

Abstract

Cobalt-mediated radical polymerizations (CMRPs) have been initiated by the radical decarboxylation of tetrachlorophthalimide activated esters. This allows for the controlled radical polymerization of activated monomers across a broad temperature range with a single cobalt species, with the incorporation of polymer end groups derived from simple carboxylic acid derivatives and termination with an organozinc reagent. This method has been applied to the synthesis of a polymer/graphene conjugate and a water-soluble protein/polymer conjugate, demonstrating the first examples of CMRP in graphene and protein conjugation.
Keywords: decarboxylative coupling, polymer end groups, radical decarboxylation, activated ester, conjugation
Introduction
Cobalt-mediated radical polymerization (CMRP) is a powerful method for the synthesis of polymers with a controlled molecular weight and dispersity. The CMRP method relies on the labile nature of the Co(III)–carbon bond under thermal and photochemical conditions, allowing for polymerization of unsaturated monomers to occur by reversible deactivation or degenerative chain transfer mechanisms, depending on the monomer and Co complex used.1
Due to the facile homolytic cleavage of the Co–C bond,2 rates of CMRPs can approach those of uncontrolled free radical polymerizations.1a This makes CMRP particularly well suited to the polymerization of less activated monomers (LAMS)3 such as vinyl esters4 and amides,5 ethylene6 and 1-octene,7 and perfluoroalkylethylenes.8
The high reactivity and lower temperatures associated with many CMRP applications have limited the range of suitable radical initiators that can be used (Scheme 1). The controlled polymerization of vinyl acetate (VA) was first demonstrated using Co(acac)2 and the diazo initiator V70 at 30 °C.4a This method generates the reactive organocobalt(III) species in situ, although the requirement to store and transport V70 at −20 °C limits its broad application.
Scheme 1. Initiation of Cobalt-Mediated Radical Polymerizations.

Low-temperature initiation has also been achieved using photoinitiators, allowing for the controlled polymerization of acrylate, vinyl amide, and vinyl ketone monomers at 0 °C under UV or visible light irradiation.5,9 In addition, a redox initiation system employing ascorbic acid and benzoyl peroxide provides a scalable method for low-temperature initiation.10
Along with control of the molecular weight and dispersity of a polymer, the ability to control the polymer end groups is essential for many applications. In CMRP, the α-end group is derived from the initiating radical, or from the cleavage of the initial Co–C bond in the case of organocobalt(III) complexes. This has been exploited to introduce functional end groups through the synthesis of halomethyl–cobalt complexes, allowing for further elaboration of the polymer by azide substitution and a copper-catalyzed alkyne cycloaddition reaction.11
Due to the versatile reactivity of organocobalt(III) complexes, a number of methods have been developed for ω-end group functionalization in CMRP. Radical trapping reagents such as nitroxides11 and thiols have been used extensively to functionalize polymers and remove cobalt from the resulting materials. Additions to C6012 and carbon nanotubes13 have also been demonstrated, along with polymer–polymer coupling via addition to dienes.14
Polymer end group control is essential for the development of biological medicines based on polymer/protein conjugates. These hybrid materials can impart longer half-lives in the bloodstream compared with unfunctionalized proteins, although all FDA approved conjugates to date have utilized poly(ethylene glycol) (PEG).15 The exploration of alternatives to simple PEGylation of protein therapeutics will provide advancements in the design of polymers which improve pharmacokinetics (PK) or the rational design of novel functional materials.16
Recently, the decarboxylation of carboxylic acids and esters has emerged as a practical method for producing alkyl radicals in small-molecule cross-coupling chemistry.17 In 2016, Baran demonstrated that single-electron transfer from a NiI intermediate could trigger the radical decarboxylation of N-hydroxyphthalimide (NHP) esters,18 resulting in the formation of an alkyl–NiIII complex which could undergo coupling with an organozinc reagent.19 This method has subsequently been extended to the coupling of redox-active esters (RAEs) with a variety of partners, including aryl and vinyl halides,20 aryl boronates,21 and alkynes.22 In 2018, Wang and co-workers showed that cobalt is also an effective catalyst for the coupling of NHP esters with organozinc reagents, proceeding via the single-electron transfer from a CoI intermediate.23
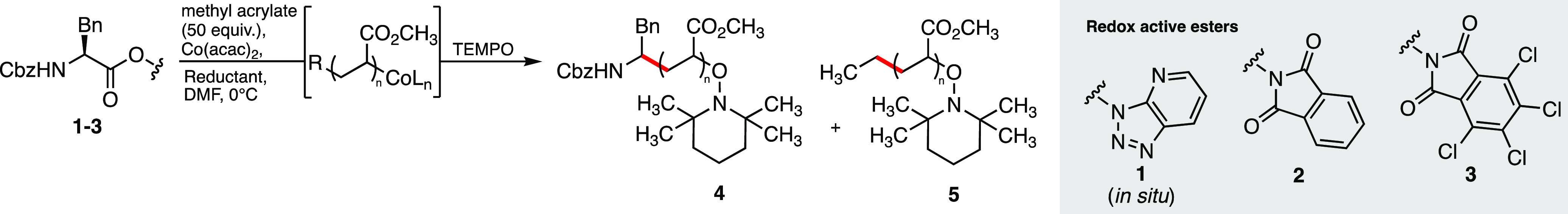
Based on this precedent, we hypothesized that the in situ generation of a CoI species would allow for single-electron transfer and radical decarboxylation of a redox-active ester, producing both the initiating radical derived from the ester functional group and the required CoII complex to mediate radical polymerization. We selected benzyloxycarbonyl (Cbz) protected phenylalanine as an appropriate starting material for the synthesis of redox-active esters 1–3, containing azabenzotriazole, phthalimide, and tetrachlorophthalimide24 activating groups, respectively.
We began our polymerization experiments employing equimolar amounts of the redox-active ester (RAE) and Co(acac)2, with 50 equiv of methyl acrylate in DMF. 1 equiv of ZnEt2 was then added, to reduce the CoII to CoI and initiate the radical decarboxylation. The ester 1 was generated in situ by reaction of the carboxylic acid with HATU (Figure 1), showing 64% conversion to the activated ester by 1H NMR analysis. Under these conditions, only 14% monomer conversion was seen after 18 h. This was almost identical to the control experiment containing no activated ester, suggesting that 1 was not undergoing the desired radical decarboxylation (Table 1, entries 1 and 2).
