

ABSTRACT
The last decade has seen mRNA modification emerge as a new layer of gene expression regulation. The Fat mass and obesity-associated protein (FTO) was the first identified eraser of N6-methyladenosine (m6A) adducts, the most widespread modification in eukaryotic messenger RNA. This discovery, of a reversible and dynamic RNA modification, aided by recent technological advances in RNA mass spectrometry and sequencing has led to the birth of the field of epitranscriptomics. FTO crystallized much of the attention of epitranscriptomics researchers and resulted in the publication of numerous, yet contradictory, studies describing the regulatory role of FTO in gene expression and central biological processes. These incongruities may be explained by a wide spectrum of FTO substrates and RNA sequence preferences: FTO binds multiple RNA species (mRNA, snRNA and tRNA) and can demethylate internal m6A in mRNA and snRNA, N6,2′-O-dimethyladenosine (m6Am) adjacent to the mRNA cap, and N1-methyladenosine (m1A) in tRNA. Here, we review current knowledge related to FTO function in healthy and cancer cells. In particular, we emphasize the divergent role(s) attributed to FTO in different tissues and subcellular and molecular contexts.
KEYWORDS: FTO, N6-methyladenosine, RNA modification, cancer, epitranscriptomics, transcription
INTRODUCTION
The discovery of the Fat mass and obesity-associated protein (FTO) originated from a fused-toe (Ft) mouse mutation that lacks several hundred kb off chromosome 8 [1]. The Fatso (FTO) gene was cloned and studied in mouse. Based on its expression throughout embryonic development and the Ft mouse phenotype, FTO was assumed to be linked to programmed cell death and craniofacial development. Almost a decade later, genome-wide association studies established a strong correlation between single-nucleotide polymorphisms of the FTO gene and human obesity [2–5]. It was then renamed the ‘Fat mass and Obesity-associated’ (FTO) gene. This connection was strengthened by a study on FTO KO mice in which the loss of Fto triggers postnatal growth retardation and decreased body mass [6]. Contrastingly, overexpression of Fto promotes food intake and results in increased body and fat mass [7]. However, the real function of Fto in obesity remains controversial to date. While the Fto single-nucleotide polymorphism does not affect Fto itself, it promotes the expression of the neighbouring IRX3 gene. An IRX3-dependent mechanism is supported by IRX3 knockout mice, which tend to be leaner, protected from diet-induced obesity and whose metabolism is disrupted (Smemo et al., Nature 2014). Genetic variants in the FTO gene are also associated with several pathologies such as metabolic disorders [8–11], neurological diseases [12] and cancers [13,14]. Altogether, these phenotypic observations have attracted interest from the research community and led to substantial investment in identifying the physiological substrate(s) of Fto as well as the downstream molecular mechanisms.
Fto was initially shown to catalyse oxidative demethylation of 3-methyl thymine on synthetic single-stranded DNA (ssDNA) and 3-methyl uracil of synthetic single-stranded RNA (ssRNA) in vitro [15]. Shortly afterwards, the N6-methyladenosine (m6A) mark, decorating messenger RNA (mRNA), was identified as the main physiological substrate of FTO [16]. Identified a few decades earlier, m6A was shown to regulate every step of mRNA processing: splicing, stability and translation [17]. The discovery of reversible modification in mRNA was a major breakthrough in the field of RNA modification as it gave birth to ‘epitranscriptomics’. Nevertheless, since its discovery, the purported functions of FTO in living cells have been repeatedly challenged.
So far, FTO has been shown to target five distinct methylation types, in various RNA species as well as in DNA. However, the individual FTO targets were identified in distinct experimental settings and/or cell models, lending towards the possibility of a context-dependent specificity of FTO towards its substrates. The aim of this review is to provide an overview of our current knowledge regarding FTO function and activity across cell lines and tissues. After a global presentation of known FTO targets, we discuss the molecular mechanisms that could be involved in fine-tuning substrate selection. Finally, we present evidence supporting the importance of tissue context for FTO function, especially in cancer.
1. Targets of FTO
FTO is a homolog of the AlkB Family of Fe(II)/α-Ketoglutarate (α-KG)-dependent Dioxygenases. This class of ubiquitous DNA repair enzymes removes alkyl adducts from nucleobases through oxidative dealkylation [18]. FTO has an amino-terminal AlkB-like domain and a carboxy-terminal domain that interact with each other, as well as an extra loop that distinguishes it from other AlkB members [19]. FTO was originally reported to catalyse the demethylation of various RNA and DNA substrates [15] (Table 1). Nevertheless, recent studies point towards m6A modification in mRNA as its most relevant biological substrate [16]. These discoveries stem from recent progress in detection techniques such as high-throughput sequencing and mass spectrometry [20].
Table 1.
Targets of FTO by RNA species and with associated evidence (experimental context)
| Modification | DNA/RNA species | Evidence |
|---|---|---|
| 3meU | ssRNA | In vitro |
| 3 meT | ssDNA | In vitro |
| m6A | pre-mRNA, snRNAs | In vitro; living cells |
| m1A | tRNA | In vitro; living cells |
| m6Am | mRNA; snRNAs | In vitro, living cells |
| m6A | rRNA | No evidence |
| Intronic m6A | Intron | Many Fto binding but no evidence yet |
1.1. FTO targets 3-methyl-thymine and 3-methyl-Uracil in vitro
The first two targets of FTO were identified more than a decade ago as the 3-meThymine (3-meT) and the 3-meUracil (3meU) [15]. The authors synthesized in vitro methylated single-stranded DNAs and RNAs and incubated them in the presence of purified mouse or human FTO proteins. High-performance liquid chromatography (HPLC) chromatograms of the modifications revealed reduced ssDNA 3-meT and ssRNA 3meU but stable ssDNA 3-meC nor 5mdc [21]. While FTO can demethylate 3-meT in ssDNA in vitro, it has negligible activity towards double-stranded DNA [15]. So far, FTO-mediated 3 meT demethylation in living cells has not been reported.
1.2. FTO targets N6-methyladenosine in various RNA species
More recently, m6A modification, which functions to shape RNA splicing, stability and translation, has been identified as the major substrate of FTO. Jia G. et al. used HPLC to evaluate the effect of FTO on m6A-modified synthetic oligonucleotides: ssRNA and ssDNA [16]. FTO treatment decreased m6A level in ssRNA but not in ssDNA. These observations were substantiated by in cellulo experiments performed in HeLa and 293 T cells, where silencing of FTO increased m6A level in mRNA while FTO overexpression resulted in the opposite effect [16]. In addition to mass spectrometry, methylated RNA immunoprecipitation and sequencing (m6A-seq) was developed to identify FTO targets at genome scale. In various system, FTO activity has been shown to regulate m6A level in hundreds of mRNAs [22–25]. In a cell type-dependent manner, FTO binds to some sequences containing a consensus motif RRACH or DRACH (R = G or A; D = A or G or T; H = A, C, or U), which was thus dubbed the m6A consensus motif [26]. However, m6A-seq require the usage of an antibody whose specificity can be questioned. m6A antibody cross-reacts with m6Am, a subtype of m6A modification that locates at the 5ʹ end of mRNA. Hence, m6Am modification at alternative transcription start sites could be mistaken for internal m6A modification (Boulias et al., Mol Cell 2019, 10.1016/j.molcel.2019.06.006). Despite the bias, other approaches have been used to confirm the effect of Fto on m6A methylation.
