

Significance
Plant genomes encode hundreds of genes controlling the detection, signaling pathways, and immune responses necessary to defend against pathogens. Pathogens, in turn, continually evolve to evade these defenses. Small RNAs, such as microRNAs (miRNAs), are one mechanism used by pathogens to overcome plant defenses and facilitate plant colonization. Mounting evidence would suggest that beneficial microbes, likewise, use miRNAs to facilitate symbiosis. Here, we demonstrate that the beneficial fungus Pisolithus microcarpus encodes a miRNA that enters plant cells and stabilizes the symbiotic interaction. These results demonstrate that beneficial fungi may regulate host gene expression through the use of miRNAs and sheds light on how beneficial microbes have evolved mechanisms to colonize plant tissues.
Keywords: effector, small RNA, symbiosis, mycorrhizae, plant defense
Abstract
Small RNAs (sRNAs) are known to regulate pathogenic plant–microbe interactions. Emerging evidence from the study of these model systems suggests that microRNAs (miRNAs) can be translocated between microbes and plants to facilitate symbiosis. The roles of sRNAs in mutualistic mycorrhizal fungal interactions, however, are largely unknown. In this study, we characterized miRNAs encoded by the ectomycorrhizal fungus Pisolithus microcarpus and investigated their expression during mutualistic interaction with Eucalyptus grandis. Using sRNA sequencing data and in situ miRNA detection, a novel fungal miRNA, Pmic_miR-8, was found to be transported into E. grandis roots after interaction with P. microcarpus. Further characterization experiments demonstrate that inhibition of Pmic_miR-8 negatively impacts the maintenance of mycorrhizal roots in E. grandis, while supplementation of Pmic_miR-8 led to deeper integration of the fungus into plant tissues. Target prediction and experimental testing suggest that Pmic_miR-8 may target the host NB-ARC domain containing transcripts, suggesting a potential role for this miRNA in subverting host signaling to stabilize the symbiotic interaction. Altogether, we provide evidence of previously undescribed cross-kingdom sRNA transfer from ectomycorrhizal fungi to plant roots, shedding light onto the involvement of miRNAs during the developmental process of mutualistic symbioses.
Noncoding small RNAs (sRNAs) regulate vital biological processes in a wide range of organisms by regulating gene expression in a highly specific fashion. Despite their short length (usually 20 to 30 nucleotides long), these sRNAs target messenger RNA by complementary base pairing, in which they alter protein production through posttranscriptional gene silencing and translational gene silencing (1). In exceptional cases, they may also activate gene expression (2). Among the different classes of sRNA, microRNAs (miRNAs) are one of the most well-characterized. Precursor miRNAs form self-complementary, hairpin-like stem loop structures [70 to 200 nucleotides long; (3)] that are then diced into short, mature miRNA by DICER-like (DCL) proteins [21 to 24 nucleotides long; (4)]. These miRNAs can then interfere with the expression of their target transcript by cleavage or inactivation when incorporated into an RNA-induced silencing complex together with the ARGONAUT protein AGO1 (5). sRNAs, including miRNAs, have been identified as playing endogenous roles in regulating physiological processes in various fungi, including saprotrophic species [e.g., Neurospora crassa; (6)], pathogenic fungi [e.g., Magnaporthe oryzae; (7)], and mutualistic fungi [e.g., Rhizophagus irregularis; (8)]. Another class of mutualistic fungi, the ectomycorrhizal (ECM) fungi, are also presumed to produce miRNAs, as they possess the necessary components for the sRNA synthesis pathway, including AGO proteins, DCL proteins, and RNA-dependent RNA polymerases (6, 9–12).
ECM fungi form essential, nutrient-acquiring symbioses with the roots of most temperate and boreal forest trees (13). Evolved from multiple lineages of saprotrophic ancestors, ECM fungi exhibit conserved genomic alterations that are thought to have led to their ability to colonize host tissues (14). One mechanism in particular includes the encoding of effector-like signaling proteins (15). To date, only a small number of these have been characterized, but their base mode of action appears to be to alter host signaling and metabolism to foster the early stages of symbiosis (16–20). sRNAs may be another signaling molecule used by ECM fungi during symbiosis. Proof of miRNAs regulating fungal–plant interactions have been experimentally shown in pathogenic models, whereby small interfering RNAs (siRNAs) produced by the fungal pathogen Botrytis cinerea were found to be exported into Arabidopsis leaves, binding to the host AGO1 protein and suppressing host immunity by targeting MAPK genes (21). Emerging evidence suggests similar cross-kingdom sRNA transfer, and silencing occurs in other plant pathosystems involving various fungal pathogens [e.g., Verticillium dahliae, Blumeria graminis, and Scleotinia sclerotiorum; (22–25)]. Besides fungal pathogens, a recent report also demonstrated that the sRNAs produced by mutualistic rhizobia regulate nodulation by hijacking the host AGO1-dependent RNA interference machinery (26). Furthermore, in silico analyses predict that sRNA produced by mutualistic arbuscular mycorrhizal (AM) fungi may target gene expression in host plants (18, 27). Despite the lack of experimental characterization of the miRNAs, this latter study supports the concept that cross-kingdom sRNA silencing is more broadly applicable beyond pathogenic models to mutualistic plant–fungal interactions.
In this current study, we made use of the mutualistic ECM fungus Pisolithus microcarpus and its interaction with the roots of the host plant Eucalyptus grandis to better understand the putative role of fungal, miRNA-mediated gene regulation during ECM symbiosis. Genome mining and sRNA sequencing (sRNA-seq) was performed at different developmental stages of mycorrhizal root tip formation to identify fungal miRNAs differentially regulated during ECM colonization. Furthermore, we tested the potential existence of cross-kingdom sRNA transfer between P. microcarpus and E. grandis. We give evidence here supporting the importance of certain miRNAs in cross-kingdom signaling during ECM symbiosis establishment.
