
Figure 1.
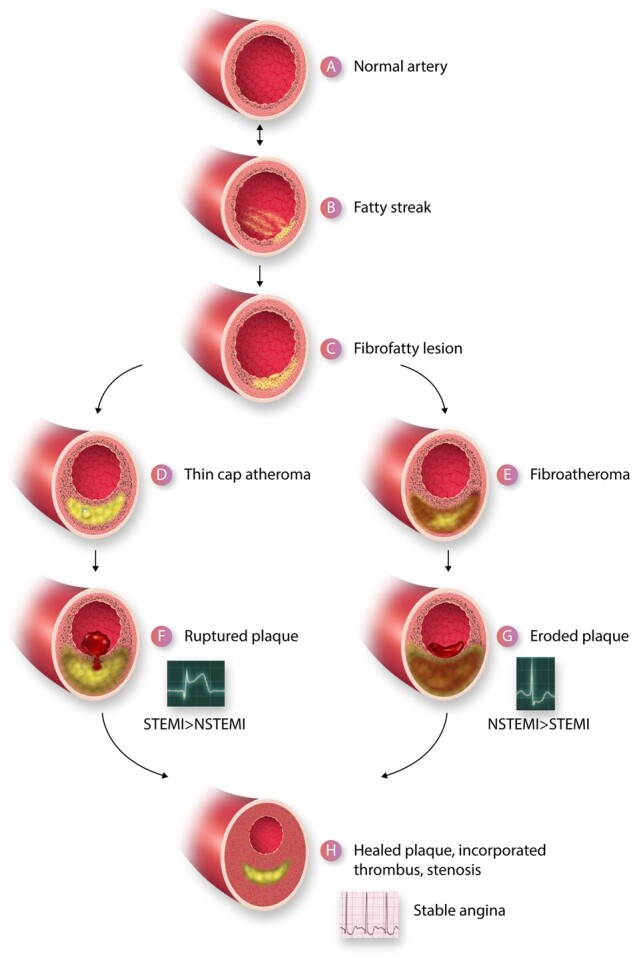
A depiction of the life cycle of an atherosclerotic plaque. (A) Depicts a normal artery. (B) Portrays the initiation of a fatty streak with mononuclear phagocyte recruitment and foam cell formation. The fatty streak may be reversible. (C) A fibrofatty lesion forms when smooth muscle cells lay down extracellular matrix in the intima that enrobes the foam cells. Some smooth muscle cells also accumulate lipid and take on the appearance and markers of foam cells derived from leucocytes of the myeloid series. With further evolution of the plaque, foam cells and smooth muscle cells undergo death and extracellular lipid accumulates from the debris of dying or dead cells and accumulated lipoproteins, forming a lipid core forms in many plaques. A fibrous cap typically forms above the lipid core. Due to decreased synthesis and increased breakdown of extracellular matrix macromolecules such as collagen, this fibrous cap thins creating a thin-capped atheroma (D). Other plaques may evolve to accumulate more matrix and less lipid. For example, the typical substrate of a plaque that has undergone thrombosis due to superficial erosion lacks an organized lipid core, but contains abundant proteoglycan and glycosaminoglycans. This type of plaque does not have a thin fibrous cap and may harbour many smooth muscle cells (E). The thin-capped fibroatheroma can rupture and provoke thrombosis (F). The more fibrous atheromata can undergo erosion (G). The thrombus that complicates eroded plaques is generally an eccentrically located sessile mural thrombus that is more platelet-rich (white thrombus) that the fibrin and erythrocyte rich red thrombus typically provoked by plaque rupture. The ruptured plaque more often gives rise to an ST segment elevation myocardial infarction (STEMI) than a non-ST segment elevation myocardial infarction (NSTEMI) whereas superficial erosion more often gives rise to NSTEMI than STEMI. Regardless of the mechanism of plaque disruption, the resultant thrombi provoke a wound healing response due to elaboration of mediators such as PDGF and TGF-β that augment extracellular matrix synthesis and promote smooth muscle cell migration. Thrombin itself is a mitogen for smooth muscle cells. The incorporation of thrombus provides a provisional matrix replicating within the previously disrupted intima recapitulating the well-known stages of wound healing. The accumulation of extracellular matrix in the healing plaque can lead to increasing luminal stenosis. Such healed, layered plaques tend to have a thicker fibrous cap, less likely to rupture, but these stenotic lesions can give rise to chronic stable angina as depicted by the electrocardiogram characteristic of a positive stress test (H).