Transcriptome-wide analysis of FTO binding sites uncovered its remarkable ability to bind a wide range of m6A-containing RNAs, such as pre-mRNA, mature mRNA, transfer RNA (tRNA), as well as various other non-coding RNA (ncRNA), suggesting potent activity towards those species [16]. This initial observation was further refined by experiments combining FTO silencing with purification of RNA species followed by m6A quantification by mass spectrometry. This approach confirmed that FTO efficiently targets m6A in 16–28 nucleotides long RNAs [27] and U6 snRNAs [28]. Yet, quantification of global m6A in 18S and 28S ribosomal RNAs (rRNAs) was not impaired by FTO silencing, suggesting that rRNA is not a substrate of FTO [28].
There is no clear evidence that could establish a direct connection between FTO and m6A demethylation in the intronic regions. However, 75% of FTO binds to introns, whereas only 7% of m6A is found in the intronic regions of pre-mRNA at steady state [29]. This discrepancy suggests that FTO binding is not biased by the global high proportion of introns over exons. Furthermore, m6A is elevated in introns of nascent pre-mRNA compared to steady-state pre-mRNA [30]. Taken together, these observations suggest that the low proportion of m6A in introns could result from a co-transcriptional demethylation activity mediated by FTO.
To sum up, FTO displays an m6A demethylase activity towards mRNAs, U6 small RNAs, and probably micro-RNAs (miRNA) but not rRNA. Moreover, while FTO seems to preferentially target intronic regions, this activity remains to be confirmed.
1.3. FTO targets m6Am in mRNA and small RNAs
m6Am modification is a specific m6A methylation that occurs on the 2ʹ-O methylAdenosine (Am) residue adjacent to the 7-methylguanosine (m7G) mRNA cap (only in transcripts whose +1 nucleoside is an A). Due to its particular location, m6Am has an expected role in regulating mRNA stability and/or translation. A first study by Mauer et al. has shown that m6Am, and not m6A, is the preferred cellular substrate for FTO both in vitro and in vivo [31]. Interestingly, FTO-mediated m6Am demethylation occurs preferentially on m7G-capped RNAs rather than uncapped RNAs, implying that m7G is required for FTO targeting [31]. In cellulo studies have quantified global m6Am level by 2D-TLC (Thin Layer Chromatography) or mass spectrometry and confirmed in vitro experiments: m6Am was increased in FTO KO cell lines and decreased following FTO overexpression [28,31–33]. m6Am was also mapped transcriptome-wide using miCLIP technology. miCLIP is based on m6A immunoprecipitation and further sequencing of methylated RNA fragments, to map m6A at nucleotide resolution [34]. This method allows m6Am residues at the mRNA cap, to be distinguished from internal m6A sites, which are mostly in the 3ʹUTR region. As expected by the authors, FTO KO significantly increased the number of m7G-m6Am sites [31]. Consistently, CLIP-seq experiment revealed an enrichment of FTO binding at the 5ʹ end of mRNA [29], therefore establishing a direct connection between m6Am level and FTO activity.
Like mRNAs, small RNAs can have an m7G-m6Am cap structure. By applying miCLIP from FTO KO cell lines. Mauer J. et al. revealed enrichment of m6Am at the 5ʹ end of several small RNAs including U1, U2, U4 and U5 small RNAs [35]. FTO-mediated demethylation of m6Am on U1 and U2 small RNAs was consolidated by 2D TLC and mass spectrometry. Interestingly, U2 snRNAs possess internal m6Am in addition to cap-m6Am. Jiangbo Wei et al. applied mass spectrometry analysis of nuclease-treated RNA modifications w/o RNA decapping in order to discriminate m7G-m6Am from internal m6Am [28]. Using this approach, they discovered that FTO preferentially demethylates cap-m6Am in U1 snRNA, whereas it targets internal m6Am in U2 snRNAs.
1.4. FTO targets m1A in tRNA
In a recent study, the CLIP-seq analysis revealed that a small fraction of FTO binds to tRNA [28]. In vitro experiments were performed in order to identify the FTO substrate in this highly modified RNA species. FTO incubation with tRNA does not affect m1G, m7G, m5C, m2G or m2,2G methylation. However, it decreases m1A methylation, suggesting that FTO could catalyse m1A demethylation in tRNAs. The biological relevance of this observation was confirmed in both living cells and mouse brain: FTO overexpression reduces m1A level in tRNA from HEK293T cells [28], whereas FTO KO increases m1A level in tRNAs extracted from mouse brain [28].
In vitro assays underscore the importance of structural context: FTO fails to target m1A in linear transcripts, whereas it efficiently demethylates m1A when present in a loop structure [28]. Eukaryotic tRNA has three potential m1A modifications, at position A9, A22 and A58 [28]. However, only m1A58 is localized in the large stem loop of tRNA, suggesting that FTO may preferentially target m1A58 rather than m1A9 or m1A22. Nevertheless, primer extension experiments will be required to confirm the activity of FTO towards m1A58 in living cells. Remarkably, while m1A58 is ubiquitous in tRNAs, CLIP analysis revealed that FTO only associates with 10 tRNA species, in particular, to tRNAGlu(CUC) (45%) and tRNAHis(RUG) (32%), suggesting some sort of specificity [28].
1.5. Compartment-specific activity of FTO
Methylases and demethylases can target different RNA species and/or modifications according to their subcellular localization [36,37]. For example, the m1A methylase TRM61 targets tRNA in the nucleus [38,39], but it targets mRNA in the cytoplasm [40]. The eraser ALKBH1 also provides a striking example of compartment-specific activity; ALKBH1 removes m1A of tRNA in the cytoplasm [41] and targets m5C of tRNA in mitochondria [42]. Like many other RNA modifying enzymes, FTO has different substrate preferences, depending on the subcellular context (Fig. 1).
Figure 1.
