

Abstract
Free amino acids are one of the main chemical components in tea, and they contribute to the pleasant flavor, function, and quality of tea, notably the level of theanine. Here, a high-density genetic map was constructed to characterize quantitative trait loci (QTLs) for free amino acid content. A total of 2688 polymorphic SNP markers were obtained using genotyping-by-sequencing (GBS) based on 198 individuals derived from a pseudotestcross population of “Longjing 43” × “Baijiguan”, which are elite and albino tea cultivars, respectively. The 1846.32 cM high-density map with an average interval of 0.69 cM was successfully divided into 15 linkage groups (LGs) ranging from 93.41 cM to 171.28 cM. Furthermore, a total of 4 QTLs related to free amino acid content (theanine, glutamate, glutamine, aspartic acid and arginine) identified over two years were mapped to LG03, LG06, LG11 and LG14. The phenotypic variation explained by these QTLs ranged from 11.8% to 23.7%, with an LOD score from 3.56 to 7.7. Furthermore, several important amino acid metabolic pathways were enriched based on the upregulated differentially expressed genes (DEGs) among the offspring. These results will be essential for fine mapping genes involved in amino acid pathways and diversity, thereby providing a promising avenue for the genetic improvement of tea plants.
Introduction
Tea, a prevalent and healthy beverage because of its excellent value and high palatability, is processed from the tender shoots of tea plants [1, 2]. Currently, tea is widely consumed worldwide for its extensive variety of bioactive compounds, such as amino acids, catechins, polyphenols, caffeine, and terpenes [3–5]. Free amino acids (AAs) comprise more than 20 components, including protein and nonprotein amino acids [6], which increase tea quality and the beneficial health effects for humans [7, 8], thereby attracting increasing attention worldwide. Of them, glutamate (Glu), glutamine (Gln), aspartic acid (Asp) and especially L-theanine (Thea) are crucial and abundant amino acids.
Furthermore, free amino acids are among the most critical sources of energy and serve as precursors in the biosynthesis of numerous important secondary metabolites such as phytohormones, chlorophyll and creatine [9]. The synthesis of Glu and Gln is linked to the nitrogen cycle to help plants utilize and assimilate nitrogen and improve their nitrogen balance [10]. Glu is a significant driving factor in the citric acid cycle via Gln conversion to α-ketoglutarate, which is an integral component of the citric acid cycle [11]. Theanine (N-ethyl-γ-L-glutamine), a specialized nonprotein amino acid in tea plants, accounts for up to ∼50% of the total free amino acids in fresh leaves and is synthesized from Glu and ethylamine in tea roots and subsequently transported to aboveground tissues through the xylem [12, 13]. Concomitantly, Thea was verified to promote the umami taste of tea infusions by counteracting the astringent and bitter tastes derived from other metabolites, such as catechins, theobromine and caffeine [14]. Moreover, Thea can promote the release of dopamine and serotonin in the human brain to enhance relaxation, concentration, and learning ability, as well as prevent obesity and cardiovascular disease and improve human immunity [15–17]. Thea and Gln are homologs due to their similar molecular structures, properties, and functions among diverse plant species [18]. On the basis of the important role of free amino acids in tea function and quality, breeding elite cultivars with high amounts of amino acids has become a vital target for tea breeders . Thus, it is important to elucidate the genetic basis of the amino acid pathway in tea plants and provide useful resources to improve tea quality. In comparison to other plants, Cheng et al., [19] using a radioisotope marked method, revealed that the accumulation of amino acids in tea plants was not due to an increase in the biosynthesis of products but to a low level of Thea decomposition. Current research has dissected the genetic mechanism of target compounds in tea plants from a reverse genetics perspective, owing to the lack of genetic resources for metabolic and genetic mapping. Consequently, the underlying functional genes for target traits affected by both genetic and environmental factors remain largely unknown and unverified, which limits tea breeding prospects.
China, regarded as the origin of tea plants [20, 21], has a broad range of natural tea germplasm resources, including normal green and variable color tea cultivars, such as yellow, purple, and white, which are classified as special resources. Albino cultivars, which exhibit abundant white and yellow colors in young leaves, especially in the spring season, and have ornamental value, are characterized by high amounts of free amino acids and low catechins, resulting in pleasant flavor and enticing a wide range of tea drinkers [22, 23]. “Baijiguan” (“BJG”), a popular light-sensitive albino tea landrace, has heritable white and yellow tender shoots and produces tea with a pleasant flavor [24]. Hence, it is necessary to breed elite cultivars such as albino tea plants to meet the increasing demand and promote revenues for tea growers and the industry. Traditional tea breeding approaches for genetic improvements are a time-consuming and labor-intensive procedure due to the long juvenile stage [25], especially when studying amino acid traits, which are controlled by multiple genes or quantitative trait loci (QTLs). Linkage map construction associated with QTL mapping can elucidate and characterize the genes responsible for complex and significant agronomic traits, such as sterility, yield, disease, and quality [26–29]. These results enhance the development of conventional plant breeding and provide powerful insight into new approaches to enhance the efficiency and precision of breeding via molecular markers. These approaches have facilitated the genetic improvement of tea by dissecting flavor genes. In recent years, a considerable number of QTLs for important agronomic traits have been successfully detected and characterized in model plants and crops, including rice [30], maize [31], and wheat [32]. However, among tea plants, it is far more difficult to obtain suitable mapping populations for QTL mapping due to its long growth cycle and self-incompatibility.
Single nucleotide polymorphisms (SNPs), which are efficient molecular markers, are widely used in genetic mapping and allele identification and are fundamental for functional gene identification and breeding improvement [33–35]. Here, we constructed a high-density SNP linkage map for a cross between a distinctive albino cultivar “BJG” and an elite green cultivar “Longjing 43” (“LJ43”), the F1 population has transgressive segregation with higher amount of free amino acids than the parents. Furthermore, we used the F1 full-sib family to detect QTLs responsible for the free amino acid content in tender shoots, which demonstrates the genetic mechanism of variation in amino acids in tea plants. Our study not only provides a valuable foundation for exploring and deciphering the amino acid pathways in tea plants but also elucidates the structure and function of these vital genes to provide useful resources for tea breeding.
Results
Sequencing data and polymorphism analysis
A total of 114.48 G of raw reads were derived from the sequencing assembly of tea plants, and the average amount of data across the 189 progenies was 681.41 M (Q20 ≥ 90%, Q30 ≥ 85%). The results showed that there were 33 678 450 female parent tags with an average read coverage of ~58-fold, 31 321 308 male parent tags with an average read coverage of ~57-fold, and F1 individuals with an average read coverage ~12-fold. The average matching rate in F1 offspring was 84.38%, with 25 771 268–184 474 tags that could be used for subsequent mutation detection and correlation analysis.