Results
Identification and Differential Expression Profiling of Pisolithus miRNAs during Root Colonization.
The P. microcarpus reference genome and sRNA-seq were used to identify fungal miRNAs and examine their expression patterns during ECM colonization. Prediction of miRNA using the ShortStack application identified 11 fungal miRNAs [Table 1; (28)]. These fungal miRNAs exhibited significant differential expression patterns between the different time points of ECM colonization considered, six of which were induced by the colonization process (Pmic_miR-1, Pmic_miR-5, Pmic_miR-6, Pmic_miR-7, Pmic_miR-8, and Pmic_miR-10; Fig. 1 A and B), expression patterns that were verified using qPCR (SI Appendix, Fig. S1). Expressions of some miRNAs were consistent across the time points taken; Pmic_miR-1 was consistently induced throughout mycorrhizal root tip development, compared to axenically grown fungal colonies, while Pmic_miR-6 was most highly induced during the early stages of colonization and steadily reduced in expression in the latter stages of colonization (Fig. 1 A and B). While most of the other identified miRNAs were repressed during colonization, the expression of Pmic_miR-8 was initially repressed followed by induction at the 2-wk time point, when ingrowth of the mycorrhizal fungus into the root apoplastic space has occurred.
Table 1.
Summary of the putative fungal miRNAs identified with the P. microcarpus 441 genome
| miRNA name | Locus (chromosome:start-end) | Strand | Predicted precursor length (nt) | Mature miRNA sequence (5′-3′) |
| Pmic_miR-1 | Pis_scaffold_12:478925–479041 | — | 117 | GUUGAGCCUGAUACAGUAGCCU |
| Pmic_miR-2 | Pis_scaffold_16:167685–167760 | + | 76 | UCACUUUCCGAGCUUGUAGUCCAACU |
| Pmic_miR-3 | Pis_scaffold_27:55536–55633 | — | 98 | CGGGUCUGAUGAUGCAGAGUCGGGC |
| Pmic_miR-4 | Pis_scaffold_5:542437–542560 | + | 124 | UAUGGGCUGGCCUAGGCAUGG |
| Pmic_miR-5 | Pis_scaffold_57:72308–72667 | + | 360 | UACCGAUCUGCCUGUGGGACA |
| Pmic_miR-6 | Pis_scaffold_134:66475–66733 | — | 259 | AACGGAUAGAUCAAUAUUAAGACC |
| Pmic_miR-7 | Pis_scaffold_106:26653–26727 | + | 75 | UCCAGCAAGGGGGAAUGGGCU |
| Pmic_miR-8 | Pis_scaffold_237:46412–46486 | — | 75 | UAUUCUCUCCUUGACUUCCC |
| Pmic_miR-9 | Pis_scaffold_502:13132–13222 | + | 91 | UGAACGUGCGUCGUCCAUGCCC |
| Pmic_miR-10 | Pis_scaffold_74:80881–80965 | . | 85 | UCAUCCUCGAACAAGACCUG |
| Pmic_miR-11 | Pis_scaffold_343:1277–1351 | + | 75 | CUUUAUCUCUCGAACGCUGCUGGA |
Fig. 1.

P. microcarpus miRNAs are differentially regulated during ECM symbiosis developments with E. grandis roots certain of which can be recovered and localized in the cells of E. grandis. Heatmaps showing the fold change in expression of the 11 predicted fungal miRNAs in P. microcarpus isolate SI14 (A) and isolate SI9 (B) across a time course of colonization with E. grandis roots. The color scale represents the normalized Log2 fold change of each miRNA at each timepoint of colonization compared to axenically grown fungal cultures (n = 3 to 4). (C) Diagram of Petri dish setup for presymbiotic exposure of the two organisms. The fungus is grown on top of a cellophane membrane and then transferred to rest over top of E. grandis roots. (D) Scanning electron micrograph of a cross-section of the cellophane membrane utilized, showing microscopic pores running throughout the material. (Scale bar, 40 μm.) (E) Bar graph depicting normalized read counts of four putative fungal miRNAs detected in E. grandis roots after presymbiotic interaction with P. microcarpus (presymbiosis; gray bars) but below detection limit in sRNA-seq libraries of E. grandis axenically grown roots where no fungus is present (axenic; white bars; see also SI Appendix, Fig. S3 for verification of results by qPCR). (F–O) ISH localization of Pmic_miR-8 in E. grandis root cells and controls using either a fluorescent anti-Pmic_miR-8 or scrambled sequence probe (blue signal in images) whereby the nuclei of root cells were visualized with propidium iodide (magenta signal in images). The specific treatments are as follows: probe-free ISH of presymbiotic roots (F); ISH of Pmic_miR-8 in E. grandis cells grown axenically (G); ISH of presymbiotic roots using a scrambled probe (H); ISH of presymbiotic roots using a Pmic_miR-8 probe with a positive signal for the target found in punctate structures within the cell (arrows) as well as more diffusely throughout the cell (I); ISH of presymbiotic roots treated with RNase prior to fixation to remove any RNA adherent to the cell surface and then probed with a scrambled probe (J); ISH of presymbiotic roots treated with RNase prior to fixation using a Pmic_miR-8 probe with a positive signal for the target found in punctate structures within the cell (arrows) (K); ISH with a scrambled probe of presymbiotic roots treated with RNase prior to, and post, fixation to remove any RNA adherent to the cell surface and within the cell postpermeabilization (L); ISH of presymbiotic roots treated with RNase prior to, and post, fixation to remove any RNA adherent to the cell surface and within the cell postpermeabilization and then probed with a Pmic_miR-8 probe (M); ISH of presymbiotic roots treated to induce plasmolysis using a scrambled probe (N); and ISH of presymbiotic roots treated to induce plasmolysis using a Pmic_miR-8 probe with a positive signal for the target found in larger concentrations within the cell (arrow head) (O). All images were acquired using an inverted Leica TCS SP5 laser-scanning confocal microscope with the same settings. (Scale bar, 20 µm for F–O.) All images are representative of four biological replicates.