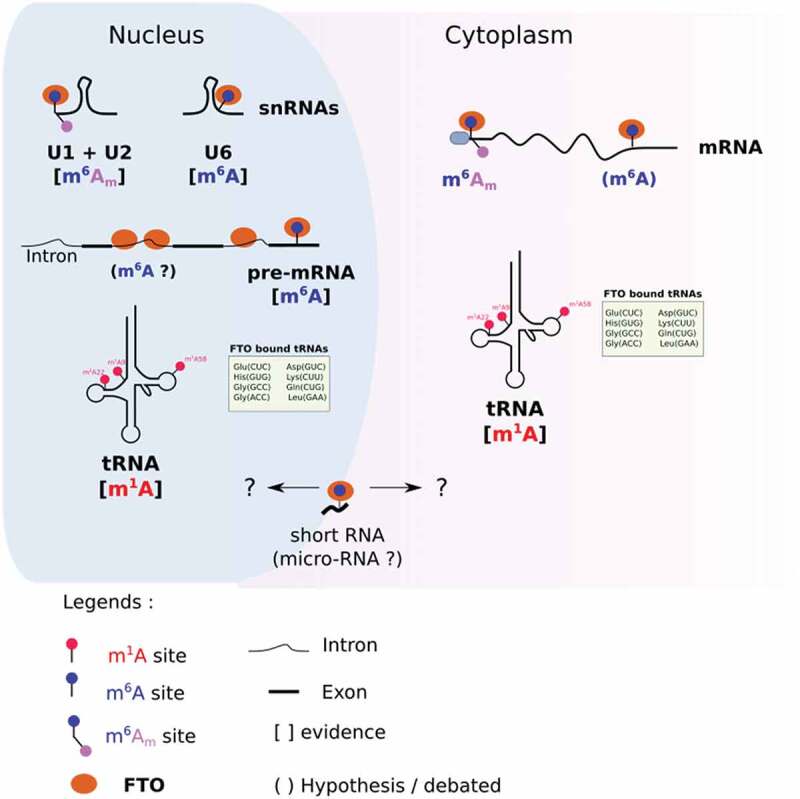
Compartment specific targets of FTO. In the nucleus, FTO demethylates m6Am in U1 and U2 snRNAs, m6A in U6 snRNAs and pre-mRNAs, and m1A in selected tRNAs. In the cytoplasm, FTO targets m6Am (and potentially m6A) on mRNA. It targets m1A in a subset of tRNAs. Compartment-specific m6A demethylation of short RNAs (20 nucleotides long) remains undocumented.
Various approaches have been employed to study how the activity of FTO depends on its localization. Mauer J. and colleagues fused FTO with a Nuclear Export Signal to promote its cytoplasmic translocation in HEK293T cells [31]. Then, they quantified both m6A and m6Am in mRNA by the means of two-dimensional TLC. Increased expression of cytoplasmic FTO significantly reduced m6Am/m6A, demonstrating that cytoplasmic FTO preferentially targets m6Am in mRNA. In another study, Jiangbo Wei et al. employed a biochemical procedure to isolate cell compartments, nucleus and cytoplasm, after silencing FTO in HEK293T cells [28]. From each compartment, they purified tRNA, small RNA and mRNA and analysed RNA modifications by mass spectrometry. They confirmed that FTO silencing increases cytoplasmic – but not nuclear – m6Am in mRNA. By contrast, only the nuclear fraction of mRNA had increased m6A after FTO depletion. Furthermore, m6A-seq experiments from nascent RNA, nuclear and cytoplasmic steady-state mRNA support the hypothesis of nuclear specific m6A demethylation43]. While m6A sites tend to be more numerous in nascent RNA than in steady-state nuclear RNA, the differences in m6A mapping become negligible when comparing nuclear versus cytoplasmic [43] mRNA. This observation strongly suggests that m6A demethylation occurs co-transcriptionally in the nucleus rather than in the cytoplasm.
Towards tissue-specific function?
Whether FTO targets preferentially m6A or m6Am has been debated for several years. In vitro demethylation assays demonstrated that FTO is a hundred times more efficient at demethylating m6Am than m6A [31]. However, a mere buffer solution cannot recapitulate the tremendous compositional complexity of the cellular environment. Furthermore, changes of molecular environment from one subcellular compartment to another could easily alter the enzyme’s behaviour44. In cellulo studies on FTO offer a striking illustration of this. First, FTO appears to have more impact on m6A than m6Am in living cells, especially in leukaemic cells45,46] . Second, FTO substrate preference may depend on subcellular context [28,36]; nuclear FTO would preferentially demethylate m6A, while cytoplasmic FTO would rather target m6Am. Therefore, conflicting results about FTO in vivo targets may result from the use of different cell models. For instance, several studies describing FTO activity were performed in HEK293T cells where FTO is strictly nuclear and does not affect m6Am 47. By contrast, in colon cancer cells, where FTO is both cytoplasmic and nuclear, FTO silencing significantly increases the level of m6Am [32]. Hence, regulation of FTO localization may be the key to regulating substrate preferences.
While more accurate than in vitro experiments, in cellulo studies do not necessarily reflect the complex reality of living tissue. In fact, FTO targets identified in a tissue context can differ from the ones identified from mere cell lines. For example, mapping of m6A at nucleotide resolution in FTO KO mouse brain revealed a strong enrichment of m6Am over internal m6A [48]. Moreover, the relative impact on each target might be different from the one established based on cell line data. In cultivated cells, the demethylase activity of FTO silencing on m1A level is marginal [28]. By contrast, FTO KO in mouse brain tissue has more impact on m1A than m6A or m6Am. While a compensatory mechanism regulating the m6A/m6Am dynamic cannot be ruled out, this prima facie evidence suggests that FTO preferentially targets m1A in mouse brain.
Discrepancies between cell lines across research reports raise the question of FTO tissue specificity.
Jun Liu and colleagues compared the expression of m6A/m6Am writers and erasers with the level of m6A and m6Am across 54 human tissues and 16 mouse tissues [49]. In both human and mouse, the m6A writers METTL3-METTL14 and the eraser ALKBH5 were correlated with m6A. As expected, the m6Am writer PCIF1 was correlated with m6Am. However, FTO correlated with neither m6A nor m6Am level. This observation supports the possibility that FTO has tissue-specific target preferences.
2. Determinants of multi-substrate specificity of FTO
2.1. Structural basis for FTO catalytic activity
Solving the structure of FTO was a crucial step towards a better understanding of its interaction with methylated RNA substrates. Several groups have tackled the challenging task of crystallizing FTO in a complex with nucleic acids [19,50. FTO is structurally composed of two main domains: the N-terminal domain from residues 32 to 326, and the C-terminal one from residues 327 to 498. Of note, the N-terminal 32 residues encode FTO’s nuclear localization signal that localizes FTO to the nucleus. However, many other signal sequences throughout the FTO reading frame also affect its localization, as will be touched on later.
Inside the N-terminal domain, several residues are important for substrate recognition. Two ‘pincers’ composed of residues K216, K88 and K316 allow FTO to interact with RNA50. Residue S229 could be involved in FTO specificity since mutation of this residue decreases FTO demethylation activity towards m6A but not towards m6Am [50]. The C-terminal domain of FTO does not contain essential residues for RNA interaction or FTO activity. Noteworthy, comparative studies have shown that FTO’s C-terminal domain is unique compared to the other ALKBH family members [44, 51. The versatile activity of FTO is likely due to this domain, through its ability to associate with molecular partners.