Marker screening and genotypic analysis
After removing abnormal bases, those with low integrity, and those with segregation distortion based on the chi-square test (P < 0.001), 30 971 informative SNP markers were obtained from 1 656 000 SNP markers and used for linkage analysis. There were 590 259 polymorphic markers based on the further genotypic analysis of the PP and MP parents, and five types of markers were available in both the parents and F1 populations, including “hk × hk”, “lm × ll”, “nn × np”, “ef × eg” and “ab × cd”, as presented in Fig. S1.
Genetic map
A total of 30 971 SNP markers were used for linkage analysis. As a result, 2688 SNP markers were finally mapped into 15 linkage groups (LGs), corresponding to the haploid chromosome number of tea plants. After linkage analysis, all these markers were placed on the genetic map and were distributed among 2689 loci on 15 linkage groups, as described in Table 1. The linkage map covered a total genetic distance of 1846.32 cM, with an average locus spacing of 0.69 cM. Individually, the linkage groups ranged in size from 93.41 cM to 171.28 cM, as shown in Fig. 1.
Table 1.
Information and statistics of the genetic map.
| Linkage group | SNP number | Genetic distance (cM) | Average distance (cM) |
|---|---|---|---|
| LG 01 | 202 | 171.28 | 0.85 |
| LG02 | 232 | 128.29 | 0.55 |
| LG03 | 127 | 101.23 | 0.8 |
| LG04 | 184 | 120.09 | 0.65 |
| LG05 | 124 | 136.05 | 1.1 |
| LG06 | 95 | 119.87 | 1.26 |
| LG07 | 178 | 102.12 | 0.57 |
| LG08 | 183 | 123.35 | 0.67 |
| LG09 | 157 | 103.69 | 0.66 |
| LG10 | 202 | 124.86 | 0.62 |
| LG11 | 239 | 106.1 | 0.44 |
| LG12 | 157 | 93.41 | 0.59 |
| LG13 | 142 | 104.92 | 0.74 |
| LG14 | 236 | 150.43 | 0.64 |
| LG15 | 230 | 160.63 | 0.7 |
| Total | 2688 | 1846.32 | 0.69 |
Figure 1.
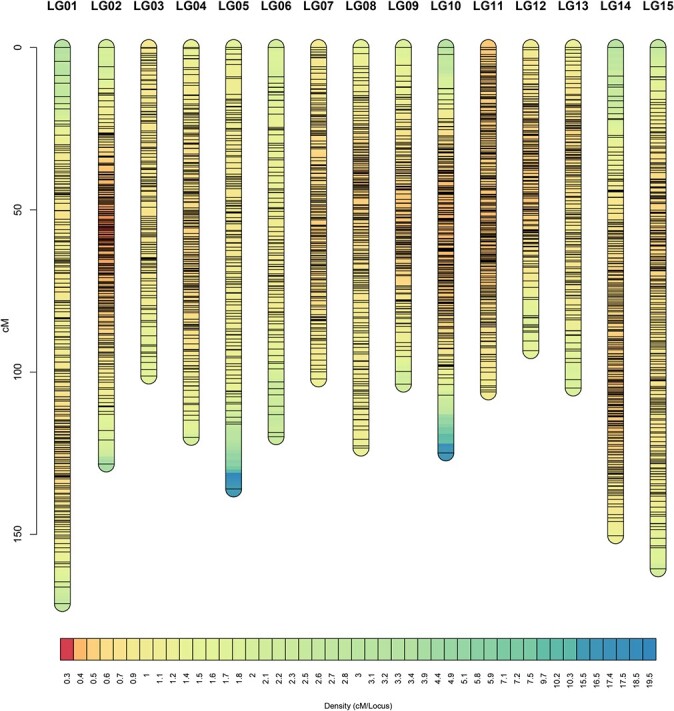
Marker distribution across the linkage map. The x-axis, y-axis, and lines in the column represent the linkage group, genetic distance, and unique marker, respectively.
Phenotypic data collection
The cardinal components of free amino acids, including Thea, Asp, Glu, Gln, and Arg, investigated in F1 progeny and both parents over two years are presented in Table 2. Moreover, the mean value, skewness, kurtosis, and coefficient of variation for each free amino acid were calculated. The two parents differed greatly in all traits investigated, and Thea was the most abundant amino acid compound. Interestingly, the paternal parent “BJG” had a higher AA content, which coincided with a higher composition of Thea, Asp, Glu, Gln and Arg than that of the female parent “LJ43” in 2018, especially Thea (Table S1). However, “BJG” had a lower AA content and composition of key amino acids than “LJ43” in 2019. Therefore, free amino acid accumulation is a sophisticated process that can be impacted by complicated and variable environmental conditions, such as nitrogenous fertilizer addition and climatic conditions, [36–38] contributing to the unstable QTLs for free amino acid content in different years. The average levels of total AA, Thea, Asp, Glu, Gln, and Arg across years were 2.15%, 1.42%, 0.11%, 0.20%, 0.18%, and 0.05% for “LJ43” and 2.09%, 1.27%, 0.12%, 0.26%, and 0.16% for “BJG”, respectively.
Table 2.
Phenotypic variation of the accumulation of individual free amino acids in the “BJG” and “LJ43” parents and F1 progeny.
