

Abstract
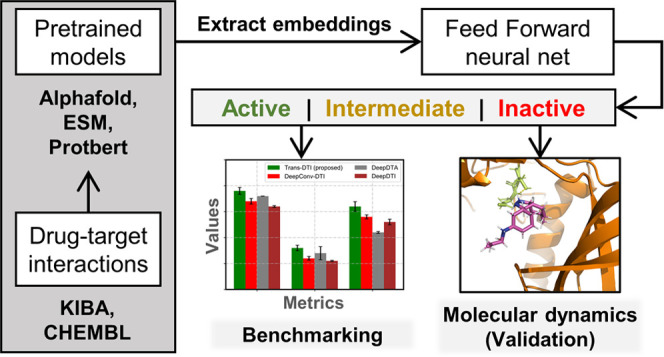
The identification of novel drug–target interactions is a labor-intensive and low-throughput process. In silico alternatives have proved to be of immense importance in assisting the drug discovery process. Here, we present TransDTI, a multiclass classification and regression workflow employing transformer-based language models to segregate interactions between drug–target pairs as active, inactive, and intermediate. The models were trained with large-scale drug–target interaction (DTI) data sets, which reported an improvement in performance in terms of the area under receiver operating characteristic (auROC), the area under precision recall (auPR), Matthew’s correlation coefficient (MCC), and R2 over baseline methods. The results showed that models based on transformer-based language models effectively predict novel drug–target interactions from sequence data. The proposed models significantly outperformed existing methods like DeepConvDTI, DeepDTA, and DeepDTI on a test data set. Further, the validity of novel interactions predicted by TransDTI was found to be backed by molecular docking and simulation analysis, where the model prediction had similar or better interaction potential for MAP2k and transforming growth factor-β (TGFβ) and their known inhibitors. Proposed approaches can have a significant impact on the development of personalized therapy and clinical decision making.
1. Introduction
Identification of novel drug–target interactions (DTIs) is generally a stagnant, labor-intensive, and precarious process. A conventional drug discovery and development pipeline can burn through a billion USD, and more importantly, around 14 years.1,2 Assay-based protocols in a drug discovery workflow follow several steps, including lead identification, optimization, screening, and characterization, eventually escalating the financial and temporal burden. Alternatively, computational methods have gathered pace for their utility in predicting novel drug–target interactions and aiding the process of drug discovery.3,4 Although traditional methods beat in silico alternatives in terms of reliability and robustness, experimental characterization of every possible drug–target is not practical owing to its low-throughput nature.
Traditional DTI prediction workflows can be categorized into three classes: (i) ligand-based approaches, (ii) docking-based approaches, and (iii) chemogenomic approaches.5−7 In DTI prediction, computational approaches are divided into three major groups. Molecular similarity serves as a deciding criterion for ligand-based approaches.8 However, due to insufficient data regarding various targets, this approach can be erroneous. Similarly, docking-based approaches rely on molecular structures and sophisticated algorithms/software to simulate interactions between the drug–target pair under consideration. The biggest bottleneck of such an approach is the nonavailability of quality three-dimensional (3D) protein structures.9 Experimental techniques for solving a protein’s crystal structure are time-taking and labor-intensive processes. For instance, solving the 3D structure for targets like G protein-coupled receptors (GPCRs) is still challenging.10 Therefore, docking-based approaches can only cover a fraction of the entire DTI spectrum. Alternatively, chemogenomic approaches try to evade the drawbacks of the aforementioned methods by concurrently employing the information of drug and target to establish their association.
Advances in sequencing technologies have enabled the collection of vast amounts of biological data.11 Data at such a scale have presented a golden opportunity for developing powerful sequence-based approaches for modeling the protein structure and functions, eventually aiding DTI prediction. Similar to grammatical rules responsible for the working of natural languages, biological sequences hold semantic and syntactical information that govern their functioning, mechanism of action, and utility in the central dogma of molecular biology. Extracting such semantic information from biological sequences can help understand the biochemical properties of novel sequences and estimate interactions among entities without explicitly formalizing their biophysical or biochemical mechanisms.12−14 There have been multiple attempts at formalizing an efficient workflow capable of extracting efficient representations of molecular data. SPVec, DTI-CDF, and DTI-MLCD are some of the many attempts in this direction.15−17 Self-supervised deep language modeling has recently been employed for biological sequences. More specifically, MolTrans and TransformerCPI are a couple of transformer-based methods developed for DTI estimation and have demonstrated improved performance in comparison to conventional methods.18,19 In particular, such encoder-based models have shown great potential for modeling protein sequences. Models like Bidirectional Encoder Representations from Transformers (BERT) rely on efficient network architectures that are specifically designed for sequence data and are pretrained with massive data sets.20,21 For instance, the attention-based transformer is one such model that has depicted immense effectiveness in a range of benchmark data sets.22,23
To incorporate advances in transformer-based protein and drug embedding methods, we have developed TransDTI, a modular framework that incorporates molecular embeddings from various language models and estimates DTI for a given drug–target pair. To evaluate the effectiveness of TransDTI, we have compared 10-fold cross-validated results with existing methods like DeepConvDTI, DeepDTA, and DeepDTI.24−26 Moreover, the predicted DTIs were analyzed through a molecular dynamics (MD) workflow to establish the effectiveness of the model estimates.
