

Abstract
BACKGROUND:
Heterozygous truncating variants in titin (TTNtv) cause 10–20% of idiopathic dilated cardiomyopathy (DCM). Though variants which disrupt canonical splice signals (i.e. GT, AG) at exon-intron junctions are readily recognized as TTNtv, the effects of other nearby sequence variations on splicing and their contribution to disease is uncertain.
METHODS:
Rare variants of unknown significance (VUS) located in the splice regions of highly expressed TTN exons from 203 DCM cases, 3329 normal subjects, and clinical variant databases were identified. The effects of these variants on splicing were assessed using an in vitro splice assay.
RESULTS:
Splice-altering VUS were enriched in DCM cases over controls and present in 2% of DCM patients (p=0.002). Application of this method to clinical variant databases demonstrated 20% of similar VUS in TTN splice regions affect splicing. Non-canonical splice-altering variants were most frequently located at position +5 of the donor site (p=4.4e-7) and position −3 of the acceptor site (p=0.002). SpliceAI, an emerging in-silico prediction tool, had a high positive predictive value (86–95%) but poor sensitivity (15–50%) for the detection of splice-altering variants. Alternate exons spliced out of most TTN transcripts frequently lacked the consensus base at +5 donor and −3 acceptor positions.
CONCLUSION:
Non-canonical splice-altering variants in TTN explain 1–2% of DCM and offer a 10–20% increase in the diagnostic power of TTN sequencing in this disease. These data suggest rules that may improve efforts to detect splice-altering variants in other genes and may explain the low percent splicing observed for many alternate TTN exons.
Keywords: Titin, Splicing, Splice-Altering, Variants of Unknown Significance, Cardiomyopathy, Genetics, Gene Expression and Regulation, Cardiomyopathy
Introduction
Dilated cardiomyopathy (DCM) is a progressive disease of cardiac muscle characterized by systolic dysfunction, ventricular enlargement, and heart failure.1–3 DCM can arise from a wide variety of insults to the myocardium, or be idiopathic, occurring in the absence of cardiovascular or systemic conditions. Although some cases of idiopathic DCM are known to be caused by genetic variants, the actual contribution of inherited or de novo variants to overall disease burden is uncertain. Current estimates suggest that 30–50% of idiopathic cases may have a gene mutation accounting for their disease.3–6
Next-generation sequencing of clinical cohorts has revealed that heterozygous loss-of-function variants in titin (TTN), the largest human protein, are the leading genetic cause of idiopathic DCM (Figure 1A).1,6–8 TTN truncating variants (TTNtv), which include nonsense, frameshift, splicing, and copy number variants that damage the structure of the 35991 amino acid polypeptide, account for ~15% of sporadic and ~25–30% of familial cases.7,9 Identification of pathogenic, disease-causing TTNtv can clarify the diagnosis in DCM patients without a definitive etiology and stratify risk in closely related family members. However, genetic testing often yields rare synonymous, missense, or intronic TTN variants with uncertain pathogenic effects. Current methods of variant interpretation categorize variants commonly seen in the general population (allele frequency > 1 × 10−4) as unlikely to cause disease.10,11 Additionally, of the 364 TTN exons, approximately half are alternative exons that are not included (measured by “Percent Spliced In” or PSI) in most mature TTN mRNAs.1,8,12 Variants located in these regions of TTN are also clinically classified as benign. Distinguishing the clinical consequences of the remaining rare variants of unknown significance (VUS) in highly expressed exons remains a considerable challenge.
Figure 1:
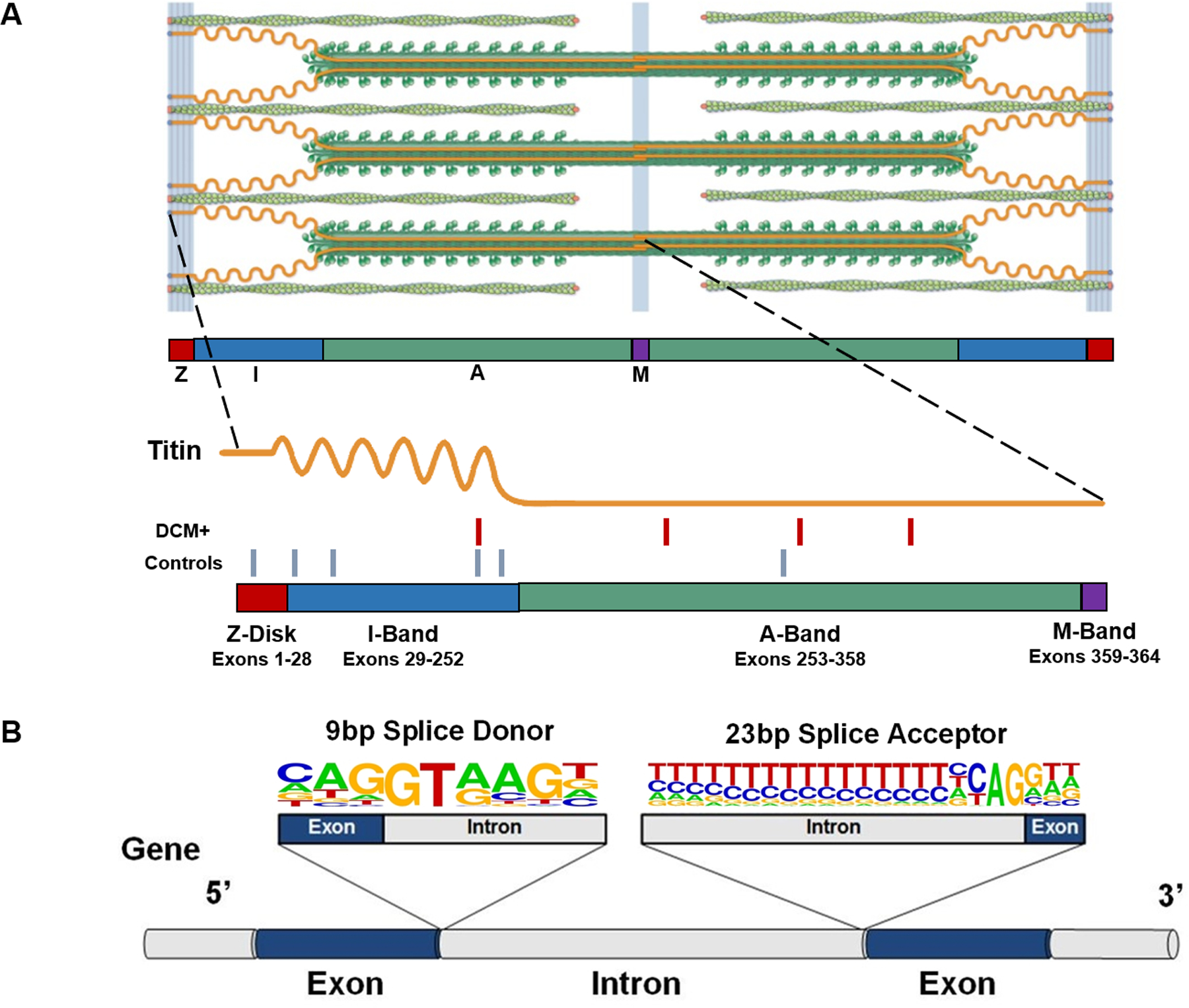