| F1 population | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| trait | year | LJ43 | BJG | min | max | mean ± SD | CV (%) | kurtosis | skewness |
| AA | 2018 | 1.51 | 2.41 | 1.67 | 6.33 | 3.75 ± 0.92 | 24.55 | 0.32 | -0.37 |
| 2019 | 2.79 | 1.77 | 2.06 | 5.92 | 3.56 ± 0.78 | 21.55 | 0.52 | -0.12 | |
| Thea | 2018 | 1.01 | 1.48 | 4.11 | 1.09 | 2.29 ± 0.56 | 24.43 | 0.4 | -0.04 |
| 2019 | 1.83 | 1.05 | 1.15 | 3.44 | 2.12 ± 0.43 | 20.46 | 0.34 | -0.28 | |
| Asp | 2018 | 0.04 | 0.12 | 0.05 | 0.47 | 0.20 ± 0.08 | 40.45 | 0.68 | 0.27 |
| 2019 | 0.17 | 0.11 | 0.07 | 0.36 | 0.20 ± 0.05 | 26.56 | 0.52 | 0.15 | |
| Glu | 2018 | 0.13 | 0.19 | 0.11 | 0.53 | 0.29 ± 0.08 | 28.43 | 0.37 | -0.12 |
| 2019 | 0.27 | 0.14 | 0.14 | 0.5 | 0.29 ± 0.07 | 23.07 | 0.4 | -0.02 | |
| Gln | 2018 | 0.16 | 0.3 | 0.09 | 1.21 | 0.38 ± 0.18 | 47.24 | 1.11 | 1.85 |
| 2019 | 0.2 | 0.22 | 0.07 | 0.9 | 0.30 ± 0.14 | 47.36 | 1.21 | 1.83 | |
| Arg | 2018 | 0.02 | 0.16 | 0.04 | 1.11 | 0.31 ± 0.19 | 60.81 | 0.99 | 0.82 |
| 2019 | 0.07 | 0.15 | 0.01 | 0.92 | 0.24 ± 0.17 | 69.56 | 1.4 | 2.5 | |
High coefficients of variation (CVs) for free amino acid content were found for the F1 progeny listed in Table 3, with a similar CV between AA and Thea. Nevertheless, the CV of Arg was the highest, which revealed that the content of Arg matched a discrete distribution. The CVs for Arg and Gln were significantly higher than those for AA, Thea, Asp, and Glu. At the same time, the phenotypic values within the progeny across years varied from 1.61% to 6.33% for AA, 1.09% to 4.11% for Thea, 0.05% to 0.47% for Asp, 0.11% to 0.53% for Glu, 0.07% to 1.21% for Gln, and 0.01% to 1.11% for Arg. Therefore, each amino acid revealed a continuous distribution with transgressive segregation (Fig. 2), indicating characteristics of quantitative traits regulated by multiple genes.
Table 3.
Pearson’s correlations between the individual free amino acid compounds.
| AA | Thea | Asp | Glu | Gln | |
|---|---|---|---|---|---|
| Thea | 0.900** | ||||
| Asp | 0.601** | 0.419** | |||
| Glu | 0.654** | 0.468** | 0.761** | ||
| Gln | 0.716** | 0.486** | 0.426** | 0.461** | |
| Arg | 0.766** | 0.603** | 0.503** | 0.492** | 0.512** |
Figure 2.

Distribution patterns of the total amino acid content (AA) and the contents of theanine (Thea), aspartic acid (Asp), glutamate (Glu), glutamine (Gln) and arginine (Arg) in the “BJG” × “LJ43” F1 progeny. Parental values are indicated with solid (“BJG”) and empty (“LJ43”) arrows. The cyan and green colors represent the values for 2018 and 2019, respectively.
AA represents total free amino acids; Thea represents theanine; Asp represents aspartic acid; Glu represents glutamate; Gln represents glutamine; and Arg represents arginine.
Pearson’s correlations among free amino acid contents across years are illustrated in Table 3. There was a significantly positive correlation between AA and Thea (r = 0.900, p < 0.001). Moreover, Asp was more highly correlated with Glu (r = 0.761, p < 0.001) than with the others (r from 0.419 to 0.601, p < 0.001). Both Gln and Arg showed a significant positive correlation with AA (r = 0.716 and 0.766, p < 0.001).
Free amino acid content QTL analysis
A total of 18 QTLs for free amino acid content were detected by the whole-genome scan and distributed across four linkage groups, LG03, LG11, LG13, and LG14, as shown in Table 4. The confidence intervals ranged from 0.18 to 48.16 cM over both years (Fig. 3). Moreover, the average PVE per trait QTL was 16.7% for 2018 and 17.4% for 2019. The LOD peaks were at 8.5 cM in 2018 (with 20.9% PVE) and 7.7 cM in 2019 (with 23.7% PVE). Although the number of QTLs per trait ranged from one to three linkage groups, the three stable QTLs for Thea, Glu, and Asp were validated on LG03 over both years.
Table 4.
Overview of QTLs associated with free amino acid compound accumulation detected in the “BJG” × “LJ43” F1 progeny.
| Trait | Year | QTL | LG | Peak (cM) | Range (cM) | LOD | Nearer marker | PVE (%) |
|---|---|---|---|---|---|---|---|---|
| Thea | 2018 | qThea-3.1 | LG03 | 8.5 | 0.18-12.95 | 6.31 | Im3210 | 20.9 |
| qThea-3.2 | LG03 | 25.35 | 24.35-25.69 | 4.23 | Im6424 | 14.5 | ||
| qThea-3.3 | LG03 | 41.25 | 38.64-42.48 | 4.86 | Im6161 | 16.5 | ||
| qThea14.1 | LG14 | 34.19 | 33.19-35.69 | 4.74 | np6998 | 16.2 | ||
| qThea-14.2 | LG14 | 45.5 | 43.94-48.16 | 4.19 | np242 | 14.4 | ||
| 2019 | qThea-3.1 | LG03 | 8.5 | 8.21-8.5 | 3.56 | Im3210 | 11.8 | |
| Glu | 2018 | qGlu-3.1 | LG03 | 9.5 | 8.21-13.95 | 5.4 | hk14774 | 18.2 |
| qGlu-3.2 | LG03 | 25.35 | 24.34-26.2 | 3.79 | Im6424 | 13.1 | ||
| qGlu-3.3 | LG03 | 41.79 | 38.64-42.48 | 4.45 | Im3494 | 15.2 | ||
| 2019 | qGlu-3.1 | LG03 | 12.95 | 3.03-15.49 | 5.62 | Im2625 | 17.9 | |
| qGlu-3.1 | LG03 | 40.09 | 37.94-50.25 | 7.34 | Im6160 | 22.7 | ||
| Gln | 2018 | qGln-11.1 | LG11 | 26.82 | 26.37-26.82 | 5.64 | Im1794 | 18.9 |
| qGln-13.1 | LG13 | 31.54 | 31.54 | 5.18 | Im2270 | 17.5 | ||
| 2019 | qGln-3.1 | LG03 | 8.5 | 0.18-13.95 | 4.15 | Im3210 | 13.6 | |
| Asp | 2018 | qAsp-3.1 | LG03 | 12.82 | 0.18-13.95 | 5.65 | Im6599 | 18.9 |
| qAsp-3.2 | LG03 | 25.69 | 25.35-27.87 | 5.24 | Im6424 | 17.7 | ||
| qAsp-3.3 | LG03 | 38.64 | 37.35-41.79 | 4.58 | hk9022 | 15.6 | ||
| 2019 | qAsp-3.1 | LG03 | 12.82 | 1.18-14.93 | 7.7 | Im6599 | 23.7 | |
| qAsp-3.3 | LG03 | 40.09 | 34.82-45.79 | 5.59 | Im6160 | 17.9 | ||
| Arg | 2018 | qArg-3.1 | LG03 | 12.82 | 3.03-13.95 | 5.4 | Im6599 | 18.2 |
| qArg-3.2 | LG03 | 25.35 | 24.34-26.2 | 3.97 | Im6424 | 13.7 | ||
| 2019 | qArg-14.1 | LG14 | 35.69 | 34.69-36.34 | 4.42 | Im6287 | 14.4 |
Figure 3.