2. Results
With comparative analysis against baseline and current methods, a model architecture with 10 models is proposed in this study. The seed architecture over which all models were built is described in Figure S1. The proposed models accurately predicted the interactions in test sets and demonstrated a high level of effectiveness, as quantified by multiple performance metrics. The general schematic of the entire workflow is compiled as Figure 1.
Figure 1.

General overview of the overall methodology. (A) Primary DTI data were collected, processed, and screened. (B) Embeddings were generated for protein and drug sequences using multiple language model-based methods. (C) Extracted embeddings were used to train a fully connected feedforward deep neural network for classification and regression task. (D) Molecular docking and dynamics workflow for validating the activity of predicted drug–target interactions.
2.1. Performances of Methods under Evaluation and Selected Hyperparameters
Following a standardized protocol, various aspects of the deep learning model were tuned: the learning rate, number of hidden layers, dropout characteristics, and activation functions. As described earlier, classification and regression models were trained separately that were evaluated on their respective metrics. A modular platform was built so that different types of embeddings could be tested on the same architectural parameters. The entire model architecture is depicted in Figure S1. The effect of using different families of sequence embedders, namely, ESM, ProtBert, and AlphaFold, was examined. On average, ESM family models gave a Matthew’s correlation coefficient (MCC) of ∼0.71 and an R2 of ∼0.77. On similar lines, models built on ProtBert and AlphaFold showed slightly better MCC and R2 of ∼0.72 and ∼0.76, respectively. These values significantly outperformed sequence and CTD-based methods. Similar trends were observed in the case of other metrics like the area under precision recall (auPR) and the area under receiver operating characteristic (auROC).
2.2. Comparison of Performance with Other Molecular Descriptors and Existing Models
Independent test data sets were employed to compare the performance of the proposed method with other molecular descriptor (sequence and CTD)-based workflows. The results showed that the protein embedding families of models consistently outperformed the conventional approaches for all evaluation metrics (Figure 2A,B). Further, a model built using CTD features performed better than the one trained using only one-hot sequence encodings. It may be interpreted that CTD features have a higher level of predictive capacity when compared with one-hot encoded sequence features (Table 1).
Figure 2.

Comparison of performances among all of the methods under observation. (A) MCC and (B) R2 statistics of all of the methods under consideration indicate the effectiveness of TransDTI over baseline methods in predicting the type of interaction for a given drug–target pair. (C) Comparison with existing methods also validates the effectiveness of the proposed method. It should be noted that the AlphaFold model was employed for the comparisons. (D) auROC and auPR statistics for the three classes are depicted for the proposed models.
Table 1. Tenfold Cross-Validated Performance Metrics for the Various Models under Considerationab.
| auROC |
auPR |
|||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | I | II | III | MCC | R2 | |
| sequence | 0.8152 (0.0135) | 0.8413 (0.0080) | 0.8155 (0.0075) | 0.6351 (0.0215) | 0.8569 (0.0267) | 0.7803 (0.0129) | 0.6587 (0.0097) | 0.7085 (0.0127) |
| CTD | 0.8461 (0.0156) | 0.8894 (0.0102) | 0.8376 (0.0088) | 0.6558 (0.0298) | 0.8733 (0.0115) | 0.8021 (0.0108) | 0.6865 (0.0120) | 0.7259 (0.0136) |
| ESM1 | 0.9305 (0.0037) | 0.9593 (0.0023) | 0.9210 (0.0028) | 0.7066 (0.0107) | 0.9563 (0.0029) | 0.8798 (0.0055) | 0.7136 (0.0043) | 0.7653 (0.0058) |
| ESM1v | 0.9300 (0.0035) | 0.9594 (0.0021) | 0.9211 (0.0024) | 0.7061 (0.0128) | 0.9568 (0.0023) | 0.8811 (0.0058) | 0.7140 (0.0065) | 0.7722 (0.0079) |
| ESM1b | 0.9305 (0.0035) | 0.9598 (0.0021) | 0.9212 (0.0026) | 0.7074 (0.0120) | 0.9574 (0.0024) | 0.8806 (0.0059) | 0.7152 (0.0062) | 0.7704 (0.0062) |
| ProtBert | 0.9304 (0.0036) | 0.9603 (0.0023) | 0.9218 (0.0024) | 0.7052 (0.0120) | 0.9581 (0.0027) | 0.8823 (0.0054) | 0.7151 (0.0054) | 0.7657 (0.0068) |
| ProtBert-BFD | 0.9291 (0.0030) | 0.9599 (0.0023) | 0.9207 (0.0032) | 0.7023 (0.0113) | 0.9578 (0.0025) | 0.8797 (0.0066) | 0.7144 (0.0074) | 0.7663 (0.0058) |
| ProtAlbert | 0.9305 (0.0039) | 0.9596 (0.0022) | 0.9212 (0.0027) | 0.7047 (0.0124) | 0.9572 (0.0027) | 0.8812 (0.0052) | 0.7161 (0.0070) | 0.7700 (0.0083) |
| ProtT5XL | 0.9305 (0.0031) | 0.9603 (0.0017) | 0.9219 (0.0020) | 0.7062 (0.0132) | 0.9580 (0.0023) | 0.8809 (0.0055) | 0.7175 (0.0057) | 0.7713 (0.0042) |
| ProtT5XLBFD | 0.9298 (0.0045) | 0.9601 (0.0017) | 0.9214 (0.0025) | 0.7043 (0.0139) | 0.9572 (0.0021) | 0.8807 (0.0046) | 0.7159 (0.0061) | 0.7694 (0.0064) |
| ProtXLNet | 0.9299 (0.0038) | 0.9599 (0.0019) | 0.9207 (0.0026) | 0.7031 (0.0138) | 0.9577 (0.0021) | 0.8809 (0.0070) | 0.7131 (0.0044) | 0.7668 (0.0070) |
| AlphaFold | 0.9378 (0.0036) | 0.9668 (0.0021) | 0.9289 (0.0025) | 0.7096 (0.0108) | 0.9593 (0.0023) | 0.8914 (0.0054) | 0.7141 (0.0069) | 0.7787 (0.0072) |
In addition to comparison with conventional descriptor-based models, the performance of TransDTI was also compared against a few recently developed DTI prediction models, namely, DeepDTI, DeepDTA, and DeepConvDTI.24−26 While DeepDTI employs deep belief networks (DBNs) to process amino acid, dipeptide, and tripeptide compositions, and drug topological fingerprints estimate DTIs, DeepDTA and DeepConvDTI employ protein sequences and drug SMILES to estimate affinity between the given drug–target pair.