2.2. Regulation by protein partners
Through its C-terminal part, FTO interacts with protein partners that regulate its subcellular localization as well as its functions (Fig. 2). In living cells, the use of RNA probes containing four repeats of the m6A consensus sequence GGACU failed to recruit FTO, suggesting the involvement of a third party52. Recently, a protein partner of FTO that could promote m6A demethylation has been identified53]. SFPQ is a splicing factor that co-localizes with FTO in nuclear speckles [53]. Co-immunoprecipitation assays have demonstrated that SFPQ interacts with the C-terminal domain of FTO53. Transcriptome-wide analysis of FTO and SFPQ binding sites revealed that FTO and SFPQ bind RNA in a proximity 53. SFPQ overexpression decreases m6A in FTO mRNA targets in various cellular models. In particular, CLIP and m6A-IP-qPCR experiments show that increased expression of SFPQ favours the binding of FTO to myc mRNA and its subsequent m6A demethylation.
Figure 2.
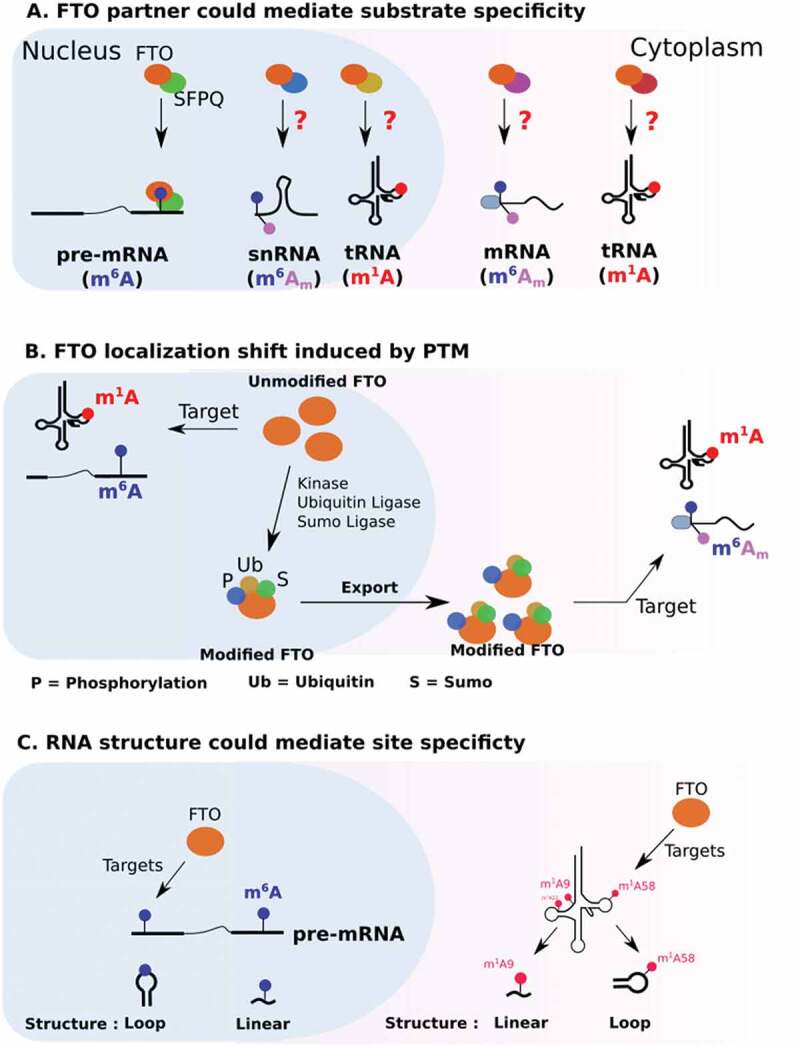
Potential mechanisms regulating FTO substrate specificity.
Interestingly, only 20% of FTO binds in proximity to SFP, suggesting that the remaining pool of FTO associates with other molecular partners 53. Protein partners that would promote FTO interaction with either m6Am or m1A sites remain to be identified. While m6Am is a structural component of the mRNA cap, FTO recruitment on this modification does not seem to involve the translation initiation machinery. Along these lines, FTO does not co-localize with stress granules. These are cytoplasmic aggregates mostly composed of stalled translation initiation complexes54.
TRMT10A, an S-adenosylmethionine-dependent methyltransferase that methylates guanosine in tRNAs at the ninth position, has also been suggested to interact with FTO and to steer its specificity towards specific m6A sites55. In this study, the authors suggested that TRMT10A coordinates the methylation status of tRNA and mRNA, to facilitate the translation of transcripts regulated by FTO.
2.3. Regulation of FTO by PTM
According to phosphosite plus (https://www.phosphosite.org), the FTO sequence harbours several potential post-translational modifications (PTMs): phosphorylation, sumoylation, ubiquitination and acetylation (Table 2). PTMs can affect protein activity, protein–protein interactions and subcellular localization56. In particular, the FTO activity is greatly influenced by its distribution, be it nuclear or cytoplasmic. Mutation of FTO lysine 216 has been shown to disrupt its nuclear localization and cause its accumulation in the cytoplasm. Three potential PTMs could be deposited at this position: ubiquitylation, sumoylation and acetylation. Interestingly, inhibiting ubiquitination with PYR-416 does not disrupt nuclear localization, but instead leads to the accumulation of a sub-pool of FTO in the perinuclear region 57. Sumoylation of K216 also affects FTO localization and regulates its nuclear transport as well as its turnover58. This modification involves a direct interaction with RanBP2, a component of the SUMO E3 ligase complex (RanBP2/RanGAP1*SUMO1/Ubc9), which localizes at the cytoplasmic side of nuclear pores and is a docking site in nucleocytoplasmic transport59. Collectively, these studies indicate that ubiquitination regulates the nucleocytoplasmic shuttling of FTO.
Table 2.
Post-translational modification sites along the FTO sequence (according to UniProt and PhosphoSite)
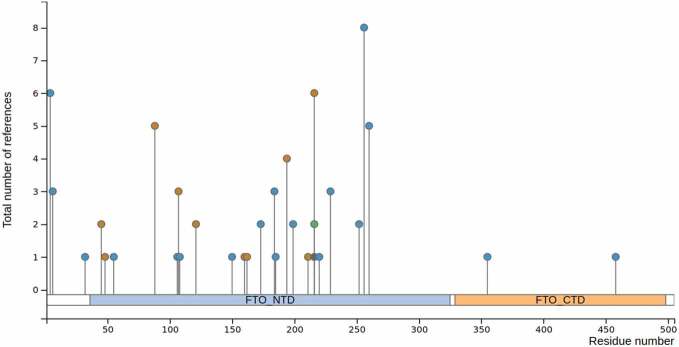 | |||
| Modification | Position | Domain | Function |
| Phosphorylation | Y106 | NTD | Binding to ssDNA in vitro |
| Phosphorylation | T150 | NTD | Localization |
| Phosphorylation | K216 | NTD | Localization |
| Ubiquitylation | K216 | NTD | Localization/Turnover |
| Sumoylation | K216 | NTD | Localization/Turnover |
| Phosphorylation | S229 | NTD | Substrate specificity in vitro |
| Phosphorylation | T4, T6, T32 | NLS | Unknown |
| Phosphorylation | S55, Y108, S173, S184, Y185, Y199, Y220, S248, P252, S256, S260 | NTD | Unknown |
| Phosphorylation | S355, S458 | CTD | Unknown |
| Acetylation | K216 | NTD | Unknown |
| Ubiquitylation | K45, K48, K88, K107 | NTD | Unknown |
PTMs have also been identified inside FTO’s predicted NLS. For instance, threonine 4 and threonine 6 can be phosphorylated. However, mutation of any of these threonines does not impair nuclear localization in an overexpression system, suggesting that they do not have an impact on FTO localization60]. By contrast, phosphorylation of T150, located inside the catalytic domain of FTO, regulates its subcellular distribution; the FTO T150A mutant has enhanced nuclear localization, whereas the T150E mutation inhibits nuclear translocation.