Figure 1.

Coupling agents used. HATU = (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate. CITU = 1,1,3,3-tetramethyl-2-(4,5,6,7-tetrachloro-1,3-dioxoisoindolin-2-yl)isouronium hexafluorophosphate.
Table 1. Optimization of Polymerization Conditions with Redox-Active Esters.
| entry | RAE | reductant | ratio RAE:Co:reductant | conversiona (%) | ratio 4:5 | Đb | Mn,theory (g/mol) | Mn,GPC (g/mol) | Mn,NMR (g/mol) | fc |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | none | ZnEt2 | 0:1:1 | 13 | ||||||
| 2 | 1 | ZnEt2 | 1:1:1 | 14 | ||||||
| 3 | 2 | ZnEt2 | 1:1:1 | 27 | ||||||
| 4 | 3 | ZnEt2 | 1:1:1 | 98 | 1:8 | 1.36 | 4600 | 19 600 | 95 600 | 0.05 |
| 5 | 3 | ZnEt2 | 1:3:3 | 98 | 1:2.4 | 1.15 | 4600 | 9300 | 21 600 | 0.21 |
| 6d | 3 | Zn | 0:1:1 | 0 | ||||||
| 7e | 3 | Zn | 1:3:3 | 88 | 4 only | 1.26 | 4100 | 8100 | 8300 | 0.49 |
| 8e | 3 | Zn | 1:3:1 | 88 | 4 only | 1.18 | 4100 | 8900 | 8100 | 0.51 |
Determined by 1H NMR spectroscopy.
Determined by GPC.
f = Mn,theory/Mn,NMR.
Run at room temperature overnight.
Run at 0 °C for 2 h and then 3–4 °C for 2 h.
While the use of isolated ester 2 resulted in a slightly increased conversion of 27%, we were pleased to see that ester 3 gave 98% monomer conversion after 3 h at 0 °C (entries 3 and 4). Following the polymerization step, the resulting polymer was functionalized by the addition of 2,2,6,6-tetramethylpiperidin-1-yloxyl radical (TEMPO) and isolated by precipitation.
An analysis of this polymer (entry 4) by gel permeation chromatography (GPC) showed a moderate dispersity (Đ = 1.36) and a molecular weight of 19 600. Conducting this polymerization with 3 equiv of cobalt and ZnEt2 (entry 5) gave an excellent dispersity of 1.15 and a reduced molecular weight of 9300. The 1H NMR spectra of this polymer showed the aromatic signals associated with the ester-derived end group (polymer 4), although the integration of this signal was lower than expected, suggesting that end group incorporation was incomplete, possibly due to the presence of other initiating radicals not derived from the activated ester.
An examination of the 1H NMR spectra of 4 (entry 5) showed a small triplet at δ 0.85 ppm, consistent with the incorporation of an ethyl group derived from ZnEt2 (polymer 5, see the Supporting Information, Figure S24). The presence of this signal, along with the 13% monomer conversion observed in the absence of the redox-active ester (Table 1, entry 1), led us to suggest that this background polymerization is initiated by an ethyl radical, possibly generated by the reaction of ZnEt2 with trace oxygen.25 A 1:2.4 ratio of Cbz-phenylalanine to ethyl end groups was observed by integration of the NMR spectra.
An analysis of this polymer (entry 5) by MALDI/TOF mass spectroscopy showed two major species, the first consistent with the incorporation of the phenylalanine-derived end group (polymer 4) and fragmentation of the TEMPO group and the Cbz imide26 (6251.56 calcd. for n = 70, found 6251.58, [M + Ag]+) and the second consistent with the incorporation of the ethyl end group (polymer 5) and loss of TEMPO (6179.53 calcd. for n = 70, found 6178.50, [M + H2O + Ag]+) (see the SI, Figure S58).
In an effort to avoid the competing initiation by the ethyl radical, we substituted the ZnEt2 reducing agent for Zn powder. In the absence of ester 3, this produced no detectable polymerization (entry 6). When a 1:3:3 ratio of 3:RAE:Co was used (entry 7), an excellent conversion and molecular weight were observed, while a 1:3:1 ratio (entry 8) gave similar results, but with slightly improved dispersity (Đ = 1.18). While this dispersity is from a polymer that has been precipitated, a GPC analysis during kinetic studies (see the SI, Figure S1) showed a dispersity of 1.29, suggesting that precipitation has only a small influence on dispersity.
A comparison of the molecular weight of this polymer (determined by 1H NMR end group analysis) with the theoretical Mn gave an initiation efficiency (f) of 0.51, where f = Mn,theory/Mn,NMR. This indicates that 51% of the functional groups derived from the decarboxylation of 3 have been successfully incorporated into the polymer. As such, the conditions in entry 8 were adopted as our standard conditions for further study.
The 1H NMR analysis of polymer 4 (Table 1, entry 8) clearly showed the presence of aromatic, benzylic, and TEMPO-derived protons, consistent with the incorporation of the desired end groups (Figure 2). An integration of the NMR indicates a 1:1 ratio of the α- and ω-end groups. Further evidence was provided by a DOSY NMR analysis, which showed the diffusion coefficients associated with the aromatic NMR signal and the TEMPO-derived signal of 5.17 × 10–10 and 5.12 × 10–10 m2 s–1, respectively, compared with 5.08 × 10–10 m2 s–1 for the signals associated with the polymer backbone. In a control experiment, the diffusion constant for the unreacted ester 3 was found to be 9.09 × 10–10 m2 s–1 (see the SI, Figure S55), indicating that the end group NMR signals in polymer 4 are not associated with small-molecule impurities. Polymer 4 was also analyzed by electrospray ionization mass spectrometry (ESI-MS) after quenching with TEMPO at low conversion (13%), showing a single major species with intact α- and ω-end groups (1293.61 calcd. for n = 10, found 1293.60, [M + Na]+) (see the SI, Figure S59).
Figure 2.