Pisolithus Fungal miRNAs Can Be Recovered in E. grandis Root Cells during Presymbiosis, prior to Physical Contact.
As certain sRNAs have previously been demonstrated to be secreted from fungal hyphae and taken up into plant cells (21), we similarly tested if we could recover P. microcarpus miRNAs in the root cells of E. grandis. Using a presymbiotic interaction experimental setup that physically separates the host roots and fungal mycelial tissues but contains pores that allow for the transfer of signaling molecules (Fig. 1C and SI Appendix, Fig. S2A), we found that ∼20% of the aligned reads sequenced from the root tissue (679,733 reads in total; Dataset S1) were encoded by the P. microcarpus genome instead of E. grandis. It is unlikely that these sequences are due to fungal contamination as the membrane used to separate the two organisms was not ruptured by the fungus (Fig. 1D and SI Appendix, Fig. S2 B and C). This indicated the possibility that a significant amount of P. microcarpus sRNAs are secreted and can be transferred into the cells of a host during root colonization. The majority of these fungal sequences (643,534 reads in total) represent 4,370 sRNA-forming loci in P. microcarpus genome (Dataset S2), accounting for 94.7% of all fungal reads. According to the ShortStack sRNA analysis result, most of these sRNA-forming loci (3,993 clusters) produce sRNA between 20 and 24 nucleotides in length and potentially belong to sRNA classes other than miRNAs [Dataset S3; (29)]. Within these sequence clusters, we also found four sequences corresponding to the aforementioned, putative, fungal miRNAs: Pmic_miR-2, Pmic_miR-3, Pmic_miR-8, and Pmic_miR-9 (Fig. 1E). The presence of these transcripts was independently verified using qPCR analysis of RNA extracted from presymbiotic roots (SI Appendix, Fig. S3). Based on its high accumulation in plant tissues, we sought to localize Pmic_miR-8 in plant tissues using fluorescence in situ hybridization (ISH) assays. No immunolocalization signal was detected in presymbiotic E. grandis root cells when a no-probe control was performed (Fig. 1F) nor was there any signal identified in axenically grown E. grandis roots when an anti–Pmic-miR-8 probe was used (Fig. 1G). Pmic-miR-8 was localized, however, as both a faint haze and as punctate structures in the cells of E. grandis roots after presymbiotic contact with the fungus (Fig. 1I). In contrast, no fluorescent signals were detectable in similarly treated roots when a scrambled probe was used (Fig. 1H). To ensure that the immunolocalization signal was not due to accumulation of Pmic-miR-8 on the outside of the roots, E. grandis roots in presymbiotic contact with P. microcarpus were treated with ribonuclease (RNase) for 15 min prior to fixation and immunolocalization. These roots also exhibited the same Pmic-miR-8 localization (Fig. 1K). This signal disappeared, however, when the tissues were treated with RNase postfixation and cell permeabilization (Fig. 1M). To further confirm that Pmic_miR-8 was in the cytoplasm, we plasmolysed another subset of the same roots prior to fixation and immunolocalization and found Pmic-miR-8 localization in larger, diffuse areas within the cell, supporting the finding that Pmic-miR-8 is within the cytoplasm of the plant cell (Fig. 1O). Overall, the detection of fungal sRNA in E. grandis root cells demonstrates the potential existence of sRNA trafficking from the fungus to the host in ECM interactions.
Fungal miRNA Pmic_miR-8 Stabilizes ECM Colonization.
The expression pattern of Pmic_miR-8 during colonization prompted further characterization of the effect this fungal miRNA has on ECM fungi–root interaction. Four synthetic sRNAs were generated and used to artificially modulate the levels of Pmic_miR-8 in vitro in two ways: 1) double-stranded (ds) and single-stranded, mature Pmic_miR-8 RNA was used to increase the overall level of this miRNA in tissues during colonization and 2) antisense (as) Pmic_miR-8 with a Zen-modification (asZEN) and asPmic_miR-8 designed with a bulge (asBulge) at bases 10 and 11 were used to “inhibit” the action of the mature Pmic_miR-8 miRNA molecules through locking the mature miRNA in a ds form unable to be loaded into the ARGONAUT complex. As controls to these treatments, we used scrambled miRNA (ssScrambled) and a scrambled miRNA inhibitor (asScrambled) sequence as well as ssPmic_miR-8*, which is the complementary sequence to the mature miRNA and not expected to be active. The application of ss and dsPmic_miR-8 led to increases in detectable Pmic_miR-8 transcripts in colonized root tissues (P < 0.05; Fig. 2A and SI Appendix, Fig. S4A), while Pmic_miR-8 inhibitor treatments led to a trend toward reduced recovery of Pmic_miR-8 sequences (Fig. 2A and SI Appendix, Fig. S4A). All control treatments, as well as supplementation of either ssPmic_miR-8 or dsPmic_miR-8, led to the normal formation of mycorrhizal root tips (Fig. 2B). Treatment with either the asZen or asBulge inhibitors, however, resulted in regrowth of the root after the initial formation of a mantle in >20% of roots (Fig. 2C, white arrows). We considered these latter structures to be senesced mycorrhizal root tips. Statistical analysis of mycorrhizal root tip formation, and of colonized root senescence, found that treatment with either Pmic_miR-8 inhibitors led to a significant reduction in colonized root tips (Fig. 2D; P < 0.05). This reduction in colonized roots was almost completely due to the increase in roots that continued to grow after the initial formation of a fungal mantle.
Fig. 2.

