of extracellular acidification in mpH/min, normalized to cell protein in individual wells determined by the Bradford protein assay (Bio-Rad). The rates of O2 were determined under basal conditions and the addition of oligomycin (1.5 μM), carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (0.5 μM), rotenone (1 μM) and antimycin A (1 μM), as detailed elsewhere (79,80). Additional details relating to theSeahorseXF-96 analyzer can also be found at http://www.seahorsebio.com/ (81).
Mitochondrial respiratory chain complex I to IV activity assay
The enzymatic activities of complex I, II, III and IV were assayed as detailed elsewhere (82,83). Complex I (NADH dehydrogenase) activity was determined as the rotenone-sensitive NADH oxidation at 340 nm, using the coenzyme Q analog 2,3-dimethyl-5-methyl 6-n-decyl-1,4-benzomethyluinone as an electron acceptor. The activity of complex II (succinate dehydrogenase) was analyzed by tracking the secondary reduction of 2,6 dichlorophenolindophenol by ubiquinone 2 at 600 nm. Complex III (cytochrome bc1 complex) activity was determined by measuring the reduction of cytochrome c at 550 nm with reduced decylubiquinone. Complex IV (cytochrome c oxidase) activity was measured by monitoring the oxidation of reduced cytochrome c as a decrease of absorbance at 550 nm. Citrate synthase activity was analyzed by the reduction of 5,5'5 synthase activity was analyzed by the reduction of 5ed by monitoring and oxaloacetate. Complex I to IV activities were normalized by citrate synthase activity and then used in the analysis.
NMR Plasma Sample Preparation
All the plasma test samples were thawed on ice on the day of the data collection. Once thawed, samples were aliquoted onto pre-washed 3 kDa spin filters (~100 μL plasma/filter) (NANOSEP 3K, Pall Life Sciences), and centrifuged 10,000x gn for 90 min at 4 °C. The 85 μl of plasma filtrate was mixed with 115 μl of NMR buffer containing 100 mM phosphate buffer in D2O, pH 7.3, which included 1.0 mM TMSP (3-Trimethylsilyl 2,2,3,3-d4 propionate). The final TMSP concentration was 0.575 mM in NMR sample.
NMR Spectroscopy data acquisition and processing.
The experiments were conducted using 180 μl samples in 103.5 mm x 3 mm NMR tubes (Bruker). One-dimensional 1H NMR spectra were acquired on a Bruker Avance II 600 MHz spectrometer. All data were collected at a calibrated temperature of 298 K using a three-pulse sequence based on the noesypr1d (or noesygppr1d) pulse sequence in the Bruker pulse sequence library. This pulse sequence provided water-suppression with good baseline characteristics. Experiments were run with 4 dummy scans (DS) and 256 acquisition scans (NS) with an acquisition time (AQ) of 2.7 s and a relaxation delay (D1) of 3.0 s for a total repetition cycle (AQ+D1) of 5.7 s. The NOESY mixing time was 10 ms. The spectral width was 20 ppm, and 64 K real data points were collected. Under automation control, each sample analysis took about 10 min for setup and 30 min for acquisition. During the 10 minute setup time, the temperature was monitored for equilibration. All FIDs were subjected to an exponential line-broadening of 0.3 Hz. Upon Fourier transformation, each spectrum was manually phased, baseline corrected, and referenced to the internal standard TMSP at 0.0 ppm for polar samples using Topspin 3.5 software (Bruker Analytik, Rheinstetten, Germany). For two dimensional 1H-1H total correlation spectroscopy (TOCSY) data, a relaxation delay equal to 2 s, isotropic mixing time of 80 ms with a B1 field strength of 10 kHz were used for 2048 data points with 128 scans per increment were acquired with spectral widths of 14 ppm. In addition, 2D 1H-13C heteronuclear single quantum coherence (HSQC) data, a relaxation delay equal to 1.5 s was used between acquisitions and a refocusing delay of 3.45 ms was implemented. The 2048 data points with 128 scans per increment were acquired with spectral widths of 11 ppm in F2 and 180 ppm in F1 (13C).