1H NMR spectra of polymer 4 (Table 1, entry 8). Molecular weights (Mn) given in g/mol.
The residual metal content of polymer 4 was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) following precipitation. This indicated initial cobalt and zinc concentrations of 800 and 400 ppm, respectively. Following treatment of the polymer with basic alumina overnight and subsequent filtration (see the SI, Scheme S10), the cobalt and zinc contents were reduced to below the limit of detection for this method (0.005 ppm for Co and 0.001 ppm for Zn corresponding to sample concentrations of <2 ppm Co and <0.5 ppm Zn).
We next examined the ability of our method to initiate polymerizations from primary, secondary, and tertiary radicals derived from functionalized carboxylic acid derivatives (Figure 3). To demonstrate the effectiveness of primary radicals, a side-chain activated glutamic acid derivative and a biotin group were successfully used to initiate polymerization, giving polymers 6 and 7, respectively. Conversions of 61% and 81% were observed, with good dispersities. To further demonstrate the effectiveness of amino-acid precursors, several amino acids were tested using a one-pot procedure where the carboxylate was activated in situ using the coupling agent CITU (Figure 1).24 Fmoc protected tBu serine and methionine were successfully used, giving polymers 8 and 10, while Fmoc-serine with a free −OH gave no polymerization (9). In addition, N-acyl tryptophan was utilized to give polymer 11, albeit with a reduced monomer conversion of 30%. This demonstrates that redox sensitive amino acids (Met and Trp) can be effectively used in this reaction, while nucleophilic residues (e.g., Ser) require side-chain protection.
Figure 3.

Functionalized polymers synthesized from redox-active esters. Molecular weights (Mn) given in g/mol.
Following the successful use of protected amino acids to initiate polymerization, we next examined several peptides, using a one-pot procedure where the carboxylate was activated in situ using the coupling agent CITU (Figure 1).24 Polymer 13 was synthesized with excellent monomer conversion and dispersity, indicating that peptides with unreactive residues were compatible with this reaction. Polymer 14, containing an unprotected tyrosine residue, was also successfully synthesized. Finally, polymer 15 was synthesized from a peptide activated by decarboxylation of an unprotected glutamate residue side chain, rather than the C-terminus. This gave a lower conversion (41%) than the previous two peptides, along with a slightly higher dispersity of 1.56.
A kinetic analysis of the polymerization to form 4 at 0 °C showed a linear dependence of Ln([M]0/[M]) versus time following an induction period of approximately 50 min (Figure 4A). A linear increase in molecular weight is observed with increasing conversion (Figure 4B). These data indicate that irreversible chain termination is not significant under these conditions and that this method shows the characteristics of a living polymerization. This was also demonstrated through chain extension experiments, where polymerization under our standard conditions gave 88% monomer conversion. This was followed by the addition of 100 equiv of MA before stirring at 6 °C for 16 h (Figure 4C). GPC analysis of the polymers before and after chain extension showed a clear shift toward higher-molecular-weight material, indicating that the organo–cobalt complex at the ω-end of the polymer is still active after the initial polymerization period. There was, however, a limit to this stability, as chain extension after an initial period of 16 h at 6 °C gave a bimodal distribution of polymers with Mn values of 9500 and 170 600. This indicates that chain termination events can be significant when the polymerization is left at higher temperatures for prolonged periods of time.
Figure 4.

(A) Plot of Ln([M]0/[M]t) vs time. (B) Plot of Mn vs monomer conversion. (C) GPC analysis of polymer before and after chain extension. Molecular weights (Mn) given in g/mol.
The origin of this chain termination event was investigated by synthesizing a low-molecular-weight polymer using 10 equiv of methyl acrylate and then allowing it to terminate by stirring at room temperature overnight. The resulting material was characterized by electrospray ionization mass spectrometry (ESI-MS; SI, Figure S60), which indicated that termination is occurring by abstraction of a hydrogen atom. Conducting this experiment in d6-DMF did not result in deuterium incorporation into the product, indicating that the solvent is not the hydrogen atom source in this process.
CMRP has been demonstrated to be an effective method for the polymerization of dimethylacrylamide (DMA), although Co–porphyrin, Co–salen, or amino–bis(phenolate) complexes are typically required for controlled polymerization.27 Given our ability to initiate CMRP at low temperatures, we examined the polymerization of DMA using Co(acac)2, in the hope that lower reaction temperatures would allow for controlled polymerization. This reaction proceeded rapidly under our optimized conditions at 0 °C, although a poor dispersity was observed (Đ = 1.99).
We were pleased to observe that lowering the reaction temperature to −42 °C resulted in a high level of monomer conversion, while delivering polymer 16 with excellent dispersity (Đ = 1.24) (Scheme 2A).
Scheme 2. Synthesis of Polydimethylacrylamide and Polymer Termination with Organozinc.

Molecular weights (Mn) given in g/mol. Coupling efficiency = percentage of ω-end groups relative to α-end groups, determine by 1H or 19F.
Based on the cobalt-catalyzed cross-coupling reaction reported by Wang,23 we hypothesized that it should be possible to intercept the organo–cobalt complex at the ω-end of the polymer with an organozinc reagent, resulting in the formation of a new C–C bond (Scheme 2B). To test this, the activated ester 3 was used to initiate polymerization under our standard conditions; however, the reaction was terminated with 6 equiv of diarylzinc in place of TEMPO. An analysis of polymer 17 by 1H NMR, DOSY NMR, and ESI-MS (SI, Figure S61) indicated the clear incorporation of the para-methoxy phenyl group with >90% incorporation relative to the α-end group determined by 1H NMR. The fluorine-containing substrate 18 was also used in this study, and the polymerization terminated with a para-trifluorophenyl group to give polymer 19. This allowed for the ratio of α- to ω-end groups to be determined by 19F NMR analysis. This showed a coupling efficiency of 56%, likely due to the reduced nucleophilicity of the fluorinated aryl group. Reducing the amount of the organozinc reagent to 3 equiv gave a reduced incorporation of 33%.