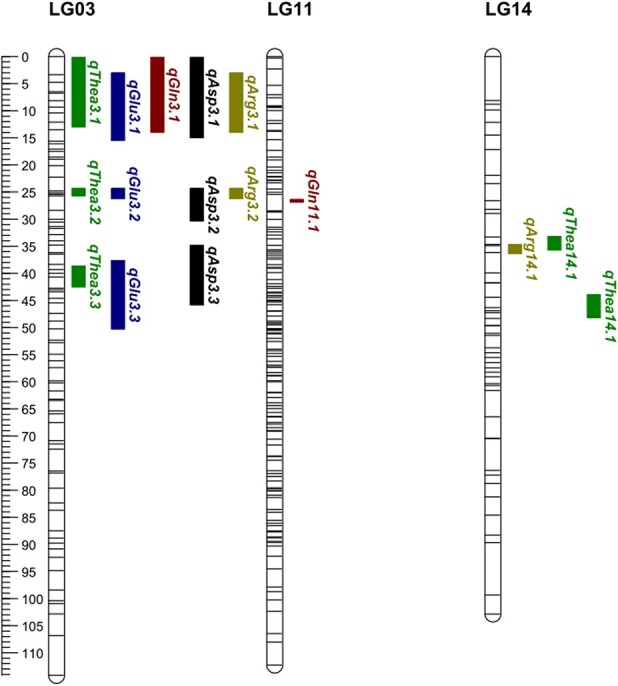
Distribution of QTLs associated with free amino acid accumulation identified in the “LJ43” × “BJG” F1 population. Significant QTLs for each trait are shown to the right of linkage groups and are represented by bars of different colors (Thea, green; Glu, blue; Gln, brown; Asp, black; and Arg, gray).
Five QTLs controlling Thea were identified on two linkage groups, with an average PVE of 16.5% for 2018 and 11.8% for 2019. Importantly, qThea-3 and qThea-14 were located on LG03 and LG14, with total PVEs of 51.9% and 30.6% for 2018 and 11.8% for 2019, respectively. The highest PVE exhibited by these QTLs was 20.9% for qThea-3.1, near locus Im3210, which was also detected in 2019. At the same time, the confidence interval ranged from 0.18 to 42.48 cM over both years, and the overlapping region ranged from 24.35 to 25.69 cM. The LODs of qThea-14.1 and qThea-14.2 presented were at positions 34.19 cM and 45.5 cM, respectively, with 16.2% and 14.4% PVE. The nearest loci, np6998 and np242, were located on LG14 with similar PVEs and LODs. Therefore, it was speculated that qThea-14.1 and qThea-14.2 may be the same QTL.
Specifically, the stable QTL associated with Glu, qGlu3, was identified on LG03 with an average PVE of 15.5% for 2018 and 20.3% for 2019. The confidence interval varied from 3.03 to 50.25 cM over both years, and the highest LOD was 7.34 near the MarkerIm6160 locus. The LOD peak was present at position 41.79 cM in 2018 (with 22.7% PVE) and position 9.5 cM in 2019 (with 18.2% PVE). qGln-11.1, qGln-13.1, and qGln-3.1 were located on LG11, LG13 and LG03, with PVEs of 18.9, 17.5, and 13.6, respectively. The nearest loci were Im1794, Im2270, and Im3210, and the LOD peak was 5.64, ranging from 26.37 to 26.82 cM.
Three QTLs controlling Asp (qAsp-3.1, qAsp-3.2, and qAsp-3.3) were all identified on LG03 across both years, as presented in Fig. 5. The total PVE for all QTLs was 52.5% for 2018 and 41.6% for 2019, and the confidence interval ranged from 0.18 to 45.79 cM over both years, with an overlapping region ranging from 13.95 to 14.93 cM. The LOD peaks for qAsp-3.1 and qAsp-3.3 were both in 2019 at 7.70 and 5.59 cM and were near loci Im6599 and Im6160, respectively, as shown in Table 4. Interestingly, the LOD peak positions of qAsp-3.1 in 2018 and 2019 were both at 12.82 cM, close to locus Im6599, with 18.9% and 23.7% PVE, respectively. Therefore, the QTL associated with Asp was stable, revealing that qAsp-3.1 may contribute strongly to the trait. Three QTLs controlling Arg were identified on two linkage groups (LG03 and LG14), with average PVEs of 15.95 for 2018 and 14.4 for 2019. The confidence interval varied from 3.03 to 36.34 cM over both years. The LOD peaks at 5.4 and 4.42 cM in 2018 and 2019, respectively, were located at positions 12.82 and 35.69 cM near loci Im6599 and Im6287, respectively.
Figure 5.