(A) The giant protein titin is a key component of the cardiac sarcomere. A standard cardiac sarcomere is composed of thick filaments (dark green), thin filaments (light green), and a filament composed of the giant protein titin (orange). A single full-length titin molecule spans the Z-disk, I-band, A-band and M-band regions, serving as a spring that preserves the precise structural integrity of the sarcomere. Being the largest human protein, the TTN gene contains 364 exons, corresponding with 726 exon-intron junctions, each of which need to be correctly recognized and processed to create a functional full-length polypeptide. The sarcomere image is adapted from Herman et al.7 with permission. The location of 4 non-canonical splice-altering variants identified in 203 DCM cases (red hash marks) and 6 non-canonical splice altering variants in 3329 controls (blue hash marks) is indicated on the schematic. (B) Splice donor and splice acceptor sites are defined by conserved 9 bp and 23 bp sequences at exon-intron junctions. A gene segment containing two exons (blue) and flanking introns (grey) with consensus 9 bp splice donor and 23 bp splice acceptor sequences is shown. The nucleotide letter sizes denote usage of that base in splice sites across the genome. Dinucleotides GT (donor site) and AG (acceptor site) are invariant, and variants that disrupt these canonical splicing residues are readily classified as TTNtv. Variants located in the other positions of these sequences may also affect splicing, but are often left labeled as VUS. TTN = Titin; bp = base pair; TTNtv = titin truncating variant; VUS = variants of unknown significance.
VUS that disrupt splicing are generally under recognized because methods for identifying splice-altering variants remain imperfect.13,14 While many residues may help guide intron splicing, nucleotides flanking the 5’ exon (donor site) and 3’ exon (acceptor site) are most frequently implicated (Figure 1B). The splice donor site comprises 9 base pairs (bp) that span the 5’ exon-intron boundary and encodes the GT dinucleotide found at the 5’ end of virtually all introns. The 23 bp splice acceptor site spans the 3’ intron-exon boundary and contains an invariant AG dinucleotide in this sequence at the 3’ end of the excised intron.15 Variants in TTN that disrupt the GT or AG dinucleotides of high PSI exons are routinely characterized as likely pathogenic or pathogenic TTNtv.1,6,8,12,16 Variants that occur within the remaining 7 bp of the splice donor site, or 21 bp of the splice acceptor site, also have the potential to disrupt splicing. However, due to time and cost restraints of earlier versions of in vitro splicing assays, these nearby sequence variations have been commonly classified as VUS.17,18
We previously described a method for identifying the variants within the splice donor and splice acceptor regions that disrupt normal splicing (Supplemental Figures I-III)13,19 and hypothesized that an analysis of VUS near TTN splice sites would expand the spectrum of pathogenic mutations that cause disease and define other VUS that are likely benign. Further, due to the large number of TTN exons and the hundreds of splice sites utilized in normal processing, we hypothesized that any patterns present in the data could provide insights into splicing biology and thereby inform variant interpretation for other disease genes.
Methods
Full details of the methods used in this study can be accessed online in the Supplemental Material. All study participants provided informed written consent and protocols were approved by the institutional human ethics committees as previously described for UK cases7 and for all other sites.20 All data and supporting materials have been provided with the published article and are available in the online supplement.
Results
Burden of Splice-Altering VUS in TTN Among DCM Cases and Controls
To compare the relative burden of TTN splice-altering VUS in DCM patients to that of ‘normal’ subjects, we analyzed TTN variants identified from 203 DCM cases (30% familial, 59% male, mean age at diagnosis 35 ± 18 years) and 3329 individuals with Alzheimer’s disease who were unlikely to have DCM. In total, 59.6% of subjects in the DCM cohort and 39.2% of subjects in the control cohort had a rare TTN variant detected by gene sequencing. As expected, TTNtv (i.e. nonsense, frameshift, and essential GT/AG splice disrupting variants) in high PSI exons were significantly enriched in individuals with DCM (23.1% vs. 0.5%, p=3.7e-47, Fisher’s exact test, Figure 2). The characteristics of these TTNtv are detailed in the Supplemental Material (Supplemental Table I). The rate of TTNtv seen in controls was similar to that seen in gnomAD v2, where 536 TTNtv in high PSI exons are noted among 141,456 unique individuals (0.4%).
Figure 2. Burden of splice-altering VUS in TTN Among DCM cases and controls.