We next examined the use of our methodology for the grafting of polymers to graphene. Detrembleur and co-workers have previously shown that poly(vinyl acetate) synthesized by CMRP and end-capped by a Co(acac)2 complex can be grafted to carbon nanotubes.13 The addition of water to this material facilitates Co–C bond homolysis and liberation of a free radical. The addition of the polymer radical to carbon nanotubes results in a conjugate material with 20% polymer content by weight. A similar approach has also been used for the addition of poly(vinyl acetate) to C60, resulting in conjugates with potential applications in photodynamic therapy.12
Graphene polymer composite materials have shown superior mechanical, thermal, electrical, and gas permeability properties compared with polymers alone.28 In addition, composite materials where the graphene and polymer are covalently linked have shown improved properties over noncovalent blended materials. To our knowledge, CMRP has not been previously demonstrated for the synthesis of graphene/polymer composite materials.
In order to prepare a graphene–PMA composite using our CMRP methodology, we performed the polymerization from initiator 3, according to our optimized procedure (Figure 5A). Following the completion of polymerization, the solution of cobalt-functionalized polymer was transferred to a flask containing graphene (20 mg, 0.6–1.2 nm thickness, 400–1000 m2 g–1 surface area), and the resulting suspension was stirred at room temperature overnight. The resulting graphene composite was isolated by centrifugation and washed exhaustively with DMF, H2O, MeOH, and EtOAc to remove any unbound polymer.
Figure 5.
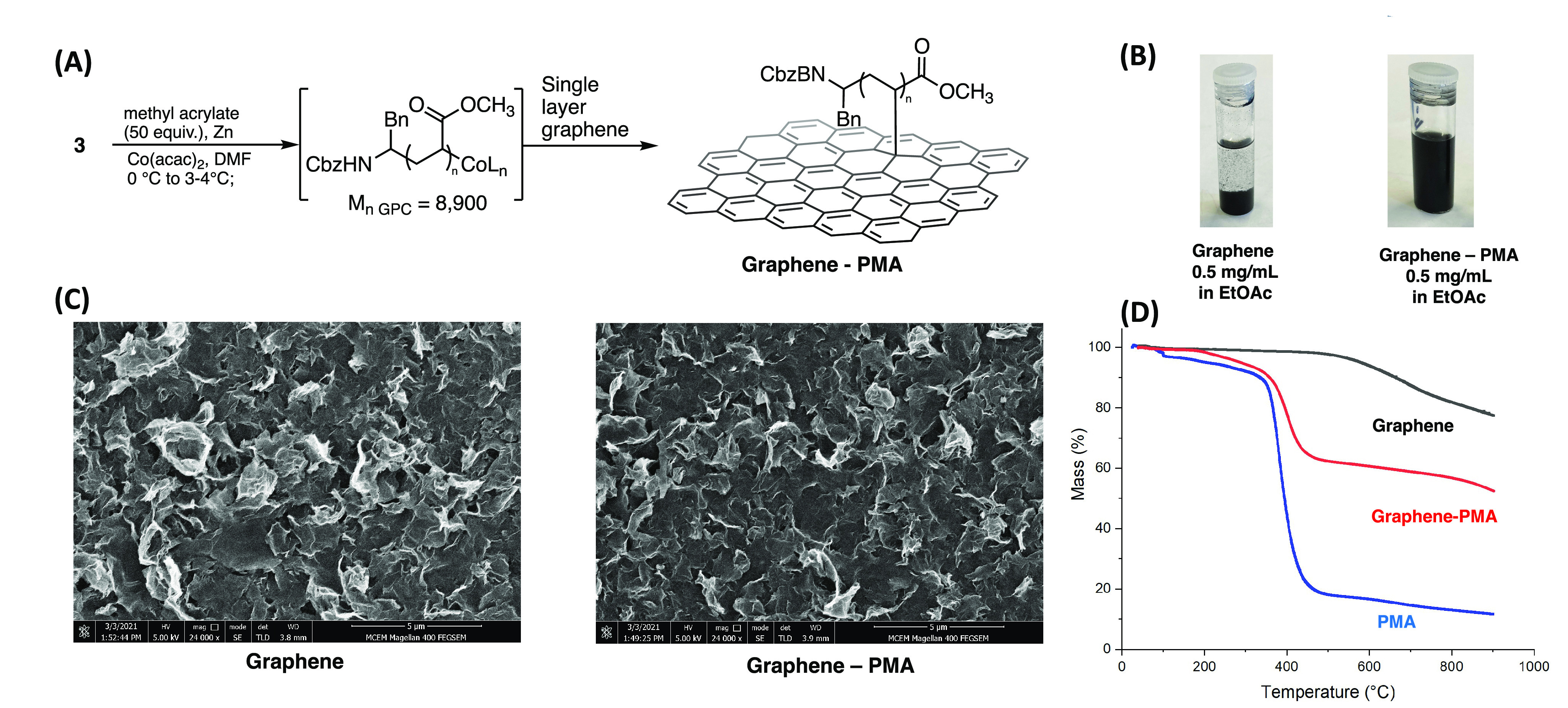
(A) Synthesis of a graphene/polymer conjugate. Molecular weights (Mn) given in g/mol. (B) Suspension of graphene and graphene–PMA conjugate in EtOAc (0.5 mg/mL), 5 min after shaking. (C) SEM images of graphene and graphene–PMA conjugate. (D) TGA analysis of graphene, bulk PMA, and graphene–PMS.
A successful functionalization of the graphene could be seen by observing the dispersion of the graphene–PMA material in organic solvents (Figure 5B). Compared with pristine graphene, a 0.5 mg/mL dispersion of graphene–PMA in EtOAc showed a high degree of dispersibility 5 min after shaking. An analysis of the graphene–PMA conjugate by scanning electron microscopy (SEM) showed a similar morphology compared with the pristine graphene material (Figure 5C). Finally, an analysis of the graphene–PMA conjugate by thermal gravimetric analysis (TGA) showed a significant mass loss at ∼400 °C, consistent with the thermal decomposition of PMA (Figure 5D). Based on the thermogravimetric (TGA) analysis, a polymer content of 30% was calculated for this material.