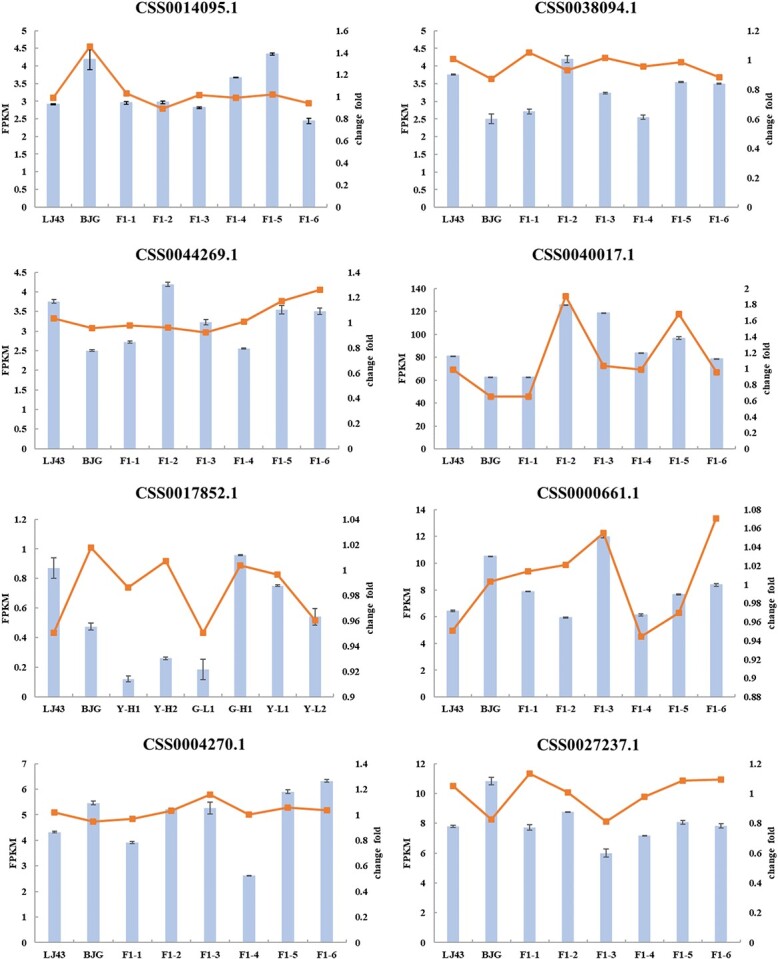
Verification of the relative gene expression levels by qRT–PCR analysis.
KEGG enrichment analysis of the DEGs
A set of 1555 DEGs, including 901 upregulated and 654 downregulated genes, was observed from the analyses of F1 offspring (Fig. 4a). To further investigate the functional profile of the identified DEGs, KEGG enrichment pathway analysis was conducted. Previously, analysis of KEGG enrichment of genes in yellow- and green-leaf progenies or their parents revealed that the photosynthetic antenna protein was the most significantly enriched [22]. However, the identification of upregulated DEGs in the F1 individuals illustrated that “riboflavin metabolism”, “tyrosine metabolism” and “arginine biosynthesis” were the most notably enriched pathways among the top 20 enriched items (Fig. 4b). Furthermore, several metabolic pathways involving amino acid components were enriched for the upregulated DEGs, which is consistent with the results of QTL mapping for genes related to amino acid transport and metabolism in the F1 population based on the genome database of tea plants.
Figure 4.

Volcano plots of DEGs (a) and the top 20 enriched KEGG pathways (b) among the free amino acid QTLs in the segregating population.
Gene expression profiling
Candidate genes that were functionally related to the free amino acid pathway were chosen for qRT–PCR analysis. Consequently, the expression profiling based on the number of mapped genes from RNA-seq (FPKM) was generally in accordance with the qRT–PCR results (Fig. 5), notably those for amino acid transporter genes. The primers designed are listed in Table S2. Furthermore, we analyzed the expression profiles of candidate genes mapped in mixed pooled samples with high and low amounts of free amino acids (AAH and AAL). As shown in Fig. 6b, the expression pattern of the parents and F1 generation with respect to AAH was significantly different that for AAL. Specifically, upregulated expression was observed in terms of nitrate transporter genes. At the same time, several downregulated genes were also found among AAH offspring at that sampling point. Pearson’s correlation analysis (Fig. 6c) illustrated that amino acid transporters were positively correlated with AA, Thea, Asp, Glu, Gln, and Arg contents. Collectively, these data suggest that it may be more difficult to elucidate the mechanism of genes affecting amino acid content simply from a transcriptional perspective. More importantly, we should pay attention to the biosynthesis and transport of amino acids to verify the function of candidate genes.
Figure 6.

Candidate gene expression profiling analysis. a Parent plants and the F1 progeny. b Expression profiles of parents and the F1 generation in mixed pooled samples. c Pearson’s correlation analysis between the amount of amino acids and the expression of candidate genes.
Discussion
A saturated genetic map plays a crucial role in the understanding of genetic mechanisms underlying horticulturally important traits via quantitative trait loci (QTL) analysis and will further facilitate molecular breeding [39]. To date, many genetic maps have been constructed by integrating dominant marker systems and have successfully identified QTL in perennial woody plants. For instance, a total of 138 QTLs for 57 flavonoids in four tissues were identified, and a major gene encoding flavanone 3-hydroxylase (F3H) was functionally verified in a Citrus reticulata × Poncirus trifoliata population [40]. In tea plants, after the first genetic map was constructed by Tanaka et al. [41], several genetic maps were reported [42]. Most recently, Xu et al. [43] genotyped F1 individuals from “LJ43” × “BHZ” using 2b-RAD sequencing and identified QTLs for catechin content. However, to our knowledge, the parents that were previously used all belong to conventional varieties, and albino segregating populations are lacking.
Here, we initially constructed an F1 albino population adopting an alternative “pseudotestcross” mapping strategy derived from an intervarietal cross between the albino landrace “BJG” and the elite green cultivar “LJ43”. A large number of F1 individuals (“BJG” × “LJ43”) were genotyped using genotyping-by-sequencing. The highest density genetic map with an average interval of 0.69 cM between adjacent markers was successfully divided into 15 linkage groups ranging from 93.41 cM to 171.28 cM.
Owing to the high self-incompatibility and low seed yield of tea plants, it is far more difficult to construct a suitable population for genotyping and QTL analysis, and selecting optimal parents and genetically superior individuals among their progeny is a vital prerequisite for translating trait loci into diagnostic genetic markers. Free amino acid contents are quantitative traits, among which the F1 population represents the average of the parents used for crossing, particularly conventional cultivars [44]. In this study, we obtained transgressively segregating albino resources for examining amino acid content, and these individuals had higher values than the parents. A total of 16 QTLs for five traits related to free amino acids distributed on 4 LGs were identified. At multiple positions, we found that the QTLs associated with Glu, Gln and Asp were combined with QTLs for Thea on LG03. Importantly, the critical interval of candidate genes related to amino acid metabolites was mined (Table S3), including cationic amino acid transporter (CAT), amino acid permease (AAP) and glutamate dehydrogenase (GDH). Among them, CAT and AAP play an important role in amino acid transport, participating in theanine transport during root-to-shoot delivery [9, 45]. Therefore, the amino acid biosynthetic and transport pathways were of interest for further analysis since theanine is a vital product of amino acid metabolism. Our study is a powerful tool for elucidating complex traits and map-based cloning of functional genes related to the amino acid content in tea plants, which will enhance marker-assisted selection during the breeding of horticultural plants. Further studies are needed to confirm that these candidate genes are linked to the underlying functions of amino acid biosynthesis and transport.