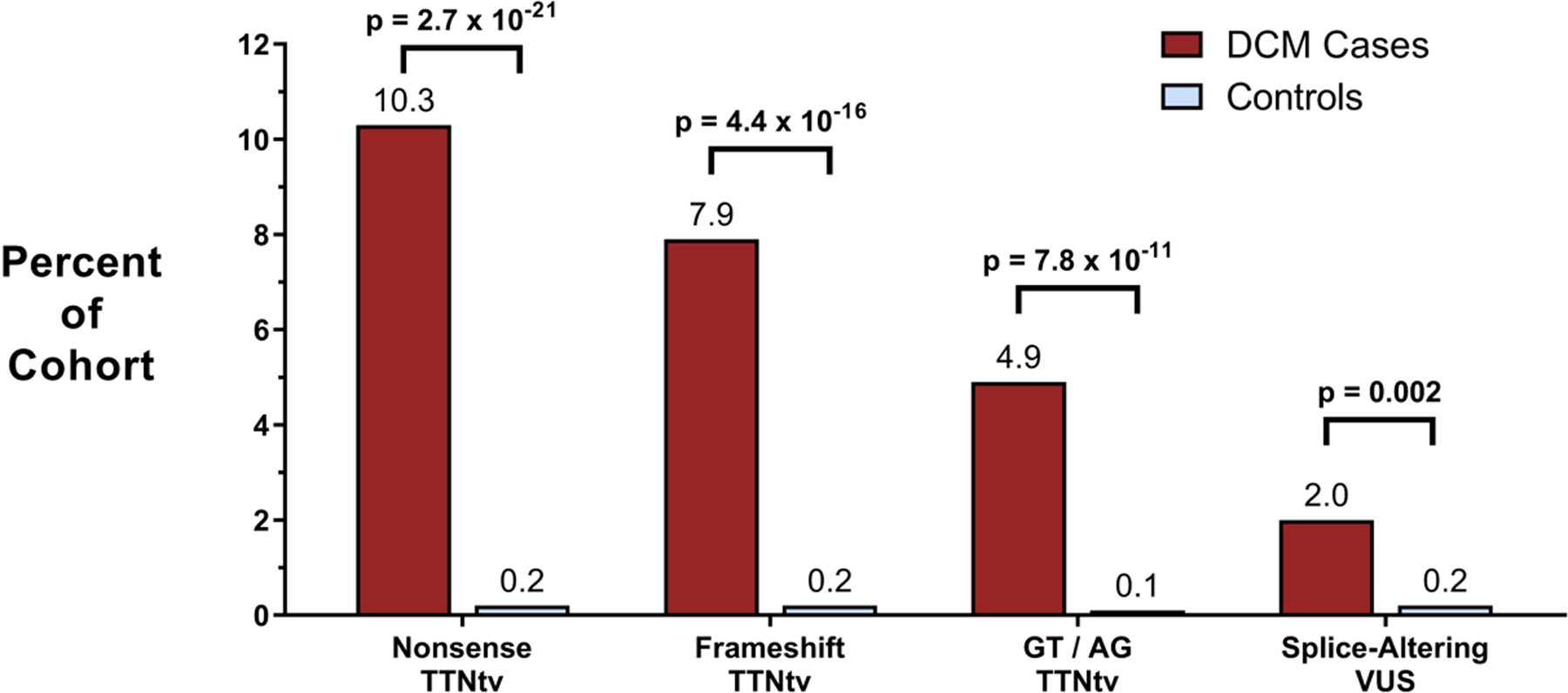
Cohorts of cases with DCM (n=203) and controls without DCM (n=3329) underwent gene sequencing of TTN. The DCM cohort had significantly more nonsense, frameshift, and canonical splice signal TTNtv than the control cohort in highly expressed exons (23.1% vs. 0.5%, p=3.7e-47), consistent with prior studies of TTNtv. In addition to these established TTNtv, a splice region VUS was present in 3.4% of cases and 1.1% of controls. Functional assessment of each VUS using the minigene assay demonstrated that splice-altering VUS were present in 2.0% of DCM cases and 0.2% of control subjects (p=0.002).
Among the remaining rare variants in these cohorts, 7 variants in cases and 36 variants in controls were located within the splice donor site or splice acceptor site of highly expressed exons, indicating that 3.4% of cases (n=7) and 1.1% of controls (n=36) possessed a rare TTN VUS near a known splice site. All 43 variants were computationally prioritized using MaxEnt scoring (an in silico splice prediction tool) and functionally assessed for splicing effects using pairs of minigenes in a cellular assay as described in the Supplemental Methods. After assessing all candidate variants in the minigene assay, 4 splice-altering VUS were identified in cases whereas 6 were found in the control cohort (Supplemental Table II). Hence, splice-altering VUS were present in 2.0% of DCM cases and 0.2% of control subjects (p=0.002, Fisher’s exact test, Figure 2).
Reclassification of Splice Region VUS in Two Databases of Human Genetic Variation
To better annotate the thousands of TTN VUS reported from clinical genetic testing, we applied our stepwise bioinformatic and functional analysis to identify additional splice-altering variants in two large databases of human genetic variation. As of 2017, the Laboratory of Molecular Medicine of Partners Healthcare had reported 6,882 unique TTN variants from clinical genetic testing of 2410 subjects. At the same time, 3,070 variants had been publicly reported by ClinVar, an NIH-sponsored database of human variants associated with clinical phenotypes, though the number of subjects from which these variants were obtained was not available. Rare TTN variants that were located within the splice donor or splice acceptor regions, but not in the canonical AG or GT sequences, of high PSI exons were identified for further study. After applying all selection criteria, 201 variants (2.0% of all TTN variants reported) were identified as rare, candidate variants. Review of the clinical significance of these variants revealed that many of these variants were classified as VUS (n=167). A smaller fraction had been classified as likely benign (n=31) or benign (n=3).
All 201 rare splice region variants from high PSI exons in TTN were bioinformatically sorted and assessed for splicing effects through the minigene assay (Supplemental Table III). Assessment of this variant set identified 45 splice-altering variants (33 LMM, 12 ClinVar); the remaining 156 splice region variants had no effect on mRNA processing (Table 1). Therefore, of the 201 rare splice region variants reported in these two clinical databases at non-canonical positions, 22.4% were determined to be likely pathogenic while 77.6% were found to be likely benign. This represented 0.5% and 1.6% of all TTN variants reported in these databases.