Beyond localization, it is still unclear whether any FTO modification regulates its enzymatic activity. For example, the K216R mutation not only promotes the shuttling of FTO towards the cytoplasm but also decreases m6A mRNA level58. By contrast, m6A demethylase activity of FTO was previously reported to occur in the nucleus [28,31]. Both sides could be reconciled by considering that K216 directly interacts with the RNA base [33]. As such, one cannot exclude that any PTM or mutation at this position would alter the FTO affinity towards its substrate.
Other residues that play key roles in the FTO function can be post-translationally modified. Take, for example, the case of Y106, which is part of the catalytic pocket and whose mutation impairs FTO binding to ssDNA. Y106 can be phosphorylated in leukaemic cell lines (Table 2), but its function remains elusive. To take a further example, Serine 229 is involved in FTO specificity towards RNA substrates. As such, the FTO S229A mutation slightly decreases the demethylation of m6A without impairing m6Am demethylation [33]. Phosphorylation of this residue has been reported in various cancer cell lines, but the function of the phosphor adduct is not yet known. To sum up, various PTMs have been reported to occur on several residues along the FTO protein sequence. However, their impact at the molecular level and their exact function in a physio-pathological context remain to be uncovered.
2.4. RNA context
As well as localization and protein partners, the activity of FTO is also dependent on RNA context. A striking example comes from m1A targeting, as previously discussed in the context of tRNA (see 1.4). In vitro, FTO demethylates m1A on hairpin loop RNA probes but not on linear RNA probes [28]. Besides secondary structure, sequence context is also important for m6A demethylation. An in vitro demethylation assay on 5 nucleotide-long m6A probes revealed that the FTO activity can vary up to two-fold depending on the sequence context [33]. Furthermore, as for m1A, FTO is more efficient at targeting m6A in the context of a large loop. To demonstrate this, the authors employed probes containing a methylated GGACU motif, embedded in either a large or a closed loop, and incubated them with FTO in vitro. FTO’s m6A demethylase activity was higher when m6A was localized in a large loop than when in a close loop or in linear RNA [33]. This predilection for targeting methylated sites in structured regions has not been demonstrated in vivo yet, and counterintuitively, m6A is preferentially localized in poorly structured regions 61]. How can we reconcile these two apparently contradictory observations? First, it has been proposed that m6A unfolds RNA structure through the recruitment of RNA helicase-containing m6A reader YTHDC261]. Second, m6A deposition can also disrupt A-U interactions to loosen RNA structure62,63 . Third, the connection between m6A and RNA structure might be a result of FTO action; preferential targeting of m6A in loop structures could be what is enriching m6A in unfolded regions.
3. ‘Context-dependent’ function of FTO in cancer
3.1. Pro-oncogenic role in cancer and therapeutic perspectives
Based on the current literature, the activity of FTO can have either pro-oncogenic or anti-oncogenic consequences depending on cancer type (Fig. 3). The pro-oncogenic role of FTO has been well described in acute myeloid leukaemia (AML) [25,46,47] and melanoma64. FTO expression is higher in AML with rearrangement of the mixed-lineage leukaemia (MLL) gene [25]. In these cancer cells, the ectopic expression of FTO promotes proliferation and colonogenic capacities, while it reduces apoptosis [25]. Silencing of FTO has the opposite effect, which concurs with a pro-oncogenic function of FTO in AML. Based on this conclusion, a therapeutic strategy based on FTO targeting has been elaborated to kill leukaemic cells65. The authors employed R-2-hydroxyglutarate (R-2HG), a metabolite produced by isocitrate dehydrogenase 1/2 (IDH1/2) enzymes. R-2HG is capable of inhibiting various alpha-ketoglutarate-dependent enzymes. As expected by the authors, the 2-HG treatment reduced proliferation and leukaemogenesis in an FTO-dependent manner65. As R-2HG is a broad inhibitor of alpha-ketoglutarate enzymes, subsequent studies explored the possibility of developing specific FTO inhibitors. Huang et al. identified meclofenamic acid as a highly specific inhibitor of FTO 66]. Meclofenamic acid is a non-steroidal, anti-inflammatory drug that competes with FTO for binding m6A sites. More recently, the same group has developed and functionally screened several derivatives of this drug and identified one of them, named FB23, as 140-fold more potent than the parent compound at inhibiting FTO-mediated demethylation [47]. A derivative of FB23, FB23-2, abrogates in vivo leukaemia progression and prolongs survival of leukaemic mice. On the same trend, Rui Su et al developed two FTO inhibitors for targeting leukaemic stem cells [46]. These compounds slow down AML and improve survival in a patient-derived xenograft mouse model. Importantly, FTO inhibitors are able to kill leukaemic cells without significantly affecting normal cells. Nevertheless, while FTO is a promising target in mouse models, FTO expression in men does not necessarily correlate with patient survival67 and another study found no effect of Fto on AML human cell lines [68] . Hence, further studies will be required to evaluate the relevance of targeting FTO for treating AML as well as other cancers.
Figure 3.
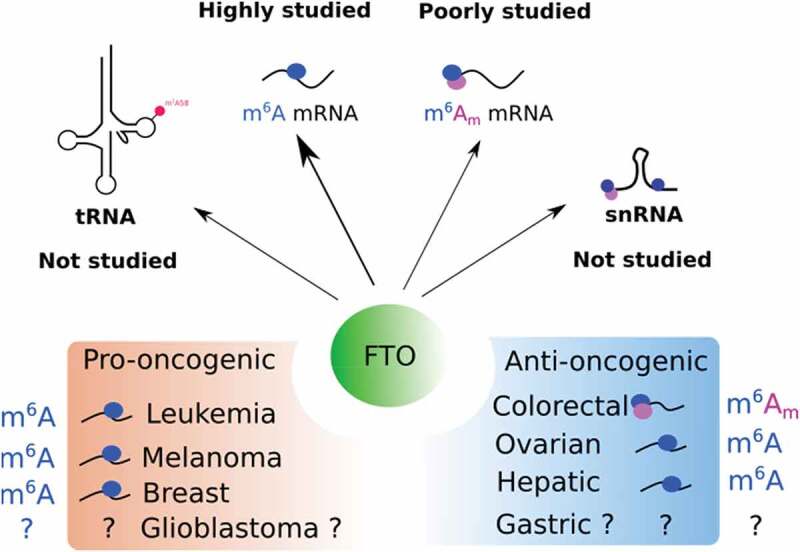
Function of FTO in cancers. The top panel illustrates the targets of FTO that have been studied or not in cancer. The bottom panel shows the pro- and anti-oncogenic function of FTO with associated targets.