Supplementation and inhibition of P. microcarpus miR-8 alters colonization of E. grandis roots. (A) Relative Pmic_miR-8 levels in E. grandis mycorrhizal root tips treated with either mature, single-stranded Pmic_miR-8 (white bar), mature as Pmic_miR-8* (striped bar), or ds Pmic_miR-8 (light gray bar), as compared to treatment with a scrambled miRNA. We also tested E. grandis mycorrhizal root tips treated with either a single-stranded ZEN-tagged as Pmic_miR-8 inhibitor (i.e., repression; black bar) or a single-stranded as Pmic_miR-8 with designed bulge mismatch at nucleotides 10 to 11 (dark gray bar), as compared to a scrambled inhibitor sequence. All values are the result of three biological replicates, ±SE; * indicates significant difference from scrambled control (P < 0.05; Student’s t test). (B) Representative image of P. microcarpus mycorrhizal root tips on E. grandis showing a typical mycorrhizal phenotype. (C) Representative image of P. microcarpus mycorrhizal root tips on E. grandis treated with the Pmic_miR-8 inhibitor in the final week of fungal colonization. White arrows indicate mycorrhizal root tips that have senesced, and the root has recommenced growth. (D) Percent of E. grandis root tips in contact with P. microcarpus that are either colonized by the fungus (black bars) or that exhibit the beginning of colonization followed by senescence (light gray bars). Values are shown for either no spray treatment (No Treatment) or spray treated with scrambled miRNA (ssScrambled); as single-stranded, mature Pmic_miR-8 (ssmiR-8*); sense ds, mature Pmic_miR-8 (dsmiR-8); sense single-stranded, mature Pmic_miR-8 (ssmiR-8); scrambled as inhibitor (asScrambled); single-stranded, ZEN-tagged as Pmic_miR-8 inhibitor (asZEN); and single-stranded as Pmic_miR-8 with designed bulge mismatch at nucleotides 10 to 11 (asBulge). All values are based on six biological replicates, ±SE; */▵ indicates significant difference from scrambled control (P < 0.05; Student’s t test).
We next performed a microscopic analysis of the ingrowth of P. microcarpus into the root apoplastic space treated with the different synthetic sRNAs (i.e., the formation of the “Hartig net” across whose surface area contact with the plant cells nutrients are exchanged; Fig. 3B). We focused on sections of roots that exhibited a mantle and found that no treatment consistently halted the ingrowth of P. microcarpus into the root apoplastic space (Fig. 3 B–H). However, significant treatment effects were observed on the degree of fungal penetration. Supplementation of Pmic_miR-8 resulted in a significant increase in Hartig net depth when compared to control-treated tissues (Fig. 3I; P < 0.05). Conversely, inhibition of Pmic_miR-8 resulted in significantly less well-developed Hartig nets in the roots analyzed (Fig. 3I; P < 0.05).
Fig. 3.

Supplementation and disruption of Pmic_miR-8 alters growth of the fungus into the apoplastic space of E. grandis roots and expression of host genes. (A) Representative transverse cross-section of an axenically grown E. grandis lateral root. P. microcarpus mycorrhizal root tip on E. grandis when sprayed for 1 wk with the following: scrambled control RNA with the Hartig net (HN) penetration marked (B); as single-stranded, mature Pmic_miR-8* (C); sense ds, mature Pmic_miR-8 (D); sense single-stranded, mature Pmic_miR-8 (E); scrambled, ZEN-tagged miRNA inhibitor (F); single-stranded, ZEN-tagged as Pmic_miR-8 inhibitor (G); and single-stranded as Pmic_miR-8 with designed bulge mismatch at nucleotides 10 to 11 (H). All images are representative of six biological replicates. (Scale bar, 10 μm.) (I) Average measured HN depth of P. microcarpus in E. grandis roots with either no spray treatment (No Treatment) or miRNA sense scrambled (ssScrambled); as single-stranded, mature Pmic_miR-8* (ssmiR-8*); ds, mature Pmic_miR-8 (dsmiR-8); sense single-stranded, mature Pmic_miR-8 (ssmiR-8); scrambled as inhibitor (asScrambled); single-stranded, ZEN-tagged as Pmic_miR-8 inhibitor (asZEN); and single-stranded as Pmic_miR-8 with designed bulge mismatch at nucleotides 10 to 11 (asBulge). n = 6, ±SE; * indicates significant difference from scrambled control (P < 0.05; Student’s t test). (J) qPCR of relative expression of the top three putative Pmic_miR-8–targeted gene transcripts (based on in silico prediction) in E. grandis mycorrhizal root tips either treated with sense ds, mature Pmic_miR-8 (light gray bars); sense single-stranded, mature Pmic_miR-8 (white bars); single-stranded, ZEN-tagged as Pmic_miR-8 inhibitor (black bars); and single-stranded as Pmic_miR-8 with designed bulge mismatch at nucleotides 10 to 11 (dark gray bars). n = 3 ± SE. (K) qPCR of relative expression of three putative Pmic_miR-8–targeted gene transcripts (based on homology to Eucgr.E03170) in E. grandis mycorrhizal root tips treated and colored as in J. n = 3 ± SE; * indicates significant difference from scrambled control (P < 0.05; Student’s t test).
Pmic_miR-8 Shows Complementarity to Host NB-ARC Domain-Containing Transcripts.
To elucidate the putative pathway(s) in the host plant targeted by Pmic_miR-8, further in silico analyses were performed on a complementary set of RNA-seq data that was generated using the same tissues as for sRNA-seq along the ECM colonization timeline in E. grandis. Using target prediction based on sequence complementarity and correlation analysis, we identified seven genes on the E. grandis genome as potential targets of Pmic_miR-8 (expectation value <3; Dataset S4) with three highly likely targets (Table 2). These latter targets are orthologous to genes related to DNA endonucleases, protein kinases, and an NB-ARC–containing protein in Arabidopsis. A similar analysis was run to identify putative targets within the fungus, but no sequence loci were identified with an appropriate expectation value (i.e., no hits <3; Dataset S5). Similar to Pmic_miR-8, Pmic_miR-3 had a putative E. grandis target transcript, while the other secreted miRNAs did not (i.e., Pmic_miR-2 and Pmic_miR-9; Dataset S4). Our analyses identified 19 potential targets endogenous to the fungal genome for these four miRNAs (Dataset S5). We also sought to identify the potential targets of the other Pisolithus miRNAs in both the host and fungal genomes (Datasets S4 and S5). For the nonsecreted miRNA Pmic_miR-1, Pmic-miR-6, Pmic-miR-7, Pmic-miR-10, and Pmic-miR-11, we identified very few endogenous target sequences (Dataset S5) while we were able to isolate several putative target sequences in the E. grandis genome (Dataset S4).