Table 1: Functional assessment of all splice region VUS reported in two clinical genetic testing databases.
Review of all variants submitted to the Laboratory of Molecular Medicine of Partners Healthcare and/or reported in ClinVar revealed that 201 rare variants (2.0% of all TTN variants reported) were located near TTN splice sites. Many of these were categorized as variants of unknown significance. Functional assessment of this entire variant set using an in vitro assay identified 45 splice-altering variants; the remaining 156 splice region variants had no effect on mRNA processing and are more likely to be benign.
| Splice Donor | Splice Acceptor | Total | |
|---|---|---|---|
| Total Variants Assessed | 75 | 126 | 201 |
| Variant Type | |||
| Synonymous | 7 | 10 | 17 |
| Missense | 33 | 22 | 55 |
| Intronic | 35 | 94 | 129 |
| Minigene Assay Result | |||
| Splice-Altering Variants | 25 | 20 | 45 |
| Benign Variants | 50 | 106 | 156 |
Across both cohorts, 75 GT/AG disrupting variants in high PSI exons had already been labeled as likely pathogenic or pathogenic. The identification of 45 additional splice-altering variants increased the total number of splicing TTNtv by 60%. Among the subset of variants obtained through LMM, 33 splice-altering variants, previously characterized as VUS, were present among 2410 individuals (1.4%).
Location of Splice-Altering Variants
Splice-altering TTN variants were non-randomly distributed within donor and acceptor sites (Figure 3). Of the 45 likely pathogenic TTN splice-altering variants identified in the cell based splicing assay, 25 variants were located within a 5’ donor site and 20 variants were located within a 3’ acceptor site. Given the relative length of donor and acceptor sequences, pathogenic variants were more likely to disrupt the donor site than the acceptor site (p=1.6e-04, binomial test). Among 25 splice-altering variants in the donor site, 9 perturbed the +5 position, significantly more than expected by chance (p=0.006, binomial test). Similarly, 7 of 25 splice-altering donor site variants were located at the last nucleotide of the upstream exon (donor position −1, p=0.078) (Figure 3A). In the acceptor site, most splice-altering variants were located at the −3 position (n=3, p=0.067) or the first base of the downstream exon (position +1, n=3, p=0.067; Figure 3B).
Figure 3. Location of splice-altering variants in TTN, MYPBC3, and LMNA.
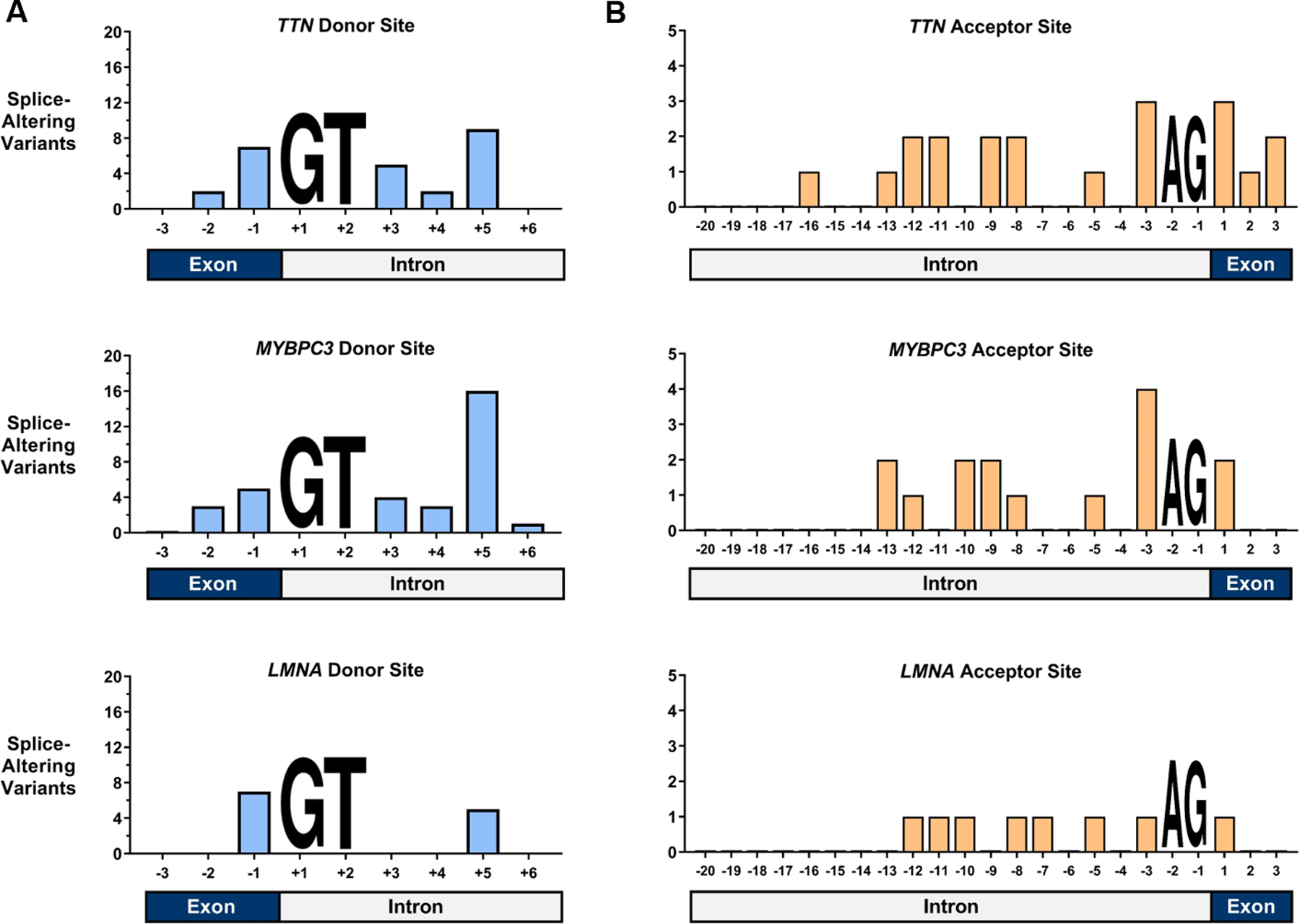
The total counts of splice-altering variants identified by direct in vitro analysis are plotted against location within the splice donor and acceptor sites. As variants that disrupted “GT” at the +1 and +2 positions of the donor site and “AG” at the −1 and −2 positions of the acceptor site were excluded from this analysis, these positions are marked with nucleotide placeholders. (A) In TTN, MYBPC3, and LMNA splice donor sites, splice-altering variants were most commonly located at the +5 position and the last base of the exon (B) In TTN, MYBPC3, and LMNA splice acceptor sites, splice-altering variants were most commonly found at the −3 position and at the first base of the downstream exon.