The oncogenic effect of FTO has also been described in melanoma, breast cancer, lung cancer, oral squamous cell carcinoma (OSCC), and glioblastoma. In melanoma, FTO silencing reduces cell proliferation in vitro and in vivo [64] . By contrast, ectopic expression of FTO favours melanoma cell proliferation and reduces the efficiency of immunotherapy64. In glioblastoma cell lines, pharmacological inhibition of FTO reduces tumour growth and cripple stem-like properties 69. In breast cancer, FTO transcript level is higher in tumours than in healthy tissue [24]. FTO silencing blocks cell proliferation and clonogenic abilities and induces apoptosis of breast cancer cells. In lung cancer, FTO overexpression promotes cell growth, migration and metastasis, in an E2F1-dependent manner. Concomitantly, the level of m6A level in E2F1 mRNA is reduced upon FTO overexpression, suggesting that FTO’s function in lung cancer depends on its m6A demethylase activity70. Moreover, FTO mediated m6A demethylation is oncogenic in OSCC. In this cancer type, FTO regulates autophagy and tumorigenesis by targeting m6A on the transcript encoding the eukaryotic translation initiation factor gamma 1 (eIF4G1). FTO silencing promotes the downregulation of eIF4G1 (through YTHDF2 binding) along with enhanced autophagic flux and inhibition of tumorigenesis71.
3.2. Anti-oncogenic function of FTO
In some tissues, FTO exhibits anti-oncogenic functions. In colon cancer cells, for instance, FTO silencing promotes the acquisition of stem-like features: sphere formation in suspension culture, tumour initiation and chemoresistance in xenografted nude mice [32]. Remarkably, the analysis of tumour microarrays from colorectal cancer patients shows that FTO expression remains relatively constant throughout tumour onset and progression [32]. However, its cellular distribution varies significantly. While FTO is strictly nuclear in healthy tissues, a large fraction relocates to the cytoplasm during the early stages of tumour progression. Considering the compartment-specific activity of FTO, any alteration of its subcellular distribution may trigger significant functional changes and impact tumour evolution. From that point of view, FTO expression and localization may be considered of equal importance for understanding its role in cancer.
Another striking example comes from hepatocellular carcinoma, where SIRT1 acetylase promotes tumour growth by downregulating the FTO expression59]. At the molecular level, SIRT1 acetylates RANBP2 that, in turn, sumoylates FTO and triggers its degradation59]. Low FTO expression is also associated with poor patient outcome in renal clear cell carcinoma72. In this study, the authors demonstrate that the m6A demethylase activity of FTO suppresses tumour growth by promoting the expression of PGC-1α, a transcriptional coactivator and central inducer of mitochondrial metabolism72. Last, downregulation of FTO is associated with poor prognosis in intrahepatic cholangiocarcinoma73. In this cancer type, FTO overexpression suppresses in vitro anchorage-independent growth as well as in vivo tumour growth 73. In gastric cancer, a reduction of FTO protein level is associated with poor prognosis suggesting an anti-oncogenic role of FTO in this cancer74. However, this discovery remains controversial, as another study found FTO to be upregulated in gastric cancer75. Investigation of the prognosis value of FTO revealed its anti-oncogenic function in lung cancer. Indeed, high FTO expression is associated with longer survival76,77]. FTO silencing promotes cell growth, cell migration and tumorigenesis in vivo, which indicates an anti-oncogenic function of FTO in lung cancer. Mechanistically, FTO silencing enhances the m6A levels in several critical genes, including Myc. Accrued m6A on Myc mRNA leads to YTHDF1 binding, which promotes Myc mRNA translation, increased glycolysis and cell proliferation 77.
Finally, a recent report associates FTO downregulation in several epithelial cancers with enhanced cancer progression features, such as growth, invasion and metastasis, as well as with worse clinical outcome78. The authors show that the FTO loss promotes the implementation of an epithelial to mesenchymal programme through the activation of the Wnt signalling pathway. Indeed, FTO-dependent m6A demethylation regulates the stability of several transcripts involved in Wnt pathway regulation.
In short, FTO function in cancer varies greatly according to tissue context and further studies are required to clarify the molecular basis of this ambiguity.
3.3. Regulation of FTO expression in cancer
The central role of FTO in tumorigenesis is reflected by the frequent alteration of its expression and/or activity in cancer79. However, few studies have investigated the molecular mechanisms governing FTO expression in cancer. In breast cancer, STAT3 binds to the FTO promoter to induce its transcription 80. FTO is also regulated at the transcriptional level in lung cancer 77]. Indeed, the FTO promoter has a LEF/TCF-binding element that allows the recruitment of the Bcat/TCF/EZH2 complex. The docking of this complex on the FTO promoter enhances H3K27me3 levels and inhibits the FTO expression. Transcriptional regulation of FTO also occurs in OSCC. Indeed, rapamycin treatment of OSCC lines decreases both FTO mRNA and FTO protein levels, suggesting that FTO is mainly regulated through transcription or mRNA stability in this model 71. In contrast, in colorectal cancer, there is no correlation between the level of FTO mRNA and protein across patient-derived cell lines from primary tumours, in metastatic tumours or in circulating tumour cells [32], implying a regulation of FTO at the post-transcriptional level. A similar observation was made in other cancers, such as gastric cancer74, where FTO protein expression is disconnected from its transcript level. Yet, the ‘cancerous’ signalling pathways involved in the post-transcriptional regulation of FTO must be identified in future studies.
DISCUSSION
The molecular basis of how FTO recognizes and selects its substrates remains a matter of debate and controversy, despite significant efforts in this research area. For example, in contrast to most cancer studies showing FTO targets m6A sites, FTO preferentially targets m6Am in colorectal cancer. One possible explanation could be that most studies focus on global m6A methylation, making no distinction between m6A and m6Am, and ignoring any potential effect on m1A. Another explanation may reside in the diversity of experimental protocols for RNA extraction and processing. Taking these parameters into consideration will be essential in future studies that will require standardized protocols and the routine use of mass spectrometry for simultaneous quantification of modified nucleosides from both total and purified RNA species. Indeed, we propose to avoid creating more uncertainties and better comprehend individual FTO target’s contribution to any FTO-related phenotype.