As a further demonstration of the applications of this methodology, we designed poly(dimethylacrylamide) 21, containing a pentafluorophenyl (PFP) ester at the α-end for the conjugation to surface lysine residues on a model protein. This functional group has been shown to selectively react with surface exposed lysine residues.29a PFP esters have been successfully incorporated into reversible addition–fragmentation chain-transfer (RAFT) and atom transfer radical polymerization (ATRP) polymer end groups for biomolecule conjugation.29b,29c The ability to incorporate PFP esters to CMRP polymers would allow the favorable properties of CMRP (low reaction temperature, diverse monomer use) to be applied in polymer–protein conjugation chemistry.
Chemical conjugation is critical for increasing the size and PK properties of therapeutic proteins, such as small mini-antibody scaffolds.30,31 Monobodies are 10 kDa binding proteins which have previously reached clinical trials using PEGylation for PK enhancement.32 This type of chemical conjugation usually requires the addition of a cysteine or glutamine residue;30 however, these mutations risk perturbing the structure and sacrificing stability when misplaced in small protein scaffolds like monobodies.34,35 Some recent PK enhancements have come from genetic fusions of albumin-binding domains or extensions of the C-terminus with PAS amino-acid repeats,33,37 although this may introduce a greater opportunity for aggregation during protein expression. We chose to conjugate an 11.6 kDa poly(dimethylacrylamide) onto lysines of the hyperstable FN3Con monobody scaffold, as the robust nature of this scaffold would be amenable to early conjugation experiments.36
The FN3Con-anti-lysozyme37 is an 11.3 kDa protein that binds the model protein lysozyme with nanomolar affinity and maintains its structure up to 87 °C. Critically, in the folded protein structure, the lysine conjugation sites are surface exposed and located on the opposite end of the molecule to the binding loops (Scheme 3). The conjugation of this protein with 50 equiv of polymer 21 for 16 h at 60 °C resulted in higher-molecular-weight conjugation products appearing as bands on SDS-PAGE (Figure 6B), with some SDS-resistant dimers of FN3Con occurring in the control sample due to the prolonged exposure to heat stress. For a further analysis, we adapted a previously established workflow for monobody conjugation analysis,38 where the protein samples undergo trypsin digestion into peptide fragments, and LC/MS is used to measure the proportionate loss of unconjugated peptides which contain the target amino acid (Figure 6A).
Scheme 3. Synthesis of the Polymer Protein Conjugate 22.
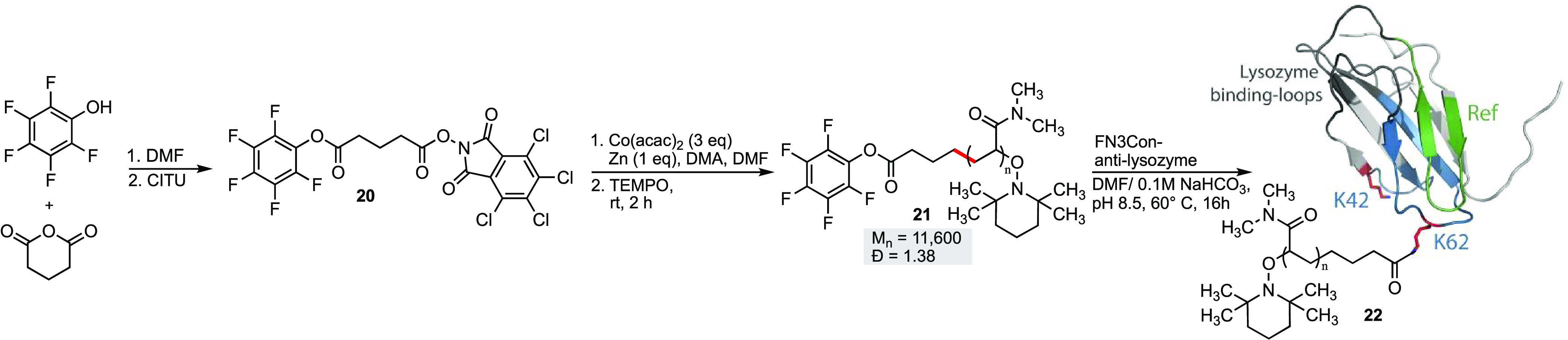
Molecular weights (Mn) given in g/mol.
Figure 6.

Conjugation experiments. (A) Amino-acid sequence of FN3Con-anti-Lys, the reference peptide for normalization of LC/MS peak area and the peptides that contain lysines for conjugation. (B) SDS-Page, after overnight incubation at 40 °C. (C) LC/MS measured a >50% decrease in the presence of either unconjugated peptide in the 20 and 30 kDa species.
We observe that the ∼20 kDa gel band of the conjugation mixture consists of mostly monomeric 11.2 kDa FN3Con-anti-lysozyme with 11 kDa 21 attached to one of the two lysines (Figure 6C). This is due to a ∼50% decrease in the presence of either unconjugated peptide fragment (Figure 6C) and also suggests that there is no preference for conjugation to one residue over the other. We also used LC/MS to investigate a very faint protein band that appeared at ∼30 kDa, which may include protein with two polymers attached (Figure 6B). Although there is a small proportion of monomer species where polymer has attached to both conjugation sites, given the >50% decrease in the K62 unconjugated peptide (Figure 6C), the results show that this band is likely to be a complex sample including some quantity of dimerized FN3Con with a single conjugate attached.
The successful conjugation of FN3Con-anti-lysozyme with polymer 21 will allow for the further study of this conjugate, with the hope that the increased size will improve the PK properties and increase the circulating half-life in vivo. This study specifically aimed at introducing a nonfunctional polymer to increase protein size; however, this chemistry can be further explored for the conjugation of functional chemical groups onto proteins of therapeutic relevance. The conjugation of a wider range of chemical groups is an avenue for improving the differentiation of the monobody scaffold.39 Additionally, the designed robustness of the FN3Con scaffold could enable earlier applications of novel functional groups than with scaffolds of lower thermal stability.40
Conclusions
We have demonstrated that the radical decarboxylation of tetrachlorophthalimide activated esters by an in situ generated CoI species can initiate CMRP. The incorporation of functional groups derived from activated esters and organozinc reagents into the α- and ω-end groups of the polymer has been demonstrated by 1H and DOSY NMR spectroscopy and mass spectrometry.