Table 2.
Summary of the three most likely host target genes of Pmic_miR-8 and their annotation of the orthologous gene in Arabidopsis
| Target gene ID | Arabidopsis ortholog | Expect value* | Alignment |
| Eucgr.K00246 | Single-stranded DNA endonuclease family protein | 1.5 |
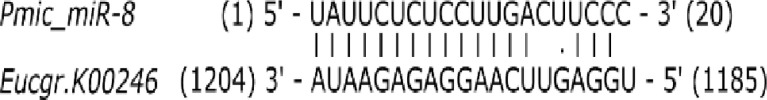
|
| Eucgr.L01882 | Single-stranded DNA endonuclease family protein | 1.5 |

|
| Eucgr.E03170 | NB-ARC domain-containing disease resistance protein | 1.5 |

|
The expectation values are an indicator of the similarity between the Pmic_miR-8 and the target candidate (50). A smaller value indicates a larger similarity and, therefore, a more probable target candidate. The Alignment column graphically presents the complementary base pairing between Pmic_miR-8 and the target candidate and is indicated by “|,” while U:G wobble pairing is indicated by “.”. All putative targets with an expectation value <3 can be found in SI Appendix, Table S4.
To identify the most likely target host gene regulated by Pmic_miR-8 from the Plant Small RNA Target Analysis Server (psRNATarget) predictions, we performed expression profiling of the three best scoring host target genes (Eucgr.K00246, Eucgr.L01882, and Eucgr.E03170; Table 2) in colonized roots treated with synthetic sRNA as outlined in the previous section. Typically, miRNAs function by reducing the expression of their target genes. Therefore, we expected that roots treated with synthetic Pmic_miR-8 would exhibit significant reductions in the target transcript with the opposing pattern in roots treated with the Pmic_miR-8 inhibitors. While this general pattern was observed in all three E. grandis genes tested, the pattern was strongest for Eucgr.E03170, with significant alterations in all treatments (Fig. 3J). This latter gene encodes conserved domains consistent with the CC nucleotide binding and leucine-rich repeat domain immune receptors (CC-NLR) class of proteins. These domains included an Rx N-terminal domain (PFAM 18,052; amino acids 3 to 91), an NB-ARC domain (amino acids 165 to 395), as well as a C-terminal LRR domain (amino acids 554 to 677). A BLASTn search found that there were other E. grandis genes encoding a sequence homologous to that found in Eucgr.E03170 that is likely to be targeted by Pmic_miR-8. qPCR analysis demonstrated that the expression of these genes was also influenced by the different treatments with Pmic_miR-8 inhibitors, with the most consistently significant impact on the expression of Eucgr.E03196 (Fig. 3K and SI Appendix, Fig. S4B). Therefore, it would appear that Pmic_miR-8 has the ability to target a number of genes within the largest class of NLRs in plants (30).
Discussion
There is strong support for the possibility of cross-kingdom RNA interference regulating host interactions with pathogenic (21–23) and mutualistic microbes (2, 8, 26, 27, 31, 32). However, we are only beginning to understand miRNAs and their roles in ECM symbioses. This is partly because of the incompatibility of ECM colonization with common model plant species, such as Arabidopsis and legumes. ECM fungi interact with the majority of woody plant species in boreal and temperate forests and are key players in soil nutrient cycling in forests globally, where they scavenge/alter soil organic matter and provide vital nutrients to their hosts (13, 33). Studying the role of miRNAs during the development of mycorrhizal roots will deepen our understanding concerning the regulatory pathways vital for this quintessential interaction. Our study provides evidence supporting the significant involvement of fungal miRNA pathways in ECM colonization of the model host plant E. grandis. Specifically, like pathogenic fungi, we provide evidence that ECM miRNAs can transfer from the fungus into host cells and that they act in a manner that stabilizes the colonization of the fungus within root tissues.
While fungal genomes encode a large number of sRNAs, the proportion of these classified as miRNAs are typically in the minority. In the two mutualistic AM genomes analyzed to date, of 702 to 3,422 predicted sRNA loci located in intragenic spaces, 10 miRNAs were annotated within the R. irregularis genome and one miRNA was annotated to the Gigaspora margarita genome (8, 27). Our discovery of 11 miRNAs within the genome of P. microcarpus, therefore, is a comparable number of sequences. Despite only a small number having been functionally characterized, a growing body of research supports the concept of s/miRNA trafficking between fungi and their host during symbiosis. sRNA secreted by fungi directly modulate host physiology to support the colonization process (21–23). While these previous studies have been in pathogenic fungi, our current work has now demonstrated that cross-kingdom sRNA trafficking occurs between ECM fungi and their plant hosts. sRNA-seq analyses identified four fungal miRNAs along with thousands other fungal, siRNA-like RNAs in E. grandis roots after presymbiotic interaction. The expression pattern of one of these miRNAs, Pmic_miR-8, during the later stages of interaction was similar to that of Botrytis cinerea small RNAs (Bc-sRNAs), which transfer from pathogenic B. cinerea to Arabidopsis (21). This implies that the expression of Pmic-miR-8 is inducible by the infection event and that it potentially plays a role in the later stage of mycorrhizal fungus–root interaction. Aside from in silico analyses, we conducted ISH assays that further supported the conclusion that Pmic_miR-8 was not plant derived but exported from P. microcarpus into E. grandis root cells. ISH assays have previously been used for demonstrating the cross-kingdom transfer of miRNA from plants to human tissue (34). Interestingly, the localization of the miRNA to both the cytoplasm and punctate structures closely mirrors the localization of ARGONAUT proteins (35, 36). Although the pathway is unknown, Pmic_miR-8 molecules may potentially be transferred through secretion of RNA-containing extracellular vesicles, as has been demonstrated in Cryptococcus neoformans, Paracoccidiodes brasiliensis, and Candida albicans (37).