Working codes were extracted for all three methods from their GitHub repositories. As the existing models were being evaluated in a binary classification setup, the model presented in this study was trained for a binary classification task using only class I (active) and class III (inactive) data points. Minor changes on the final layers were induced on the original codes to train the classification tasks for a fair comparison. The results of comparative analysis in terms of accuracy, MCC, and F1 score for the binary classification task are compiled in Figure 2C. The proposed model in this study (TransDTI) performs better than related models in all three comparison metrics. This can be attributed to the greater ability of language models to encode residue-level information in their embeddings.
2.3. Models Trained with Random Embeddings Depicted the Importance of Choosing Appropriate Evaluation Metrics
In addition to using embeddings from various methods, models were also trained on randomly generated data. As evident from Figure 3A–I, the models performed very poorly for both the tasks (classification Figure 3A–F and regression Figure 3G–I) when analyzed using metrics like R2, auPR, and MCC. However, other metrics such as class-wise accuracy and auROC deemed these random (partial or complete) models effective by giving them a higher score. Therefore, the choice of performance metrics is essential in determining the actual effectiveness of ML models. The performance of the proposed models (TransDTI) in terms of MCC and R2 reinforces the inference that embeddings generated with language models have predictive ability and can efficiently encode the semantic and structural information hidden in biological sequences.
Figure 3.

Performance of prediction models on partial random data. (A–D) Prediction losses, MAE, and R2 trends for models trained on random protein and drug embeddings. (E, F) Model performance in terms of ROC and PR curves for randomized data. R2 scores for regression models trained on (G, H) partial and (I) complete random data.
2.4. External Validation on Gold-Standard Data Sets Reinforces the Robustness of TransDTI
TransDTI demonstrated high predictive ability with KIBA data sets used for training and testing. However, model generalizability is considered to be the ultimate criterion of an effective ML workflow. Although performance was calculated using the holdout test data sets, the effectiveness of the proposed models was externally validated on gold-standard external data sets from DTI-MLCD.15 In addition to two baseline methods, three descriptor methods and two transformer-based methods (MolTrans and TransformerCPI) were included for this analysis.18,19 As evident from Table 2, TransDTI performed exceedingly well, further reinforcing its effectiveness. Out of the four protein classes in the external validation data set, TransDTI outperformed all other methods under consideration in three classes (GPCR, enzymes, and nuclear receptors). In the case of ion channels, the performance of TransDTI was not comparable with other methods, as its architecture has been optimized for a three-class classification setup and might be marginally less effective even after being retrofitted into a binary classifier for the purpose of comparison.
Table 2. External Validation on Gold-Standard Data Sets Showed the Effectiveness of TransDTI against Baseline and Competing Methodsac.
| validation set | metric | CTD | Seq | DeepDTA | DeepConvDTI | DeepDTI | MolTrans | TransforerCPI | TransDTIb |
|---|---|---|---|---|---|---|---|---|---|
| GPCR | accuracy | 0.61 | 0.57 | 0.73 | 0.68 | 0.74 | 0.72 | 0.69 | 0.77 |
| MCCd | 0.43 | 0.41 | 0.58 | 0.67 | 0.61 | 0.59 | 0.56 | 0.69 | |
| F1 score | 0.56 | 0.53 | 0.63 | 0.68 | 0.59 | 0.62 | 0.59 | 0.60 | |
| sensitivity | 0.57 | 0.52 | 0.67 | 0.67 | 0.54 | 0.55 | 0.54 | 0.58 | |
| specificity | 0.69 | 0.64 | 0.78 | 0.72 | 0.83 | 0.84 | 0.80 | 0.85 | |
| enzyme | accuracy | 0.58 | 0.53 | 0.62 | 0.57 | 0.71 | 0.69 | 0.67 | 0.75 |
| MCCd | 0.45 | 0.38 | 0.47 | 0.41 | 0.53 | 0.55 | 0.57 | 0.59 | |
| F1 score | 0.53 | 0.51 | 0.59 | 0.56 | 0.68 | 0.65 | 0.64 | 0.70 | |
| sensitivity | 0.56 | 0.50 | 0.53 | 0.53 | 0.67 | 0.61 | 0.59 | 0.68 | |
| specificity | 0.60 | 0.57 | 0.69 | 0.64 | 0.64 | 0.77 | 0.77 | 0.81 | |
| ion channel | accuracy | 0.62 | 0.56 | 0.67 | 0.59 | 0.64 | 0.65 | 0.62 | 0.63 |
| MCCd | 0.39 | 0.37 | 0.49 | 0.38 | 0.46 | 0.45 | 0.49 | 0.48 | |
| F1 score | 0.51 | 0.48 | 0.62 | 0.56 | 0.60 | 0.56 | 0.55 | 0.59 | |
| sensitivity | 0.47 | 0.44 | 0.60 | 0.52 | 0.57 | 0.51 | 0.50 | 0.57 | |
| specificity | 0.72 | 0.67 | 0.72 | 0.67 | 0.69 | 0.76 | 0.72 | 0.70 | |
| nuclear receptors | accuracy | 0.54 | 0.57 | 0.58 | 0.61 | 0.65 | 0.71 | 0.64 | 0.74 |
| MCCd | 0.34 | 0.31 | 0.35 | 0.43 | 0.47 | 0.52 | 0.48 | 0.55 | |
| F1 score | 0.51 | 0.52 | 0.53 | 0.56 | 0.62 | 0.58 | 0.50 | 0.68 | |
| sensitivity | 0.41 | 0.47 | 0.50 | 0.50 | 0.61 | 0.52 | 0.45 | 0.63 | |
| specificity | 0.65 | 0.66 | 0.67 | 0.72 | 0.69 | 0.83 | 0.77 | 0.84 |
All models are validated for a binary classification task.