Furthermore, the pathways for the upregulated DEGs between the F1 and parents based on transcriptome data included those for several metabolic pathways for amino acid components, indicating that there are potential functional genes affecting the variation in free amino acid traits in F1 individuals. Moreover, most progenies with yellow tender shoots have higher amino acid contents to a certain extent, suggesting that there is a potential relationship between amino acid metabolites and leaf color. In a previous study, we revealed that albinism in yellow-leaf tea plants was due to repressed genes encoding LHC that were linked to aberrant chloroplast development [22] which is consistent with the reported subcellular sites of L-theanine biosynthesis in tea plants [46]. Yuan et al. [47] also found that higher amounts of amino acids in albino tea cultivar shoots were closely related to chloroplast ribosomes and the synthesis and degradation of chloroplast proteins. Collectively, among the amino acid pathways studied, it is of interest to further analyze potential genes that may simultaneously regulate both amino acid content and leaf color traits, which may provide valuable information for identifying albino resources with higher amino acid contents.
In summary, the results presented here, which combine QTLs and transcriptome analysis based on an albino genetic population, identified a number of QTLs for free amino acid content, providing an important foundation for gene discovery and cloning of amino acid biosynthetic pathway genes, especially those related to theanine, in tea plants. Moreover, the study lays a foundation for marker-assisted selection in breeding to enhance the genetic improvement of tea plants by dissecting important metabolites. In the future, we will continue to adopt a strategy to fine map and identify QTL mapping intervals to further identify favorable target genes and verify their function. For example, bulked segregant analysis (BSA) uses DNA or RNA pools that are constructed based on target traits with extreme phenotypes among the segregating population to enhance the precision and resolution of QTL identification [30, 48].
Materials and methods
Plant materials and DNA extraction
The mapping population composed of 198 F1 individuals was derived from an intervarietal, controlled cross between Camellia sinensis (L.) O. Kuntze “Baijiguan” (“BJG”) and the elite green tea cultivar “Longjing 43” (LJ43), using “LJ43” as the female parent (Fig. 6a). In addition, 300 F1 individuals were used to collect phenotypic data. The male parent “BJG”, with white and yellow shoots is one a popular landrace for making oolong tea. The female parent “Longjing 43” is selected from a seedling landrace of “Longjingzhong”, which is used to make Longjing tea. All individuals were planted in the National Germplasm Hangzhou Tea Repository of TRICAAS, China. The “two and a bud” (one bud with two leaves) samples were collected from each individual and its parents for DNA extraction, following an improved CTAB method described by Dellaporta et al. [49], for use in subsequent experiments.
Library preparation and SNP marker mining
Genomic DNA from each parent and F1 progeny was incubated with MseI, T4 DNA ligase, ATP, and MseI adapter at 37°C, followed by digestion using HaeIII and NlaIII. PCR amplification was performed using a diluted restriction-ligation mixture, dNTPs, Taq DNA polymerase and MseI primers containing barcodes. After purification, the PCR products were pooled, and a genotyping-by-sequencing library was constructed for paired-end sequencing (2 × 75 bp) using the Illumina HiSeq platform. Clean data were used for cluster detection to filter tags with sequencing depths less than 20× via BWA and SAMtools [50], avoiding false-positive SNPs. Mismatches within the sequence for the same tag were considered a haplotype and regarded as one potential SNP marker, and these markers were used for subsequent genetic mapping.
Genetic map construction
Using the high-quality SNP markers and genotyping, the genetic map was constructed using JoinMap with the pseudotestcross strategy. All markers were clustered into different linkage groups (LGs) using the selected SNP based on a pairwise modified independence LOD score as a distance matrix. The distance and sequence were calculated for each marker by a regression mapping algorithm to construct an integrated genetic map using MapChart. Recombination between marker pairs belonging to the same genetic bin was calculated as zero on each LG if there was linkage.
Free amino acid extraction and identification
The contents of free amino acids in each parent and progeny were determined using an automatic amino acid analyzer (Sikam S433D) with two replicates in the spring of 2018 and 2019. The “two and a bud” of the first flush were immediately harvested and steamed at 120°C for 5 min and then dried to constant weight at 80°C for 1 hour. The Sikam S433D method was carried out following national standard GB/T30987–2014 with minor modifications. First, 0.1 g fine powder was extracted in a 90°C water bath for 30 min with 10 mL boiling water. The supernatant was collected, and the volume was made up to 10 mL with distilled water and subsequently filtered through a hydrophilic nylon membrane filter with a 0.22 μm pore size. Afterward, the solution was mixed 1:1 (v:v) with the sample diluent and stored at 4°C for testing.
The sample was detected using a lithium system with a 50 μl injection volume. The flow rate was 0.45 mL/min, and the reactor temperature was maintained at 35°C and divided into S2100 and S4300 modules. In S2100, the reaction reagent was ninhydrin solution at 0.25 ml/min, and spectral data were collected at 280 nm. In S4300, the reaction reagent was buffer A, B, C, and regeneration solution D. The detailed configuration method of the buffer and regeneration solution is shown in Table S4. The elution time was 130 min, and a gradient elution program was used (Table S5).
Free amino acid data analysis
IBM SPSS Statistics version 24.0 was used for the canonical correlation analysis of free amino acid contents in the F1 population and parents. The maximum, minimum, mean, standard deviation (SD), skewness, coefficient of variation (CV) and kurtosis in the F1 population were calculated for each trait in Excel. R 4.0.3 software was used to analyze the significant differences between the mean values of parental traits with a t test and detected the distribution and variation of free amino acids in the parents and F1 population with ggplot2.
QTL mapping
MapQTL 6.0 combined with the constructed genetic map was used for phenotypic QTL mapping with interval mapping (IM) and restricted multiple QTL model (rMQM) methods [51]. The LOD score threshold for QTL declaration was determined for the free amino acid trait across years using the permutation test (1000 replications). Based on the displacement test, if the LOD score of each linkage group exceeded the genome-wide significance threshold of 95%, then a QTL was detected. A QTL with >20% phenotypic variance explained (PVE) was identified as a major QTL.