To assess whether this distribution of splice-altering variants was unique to TTN, we extended our application of the minigene assay to identify damaging splice region variants in MYBPC3 and LMNA, two additional autosomal dominant cardiomyopathy genes. After identifying LMM and ClinVar variants within the splice region of both genes, exclusive of common and GT/AG-disrupting variants, a total of 150 variants were identified (Supplemental Table IV).
In total, 67 of 150 rare variants tested in the minigene assay altered splicing (MYBPC3: 47, LMNA: 20, Supplemental Table IV). Again, splice-altering variants in these genes were not randomly distributed across the splice site. Splice-altering variants identified in MYBPC3 donor sites and LMNA donor sites (Figure 3A) were most frequently located at the last base of the exon (position −1, n=12, p=0.027, binomial test) and at position +5 (n=21, p=1.3e-7), significantly more than expected by chance. Similarly, splice-altering variants in MYBPC3 and LMNA acceptor sites (Figure 3B) were most commonly found at the −3 position (n=5, p=0.004).
Probability of Damaging Splice Effects by Site and Position
In a meta-analysis of all 143 rare variants reported by LMM and ClinVar in splice donor regions of the three studied cardiomyopathy genes, 48.3% (69/143) were found to be splice-altering variants by the in vitro minigene assay. Variants at donor position +5 were more likely to be splice-altering than the rest of the splice region (90.9%, p=4.4e-7, binomial test). In contrast, no splice-altering variants were identified 3 bases into the donor exon (0.0%, p=4.6e-6) and variants at the donor +6 position were unlikely to alter splicing (9.1%, p=0.01). Other positions did not deviate significantly from expectation. The fraction of rare variants which affected splicing at each position was consistent with the conservation displayed at each position of the genome-wide splice consensus sequence (Figure 4A).
Figure 4. Probability of damaging splice effects by site and position.
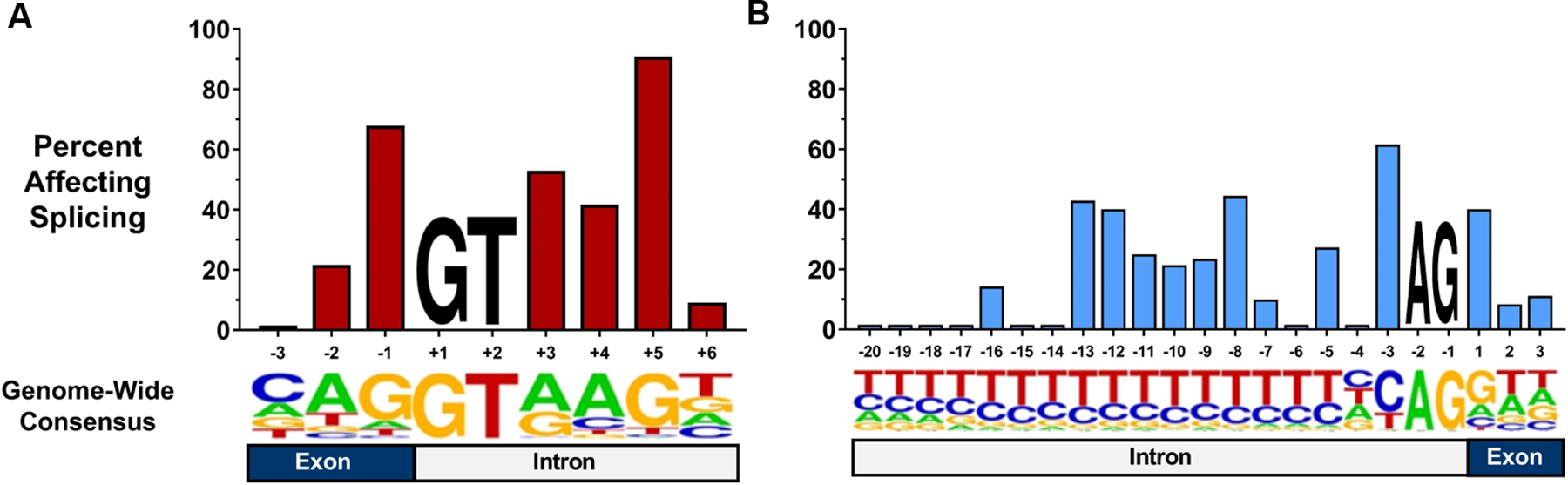
The percent of TTN, MYBPC3, and LMNA variants affecting splicing among all rare variants reported by LMM and ClinVar at the position is shown. (A) On average, 48.3% of rare variants located in a splice donor site disrupted splicing. Substitutions located at the +5 position were significantly more likely to alter splicing than this average (90.9%, p=4.4e-7). (B) In total, 20.7% of reported rare variants in splice acceptor regions affected splicing. The −3 position was more likely to contain pathogenic variants than the expectation (61.5%, p=0.002). Below each graph is a sequence logo representing the relative usage of each base at each position in splice sites across the genome. While GT and AG are invariant, other positions display variable conservation of the consensus nucleotide.