Trained on AF embeddings for GPCR and ProtT5XL embeddings for the enzyme, ion channel, and nuclear receptors.
Best performing method is marked in bold for every metric and validation set.
Scaled 0–1 for consistency with other metrics.
2.5. Molecules Predicted by the Models Are at Par with Known Inhibitors in Terms of Interacting Potential
The interacting potential of predicted compounds was compared against already known protein inhibitors to gauge the effectiveness of the proposed models. The top 20 predictions from the 10 models for the two proteins under consideration are shown in Figure 4. It can be observed that the docking scores for most of the predictions were at par (comparable) with the known inhibitor. For mitogen-activated protein kinase (MAP2k), the docking score of the known inhibitor (extracted from 5Z1D) was −4.483 kcal/mol. Dock scores for the model predictions were −6.691, −9.324, and −7.332 kcal/mol for ESM1, ProtBert, and AlphaFold, respectively. Similarly, model predictions for (transforming growth factor-β (TGFβ)) were at par with the known inhibitor (Figure 4). Random molecules from the Schrodinger decoy molecule set (functionality of Desmond) were also docked at the active site. As expected, the molecules did not exhibit comparable docking results. Moreover, predictions from DeepDTI, DeepDTA, and DeepConvDTI were also docked with MAP2k and TGFβ. The effectiveness of the competing methods was underlined in the docking results, as a significant number of predicted molecules were at par with the known inhibitor. However, as evident from Figure 4, TransDTI gave better predictions, both in terms of quality and quantity. Further, to test for statistically significant differences in the docking scores among competing models, the paired sample t-test was performed. The results are compiled in Tables S1 and S2 for TGFβ and MAP2k docking scores, respectively, which agree with previously observed trends (Figure 4).
Figure 4.

Docking statistics for known and predicted molecules with MAP2k and TGFβ. Docking scores of the predicted molecules were at par with those of the known inhibitors for (A) MAP2k and (B) TGFβ. (C) The mean docking scores for all of the predictions under consideration are summarized.
2.6. Molecular Dynamics Validates the Effectiveness of Predicted Molecules
Docked protein–ligand complexes for MAP2k and TGFβ were simulated for 100 ns. Figure S2 shows the comparative analysis of sustained interactions between known inhibitor–protein and predicted compounds–protein complexes. For MAP2k, while the complex with the known inhibitor had around four interactions on average, complexes with predicted compounds had six, seven, and four interactions for ESM1, ProtBert, and AlphaFold, respectively. Moreover, no significant fluctuations were observed in the carbon backbone throughout the simulations (Figure S3). Similar results were obtained for TGFβ and its predicted molecules. Table 3 compiles the polar and nonpolar interacting residues for all of the simulations. The average poses for MAP2k and TGFβ are depicted in Figure 5, while all polar and nonpolar interactions have been depicted in Figure 6. It is evident from the docking poses and their interacting residues that predicted molecules carry a similar signature as the known inhibitor. For instance, in the case of MAP2k, it can be observed that Asp277 serves as a consistent binding partner for all of the predicted drug molecules. This finds its relevance in identifying novel drugs for a given target by incorporating similarity characteristics of already known inhibitors indirectly. Moreover, sustained interactions (Figure 5) during the course of 100 ns simulations for both the proteins under consideration provide mechanistic confidence in the prediction given by TransDTI.