A broad range of functional groups can be included, and primary, secondary, and tertiary initiating radicals are all well tolerated.
These results demonstrate that cobalt can play three distinct roles in this polymerization reaction: facilitating the incorporation of the α-end group by radical decarboxylation, mediating the polymerization to control molecular weight and dispersity, and allowing control of the ω-end group through transmetalation and reductive elimination from organozinc reagents.
Further, we have demonstrated the application of this method to the synthesis of polymer-functionalized graphene, resulting in a composite material containing 30% PMA by mass. Finally, we have produced a proof of concept that CMRP can generate reactive polymers for conjugation to residues on a biological protein scaffold.
Acknowledgments
The authors thank the staff of Monash Centre for Electron Microscopy at Monash University for their technical assistance with SEM. This work was performed in part at the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility (ANFF).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00453.
Experimental procedures, characterization data and NMR spectra on small molecules, NMR and GPC data on all polymers, mass spectra of relevant polymers, and protein LSMC data (PDF)
Author Contributions
∇ M.A. and P.G.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Debuigne A.; Poli R.; Jerome C.; Jerome R.; Detrembleur C. Overview of cobalt-mediated radical polymerization: Roots, state of the art and future prospects. Prog. Polym. Sci. 2009, 34, 211–239. 10.1016/j.progpolymsci.2008.11.003. [DOI] [Google Scholar]; b Peng C.-H.; Yang T.-Y.; Zhao Y.; Fu X. Reversible deactivation radical polymerization mediated by cobalt complexes: recent progress and perspectives. Org. Biomol. Chem. 2014, 12, 8580–8587. 10.1039/C4OB01427H. [DOI] [PubMed] [Google Scholar]
- Demarteau J.; Debuigne A.; Detrembleur C. Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev. 2019, 119, 6906–6955. 10.1021/acs.chemrev.8b00715. [DOI] [PubMed] [Google Scholar]
- Debuigne A.; Jerome C.; Detrembleur C. Organometallic-mediated radical polymerization of ‘less activated monomers’: Fundamentals, challenges and opportunities. Polymer 2017, 115, 285–307. 10.1016/j.polymer.2017.01.008. [DOI] [Google Scholar]
- a Debuigne A.; Caille J.-R.; Jerome R. Highly Efficient Cobalt-Mediated Radical Polymerization of Vinyl Acetate. Angew. Chem., Int. Ed. 2005, 44, 1101–1104. 10.1002/anie.200461333. [DOI] [PubMed] [Google Scholar]; b Silva T. T.; Silva Y. F.; Machado A. E. H.; Maia P. I. S.; Tasso C. R. B.; Lima-Neto B. S.; Silva Sa J. L.; Carvalho-Jr V. P.; Batista N. C.; Goi B. E. Cycloalkyl-substituted salicylaldimine-nickel(II) complexes as mediators in controlled radical polymerization of vinyl acetate. J. Macromol. Sci., Part A: Pure Appl.Chem. 2019, 56, 1132–1140. 10.1080/10601325.2019.1661781. [DOI] [Google Scholar]
- Debuigne A.; Schoumacher M.; Willet N.; Riva R.; Zhu X.; Ruetten S.; Jerome C.; Detrembleur C. New functional poly(N-vinylpyrrolidone) based (co)polymers via photoinitiated cobalt-mediated radical polymerization. Chem. Commun. 2011, 47, 12703–12705. 10.1039/c1cc15471k. [DOI] [PubMed] [Google Scholar]
- Zeng T.; You W.; Chen G.; Nie X.; Zhang Z.; Xia L.; Hong C.; Chen C.; You Y. Degradable PE-Based Copolymer with Controlled Ester Structure Incorporation by Cobalt-Mediated Radical Copolymerization under Mild Condition. iScience 2020, 23, 100904. 10.1016/j.isci.2020.100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bryaskova R.; Willet N.; Degee P.; Dubois P.; Jerome R.; Detrembleur C. Copolymerization of vinyl acetate with 1-octene and ethylene by cobalt-mediated radical polymerization. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 2532–2542. 10.1002/pola.21963. [DOI] [Google Scholar]; b Kermagoret A.; Debuigne A.; Jerome C.; Detrembleur C. Precision design of ethylene- and polar-monomer-based copolymers by organometallic-mediated radical polymerization. Nat. Chem. 2014, 6, 179–187. 10.1038/nchem.1850. [DOI] [PubMed] [Google Scholar]
- Demarteau J.; Ameduri B.; Ladmiral V.; Mees M. A.; Hoogenboom R.; Debuigne A.; Detrembleur C. Controlled Synthesis of Fluorinated Copolymers via Cobalt-Mediated Radical Copolymerization of Perfluorohexylethylene and Vinyl Acetate. Macromolecules 2017, 50, 3750–3760. 10.1021/acs.macromol.7b00578. [DOI] [Google Scholar]
- a Arvanitopoulos L. D.; Greuel M. P.; King B. M.; Shim A. K.; Harwood H. J. Photochemical Polymerizations Initiated and Mediated by Soluble Organocobalt Compounds. ACS Symp. Ser. 1998, 685, 316–331. 10.1021/bk-1998-0685.ch020. [DOI] [Google Scholar]; b Detrembleur C.; Versace D.-L.; Piette Y.; Hurtgen M.; Jerome C.; Lalevee J.; Debuigne A. Synthetic and mechanistic inputs of photochemistry into the bis-acetylacetonatocobalt-mediated radical polymerization of n-butyl acrylate and vinyl acetate. Polym. Chem. 2012, 3, 1856–1866. 10.1039/C1PY00443C. [DOI] [Google Scholar]
- Bryaskova R.; Detrembleur C.; Debuigne A.; Jerome R. Cobalt-Mediated Radical Polymerization (CMRP) of Vinyl Acetate Initiated by Redox Systems: Toward the Scale-Up of CMRP. Macromolecules 2006, 39, 8263–8268. 10.1021/ma060339r. [DOI] [Google Scholar]