As for many cellular enzymes, FTO can be affected by PTMs and molecular partners at the level of distribution, activity and subsequent cell phenotype. Remarkably, besides predictable discrepancies between transcript and protein level, changes in FTO localization may arise following neoplastic transformation [32], thus skewing survival statistics. This exemplifies why localization must also be evaluated in cancer studies in addition to FTO protein level. We are optimistic that pan-cancer analyses of FTO expression and localization paralleled by the identification of FTO targets will clarify the debate regarding the impact of FTO’s subcellular localization on its target specificity and function.
Acknowledgments
Our teams are generously supported by the Occitanie Region/FEDER (PPRi, SMART project), French National Cancer Institute (INCa) (DA N°2020-116), Ligue contre le Cancer, SIRIC Montpellier Cancer (INCa-DGOS-Inserm 6045), Cancéropole GSO and LabEx NUMEV (GEM flagship project, ANR 2011-LABX-076).
Funding Statement
This work was supported by the LabEx NUMEV [GEM flagship project, ANR 2011-LABX-076]; SIRIC Montpellier cancer [INCa-DGOS-Inserm 6045]; Occitanie Region/FEDER [PPRi SMART].French National Cancer Institute (INCa) [DA N°2020-116];
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Peters T, Ausmeier K, Ruther U.. Cloning of Fatso (Fto), a novel gene deleted by the Fused toes (Ft) mouse mutation. Mamm Genome. 1999;10(10):983–986. [DOI] [PubMed] [Google Scholar]
- [2].Frayling TM, N.J. Timpson, M.N. Weedon., et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Scuteri A, S. Sanna, W.M. Chen, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3(7):e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Price RA, Li WD, Zhao H. FTO gene SNPs associated with extreme obesity in cases, controls and extremely discordant sister pairs. BMC Med Genet. 2008;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meyre D, K. Proulx, H. Kawagoe-Takaki, et al. Prevalence of loss-of-function FTO mutations in lean and obese individuals. Diabetes. 2010;59(1):311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gao X, Y.H. Shin, M. Li, et al. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One. 2010;5(11):e14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Church C, L. Moir, F. McMurray, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42(12):1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Khella MS, N.M. Hamdy, A.I. Amin, et al. The (FTO) gene polymorphism is associated with metabolic syndrome risk in Egyptian females: a case- control study. BMC Med Genet. 2017;18(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guclu-Geyik F, A. Onat, A.B. Yuzbasiogullari, et al. Risk of obesity and metabolic syndrome associated with FTO gene variants discloses clinically relevant gender difference among Turks. Mol Biol Rep. 2016;43(6):485–494. [DOI] [PubMed] [Google Scholar]
- [10].Wang H, S. Dong, H. Xu, et al. Genetic variants in FTO associated with metabolic syndrome: a meta- and gene-based analysis. Mol Biol Rep. 2012;39(5):5691–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Freathy RM, N.J. Timpson, D.A. Lawlor, et al. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes. 2008;57(5):1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mitropoulos K, E. Merkouri Papadima, G. Xiromerisiou, et al. Genomic variants in the FTO gene are associated with sporadic amyotrophic lateral sclerosis in Greek patients. Hum Genomics. 2017;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hernandez-Caballero ME, Sierra-Ramirez JA. Single nucleotide polymorphisms of the FTO gene and cancer risk: an overview. Mol Biol Rep. 2015;42(3):699–704. [DOI] [PubMed] [Google Scholar]
- [14].Lan N, Y. Lu, Y. Zhang . FTO - A Common Genetic Basis for Obesity and Cancer. Front Genet. 2020;11:559138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jia G, C.G. Yang, S. Yang, et al. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008;582(23–24):3313–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jia G, Y. Fu, X. Zhao, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roundtree IA, M.E. Evans, T. Pan, et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169(7):1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fedeles BI, V. Singh, J.C. Delaney, et al. The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: repairing Nucleic Acid Alkylation Damage and Beyond. J Biol Chem. 2015;290(34):20734–20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Han Z, T. Niu, J. Chang, et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature. 2010;464(7292):1205–1209. [DOI] [PubMed] [Google Scholar]
- [20].Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18(5):275–291. [DOI] [PubMed] [Google Scholar]
- [21].Zhu Y, G. Zhou, X. Yu, et al. LC-MS-MS quantitative analysis reveals the association between FTO and DNA methylation. PLoS One. 2017;12(4):e0175849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhao X, Y. Yang, B.F. Sun, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hess ME, S. Hess, K.D. Meyer, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16(8):1042–1048. [DOI] [PubMed] [Google Scholar]
- [24].Niu Y, Z. Lin, A. Wan, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Z, H. Weng, R. Su, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31(1):127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li Y, K. Wu, W. Quan, et al. The dynamics of FTO binding and demethylation from the m(6)A motifs. RNA Biol. 2019;16(9):1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lan MD, J. Xiong, X.J., et al. Existence of Diverse Modifications in Small-RNA Species Composed of 16-28 Nucleotides. Chemistry. 2018;24(39):9949–9956. [DOI] [PubMed] [Google Scholar]
- [28].Wei J, F. Liu, Z. Lu, et al. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell. 2018;71(6):973–985. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bartosovic M, H.C. Molares, P. Gregorova, et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3ʹ-end processing. Nucleic Acids Res. 2017;45(19):11356–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Louloupi A, E. Ntini, T. Conrad, et al. Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. 2018;23(12):3429–3437. [DOI] [PubMed] [Google Scholar]
- [31].Mauer J, X. Luo, A. Blanjoie, et al. Reversible methylation of m(6)Am in the 5ʹ cap controls mRNA stability. Nature. 2017;541(7637):371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Relier S, J. Ripoll, H. Guillorit, et al. FTO-mediated cytoplasmic m(6)Am demethylation adjusts stem-like properties in colorectal cancer cell. Nat Commun. 2021;12(1):1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li Y, D. Zheng, F. Wang, et al. Expression of Demethylase Genes, FTO and ALKBH1, Is Associated with Prognosis of Gastric Cancer. Dig Dis Sci. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Linder B, A.V. Grozhik, A.O. Olarerin-George, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mauer J, M. Sindelar, V. Despic, et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15(4):340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shi H, Wei J, He C. Where, When, and How: context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74(4):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang C, Jia G. Reversible RNA Modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genomics Proteomics Bioinformatics. 2018;16(3):155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roovers M, et al. A primordial RNA modification enzyme: the case of tRNA (m1A) methyltransferase. Nucleic Acids Res. 2004;32(2):465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Macari F, et al. TRM6/61 connects PKCalpha with translational control through tRNAi(Met) stabilization: impact on tumorigenesis. Oncogene. 2016;35(14):1785–1796. [DOI] [PubMed] [Google Scholar]