Table 3. Residues of MAP2k and TGFβ Interacting with the Known and Predicted Ligands in Their Best-Docked Poses.
| types
of interactions and residues involved (premolecular dynamic simulations) |
||||
|---|---|---|---|---|
| protein | ligand | molecular docking (kcal/mol) | H-bonds | hydrophobic, polar, and π–π stacking |
| MAP2k | CID135398122a | –4.483 | Asp277 | Met142 |
| CHEMBL287306 | –6.691 | Asp277 | Lys165 | |
| CHEMBL361297 | –9.324 | Asp277, Met215, Lys165 | Gly148, Ser276, Ser263, Leu266, Met142 | |
| CHEMBL1242568 | –7.332 | Asp277, Met215 | ||
| TGFβ | DB02010a | –9.345 | His283, Asn287, Asp281 | Val211, Asn287, Lys337 |
| CHEMBL1852770 | –7.417 | His283, Asp351, Cys350 | Glu245, Phe352, Lys232, Ala230 | |
| CHEMBL1762790 | –10.876 | Asp351, Glu245, Thr280 | Lys213, Leu260, Leu340 | |
| CHEMBL1241768 | –9.751 | His283, Asp281, Thr280 | Ala230, Phe282, Lue260 | |
Figure 5.

Interaction poses for averaged structures for all of the simulated drug–target complexes for (A–D) MAP2k and (E–H) TGFβ. It can be observed that the interacting partners for the predicted compounds are similar to the ones for the known inhibitors. Also, interactions and their interacting residues are annotated.
Figure 6.

H-bond statistics throughout the simulations for (A–D) MAP2k and (E–H) TGFβ. The interactions were consistent throughout the simulations, showing the effectiveness of the prediction models. Moreover, the predicted compounds from ESM, ProtBert, and AlphaFold showed similar interacting potential as the known inhibitor.
2.7. Lower-Dimensional Visualization of Protein Embeddings
Comparative and validation results suggested that TransDTI embeddings could capture critical aspects of protein and drug sequences that are predictive of DTIs. Therefore, protein embeddings were examined using t-distributed stochastic neighbor embedding (tSNE) to generate lower-dimensional representations. Protein ANalysis THrough Evolutionary Relationships (PANTHER) was employed for ontology analysis for identifying protein classes for a subset of data points.32 Although the transformer models were not explicitly tuned for identifying protein subclasses, some level of classification could be observed (Figure S4).
3. Discussion
This study demonstrates the utility of transformer-based language models for identifying novel DTIs by employing embeddings from ESM, ProtBert, and AlphaFold families of models. In contrast to the conventional binary classification setup for predicting DTIs, we opted for a multiclass (three) classification approach by categorizing drug activity in an intermediate range (0.01 μM < IC50 < 30 μM). This was done to mitigate the ambiguity of defining a strict threshold between active and inactive drug–target pairs in a conventional setup and provide a realistic and practical solution. Transformer models extract semantic information in NLP tasks by jointly conditioning on both left and right contexts in all layers.21 This is particularly an essential feature in context to biological sequences, which are multidirectional in nature. The inclusion of robust sequence embeddings allowed the proposed models to score well with various performance metrics (Figures S5 and S6).
As described in earlier sections, the proposed methods consistently excelled at multiple validation checks and outperformed other descriptors and earlier models in the literature. The proposed models also avoided overfitting, which is evident from the training curves (Figure S7). Furthermore, molecular docking and dynamics analysis revealed molecular insights into the effectiveness of the predicted drugs in comparison to already known inhibitors. Figure S2 depicts the representative structure from eight simulation runs for the two proteins (MAP2k and TGFβ) and their model predictions under consideration.
Ultimately, TransDTI’s integrative approach recommended a set of promising drug–target interactions that could be experimentally validated as promising leads for novel cancer therapies. Although validation of the binding energies of putative drug–target interactions can only be verified by experimental screening methods, these results indicated that the proposed models could mature into promising methods for identifying novel drug–target interactions. It should be noted that the models were developed using heterogeneous classes of proteins that included transporters, transcription regulators, and junction proteins, among others (Figure S8). It can be inferred that the heterogeneous nature of the training data contributed to the effectiveness of the models.
It was observed that although TransDTI performed relatively well, there is still scope for improvement. For instance, most protein and drug embeddings except AlphaFold are entirely dependent on protein or SMILES sequences. However, structural features ideally have critical information that could be employed for developing more generalized models. Slightly better performance of the AlphaFold-based model in the regression task could be attributed to the fact that structural templates are incorporated in its pipeline.29,33 In contrast to the BFD database that was used in the original AlphaFold workflow for generating MSAs, UniRef100 was employed in the interest of computational runtimes and hardware bottlenecks.
Although DTI predictions were evaluated by external validation and comparative analysis with existing methods and conventional descriptors, as an alternative validation strategy, the proposed models were tested to predict potential drug candidates for mitogen-activated protein kinase (MAP2k) and transforming growth factor-β (TGFβ), both of which serve as essential therapeutic targets in anticancer therapy. The docked complexes were simulated in an explicit water environment to evaluate the dynamics and sustainability of interactions for an extended duration, which cannot be reliably ascertained by just molecular docking. For instance, despite good docking scores, TGFβ-CHEMBL1241678 was found to be unstable in a 100 ns MD simulation.
4. Conclusions
This study developed an end-to-end scalable framework that could understand the intricate relationships among drug–target pairs and make inferences for interactions among given drugs and targets using transformer-based language models. Extracting the ordered information present in data sets like SMILES and protein sequences was central to the idea of TransDTI. The transformer-based language models were employed to learn representations from raw sequence data and molecular descriptors and to solve a multiclass classification and regression problem. These methods were compared extensively with baseline and existing methods on various measures of performance.
The results obtained in this study reinforced the idea of using representations that try to capture the underlying order in sequential data. Including language models served the same purpose, and as a result, it was observed that there was a significant improvement in the performance compared to the baseline methods. Moreover, TransDTI’s effectiveness was evident in the external validation setup, where they consistently outperformed the baseline models with a healthy margin.