A similar meta-analysis of all 208 rare variants reported by LMM and ClinVar in splice acceptor regions of these three genes demonstrated that 20.7% (43/208) of these variants altered splicing. Only the −3 position was more likely to contain pathogenic variants than the remainder of the splice region (61.5%, p=0.002). By contrast, splice-altering variants were rarely found at positions −20 to −14 (grouped: 2.9%, p=0.006). As with the donor site, the positions that appeared to be most intolerant of variation also displayed the greatest nucleotide conservation in the splice consensus sequence (Figure 4B).
Performance of SpliceAI in Identifying Pathogenic Splice Region Variants
We assessed the performance of SpliceAI in this dataset of TTN, LMNA, and MYBPC3 variants that had been functionally characterized for their impact on mRNA splicing [n=43 from case vs control comparison (Supplemental Table II), n=351 from clinical variant databases (Supplemental Tables III, IV); n=394 in total]. Among rare variants reported in the splice regions that did not disrupt the canonical GT/AG residues, a SpliceAI score threshold ≥ 0.2 for donor or acceptor site loss resulted in a positive predictive value (PPV) of 85.7%, a negative predictive value (NPV) of 80.9%, and a sensitivity of 49.2%. When using a more stringent SpliceAI score threshold ≥ 0.8, the PPV for identifying splice site loss variants increased to 94.7%, but was accompanied by decreases in NPV to 72.3% and overall sensitivity to 14.8% (Table 2). The PPVs and NPVs seen at these thresholds were consistent across all three genes studied (Supplemental Table V). In line with the higher frequency of splice-altering variants at certain positions, variants at the +5 position of the donor site, −3 position of the acceptor site, and last nucleotide of the donor exon had the highest average SpliceAI score (Supplemental Figure IV).
Table 2: Performance of SpliceAI among all rare variants analyzed in this study.
A total of 394 variants were analyzed using an in vitro minigene assay. When all SpliceAI score types were considered (any of the four acceptor gain, acceptor loss, donor gain, or donor loss scores were greater than the threshold), the positive predictive values (PPVs) of SpliceAI scores of 0.2 and 0.8 were 76.6% and 85.7%, respectively. Limiting this analysis to predicted splice site loss (either the acceptor loss or donor loss SpliceAI scores exceeded the threshold) led to PPVs of 84.4% and 94.7% at SpliceAI scores of 0.2 and 0.8, respectively.
|
|
SpliceAI Score ≥ 0.2 | SpliceAI Score ≥ 0.8 | ||
|---|---|---|---|---|
| All Score Types Included | Acceptor / Donor Loss Only | All Score Types Included | Acceptor / Donor Loss Only | |
| True Positive | 72 | 60 | 27 | 18 |
| False Positive | 22 | 10 | 5 | 1 |
| True Negative | 200 | 262 | 267 | 271 |
| False Negative | 50 | 62 | 95 | 104 |
| PPV | 76.6% | 85.7% | 84.4% | 94.7% |
| NPV | 80.0% | 80.9% | 73.8% | 72.3% |
Exon PSI and Nucleotide Conservation
Because variants in some positions of splice donor and splice acceptor sites conferred a bigger effect on splicing than others, we hypothesized that splice site sequences in high PSI exons might differ from those of low PSI exons. Using data from the Genotype-Tissue Expression (GTEx) project, we calculated average exon PSI in skeletal (n=451 subjects), left ventricular (n=246), and right atrial (n=217) tissue. A total of 177 exons demonstrated constitutive expression (PSI ≥ 0.9) across all tissues whereas 58 exons displayed non-constitutive expression (PSI < 0.7) in all tissue types (Figure 5A), consistent with previous observations.8,12
Figure 5. Nucleotide conservation among high and low PSI exons across multiple tissue types.
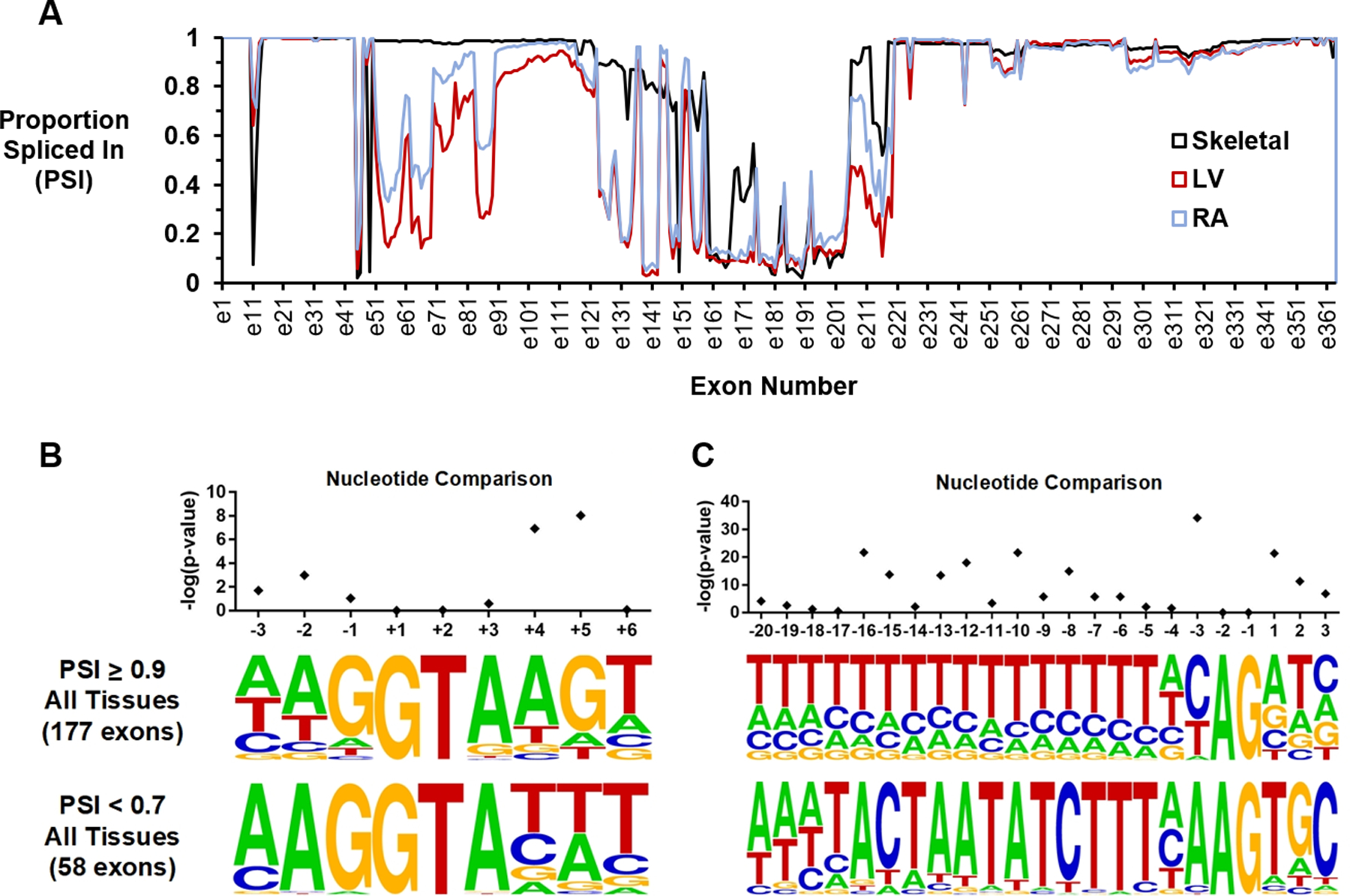
(A) Exon PSI for skeletal, left ventricular, and right atrial tissue samples is shown. One hundred and seventy-seven exons had PSI ≥ 0.90 in all three tissues, whereas 58 exons had PSI < 0.7 in all tissue types. (B) Sequence motifs for the donor sites of high and low PSI exons. The conservation of guanine at the +5 position was strongly associated with exon inclusion (p=9.1e-09). (C) Sequence motifs for the acceptor sites of high and low PSI exons. The presence of cytosine at the −3 position promoted exon inclusion (p=7.4e-35). Sequence logos were created using WebLogo (https://weblogo.berkeley.edu/logo.cgi). PSI=Proportion Spliced In.