- Debuigne A.; Caille J.-R.; Jerome R. Synthesis of End-Functional Poly(vinyl acetate) by Cobalt-Mediated Radical Polymerization. Macromolecules 2005, 38, 5452–5458. 10.1021/ma047726q. [DOI] [Google Scholar]
- Detrembleur C.; Stoilova O.; Bryaskova R.; Debuigne A.; Mouithys-Mickalad A.; Jerome R. Preparation of Well-Defined PVOH/C60 Nanohybrids by Cobalt-Mediated Radical Polymerization of Vinyl Acetate. Macromol. Rapid Commun. 2006, 27, 498–504. 10.1002/marc.200500871. [DOI] [Google Scholar]
- Thomassin J.-M.; Molenberg I.; Huynen I.; Debuigne A.; Alexandre M.; Jerome C.; Detrembleur C. Locating carbon nanotubes (CNTs) at the surface of polymer microspheres using poly(vinyl alcohol) grafted CNTs as dispersion co-stabilizers. Chem. Commun. 2010, 46, 3330–3332. 10.1039/c000125b. [DOI] [PubMed] [Google Scholar]
- Debuigne A.; Poli R.; De Winter J.; Laurent P.; Gerbaux P.; Dubois P.; Wathelet J.-P.; Jerome C.; Detrembleur C. Cobalt-Mediated Radical Coupling (CMRC): An Unusual Route to Midchain-Functionalized Symmetrical Macromolecules. Chem. - Eur. J. 2010, 16, 1799–1811. 10.1002/chem.200902618. [DOI] [PubMed] [Google Scholar]
- a Pelegri-O’Day E. M.; Lin E.-W.; Maynard H. D. Therapeutic Protein–Polymer Conjugates: Advancing Beyond PEGylation. J. Am. Chem. Soc. 2014, 136, 14323–14332. 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]; b Moncalvo F.; Martinez E. M. I.; Cellesi F. Nanosized Delivery Systems for Therapeutic Proteins: Clinically Validated Technologies and Advanced Development Strategies. Front. Bioeng. Biotechnol. 2020, 8, 89. 10.3389/fbioe.2020.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. H.; Maynard H. D. A guide to maximizing the therapeutic potential of protein–polymer conjugates by rational design. Chem. Soc. Rev. 2018, 47, 8998–9014. 10.1039/C8CS00606G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Fu H. Visible-Light Photoredox Decarboxylative Couplings. Asian J. Org. Chem. 2017, 6, 368–385. 10.1002/ajoc.201600513. [DOI] [PubMed] [Google Scholar]
- Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. Photosensitized decarboxylative Michael addition through N-(acyloxy)phthalimides via an electron-transfer mechanism. J. Am. Chem. Soc. 1991, 113, 9401–9402. 10.1021/ja00024a074. [DOI] [Google Scholar]
- Cornella J.; Edwards J. T.; Qin T.; Kawamura S.; Wang J.; Pan C.-M.; Gianatassio R.; Schmidt M.; Eastgate M. D.; Baran P. S. Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc. 2016, 138, 2174–2177. 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huihui K. M. M.; Caputo J. A.; Melchor Z.; Olivares A. M.; Spiewak A. M.; Johnson K. A.; DiBenedetto T. A.; Kim S.; Ackerman L. K. G.; Weix D. J. Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc. 2016, 138, 5016–5019. 10.1021/jacs.6b01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Qin T.; Chen T.-G.; Wimmer L.; Edwards J. T.; Cornella J.; Vokits B.; Shaw S. A.; Baran P. S. Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids. Angew. Chem., Int. Ed. 2016, 55, 9676–9679. 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Huang L.; Olivares A. M.; Weix D. J. Reductive Decarboxylative Alkynylation of N-Hydroxyphthalimide Esters with Bromoalkynes. Angew. Chem., Int. Ed. 2017, 56, 11901–11905. 10.1002/anie.201706781. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith J. M.; Qin T.; Merchant R. R.; Edwards J. T.; Malins L. R.; Liu Z.; Che G.; Shen Z.; Shaw S. A.; Eastgate M. D.; Baran P. S. Decarboxylative Alkynylation. Angew. Chem., Int. Ed. 2017, 56, 11906–11910. 10.1002/anie.201705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.-G.; Zhou C.-J.; Lin E.; Han X.-L.; Zhang S.-S.; Li Q.; Wang H. Decarboxylative Negishi Coupling of Redox-Active Aliphatic Esters by Cobalt Catalysis. Angew. Chem., Int. Ed. 2018, 57, 13096–13100. 10.1002/anie.201806799. [DOI] [PubMed] [Google Scholar]
- deGruyter J. N.; Malins L. R.; Wimmer L.; Clay K. J.; Lopez-Ogalla J.; Qin T.; Cornella J.; Liu Z.; Che G.; Bao D.; Stevens J. M.; Qiao J. X.; Allen M. P.; Poss M. A.; Baran P. S. CITU: A Peptide and Decarboxylative Coupling Reagent. Org. Lett. 2017, 19, 6196–6199. 10.1021/acs.orglett.7b03121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal J. Polymerization of olefin oxides and of olefin sulfides. J. Polym. Sci., Part A-1: Polym. Chem. 1966, 4, 1163–1177. 10.1002/pol.1966.150040516. [DOI] [Google Scholar]
- Schaiberger A. M.; Moss J. A. Optimized sample preparation for MALDI mass spectrometry analysis of protected synthetic peptides. J. Am. Soc. Mass Spectrom. 2008, 19, 614–619. 10.1016/j.jasms.2008.01.010. [DOI] [PubMed] [Google Scholar]
- a Zhao Y.; Dong H.; Li Y.; Fu X. Living radical polymerization of acrylates and acrylamides mediated by a versatile cobalt porphyrin complex. Chem. Commun. 2012, 48, 3506–3508. 10.1039/c2cc00114d. [DOI] [PubMed] [Google Scholar]; b Chen X.; Wang L.; Li Q.; Zhang J.; Wang J.; Sun J.; Zhang Y. Reversible-deactivation radical polymerizations of acrylamide mediated by cobalt complexes supported by amino-bis (phenolate) ligands. J. Macromol. Sci., Part A: Pure Appl.Chem. 2020, 57, 743–750. 10.1080/10601325.2019.1691453. [DOI] [Google Scholar]