Typically, exported microbial miRNAs are predicted to suppress host immunity and thus facilitate infection in pathogenic interactions (8, 21–23, 26, 27, 32). This promotional effect on infection was experimentally validated through overexpression of Bc-sRNAs derived from B. cinerea and in root colonization of rhizobia when using transfer RNA (tRNA)-derived sRNA fragments (21, 26). Similar to these previous studies, our results indicate that Pmic_miR-8 stabilizes root colonization during the late stages of colonization, as inhibition of the miRNA led to the host root escaping the developing, fungal mantle to continue growing. While the full mechanism by which Pmic_miR-8 achieves this is unknown, one potential pathway is through the modulation of transcript abundance of CC-NLR disease resistance proteins, which are most closely related to R proteins RGA4 and RGA2. These proteins are known to be key regulators of plant immunity, as they detect the presence of an invasive fungus through the perception of fungal avirulence proteins. Activation of RGA4- and RGA2-containing proteins during pathogenic interactions leads to plant resistance against bacterial (38), oomycete (39), and fungal pathogens (40). Therefore, the potential role of Pmic_miR-8 in the repression of a number of CC-NLR transcripts may be one of the key mechanisms by which P. microcarpus colonizes its host. The finding that disease resistance transcripts may be targeted by an ECM fungal miRNA, though, is different from the predicted plant targets of AM fungal miRNAs (8). In this latter study, the gene ontology of host proteins predicted to be affected by miRNAs included serine-type hydrolases and peptidases, as well as genes associated with the host secretory mechanism. Interestingly, we could not identify potential targets of two other secretory miRNAs in E. grandis genome (i.e., Pmic_miR-2 and Pmic_miR-9). As Pisolithus species have a broad host range, including Eucalyptus, Cistus, Quercus, Acacia, and Pinus (41, 42), these secreted miRNAs may have regulatory targets in other plant host species. On the other hand, several putative targets were identified in the E. grandis genome for fungal miRNAs undetected in E. grandis roots in our study condition (e.g., Pmic_miR-6). This may indicate that these miRNAs have regulatory roles in supporting symbiosis under different conditions (e.g., abiotic stress). Further work will be needed to determine if this is the case. Taken together, our results suggest that we have only begun to uncover the breadth of plant-based pathways that are modulated by cross-kingdom fungal miRNA signaling.
The interaction between ECM fungi and plants requires a complex mix of signaling compounds, including metabolites (43–45) and protein effectors (16, 17, 19, 20). We now add to this body of knowledge by demonstrating that ECM fungi can also modulate host function through the transfer of sRNA. In the future, it will be critical to broaden these findings to compare the miRNAs of P. microcarpus with other species/evolutionary lineages of ECM fungi to identify the key miRNAs essential for ECM symbiosis.
Materials and Methods
Detailed methods can be found in SI Appendix. In brief, following the method previously described in Wong et al. (45), E. grandis seeds were sterilized, germinated, and placed into symbiotic interaction with four isolates of the model ECM fungal species P. microcarpus [isolates SI9, SI14, R4, and R10; (46)] to identify miRNAs. A time course of colonization between P. microcarpus isolates SI14 and SI9 and E. grandis was used to profile the expression of identified miRNAs over the course of colonization (i.e., 24 h, 48 h, and 2 wk). In addition, a presymbiotic interaction setup was also prepared according to Wong et al. (45). RNA was extracted from six different conditions (E. grandis axenic control, presymbiosis, 24 h, 48 h, 2 wk, and free-living P. microcarpus mycelium) using the ISOLATE II miRNA kit (Bioline). Three to four biological replicates were extracted per condition. The sRNA fractions were sent for sRNA-seq at the Joint Genome Institute (JGI). In total, 42 sRNA libraries were generated and analyzed (Dataset S6).
Using the reference genome of E. grandis (47) and P. microcarpus 441 (15), ShortStack was run for each sequencing library in order to identify sRNA-producing loci (or “Clusters”) and to identify putative miRNA genes de novo (28, 29). The criteria for sRNA cluster and miRNA gene classification are further detailed in SI Appendix. Loci classified as miRNA in the single libraries were merged across libraries (again specifying the “–nohp” option in ShortStack). Along with these miRNA loci clusters, sequences of known miRNAs, previously identified in E. grandis roots by Lin et al. (48), were identified and removed. Differential expression analysis was done using DESeq [version 1.30.1; (49)]. Heatmaps were generated with the fold change values of differential-expressed miRNAs [│log2(fold change)│> 1; adjusted P value < 0.05] using Heatmapper (50).
E. grandis seedlings were set up into a presymbiotic interaction or left to grow axenically without fungus for 1 wk before being used for ISH. As detailed in SI Appendix, root tips were then divided into several treatments, including control, which were fixed immediately upon harvest; RNase treated for 15 min followed by fixation to remove external bound RNA; RNase treated for 15 min followed by fixation and then a secondary treatment with RNase postfixation and permeabilization to remove external and internal bound RNA; and plasmolysed with 1.5 M sucrose to concentrate internal miRNA and then fixed. Following fixation, samples were treated using 20 µg/mL proteinase K and 0.5% Triton-X in TE buffer for 60 min at 37 °C to digest cellular RNases and protein excess and to permeabilize the cells for probe entry. Samples were incubation in 0.2% glycine at room temperature for 5 min to stop protease activity and probed with LNA probe with a fluorophore modification with anti–Pmic_miR-8 or a scrambled probe as a control (35 µM, sequences detailed in SI Appendix, Table S1). Following incubation, the samples were placed through a series of high-stringency washes to remove unbound probes, followed by staining in 0.1% propidium iodide to stain nuclei. Samples were observed using an inverted Leica SP6 confocal microscope.