Metabolomics Data Analysis
Principal components analysis (PCA) was performed to determine the reproducibility of the extraction and sample stability and to look for metabolic differences. 1H NMR spectra were processed and analyzed with AMIX for PCA analysis. The spectra from 0.5 to 10.0 ppm, excluding the region of the residual water resonance (4.6 to 5.0 ppm) and methanol (3.36–3.38 ppm), were reduced by uniform binning to 995 buckets 0.01 ppm wide. Signal intensities were summed for integration, and the spectra were normalized to constant total spectral area. Prior to PCA analysis, the binned spectra were mean-centered with no scaling.
The spectral bin intensities tables were further analyzed using a univariate approach, based on bin-by-bin differences between WT and PTD spectra. Metabolites were assigned to those bins based on the chemical shifts as described below. Pairwise differences within each bin were compared using a two-tailed Welch’s t-test and ANOVA (JMP 12). In addition to the comparison of the bin intensities, metabolites’ concentration of TCA cycle metabolites was calculated based on the internal standard (Chenomx Inc. version 8.1). The concentrations of these metabolites were used for correlation analysis (MetaboAnalyst 3.0). Metabolites found in plasma were assigned based on 1D 1H, 2D TOCSY and HSQC NMR experiments. Peaks were assigned by comparing the chemical shifts and spin-spin couplings with reference spectra found in databases, such as the Human Metabolome Database (HMDB), the biological magnetic resonance data bank (BMRB), and Chenomx® NMR Suite profiling software (Chenomx Inc. version 8.1).
Reagents and Cell lines
Echinomycin were purchased from Enzo Life Sciences. Stock solutions were made in dimethyl sulfoxide (DMSO). Echinomycin was injected into wild type B6.SJL (CD45.1) mice intraperitoneally as the following: 100μg/kg/day, 5 times (every other day). Echinomycin was injected into the recipient mice transplanted with MllPTD/WT/Rx1-S291fs cells intraperitoneally as the following: from day 14 after BMT; 100μg/kg/day, every other day, total 7 times. Echinomycin was injected into the xenografted NSGS mice intraperitoneally as the following: 50μg/kg/day, 5 times (every other day). MDSL cells were acquired in 2015. MDSL cells are a gift of Dr. Kaoru Tohyama.
Statistical Analysis
Survival of mice was analyzed using the log-rank test. Statistical analyses for others were generally performed using two-tailed Student’s t-test. One-way ANOVA with multiple comparisons correction was used for comparisons among four groups in Figure 6E. P-values of < 0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Rife and A. Woeste (Cincinnati Children’s Hospital Medical Center) for experiment assistance. This work was supported by the Kyoto University Foundation (to Y. Hayashi), the MDS Foundation (Young Investigator Award to Y. Hayashi), the Cincinnati Children’s Hospital Research Foundation (to G. Huang.), the Leukemia Research Foundation (to G. Huang), the OCRA (to G. Huang.), National Institutes of Health (NIH) (R01CA166835 to S.D. Nimer, R01DK105014 to G. Huang, P50CA140158 and R01CA89341 to M.A. Caligiuri), National Natural Science Funds of China (No. 81470338 to YU. Zhang, No.81370611 to ZF. Xu, No. 81300392 to JIE. Wang, No. 81470297, No. 81770129 to G. Huang, and No. 81530008, No. 81470295 to ZJ. Xiao), CAMS Initiative Fund for Medical Sciences (2016-I2M-1-001 to ZJ. Xiao), Tianjin science and technology projects (13CYBJC42400 to YU. Zhang), CCHMC Research and Development Project through the Cystic Fibrosis Foundation (to J.P. Bridges), and the Ministry of Science and Technology, Taiwan (MOST 103-2321-B-182-015 to L-Y. Shih). The mouse BMT services were conducted by the Comprehensive Mouse and Cancer Core in Cancer and Blood Diseases Institute at Children’s Hospital Research Foundation, which is supported through the NIDDK Centers of Excellence in Molecular Hematology (P30DK090971). RNA-Seq was conducted by Genomics, Epigenomics and Sequencing Core, Department of Environmental Health, University of Cincinnati, which is supported in part through CEG grant (NIEHS P30-ES006096). NMR Spectroscopy-based analysis was conducted by the NMR-Based Metabolomics Core (NBMC) at Children’s Hospital Research Foundation. A part of RNA-Seq analyses was conducted by J. Chen and M. Medvedovic at Laboratory for Statistical Genomics and Systems Biology, Department of Environmental Health, University of Cincinnati. MDSL cells were kindly provided by Dr. Kaoru Tohyama.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007;7(2):118–29 doi 10.1038/nrc2047. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009;361(19):1872–85 doi 10.1056/NEJMra0902908. [DOI] [PubMed] [Google Scholar]