Gene expression analyses using RNA-seq and qRT–PCR
Total RNA was isolated using the RNAprep Pure Plant Kit (Tiangen Biotechnology Beijing, China) following the manufacturer’s recommendations. The first-strand cDNA was synthesized by random primers and reverse transcriptase with RNA as the template, and complete cDNA was subsequently generated using the Illumina TruSeq RNA Sample Prep Kit (USA). cDNA libraries were constructed with an insert fragment size of 450 bp, assessed with an Agilent 2100 Bioanalyzer (USA), and sequenced by the Illumina HiSeq2000 system with paired-end reads. Adapters and low-quality reads (average mass fraction lower than Q20) were trimmed by Cutadapt software [52] in accordance with default parameters. The clean reads were mapped to the Camellia sinensis var. sinensis genome [53] using Tophat2 [54]. Fragments per kilobase of exon model per million mapped reads (FPKM) were extracted to estimate the differential expression levels of protein-coding genes (p value <0.05).
The validation of gene expression was conducted by using quantitative qRT–PCR, and first-strand cDNA was conducted as described with the PrimeScript™ RT Reagent Kit (TaKaRa). The relative expression was calculated using the 2 −ΔΔCt method.
Analysis of the differentially expressed genes (DEGs)
The differential expression levels of protein-coding genes between offspring were determined based on DESeq and the ggplots2 R package with a |log2foldchang| >1 and p value <0.05, the low free amino acid content offspring were used as controls. The KEGG enrichment analysis of DEGs (Table S6) was conducted using KAAS software under the bidirectional best hit (BBH) parameter with a false discovery rate (FDR) <0.05.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32072631, U19A2030), the China Agricultural Research System of MOF and MARA, and the Chinese Academy of Agricultural Sciences through the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2017-TRICAAS) to LC.
Author contributions
C.L., M.C.L., and Y.M.Z. conceived and designed the research. H.R. and W.J.Y. conducted the experiments. H.R. and W.J.Y. analyzed the data. H.R. wrote the manuscript. C.L. revised the manuscript.
Data availability statements.
The raw sequencing data obtained here have been deposited into the NCBI Sequence Read Archive (SRA) under the BioProject accession no. PRJNA747741.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
References
- 1. Wu LY, Lv YQ, Ye Y.et al. . Transcriptomic and translatomic analyses reveal insights into the developmental regulation of secondary metabolism in the young shoots of tea plants (Camellia sinensis L.). J Agric Food Chem. 2020;68:10750–62. [DOI] [PubMed] [Google Scholar]
- 2. Ma LL, Cao D, Liu YL.et al. . A comparative analysis of the volatile components of green tea produced from various tea cultivars in China. Turk J Agric For. 2019;43:451–63. [Google Scholar]
- 3. Jiang H, Zhang M, Wang D.et al. . Analytical strategy coupled to chemometrics to differentiate Camellia sinensis tea types based on phenolic composition, alkaloids, and amino acids. J Food Sci. 2020;85:3253–63. [DOI] [PubMed] [Google Scholar]
- 4. Gramza-Michałowska A. Caffeine in tea Camellia sinensis content, absorption, benefits and risks of consumption. J Nutr Health Aging. 2014;18:143–9. [DOI] [PubMed] [Google Scholar]
- 5. Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Crit Rev Food Sci Nutr. 2003;43:89–143. [DOI] [PubMed] [Google Scholar]
- 6. Zhou P, Feng Z, Chen F.et al. . Determination of 21 free amino acids in 5 types of tea by ultra-high performance liquid chromatography coupled with tandem mass spectrometry (UHPLC–MS/MS) using a modified 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) method. J Food Compos Anal. 2019;81:46–54. [Google Scholar]
- 7. Williams JL, Everett JM, D’Cunha NM.et al. . The effects of green tea amino acid L-Theanine consumption on the ability to manage stress and anxiety levels: a systematic review. Plant Food Hum Nutr. 2020;75:12–23. [DOI] [PubMed] [Google Scholar]
- 8. Li L, Li T, Jiang Y.et al. . Alteration of local and systemic amino acids metabolism for the inducible defense in tea plant (Camellia sinensis) in response to leaf herbivory by ectropis oblique. Arch Biochem Biophys. 2020;683:108301. [DOI] [PubMed] [Google Scholar]
- 9. Dong C, Li F, Yang T.et al. . Theanine transporters identified in tea plants (Camellia sinensis L.). Plant J. 2020;101:57–70. [DOI] [PubMed] [Google Scholar]
- 10. Cheng S, Fu X, Wang X.et al. . Studies on the biochemical formation pathway of the amino acid l-Theanine in tea (Camellia sinensis) and other plants. J Agric Food Chem. 2017;65:7210–6. [DOI] [PubMed] [Google Scholar]
- 11. Tapiero H, Mathé G, Couvreur P.et al. . Glutamine and glutamate. Biomed Pharmacother. 2002;56:446–57. [DOI] [PubMed] [Google Scholar]
- 12. Oh K, Kato T, Xu HL. Transport of nitrogen assimilation in xylem vessels of green tea plants fed with NH4-N and NO3-N. Pedosphere. 2008;18:222–6. [Google Scholar]
- 13. Sasaoka K, Kito M, Onishi Y. Some properties of the theanine synthesizing enzyme in tea seedlings. Agric Biol Chem. 1965;29:984–8. [Google Scholar]
- 14. Kottawa-Arachchi JD, Gunasekare MTK, Ranatunga MAB. Biochemical diversity of global tea [Camellia sinensis (L.) O. Kuntze] germplasm and its exploitation: a review. Genet Resour Crop Evol. 2019;66:259–73. [Google Scholar]
- 15. Adhikary R, Mandal V. L -theanine: a potential multifaceted natural bioactive amide as health supplement. Asian Pac J Trop Biomed. 2017;7:842–8. [Google Scholar]
- 16. Liang YR, Liu C, Xiang LP.et al. . Health benefits of theanine in green tea: a review. Trop J Pharm Res. 2015;14:1943–4949. [Google Scholar]
- 17. Zheng GD. Anti-obesity effects of three major components of green tea, catechins, caffeine and theanine, in mice. In Vivo. 2004;18:55–62. [PubMed] [Google Scholar]
- 18. Chen Z, Wang Z, Yuan H.et al. . From tea leaves to factories: a review of research progress in L-Theanine biosynthesis and production. J Agric Food Chem. 2021;69:1187–96. [DOI] [PubMed] [Google Scholar]
- 19. Cheng S, Fu X, Liao Y.et al. . Differential accumulation of specialized metabolite L-theanine in green and albino-induced yellow tea (Camellia sinensis) leaves. Food Chem. 2019;276:93–100. [DOI] [PubMed] [Google Scholar]
- 20. Zhang W, Zhang Y, Qiu H.et al. . Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat Commun. 2020;11:3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wheeler DS, Wheeler WJ. The medicinal chemistry of tea. Drug Dev Res. 2004;61:45–65. [Google Scholar]