We compared consensus donor and acceptor splice signal sequence motifs for high and low PSI exons. There were significant differences in nucleotide distribution at several non-canonical positions. In the donor site, usage of the widely-conserved guanine base at the +5 position of the donor site was significantly more frequent among constitutive versus non-constitutive exons (67.3% vs. 8.6%, p=9.1e-09, Fisher’s exact test with Holm-Bonferroni correction, Figure 5B). In the acceptor site, high PSI exons were far more likely to contain a cytosine at position −3 than low PSI exons (62.1% vs. 6.9%, p=7.4e-35, Figure 5C). A lack of conservation of the +4 adenine in the donor site (67.3% vs. 12.1%, p=1.2e-07) and of the −16 to −8 polypyrimidine tract in the acceptor site (each p<7.5e-03) were additional features strongly associated with exon exclusion.
Discussion
Using bioinformatic prioritization and an in vitro minigene splice assay we investigated the contribution of non-canonical splice-altering variants near TTN splice signals to TTNtv cardiomyopathy. Approximately 20% of rare variants of unknown significance located in TTN splice regions prevent normal mRNA processing and currently represent a group of unrecognized pathogenic variants. We provide evidence that these splice-altering VUS are enriched in cohorts of DCM and are present in 1–2% of DCM cases. Recognition of splice-altering synonymous, missense, and intronic variants in TTN improves the yield of DNA sequence-based analysis in individuals with DCM who were previously thought to have no known genetic cause for their condition.
We have previously used the same technology to identify rare variants that cause LMNA and MYBPC3 cardiomyopathies.13,19 By combining these lists of splice-altering variants in these genes we provide insights that can be broadly applicable to variants beyond the scope of this study. Krawczak et al.21 first noted that disease-causing mutations across 38 genes tended to cluster at certain positions throughout extended splice sites. Supporting these earlier findings, our data suggest that sequence variations at many positions of the splice signals are tolerated, whereas variations which disrupt the “G” at the +5 position of the donor site and the “C” at the −3 position of the acceptor site have the highest likelihood of affecting splicing. VUS at these positions in other genes that cause disease by haploinsufficiency may well be pathogenic.
The higher impact of sequence variation at the donor +5 position and acceptor −3 positions on splicing also helps to explain the large number of alternate (low PSI) exons in TTN. Low PSI exons in TTN demonstrate a paucity of guanine residues at donor position +5, cytosine residues at acceptor position −3, and exhibit poor conservation of the polypyrimidine tract in the acceptor site. The degree to which these aspects of the reference gene sequence and other trans-acting elements direct exon usage in TTN will warrant further study. We note that splicing of many TTN low PSI exons is more complicated than splicing of most alternate exons, with RBM20, a known RNA binding protein, closely regulating this process.22–24
Lastly, we have used this list of splice-altering variants validated in the mini-gene assay to provide an assessment of SpliceAI, a recently described in silico prediction tool, for the detection of pathogenic variants near exon-intron junctions. For rare non-canonical variants with a donor or acceptor loss score ≥ 0.8, the positive predictive value for the detection of splice-altering variants was 94.7%. Though prior computational splice prediction methods have been limited by poor specificity, the high positive predictive value observed with SpliceAI in this study suggests that this method may be reliably applied to identify likely pathogenic variants, especially those predicted to cause donor or acceptor site loss. This is of considerable interest given the costly and cumbersome nature of functional assays required to confirm splice-altering variants, and the exponential accumulation of rare genetic variation from clinical DNA sequencing.