- 3.Zhou T, Kinney MC, Scott LM, Zinkel SS, Rebel VI. Revisiting the case for genetically engineered mouse models in human myelodysplastic syndrome research. Blood 2015;126(9):1057–68 doi 10.1182/blood-2015-01-624239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nimer SD. Myelodysplastic syndromes. Blood 2008;111(10):4841–51 doi 10.1182/blood-2007-08-078139. [DOI] [PubMed] [Google Scholar]
- 5.Raza A, Galili N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat Rev Cancer 2012;12(12):849–59 doi 10.1038/nrc3321. [DOI] [PubMed] [Google Scholar]
- 6.Haferlach T Molecular genetics in myelodysplastic syndromes. Leuk Res 2012;36(12):1459–62 doi 10.1016/j.leukres.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014;28(2):241–7 doi 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3(10):721–32 doi 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 9.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010;7(3):391–402 doi 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 10.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 2014;41(4):518–28 doi 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013;123(9):3664–71 doi 10.1172/jci67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015;29(7):1458–69 doi 10.1038/leu.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010;24(4):756–64 doi 10.1038/leu.2010.31. [DOI] [PubMed] [Google Scholar]
- 14.Wierenga AT, Vellenga E, Schuringa JJ. Convergence of hypoxia and TGFbeta pathways on cell cycle regulation in human hematopoietic stem/progenitor cells. PLoS One 2014;9(3):e93494 doi 10.1371/journal.pone.0093494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120(12):2454–65 doi 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges JP, Lin S, Ikegami M, Shannon JM. Conditional hypoxia inducible factor-1alpha induction in embryonic pulmonary epithelium impairs maturation and augments lymphangiogenesis. Dev Biol 2012;362(1):24–41 doi 10.1016/j.ydbio.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kroger N, Zabelina T, van Biezen A, Brand R, Niederwieser D, Martino R, et al. Allogeneic stem cell transplantation for myelodysplastic syndromes with bone marrow fibrosis. Haematologica 2011;96(2):291–7 doi 10.3324/haematol.2010.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood 2008;112(3):470–8 doi 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Yan X, Sashida G, Zhao X, Rao Y, Goyama S, et al. Stress hematopoiesis reveals abnormal control of self-renewal, lineage bias, and myeloid differentiation in Mll partial tandem duplication (Mll-PTD) hematopoietic stem/progenitor cells. Blood 2012;120(5):1118–29 doi 10.1182/blood-2012-02-412379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Chen A, Yan XM, Huang G. Disordered epigenetic regulation in MLL-related leukemia. Int J Hematol 2012;96(4):428–37 doi 10.1007/s12185-012-1180-0. [DOI] [PubMed] [Google Scholar]
- 21.Dorrance AM, Liu S, Yuan W, Becknell B, Arnoczky KJ, Guimond M, et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest 2006;116(10):2707–16 doi 10.1172/jci25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenney K, Shilatifard A. A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: linking leukemogensis to covalent modifications of chromatin. J Cell Biochem 2005;95(3):429–36 doi 10.1002/jcb.20421. [DOI] [PubMed] [Google Scholar]
- 23.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol 2014;15(4):243–56 doi 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399(6733):271–5 doi 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 25.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev 2000;14(1):34–44. [PMC free article] [PubMed] [Google Scholar]