To test for the impact of varying Pmic_miR-8 levels on colonization, E. grandis seedlings were directly inoculated with SI14 using the setup mentioned previously, and after 7 d, root systems were sprayed with nebulized, synthetic sRNA of different sequences daily for a further 7 d (100 µL 20-nM sRNA solution per plant using MAD Nasal Intranasal Mucosal Atomization Device; Teleflex; sequences found in SI Appendix, Table S1). On the 14th day of colonization (seventh day of sRNA treatment), ECM colonization rate was scored, as were the number of senesced mycorrhizal root tips. Hartig net formation was analyzed as previously described (46). Samples were also collected, and RNA transcript quantitation by qPCR (Primers found in Dataset S7) was used to verify that Pmic_miR-8 was differentially regulated as expected using a stem loop qRT-PCR approach, as described previously (51).
The sequence of Pmic_miR-8 has been searched against the nucleotide databases for E. grandis on National Center for Biotechnology Information (NCBI) using megaBLAST algorithm to confirm that Pmic_miR-8 is not of plant origin. No matching loci with a high-confidence score to Pmic_miR-8 were identified in the E. grandis genome but were identified in the P. microcarpus genome (Dataset S2). Therefore, we can be confident that this sequence is of fungal origin. Using psRNATarget [version 2.0; (52)], target sequences of Pmic_miR-8 and other miRNAs were identified within the E. grandis transcript library (Phytozome 11, version 297 v2.0) and the P. microcarpus 441 transcript library. Transcripts with an “expectation value” <3 are considered as putative target genes. Detailed methodology for this process can be found in SI Appendix. To determine which of the putative host gene(s) may be influenced at the transcriptional level by Pmic_miR-8 treatment, we took the total RNA extracted from ss/dsPmic_miR-8 or Pmic_miR-8 inhibitor-treated tissues as described in the previous paragraph, as well as the scrambled controls, and generated cDNA using the Tetro cDNA synthesis kit (Bioline), as per manufacturer’s instructions and using only the oligo-dT primer. Using the SensiFAST SYBR no-ROX qPCR kit (Bioline) following manufacturers’ instructions, we then analyzed the expression patterns of the top three putative target genes of Pmic_miR-8 based on in silico analysis (Eucgr.K00246, Eucgr.L01882, and Eucgr.E03170) or three candidates based on homology to Eucgr.E03170 (Eucgr.E03194, Eucgr.E03196, and Eucgr.E03203). Relative expression was determined using the 2-ΔΔCT method, whereby tissues treated with the scrambled sRNA (either scrambled sense or as miRNA) were used as the control tissues for tissues treated with ss/dsPmic_miR-8 or Pmic_miR-8 inhibitors, respectively.
Supplementary Material
Acknowledgments
J.W.- B. would like to thank Western Sydney University for a PhD research scholarship and the Hawkesbury Foundation for the F.G. Swain Award that supported this research study. J.M.P. would like to acknowledge the Australian Research Council for research funding (DE150100408). This research was also supported by the Laboratory of Excellence ARBRE (ANR-11-LABX-0002-01) to F.M.P. The sequencing work conducted by the US Department of Energy (DOE) JGI, a DOE Office of Science User Facility, is supported by the Office of Science of the US DOE under Contract No. DE-AC02-05CH11231. We wish to acknowledge the aid of R. Wuhrer and T. Richardson of the Western Sydney University Advanced Materials Characterisation Facility for aid in imaging and the Western Sydney Confocal Facility for access to equipment. We would also like to acknowledge the New South Wales National Parks and Wildlife Services for approving the collection of the Pisolithus fruiting bodies (Scientific License No. S13146).
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2103527119/-/DCSupplemental.
Data Availability
Sequencing data have been deposited in Mycocosm (https://mycocosm.jgi.doe.gov/mycocosm/home) and NCBI (https://www.ncbi.nlm.nih.gov/) (accession nos. are provided in Dataset S6).
References
- 1.Ku Y.-S., et al. , Small RNAs in plant responses to abiotic stresses: Regulatory roles and study methods. Int. J. Mol. Sci. 16, 24532–24554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couzigou J.-M., et al. , Positive gene regulation by a natural protective miRNA enables arbuscular mycorrhizal symbiosis. Cell Host Microbe 21, 106–112 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Reinhart B. J., Weinstein E. G., Rhoades M. W., Bartel B., Bartel D. P., MicroRNAs in plants. Genes Dev. 16, 1616–1626 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurihara Y., Watanabe Y., Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaucheret H., Vazquez F., Crété P., Bartel D. P., The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulci V., Macino G., Quelling: Post-transcriptional gene silencing guided by small RNAs in Neurospora crassa. Curr. Opin. Microbiol. 10, 199–203 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Raman V., et al. , Physiological stressors and invasive plant infections alter the small RNA transcriptome of the rice blast fungus, Magnaporthe oryzae. BMC Genomics 14, 326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silvestri A., et al. , In silico analysis of fungal small RNA accumulation reveals putative plant mRNA targets in the symbiosis between an arbuscular mycorrhizal fungus and its host plant. BMC Genomics 20, 169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemppainen M., Duplessis S., Martin F., Pardo A. G., RNA silencing in the model mycorrhizal fungus Laccaria bicolor: Gene knock-down of nitrate reductase results in inhibition of symbiosis with Populus. Environ. Microbiol. 11, 1878–1896 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Martin F., et al. , Périgord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature 464, 1033–1038 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Montanini B., et al. , Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 15, 411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raman V., et al. , Small RNA functions are required for growth and development of Magnaporthe oryzae. Mol. Plant Microbe Interact. 30, 517–530 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Tedersoo L., et al. , Fungal biogeography. Global diversity and geography of soil fungi. Science 346, 1256688 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Miyauchi S., et al. , Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 11, 5125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohler A., et al. , Mycorrhizal Genomics Initiative Consortium, Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 47, 410–415 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Pellegrin C., et al. , Laccaria bicolor MiSSP8 is a small-secreted protein decisive for the establishment of the ectomycorrhizal symbiosis. Environ. Microbiol. 