- 22. Wang JY, Chen JD, Wang SL.et al. . Repressed gene expression of photosynthetic antenna proteins associated with yellow leaf variation as revealed by bulked segregant RNA-seq in tea plant Camellia sinensis. J Agric Food Chem. 2020;68:8068–79. [DOI] [PubMed] [Google Scholar]
- 23. Ma Q, Li H, Zou Z.et al. . Transcriptomic analyses identify albino-associated genes of a novel albino tea germplasm ‘Huabai 1’. Hortic. Res. 2018;5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu Q, Chen Z, Sun W.et al. . De novo sequencing of the leaf transcriptome reveals complex light-responsive regulatory networks in Camellia sinensis cv. Frontiers in plant science. 2016;7:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L, Yu FL, Yang YJ. Germplasm and Genetic Improvement of Tea Plant. Beijing: China Agricultural Science and Technology Press; 2006:28–35. [Google Scholar]
- 26. Wei Q, Wang W, Hu T.et al. . Construction of a SNP-based genetic map using SLAF-Seq and QTL analysis of morphological traits in eggplant. Front Genet. 2020;11:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Salsman E, Wang R.et al. . Meta-QTL analysis of tan spot resistance in wheat. Theor Appl Genet. 2020;133:2363–75. [DOI] [PubMed] [Google Scholar]
- 28. Hwang S, Lee TG. Integration of lodging resistance QTL in soybean. Sci Rep. 2019;9:6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang X, Yang S, Gong J.et al. . Genomic architecture of heterosis for yield traits in rice. Nature. 2016;537:629–33. [DOI] [PubMed] [Google Scholar]
- 30. Takagi H, Abe A, Yoshida K.et al. . QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013;74:174–83. [DOI] [PubMed] [Google Scholar]
- 31. Martins LB, Rucker E, Thomason W.et al. . Validation and characterization of maize multiple disease resistance QTL. G3-genes Genom. Genet. 2019;9:2905–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soriano JM, Royo C. Dissecting the genetic architecture of leaf rust resistance in wheat by QTL meta-analysis. Phytopathology. 2015;105:1585–93. [DOI] [PubMed] [Google Scholar]
- 33. Jiang CK, Ma JQ, Liu YF.et al. . Identification and distribution of a single nucleotide polymorphism responsible for the catechin content in tea plants. Hortic. Res. 2020;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen JD, Zheng C, Ma JQ.et al. . The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Hortic Res. 2020;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palumbo F, Qi P, Pinto VB.et al. . Construction of the first SNP-based linkage map using genotyping-by-sequencing and mapping of the male-sterility gene in leaf chicory. Front Plant Sci. 2019;10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curtis TY, Powers SJ, Halford NG. Effects of fungicide treatment on free amino acid concentration and acrylamide-forming potential in wheat. J Agric Food Chem. 2016;64:9689–96. [DOI] [PubMed] [Google Scholar]
- 37. Ruan J, Ma L, Yang Y. Magnesium nutrition on accumulation and transport of amino acids in tea plants. J Sci Food Agric. 2012;92:1375–83. [DOI] [PubMed] [Google Scholar]
- 38. Ueno S, Shigematsu T, Watanabe T.et al. . Generation of free amino acids and gamma-aminobutyric acid in water-soaked soybean by high-hydrostatic pressure processing. J Agric Food Chem. 2010;58:1208–13. [DOI] [PubMed] [Google Scholar]
- 39. Souleyre EJF, Bowen JK, Matich AJ.et al. . Genetic control of alpha-farnesene production in apple fruit and its role in fungal pathogenesis. Plant J. 2019;100:1148–62. [DOI] [PubMed] [Google Scholar]
- 40. Mou J, Zhang Z, Qiu H.et al. . Multiomics-based dissection of citrus flavonoid metabolism using a Citrus reticulata x Poncirus trifoliata population. Hortic. Res. 2021;8:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka J, Taniguchi F, Hirai N.et al. . Estimation of the genome size of tea (Camellia sinensis), camellia (C. japonica), and their interspecific hybrids by flow cytometry. 87th Cong, 2d sess Senate Report, ; no 2181. 2006;2006:1–7. [Google Scholar]
- 42. Xia EH, Tong W, Wu Q.et al. . Tea plant genomics: achievements, challenges and perspectives. Hortic Res. 2020;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu LY, Wang LY, Wei K.et al. . High-density SNP linkage map construction and QTL mapping for flavonoid-related traits in a tea plant (Camellia sinensis) using 2b-RAD sequencing. BMC Genomics. 2018;19:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li XJ. QTL mapping and genetic analysis for free amino acid in tea plant, master thesis. Chinese Academy of Agricultural Sciences Dissertation. 2018.
- 45. Feng L, Yang TY, Zhang ZL.et al. . Identification and characterization of cationic amino acid transporters (CATs) in tea plant (Camellia sinensis). Plant Growth Regul. 2018;84:57–69. [Google Scholar]
- 46. Fu X, Liao Y, Cheng S.et al. . Nonaqueous fractionation and overexpression of fluorescent-tagged enzymes reveals the subcellular sites of L-theanine biosynthesis in tea. Plant Biotechnol J. 2021;19:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuan L, Xiong LG, Deng TT.et al. . Comparative profiling of gene expression in Camellia sinensis L. AnJiBaiCha leaves during periodic albinism. Gene. 2015;561:23–9. [DOI] [PubMed] [Google Scholar]
- 48. Zou C, Wang P, Xu Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J. 2016;14:1941–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dellaporta SL, Wood J, Hicks JB. A plant DNA minipreparation: version II. Plant Mol Biol Rep. 1983;1:19–21. [Google Scholar]
- 50. Li H, Handsaker B, Wysoker Aet al. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ooijen JW, et al. MapQTL 6. Software for the mapping of quantitative trait loci in experimental populations of diploid species. https://www.kyazma.nl/index.php/MapQTL/.
- 52. Kechin A, Boyarskikh U, Kel A.et al. . CutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J Comput Biol. 2017;24:1138–43. [DOI] [PubMed] [Google Scholar]
- 53. Xia E, Tong W, Hou Y.et al. . The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation of tea plants. Mol Plant. 2020;13:1013–26. [DOI] [PubMed] [Google Scholar]
- 54. Kim D, Pertea G, Trapnell C.et al. . TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data obtained here have been deposited into the NCBI Sequence Read Archive (SRA) under the BioProject accession no. PRJNA747741.