- 26.van Nederveen FH, Korpershoek E, Lenders JW, de Krijger RR, Dinjens WN. Somatic SDHB mutation in an extraadrenal pheochromocytoma. N Engl J Med 2007;357(3):306–8 doi 10.1056/NEJMc070010. [DOI] [PubMed] [Google Scholar]
- 27.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 2011;108(1):314–8 doi 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jafri M, Wake NC, Ascher DB, Pires DE, Gentle D, Morris MR, et al. Germline Mutations in the CDKN2B Tumor Suppressor Gene Predispose to Renal Cell Carcinoma. Cancer Discov 2015;5(7):723–9 doi 10.1158/2159-8290.cd-14-1096. [DOI] [PubMed] [Google Scholar]
- 29.Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 2011;89(3):213–20 doi 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005;7(1):77–85 doi 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 31.Seward DJ, Cubberley G, Kim S, Schonewald M, Zhang L, Tripet B, et al. Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat Struct Mol Biol 2007;14(3):240–2 doi 10.1038/nsmb1200. [DOI] [PubMed] [Google Scholar]
- 32.Ploumakis A, Coleman ML. OH, the Places You’ll Go! Hydroxylation, Gene Expression, and Cancer. Mol Cell 2015;58(5):729–41 doi 10.1016/j.molcel.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Johansson C, Velupillai S, Tumber A, Szykowska A, Hookway ES, Nowak RP, et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat Chem Biol 2016;12(7):539–45 doi 10.1038/nchembio.2087. [DOI] [PubMed] [Google Scholar]
- 34.Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016;537(7618):63–8 doi 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012;120(5):1130–6 doi 10.1182/blood-2012-03-415067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364(26):2496–506 doi 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 2013;210(12):2641–59 doi 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 2013;210(12):2627–39 doi 10.1084/jem.20131144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun 2014;5:4177 doi 10.1038/ncomms5177. [DOI] [PubMed] [Google Scholar]
- 40.Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004;103(6):2316–24 doi 10.1182/blood-2003-09-3074. [DOI] [PubMed] [Google Scholar]
- 41.Peng ZG, Zhou MY, Huang Y, Qiu JH, Wang LS, Liao SH, et al. Physical and functional interaction of Runt-related protein 1 with hypoxia-inducible factor-1alpha. Oncogene 2008;27(6):839–47 doi 10.1038/sj.onc.1210676. [DOI] [PubMed] [Google Scholar]
- 42.Lee SH, Che X, Jeong JH, Choi JY, Lee YJ, Lee YH, et al. Runx2 protein stabilizes hypoxia-inducible factor-1alpha through competition with von Hippel-Lindau protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J Biol Chem 2012;287(18):14760–71 doi 10.1074/jbc.M112.340232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SH, Bae SC, Kim KW, Lee YM. RUNX3 inhibits hypoxia-inducible factor-1alpha protein stability by interacting with prolyl hydroxylases in gastric cancer cells. Oncogene 2014;33(11):1458–67 doi 10.1038/onc.2013.76. [DOI] [PubMed] [Google Scholar]
- 44.Cai X, Gao L, Teng L, Ge J, Oo ZM, Kumar AR, et al. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015. doi 10.1016/j.stem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005;106(2):494–504 doi 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell 2012;151(2):289–303 doi 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell 2012;47(5):810–22 doi 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, et al. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics 2012;28(16):2209–10 doi 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res 2011;39(Database issue):D685–90 doi 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011;8(4):399–411 doi 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuoka A, Tochigi A, Kishimoto M, Nakahara T, Kondo T, Tsujioka T, et al. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia 2010;24(4):748–55 doi 10.1038/leu.2009.296. [DOI] [PubMed] [Google Scholar]