Analyzing the details of what the model is learning could be of great utility in improving the methodology further. Furthermore, the idea of using structural information of proteins for DTI prediction remains to be of immense utility, especially in the context of the recently developed AlphaFold model.34 Therefore, spatial information provided by the protein 3D structure would be incorporated in the future to enhance its effectiveness further.
5. Methods
This section describes the various elements of the entire DTI prediction workflow. The entire workflow was divided into three steps:
-
1.
Extracting protein and drug embeddings using multiple language models.
-
2.
The second phase trained a fully connected feedforward deep neural network on the extracted embeddings for predicting interaction scores as well as the interaction state.
-
3.
The final phase was aimed toward validating the model prediction using molecular docking and dynamics analysis.
A general schematic describing the various sections of the proposed workflow is shown in Figure 1.
5.1. Data Sets and Study Design
TransDTI has been designed to be compatible with regression and classification tasks with minimal architectural differences. While the regression task was straightforward, the conventional binary classification setup to predict DTIs had some inherent limitations discussed in the following sections. A large-scale kinase inhibitor bioactivity (KIBA) data set for model building and evaluation was adopted.35 The KIBA data set quantifies bioactivities of kinase inhibitors in terms of a single scoring measure derived from individual metrics like IC50, Kd, and Ki.3636 As the activity thresholds were not well defined in the KIBA data set, IC50 values for the interacting pairs were extracted directly from ChEMBL. Although a large pool (∼0.2 M) of drug–target interactions was retrieved from ChEMBL, a healthy chunk of it was filtered out due to nonstandard/missing activity values and incomplete information. Of 30 474 compounds, 961 targets and 61 624 interactions were finally screened after all of the preprocessing steps. The summary statistics of all of the data sets employed is compiled in Table 4.
Table 4. Summary of All of the Data Sets Used in the Studya.
| proteins | compounds | interactions | ||
|---|---|---|---|---|
| training | KIBA (IC50)b | 961 | 30 474 | 61 624 |
| validationc (DTI-MLCD) | enzyme | 1411 | 1777 | 7371 |
| GPCR | 156 | 1680 | 5383 | |
| nuclear receptors | 33 | 541 | 886 | |
| ion channel | 204 | 210 | 1476 |
KIBA data set was employed for training and internal validation, while the gold-standard data set from DTI-MLCD was used for external validation.
Proteins for drugs listed in the KIBA data set were extracted manually from ChEMBL.
Used as an external validation data set.
5.2. Formulation of the Problem
The proposed methodology followed a two-step process: (i) Training a multiclass classification problem on given drug–target pairs. (ii) Inferring the interactions among unknown drug–target and validating using an external data set to infer real-world performance.
A multiclass classification approach was chosen for an efficient understanding of drug–target interactions rather than the more conventional binary classification. The reason for this is that (i) most of the binary classification tasks tend to label nontested drug–target combinations as a negative data point and (ii) even in the case where one has information regarding the activity profile of the drug–target pair (in terms of IC50, Kd or Ki), a single activity threshold was not uniformly followed in the literature. Further, the conventional binary classification task had some inherent drawbacks and inadequacies. The most evident is the need for a predefined binarization threshold, which is often arbitrarily decided.
To mitigate the issues mentioned above, binding affinities were segregated into three categories based on the magnitude of their value. The entire batch of retrieved data points was categorized into three classes, namely, (i) class I: active, (ii) class II: intermediate, and (iii) class III: inactive, owing to the disadvantages of a binary formulation as described earlier. For instance, an IC50 value of <0.1 μM was considered a good indicator of an active drug–target interaction, and such drug–target pairs were marked as positive data points.37 As indicated earlier, due to no distinct separation among strong and weak binding affinities, a threshold of >30 μM was set for an interaction to be categorized as an noninteracting (negative) example. Everything in between was labeled as intermediate interactions. Following this criterion, a total of 7057 active (class I), 24 752 intermediate (class II), and 28 748 (class III) inactive interactions were fed into the classification and regression models.
5.3. Protein and Drug Embeddings
Drug embeddings were generated using SMILES transformer, a pretrained language model-based utility that learns molecular fingerprints through an unsupervised sequence-to-sequence language model employing extensive SMILES data.38
Multiple transformer protein language models were employed for generating protein embeddings in addition to conventional sequences and chemical descriptor-based features. The protein embedding could be categorized under three families, ESM, ProtBert, and AlphaFold. While the ESM family of embeddings had models like ESM1, ESM1v, and ESM1b, the ProtBert family included ProtBert, ProtBert-BFD, Prot-T5-XL, Prot-T5-XL-BFD, ProtAlbert, and ProtXLNet.27,28 As most of the mentioned language models do not explicitly provide the functionality to extract data embeddings, custom scripts were written on top of the publicly provided code for the task. Embeddings from AlphaFold were extracted by reverse engineering the open-source code on their GitHub repository and ColabFold.29,33 Similarly, embeddings for other methods were extracted using the resources provided in GitHub. Details regarding the model architecture and parameters for each model are compiled in Table 5.