21, 3765–3779 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Kang H., et al. , The small secreted effector protein MiSSP7.6 of Laccaria bicolor is required for the establishment of ectomycorrhizal symbiosis. Environ. Microbiol. 22, 1435–1446 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Plett J. M., et al. , A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr. Biol. 21, 1197–1203 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Plett J. M., et al. , Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc. Natl. Acad. Sci. U.S.A. 111, 8299–8304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plett J. M., et al. , Mycorrhizal effector PaMiSSP10b alters polyamine biosynthesis in Eucalyptus root cells and promotes root colonization. New Phytol. 228, 712–727 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Weiberg A., et al. , Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derbyshire M., Mbengue M., Barascud M., Navaud O., Raffaele S., Small RNAs from the plant pathogenic fungus Sclerotinia sclerotiorum highlight host candidate genes associated with quantitative disease resistance. Mol. Plant Pathol. 20, 1279–1297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusch S., Frantzeskakis L., Thieron H., Panstruga R., Small RNAs from cereal powdery mildew pathogens may target host plant genes. Fungal Biol. 122, 1050–1063 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Song Y., Thomma B. P. H. J., Host-induced gene silencing compromises Verticillium wilt in tomato and Arabidopsis. Mol. Plant Pathol. 19, 77–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T., et al. , Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2, 16153 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Ren B., Wang X., Duan J., Ma J., Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science 365, 919–922 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Silvestri A., et al. , Different genetic sources contribute to the small RNA population in the arbuscular mycorrhizal fungus Gigaspora margarita. Front. Microbiol. 11, 395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axtell M. J., ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 19, 740–751 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson N. R., Yeoh J. M., Coruh C., Axtell M. J., Improved placement of multi-mapping small RNAs. G3 (Bethesda) 6, 2103–2111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kourelis J., Sakai T., Adachi H., Kamoun S., RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biol. 19, e3001124 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lauressergues D., et al. , The microRNA miR171h modulates arbuscular mycorrhizal colonization of Medicago truncatula by targeting NSP2. Plant J. 72, 512–522 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Lee S.-J., Kong M., Harrison P., Hijri M., Conserved proteins of the RNA interference system in the arbuscular mycorrhizal fungus Rhizoglomus irregulare provide new insight into the evolutionary history of Glomeromycota. Genome Biol. Evol. 10, 328–343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landeweert R., Hoffland E., Finlay R. D., Kuyper T. W., van Breemen N., Linking plants to rocks: Ectomycorrhizal fungi mobilize nutrients from minerals. Trends Ecol. Evol. 16, 248–254 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Chin A. R., et al. , Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 26, 217–228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung A. K. L., Calabrese J. M., Sharp P. A., Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc. Natl. Acad. Sci. U.S.A. 103, 18125–18130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. B.Derrien et al., Degradation of the antiviral component ARGONAUTE1 by the autophagy pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 15942–15946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peres da Silva R., et al. , Extracellular vesicle-mediated export of fungal RNA. Sci. Rep. 5, 7763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Césari S., et al. , The NB-LRR proteins RGA4 and RGA5 interact functionally and physically to confer disease resistance. EMBO J. 33, 1941–1959 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y.-L., et al. , Characteristic of the pepper CaRGA2 gene in defense responses against Phytophthora capsici Leonian. Int. J. Mol. Sci. 14, 8985–9004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Césari S., et al. , The rice resistance protein pair RGA4/RGA5 recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell 25, 1463–1481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin F., Díez J., Dell B., Delaruelle C., Phylogeography of the ectomycorrhizal Pisolithus species as inferred from nuclear ribosomal DNA ITS sequences. New Phytol. 153, 345–357 (2002). [Google Scholar]
- 42.Hosaka K., Phylogeography of the genus Pisolithus revisited with some additional taxa from New Caledonia and Japan. Bull. Natl. Mus. Nat. Sci. Ser. B Bot., 35, 151–167 (2009). [Google Scholar]
- 43.Ditengou F. A., Lapeyrie F., Hypaphorine from the ectomycorrhizal fungus Pisolithus tinctorius counteracts activities of indole-3-acetic acid and ethylene but not synthetic auxins in eucalypt seedlings. Mol. Plant Microbe Interact. 13, 151–158 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Lagrange H., Jay-Allgmand C., Lapeyrie F., Rutin, the phenolglycoside from eucalyptus root exudates, stimulates Pisolithus hyphal growth at picomolar concentrations. New Phytol. 149, 349–355 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Wong J. W.-H., et al. , The influence of contrasting microbial lifestyles on the pre-symbiotic metabolite responses of Eucalyptus grandis roots. Front. Ecol. Evol. 7, 10 (2019). [Google Scholar]
- 46.Plett K. L., et al. , Intra-species genetic variability drives carbon metabolism and symbiotic host interactions in the ectomycorrhizal fungus Pisolithus microcarpus. Environ. Microbiol. 23, 2004–2020 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Myburg A. A., et al. , The genome of Eucalyptus grandis. Nature 510, 356–362 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Lin Z., et al. , Identification of novel miRNAs and their target genes in Eucalyptus grandis. Tree Genet. Genomes 14, 60 (2018). [Google Scholar]
- 49.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babicki S., et al. , Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varkonyi-Gasic E., Wu R., Wood M., Walton E. F., Hellens R. P., Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3, 12 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai X., Zhao P. X., psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited in Mycocosm (https://mycocosm.jgi.doe.gov/mycocosm/home) and NCBI (https://www.ncbi.nlm.nih.gov/) (accession nos. are provided in Dataset S6).