- 52.Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci 2008;33(11):526–34 doi 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Li X, Xu F, He Q, Wu L, Zhang Z, Chang C. Comparison of immunological abnormalities of lymphocytes in bone marrow in myelodysplastic syndrome (MDS) and aplastic anemia (AA). Intern Med 2010;49(14):1349–55. [DOI] [PubMed] [Google Scholar]
- 54.Scott CL, Soen B, Martens L, Skrypek N, Saelens W, Taminau J, et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J Exp Med 2016;213(6):897–911 doi 10.1084/jem.20151715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med 2015;212(12):2027–39 doi 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fernandez-Mercado M, Yip BH, Pellagatti A, Davies C, Larrayoz MJ, Kondo T, et al. Mutation patterns of 16 genes in primary and secondary acute myeloid leukemia (AML) with normal cytogenetics. PLoS One 2012;7(8):e42334 doi 10.1371/journal.pone.0042334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development 2005;132(1):203–13 doi 10.1242/dev.01558. [DOI] [PubMed] [Google Scholar]
- 58.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2012;44(1):23–31 doi 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinrich AC, Pelanda R, Klingmuller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood 2004;104(3):659–66 doi 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 60.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 2010;24(10):1785–8 doi 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25(9):1105–11 doi 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 2015;12(2):115–21 doi 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 2013;8(9):1765–86 doi 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 65.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003;100(16):9440–5 doi 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics 2009;25(2):211–7 doi 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Freudenberg JM, Joshi VK, Hu Z, Medvedovic M. CLEAN: CLustering Enrichment ANalysis. BMC Bioinformatics 2009;10:234 doi 10.1186/1471-2105-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016. doi 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res 2002;12(5):832–9 doi 10.1101/gr.225502. Article published online before print in April 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Y, Fear J, Hu J, Haecker I, Zhou L, Renne R, et al. Leveraging biological replicates to improve analysis in ChIP-seq experiments. Comput Struct Biotechnol J 2014;9:e201401002 doi 10.5936/csbj.201401002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kartashov AV, Barski A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol 2015;16:158 doi 10.1186/s13059-015-0720-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res 2002;12(6):996–1006 doi 10.1101/gr.229102. Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao X, Chen A, Yan X, Zhang Y, He F, Hayashi Y, et al. Downregulation of RUNX1/CBFbeta by MLL fusion proteins enhances hematopoietic stem cell self-renewal. Blood 2014;123(11):1729–38 doi 10.1182/blood-2013-03-489575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou J, Wu J, Li B, Liu D, Yu J, Yan X, et al. PU.1 is essential for MLL leukemia partially via crosstalk with the MEIS/HOX pathway. Leukemia 2014;28(7):1436–48 doi 10.1038/leu.2013.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38(4):576–89 doi 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013;41(Web Server issue):W77–83 doi 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med 2011;51(9):1621–35 doi 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 2011;6(7):e21746 doi 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider L, Giordano S, Zelickson BR, M SJ, G AB, Ouyang X, et al. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic Biol Med 2011;51(11):2007–17 doi 10.1016/j.freeradbiomed.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS One 2009;4(2):e4492 doi 10.1371/journal.pone.0004492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li L, Li B, Zhang H, Bai S, Wang Y, Zhao B, et al. Lentiviral vector-mediated PAX6 overexpression promotes growth and inhibits apoptosis of human retinoblastoma cells. Invest Ophthalmol Vis Sci 2011;52(11):8393–400 doi 10.1167/iovs.11-8139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.