- Layek R. K.; Nandi A. K. A review on synthesis and properties of polymer functionalized graphene. Polymer 2013, 54 (19), 5087–5103. 10.1016/j.polymer.2013.06.027. [DOI] [Google Scholar]
- a Pham G. H.; Ou W.; Bursulaya B.; Di Donato M.; Herath A.; Jin Y.; Hao X.; Loren J.; Spraggon G.; Brock A.; Uno T.; Geiersanger B. H.; Cellitti S. E. Tuning a Protein-Labeling Reaction to Achieve Highly Site Selective Lysine Conjugation. ChemBioChem 2018, 19, 799–804. 10.1002/cbic.201700611. [DOI] [PubMed] [Google Scholar]; b McRae S.; Chen X.; Kratz K.; Samanta D.; Henchey E.; Schneider S.; Emrick T. Pentafluorophenyl ester-functionalized phosphorylcholine polymers: preparation of linear, two-arm, and grafted polymer–protein conjugates. Biomacromolecules 2012, 13, 2099–2109. 10.1021/bm3004836. [DOI] [PubMed] [Google Scholar]; c Vanparijs N.; Maji S.; Louage B.; Voorhaar L.; Laplace D.; Zhang Q.; Shi Y.; Hennink W. E.; Hoogenboom R.; De Geest B. G. Polymer-protein conjugation via a ‘grafting to’approach–a comparative study of the performance of protein-reactive RAFT chain transfer agents. Polym. Chem. 2015, 6, 5602–5614. 10.1039/C4PY01224K. [DOI] [Google Scholar]
- Vazquez-Lombardi R.; Phan T. G.; Zimmermann C.; Lowe D.; Jermutus L.; Christ D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today 2015, 20, 1271–1283. 10.1016/j.drudis.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Crook Z. R.; Nairn N. W.; Olson J. M. Miniproteins as a powerful modality in drug development. Trends Biochem. Sci. 2020, 45, 332–346. 10.1016/j.tibs.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mamluk R.; Carvajal I. M.; Morse B. A.; Wong H.; Abramowitz J.; Aslanian S.; Lim A.-C.; Gokemeijer J.; Storek M. J.; Lee J.; Gosselin M.; Wright M. C.; Camphausen R. T.; Wang J.; Chen Y.; Miller K.; Sanders K.; Short S.; Sperinde J.; Prasad G.; Williams S.; Kerbel R.; Ebos J.; Mutsaers A.; Mendlein J. D.; Harris A. S.; Furfine E. S. Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2. MAbs 2010, 2, 199–208. 10.4161/mabs.2.2.11304. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schiff D.; Kesari S.; de Groot J.; Mikkelsen T.; Drappatz J.; Coyle T.; Fichtel L.; Silver B.; Walters I.; Reardon D. Phase 2 study of CT-322, a targeted biologic inhibitor of VEGFR-2 based on a domain of human fibronectin, in recurrent glioblastoma. Invest. New Drugs 2015, 33, 247–253. 10.1007/s10637-014-0186-2. [DOI] [PubMed] [Google Scholar]
- Zhou Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines 2017, 5, 64. 10.3390/biomedicines5040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg S. D.; Cardoso R. M. F.; Lin T.; Spinka-Doms T.; Klein D.; Jacobs S. A.; Dudkin V.; Gilliland G.; O’Neil K. T. Engineering a targeted delivery platform using Centyrins. Protein Eng., Des. Sel. 2016, 29, 563–572. 10.1093/protein/gzw054. [DOI] [PubMed] [Google Scholar]
- a Jacobs S. A.; Gibbs A. C.; Conk M.; Yi F.; Maguire D.; Kane C.; O’Neil K. T. Fusion to a highly stable consensus albumin binding domain allows for tunable pharmacokinetics. Protein Eng., Des. Sel. 2015, 28, 385–393. 10.1093/protein/gzv040. [DOI] [PubMed] [Google Scholar]; b Aghaabdollahian S.; Ahangari Cohan R.; Norouzian D.; Davami F.; Asadi Karam M. R.; Torkashvand F.; Vaseghi G.; Moazzami R.; Latif Dizaji S. Enhancing bioactivity, physicochemical, and pharmacokinetic properties of a nano-sized, anti-VEGFR2 Adnectin, through PASylation technology. Sci. Rep. 2019, 9, 1–14. 10.1038/s41598-019-39776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebski B. T.; Nickson A. A.; Hoke D. E.; Hunter M. R.; Zhu L.; McGowan S.; Webb G. I.; Buckle A. M. Structural and dynamic properties that govern the stability of an engineered fibronectin type III domain. Protein Eng., Des. Sel. 2015, 28, 67–78. 10.1093/protein/gzv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebski B. T.; Conroy P. J.; Drinkwater N.; Schofield P.; Vazquez-Lombardi R.; Hunter M. R.; Hoke D. E.; Christ D.; McGowan S.; Buckle A. M. Circumventing the stability-function trade-off in an engineered FN3 domain. Protein Eng., Des. Sel. 2016, 29, 541–550. 10.1093/protein/gzw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.; Goldberg S.; Lin T.; Dudkin V.; Widdison W.; Harris L.; Wilhelm S.; Jmeian Y.; Davis D.; O’Neil K.; Weng N.; Jian W. Bioanalytical workflow for novel scaffold protein–drug conjugates: quantitation of total Centyrin protein, conjugated Centyrin and free payload for Centyrin–drug conjugate in plasma and tissue samples using liquid chromatography–tandem mass spectrometry. Bioanalysis 2018, 10, 1651–1665. 10.4155/bio-2018-0201. [DOI] [PubMed] [Google Scholar]
- Chandler P. G.; Buckle A. M. Development and differentiation in monobodies based on the fibronectin type 3 domain. Cells 2020, 9, 610. 10.3390/cells9030610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler P. G.; Broendum S. S.; Riley B. T.; Spence M. A.; Jackson C. J.; McGowan S.; Buckle A. M. Strategies for increasing protein stability. Methods Mol. Biol. 2020, 2073, 163–181. 10.1007/978-1-4939-9869-2_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.