Table 5. Summary of All of the Protein Embedding Models Employed for Developing the Proposed Methodology.
| shorthand | layers | params | data set | embedding dim |
|---|---|---|---|---|
| ESM1 | 34 | 670M | UR50/S2018_03 | 1280 |
| ESM1b | 33 | 650M | UR50/S2018_03 | 1280 |
| ESM1v | 33 | 650M | UR90/S2020_03 | 1280 |
| ProtBert | 30 | 420M | UniRef100 | 1024 |
| ProtBert-BFD | 12 | 224M | UniRef100 | 4096 |
| ProtAlbert | 30 | 420M | BFD100 | 1024 |
| Prot-T5-XL | 24 | 3B | UniRef100 | 1024 |
| Prot-T5-XL-BFD | 24 | 3B | BFD100 | 1024 |
| ProtXLNet | 30 | 409M | UniRef100 | 1024 |
| AlphaFold | 48a | 92M | UniRef100 | 384 |
Evoformer blocks.
5.4. Model Architecture
Once the embeddings for protein and drugs were extracted, a fully connected feedforward deep neural network was developed and optimized for classification and regression tasks separately. While the underlying architecture remained the same for the two tasks, the final layer of the network was modified for their activations and shapes.
All of the hidden layers had an exponential linear unit (elu) as an activation function.
| 1 |
5.4.1. Loss and Model Optimization
For the multiclass classification problem, categorical cross-entropy was used as the loss function. It was defined as a sum of losses for each class labels coming out of a SoftMax function and is mathematically given by
| 2 |
Here, N is the number of classes (three in this case), i is the data point, yi,c is the binary target indicator [0,1], and p is the model prediction. In the case of the regression task, the mean squared error (MSE) was employed as the loss function.
As for the regression setup, the mean squared error (MSE) was employed as the loss function, which is the average of the squared differences between the actual and predicted values. It can be mathematically represented as follows
| 3 |
where n is the number of samples, Yi is the vector of predicted values, and Ŷi is the vector of actual values.
5.5. Performance Evaluation Metrics
Tenfold cross-validation (CV) was employed during model training to ensure the goodness of fit and minimize overfitting. Multiple evaluation metrics were utilized for the classification and regression task including coefficient of determination (r2), Matthew’s correlation coefficient (MCC), auROC, and auPR.
auROC, auPR, accuracy, and macro-averaged F1 score were used to evaluate all of the methods under comparison. F1 scores became necessary for the cases wherein false positives and false negatives were crucial, as in the case here. It was computed as follows
| 4 |
While accuracy does not consider the effect of imbalance in the data set, MCC and auPR are independent of imbalances and serve as better indicators of performance for the given use case. A completely random classifier would have a value of 0.5 and a perfect classifier would have a score of 1 for both the metrics. Also, both metrics ranged from 0 to 1 except MCC, which ranged from −1 to 1. In addition to the above metrics, sensitivity and specificity were also reported for the external validation analysis. Moreover, to efficiently balance the well-known bias and variance tradeoff, 10-fold cross-validation was employed.
5.6. Preparation of the Structures of the Protein and Ligands for Molecular Docking
The crystal structures of MAP2k and TGFβ are available in the Protein Data Bank (PDB) with IDs 5Z1D and 5E8X respectively, along with their known inhibitors.30,31 These structures were optimized and prepared for docking studies using the protein preparation wizard of the Schrodinger suite.39,40 Preparation of the structures included removing water molecules, adding polar hydrogen atoms, filling of missing amino acid side chains, and minimization of the structure using the OPLS3e force field.39,41
5.7. Molecular Docking of the Predicted Ligands with MAP2k and TGFβ
The key catalytic residues of MAP2k and TGFβ involved in the proteolytic activity are reported in the literature.30,31 A 20 Å grid for docking with MAP2k and TGFβ was generated, taking its catalytic residues as the centroid. The Glide module of Schrodinger was used for the extra precision (XP) flexible docking of the ligands at the generated grid sites.42,43 For MD validation, molecular interactions of the protein and its known inhibitor were compared against predictions from the proposed models. One model from each family (ESM1, ProtBert, and AlphaFold) was selected for comparison.
5.8. Explicit Water Molecular Dynamics (MD) Simulation and Its Analysis
A standardized MD protocol has been used for the analysis of docked protein–ligand complexes.44 The docked molecules were subjected to MD simulations to investigate the stability of binding and interactions between the proteins and ligands. All MD simulations were performed in the Schrodinger suite using the Desmond tool and the Maestro platform.41,45 First, the docked complexes were solvated with the TIP3P water model to build the system. It was followed by neutralizing the orthorhombic periodic boundary system by adding counterions (Na+/Cl−). The energy of the prepared systems was minimized by running 100 ps low-temperature (10 K) Brownian motion MD simulations in the NVT ensemble to remove steric clashes and move the system away from an unfavorable high-energy conformation. The systems were further relaxed using the default parameters of the “relax system before simulation” option of Desmond. The equilibrated systems were then subjected to 100 ns simulations in the NPT ensemble at a temperature of 300 K maintained using a Nose–Hoover chain thermostat, a constant pressure of 1 atm maintained using a Martyna–Tobias–Klein barostat, a time step of 2 fs, and a recording interval of 20 ps.
The generated MD simulation trajectories were visualized and analyzed using the system event analysis and simulation interaction diagram tools. The root-mean-square deviation (RMSD) of the simulated structures with reference to initial docked structures throughout the simulation run was analyzed to account for the stability of the interactions. The root-mean-square fluctuation (RMSF) was also calculated to investigate the average fluctuation in the amino acid residues of apoproteins and their complexes with ligands under consideration.