We anticipate that the data provided in this study will inform reclassification of several hundred variants in publicly available genetic databases. As clinicians rely on these public resources to determine variant pathogenicity, inclusion of our findings in each database will eventually allow these results to be applied to clinical practice. Furthermore, these data should provide an important stimulus to clinical laboratories seeking to determine the pathogenicity/non-pathogenicity of other VUS reported near TTN splice sites in the absence of functional testing. Our findings suggest that VUS that are (1) located within a 9 bp donor or 23 bp acceptor splice site, (2) have allele frequency less than 1×10−4, (3) affect an exon with PSI ≥ 0.90, and (4) have a SpliceAI score ≥ 0.80 are highly likely to be pathogenic. We note that this algorithm may only identify 15% of all non-canonical splice altering variants. A need exists for clinical genetic testing programs to implement splicing minigene-like assays to assess the functional consequences of all VUS near splice sites.
In summary, the identification of DNA variants in splice regions that affect mRNA processing of the giant gene TTN increases the burden of TTNtv in DCM, improves the yield of clinical genetic testing, and suggests rules that govern exon selection in TTN transcripts.
Supplementary Material
Acknowledgments
Funding Sources:
P.N.P. was supported by a fellowship from the Sarnoff Cardiovascular Research Foundation. K.I. was supported by the Banyu Fellowship Program and the Uehara Research Fellowship Program. A.H. was supported by The National Heart, Lung, and Blood Institute (NHLBI K8HL150284) and the American Heart Association. D.F. was supported by the Victor Chang Cardiac Research Institute, Australian Genomics, NSW Health, and National Health and Medical Research Council. This work was also supported by grants from the National Heart, Lung, and Blood Institute or the National Institutes of Health (1R01HL080494 and 1R01HL084553; C.E.S. and J.G.S.), Fondation Leducq (S.A.C., C.E.S., and J.G.S.), British Heart Foundation (S.A.C.), Medical Research Council UK (S.A.C.), NIHR Cardiovascular Biomedical Research Unit at the Brompton & Harefield NHS Foundation Trust and Imperial College Biomedical Research Centre London (P.J.B. and S.A.C.), and the Howard Hughes Medical Institute (C.E.S).
Non-Standard Abbreviations and Acronyms:
- TTNtv
titin truncating variants
- DCM
dilated cardiomyopathy
- VUS
variants of unknown significance
- PSI
percent spliced in
- bp
base pairs
- LMM
Laboratory for Molecular Medicine
- GT
invariant dinucleotide of splice donor site
- AG
invariant dinucleotide of the splice acceptor site
- PPV
positive predictive value
- NPV
negative predictive value
- GTEx
Genotype-Tissue Expression Project
Footnotes
Disclosures:
Christine E. Seidman and Jonathan G. Seidman are founders of, and own shares in, Myokardia Inc., a startup company that is developing therapeutics that target the sarcomere.
References:
- 1.Yotti R, Seidman CE, Seidman JG. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu Rev Genomics Hum Genet 2019;20:129–153. [DOI] [PubMed] [Google Scholar]
- 2.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 3.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol 2010;25:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J Am Coll Cardiol 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garfinkel AC, Seidman JG, Seidman CE. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail Clin 2018;14:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol 2020;17:286–297. [DOI] [PubMed] [Google Scholar]
- 7.Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of Titin Causing Dilated Cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franaszczyk M, Chmielewski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M, Sobieszczanska-Malek M, Pollak A, Szczygieł J, Kosinska J, et al. Titin Truncating Variants in Dilated Cardiomyopathy – Prevalence and Genotype-Phenotype Correlations. PLoS One 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJL, van Heesch S, Pua CJ, Kui M, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Patel PN, Gorham JM, McDonough B, DePalma SR, Adler EE, Lam L, MacRae CA, Mohiuddin SM, Fatkin D, et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc Natl Acad Sci U S A 2017;114:7689–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, Bayrak-Toydemir P, McDonald J, Fairbrother WG. Pathogenic variants that alter protein code often disrupt splicing. Nat Genet 2017;49:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987;15:7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015;349:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaildrat P, Killian A, Martins A, Tournier I, Frébourg T, Tosi M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol Biol 2010;653:249–257. [DOI] [PubMed] [Google Scholar]
- 18.Vreeswijk MPG, Kraan JN, van der Klift HM, Vink GR, Cornelisse CJ, Wijnen JT, Bakker E, van Asperen CJ, Devilee P. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum Mutat 2009;30:107–114. [DOI] [PubMed] [Google Scholar]
- 19.Patel PN, Gorham JM, Ito K, Seidman CE. In Vivo and In Vitro Methods to Identify DNA Sequence Variants that Alter RNA Splicing. Curr Protoc Hum Genet 2018;97:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horvat C, Johnson R, Lam L, Munro J, Mazzarotto F, Roberts AM, Herman DS, Parfenov M, Haghighi A, McDonough B, et al. A gene-centric strategy for identifying disease-causing rare variants in dilated cardiomyopathy. Genet Med 2019;21:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krawczak M, Thomas NST, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat 2007;28:150–158. [DOI] [PubMed] [Google Scholar]
- 22.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest 2014;124:3419–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Morales A, Gonzalez‐Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of Novel Mutations in RBM20 in Patients with Dilated Cardiomyopathy. Clin Transl Sci 2010;3:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004;11:377–394. [DOI] [PubMed] [Google Scholar]
- 29.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 2008;44:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009;105:219–222. [DOI] [PubMed] [Google Scholar]
- 31.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJM, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009;119:1473–1483. [DOI] [PubMed] [Google Scholar]
- 32.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019;176:535–548.e24. [DOI] [PubMed] [Google Scholar]
- 33.Rowlands CF, Baralle D, Ellingford JM. Machine Learning Approaches for the Prioritization of Genomic Variants Impacting Pre-mRNA Splicing. Cells 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, Chen M-H, Raffield LM, Tardaguila M, Huffman JE, et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell 2020;182:1214–1231.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore HM. Acquisition of normal tissues for the GTEx program. Biopreserv Biobank 2013;11:75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank 2015;13:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafer S, Miao K, Benson CC, Heinig M, Cook SA, Hubner N. Alternative Splicing Signatures in RNA-seq Data: Percent Spliced in (PSI). Curr Protoc Hum Genet 2015;87:11.16.1–11.16.14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.