- [40].Safra M, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551(7679):251–255. [DOI] [PubMed] [Google Scholar]
- [41].Liu F, et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell. 2016;167(7):1897. [DOI] [PubMed] [Google Scholar]
- [42].Haag S, et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016;35(19):2104–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ke Set al , Genes Dev 2017 m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. 2017 May 15;31(10):990–1006. 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Han Z, et al. A loop matters for FTO substrate selection. Protein Cell. 2010;1(7):616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mao Y, et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10(1):5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Su R, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell. 2020;38(1):79–96. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang Y, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35(4):677–691. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Engel, M., Eggert C., and Kaplick P.M.et al, , The Role of m(6)A/m-RNA Methylation in Stress Response Regulation. Neuron, 2018. 99(2): p. 389–403 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu, J., Li K., and Cai J.et al, , Landscape and Regulation of m(6)A and m(6)Am Methylome across Human and Mouse Tissues. Mol Cell, 2020. 77(2): p. 426–440 e6. [DOI] [PubMed] [Google Scholar]
- [50].Zhang, X., Wei L.H., and Wang Y.et al, , Structural insights into FTO's catalytic mechanism for the demethylation of multiple RNA substrates. Proc Natl Acad Sci U S A, 2019. 116(8): p. 2919–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Han, Z., Huang N., and Niu T.et al, , A loop matters for FTO substrate selection. Protein Cell, 2010. 1(7): p. 616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Edupuganti, R.R., Geiger S., and Lindeboom R.G.H.et al, , N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol, 2017. 24(10): p. 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Song, H., Wang Y., and Wang R.et al, , SFPQ Is an FTO-Binding Protein that Facilitates the Demethylation Substrate Preference. Cell Chem Biol, 2020. 27(3): p. 283–291 e6. [DOI] [PubMed] [Google Scholar]
- [54].Anders, M., Chelysheva I., and Goebel I.et al, , Dynamic m(6)A methylation facilitates mRNA triaging to stress granules. Life Sci Alliance, 2018. 1(4): p. e201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ontiveros, R.J., Shen H., and Stoute J.et al, , Coordination of mRNA and tRNA methylations by TRMT10A. Proc Natl Acad Sci U S A, 2020. 117(14): p. 7782–7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Millar, A.H., Heazlewood J.L., and Giglione C.et al, , The Scope, Functions, and Dynamics of Posttranslational Protein Modifications. Annu Rev Plant Biol, 2019. 70: p. 119–151. [DOI] [PubMed] [Google Scholar]
- [57].Russell, M.A., and Morgan N.G., Conditional expression of the FTO gene product in rat INS-1 cells reveals its rapid turnover and a role in the profile of glucose-induced insulin secretion. Clin Sci (Lond), 2011. 120(9): p. 403–13. [DOI] [PubMed] [Google Scholar]
- [58].Zhu, T., Yong X.L.H., and Xia D.et al, , Ubiquitination Regulates the Proteasomal Degradation and Nuclear Translocation of the Fat Mass and Obesity-Associated (FTO) Protein. J Mol Biol, 2018. 430(3): p. 363–371. [DOI] [PubMed] [Google Scholar]
- [59].Liu, X., Liu J., and Xiao W.et al, , SIRT1 Regulates N(6) -Methyladenosine RNA Modification in Hepatocarcinogenesis by Inducing RANBP2-Dependent FTO SUMOylation. Hepatology, 2020. 72(6): p. 2029–2050. [DOI] [PubMed] [Google Scholar]
- [60].Hirayama, M., Wei F.Y., and Chujo T.et al, , FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep, 2020. 31(1): p. 107464. [DOI] [PubMed] [Google Scholar]
- [61].Mao, Y., Dong L., and Liu X.M.et al, , m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun, 2019. 10(1): p. 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu, N., Dai Q., and Zheng G.et al, , N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature, 2015. 518(7540): p. 560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kierzek, E., and Kierzek R., The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res, 2003. 31(15): p. 4472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yang, S., Wei J., and Cui Y.H.et al, , m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun, 2019. 10(1): p. 2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].R-2HG Targets FTO to Increase m(6)A Levels and Suppress Tumor Growth. Cancer Discov, 2018. 8(2): p. 137. [DOI] [PubMed] [Google Scholar]
- [66].Huang, Y., Yan J., and Li Q.et al, , Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res, 2015. 43(1): p. 373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Begik, O., Lucas M.C., and Liu H.et al, , Integrative analyses of the RNA modification machinery reveal tissue- and cancer-specific signatures. Genome Biol, 2020. 21(1): p. 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vu, L.P., Pickering B.F., and Cheng Y.et al, , The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med, 2017. 23(11): p. 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Huff, S., Tiwari S.K., and Gonzalez G.M.et al, , m(6)A-RNA Demethylase FTO Inhibitors Impair Self-Renewal in Glioblastoma Stem Cells. ACS Chem Biol, 2021. 16(2): p. 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang, Y., Li M., and Zhang L.et al, , m6A demethylase FTO induces NELL2 expression by inhibiting E2F1 m6A modification leading to metastasis of non-small cell lung cancer. Mol Ther Oncolytics, 2021. 21: p. 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang, F., Liao Y., and Zhang M.et al, , N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene, 2021. 40(22): p. 3885–3898. [DOI] [PubMed] [Google Scholar]
- [72].Zhuang, C., Zhuang C., and Luo X.et al, , N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1alpha signalling axis. J Cell Mol Med, 2019. 23(3): p. 2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rong, Z.X., Li Z., and He J.J.et al, , Downregulation of Fat Mass and Obesity Associated (FTO) Promotes the Progression of Intrahepatic Cholangiocarcinoma. Front Oncol, 2019. 9: p. 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li, Y., Zheng D., and Wang F.et al, , Expression of Demethylase Genes, FTO and ALKBH1, Is Associated with Prognosis of Gastric Cancer. Dig Dis Sci, 2019. 64(6): p. 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Guo, C., Chu H., and Gong Z.et al, , HOXB13 promotes gastric cancer cell migration and invasion via IGF-1R upregulation and subsequent activation of PI3K/AKT/mTOR signaling pathway. Life Sci, 2021. 278: p. 119522. [DOI] [PubMed] [Google Scholar]
- [76].Wu, G., Yan Y., and Cai Y.et al, , ALKBH1-8 and FTO: Potential Therapeutic Targets and Prognostic Biomarkers in Lung Adenocarcinoma Pathogenesis. Front Cell Dev Biol, 2021. 9: p. 633927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yang, X., Shao F., and Guo D.et al, , WNT/beta-catenin-suppressed FTO expression increases m(6)A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis, 2021. 12(5): p. 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jeschke, J., Collignon E., and Al Wardi C.et al, , Downregulation of the FTO m6A RNA demethylase promotes EMT-mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nature Cancer, 2021(2): p. 611–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chen, J., and Du B., Novel positioning from obesity to cancer: FTO, an m(6)A RNA demethylase, regulates tumour progression. J Cancer Res Clin Oncol, 2019. 145(1): p. 19–29. [DOI] [PubMed] [Google Scholar]
- [80].Wang, Y., Cheng Z., and Xu J.et al, , Fat mass and obesity-associated protein (FTO) mediates signal transducer and activator of transcription 3 (STAT3)-drived resistance of breast cancer to doxorubicin. Bioengineered, 2021. 12(1): p. 1874–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]