As hydrogen bonds are crucial in determining the specificity and affinity of a drug toward its receptor, the average number of the hydrogen bonds formed during the simulation time for each protein–ligand complex was then calculated. Furthermore, to investigate the significant interactive residues of the proteins, which were contacting the ligands during the MD simulation, the polar and nonpolar interactions, as well as the occupancy time of these interactions, were calculated.
6. Implementation
All of the models were developed on the TensorFlow-Keras platform and trained with Nvidia RTX2080 GPU (8 GB vRAM) and Tesla V100 (32GB vRAM). The model parameters were optimized using grid search, and saved model weights can be found on the GitHub repository.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05203.
Paired t-test to compare docking scores of all of the models under consideration for TGFβ (Table S1); paired t-test to compare docking scores of all of the models under consideration for MAP2K (Table S2); schematic representation of the seed model architecture on which all of the proposed methods are based on (Figure S1); interaction dynamics for MAP2k and TGFβ from 100 ns simulations (Figure S2); RMSD for the simulated complexes (Figure S3); TSNE mappings for proteins (Figure S4); ROC and PR curves for all of the models under consideration in the classification task. auPR and auROC scores are also mentioned for each model (Figure S5); coefficient of determination for all of the proposed models (Figure S6); training statistics for all of the models under consideration shows excellent statistics and minimal overfitting (Figure S7); and PANTHER enrichment results for a selected group of proteins in the data for classification, molecular function, and biological processes (Figure S8) (PDF)
Author Contributions
Y.K., D.S., and S.Y. conceived and designed the computational pipeline. Y.K. and S.Y. contributed to study design, computational pipeline, and executing experiments. Y.K. prepared the first draft of the manuscript.
The authors declare no competing financial interest.
Notes
The source codes, implementation, data sets, and all prediction results for TransDTI are available at https://github.com/TeamSundar/transDTI. Moreover, exact training statistics, ROC, and PR plots for classification and R2 plots for regression and enrichment results can be found in Supporting File 1.
Supplementary Material
References
- Moses H. 3rd; Dorsey E. R.; Matheson D. H.; Thier S. O. Financial anatomy of biomedical research. JAMA 2005, 294, 1333–1342. 10.1001/jama.294.11.1333. [DOI] [PubMed] [Google Scholar]
- Myers S.; Baker A. Drug discovery--an operating model for a new era. Nat. Biotechnol. 2001, 19, 727–730. 10.1038/90765. [DOI] [PubMed] [Google Scholar]
- Brogi S.; Ramalho T. C.; Kuca K.; Medina-Franco J. L.; Valko M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612 10.3389/fchem.2020.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muster W.; Breidenbach A.; Fischer H.; Kirchner S.; Müller L.; Pähler A. Computational toxicology in drug development. Drug Discovery Today 2008, 13, 303–310. 10.1016/j.drudis.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Hendrickson J. B. Similarity in Chemistry [Review of Concepts and Applications of Molecular Similarity, by M. A. Johnson & G. M. Maggiora]. Science 1991, 252, 1189. [Google Scholar]
- Chen Y.; Zhi D. Ligand–protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins: Struct., Funct., Bioinf. 2001, 43, 217–226. . [DOI] [PubMed] [Google Scholar]
- Yamanishi Y.; Araki M.; Gutteridge A.; Honda W.; Kanehisa M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob L.; Vert J.-P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics 2008, 24, 2149–2156. 10.1093/bioinformatics/btn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yıldırım M. A.; Goh K.-I.; Cusick M. E.; Barabási A.-L.; Vidal M. Drug—target network. Nat. Biotechnol. 2007, 25, 1119–1126. 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- Opella S. J.; Marassi F. M. Structure determination of membrane proteins by nuclear magnetic resonance spectroscopy. Chem. Rev. 2004, 104, 3587–3606. 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S.; Mistry J.; Bateman A.; Eddy S. R.; Luciani A.; Potter S. C.; Qureshi M.; Richardson L. J.; Salazar G. A.; Smart A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V. I.; Johansen A. R.; Nielsen M.; Almagro Armenteros J. J.; Nielsen H.; Sonderby C. K.; Winther O.; Sonderby S. K. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 2017, 33, 3685–3690. 10.1093/bioinformatics/btx531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Wu Q.; Zhang Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346.e2–1351.e2. 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Wang F.; Tang J.; Nussinov R.; Cheng F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digital Health 2020, 2, e667–e676. 10.1016/S2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y.; Shan X.; Chen T.; Jiang M.; Wang Y. A.-O.; Wang Q.; Salahub D. R.; Xiong Y. A.-O.; Wei D. Q. DTI-MLCD: predicting drug-target interactions using multi-label learning with community detection method. Briefings Bioinf. 2021, bbaa205 10.1093/bib/bbaa205. [DOI] [PubMed] [Google Scholar]
- Chu Y.; Kaushik A. C.; Wang X.; Wang W.; Zhang Y.; Shan X.; Salahub D. R.; Xiong Y.; Wei D.-Q. DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Briefings Bioinf. 2021, 22, 451–462. 10.1093/bib/bbz152. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-F.; Wang X.; Kaushik A. C.; Chu Y.; Shan X.; Zhao M.-Z.; Xu Q.; Wei D.-Q. SPVec: A Word2vec-Inspired Feature Representation Method for Drug-Target Interaction Prediction. Front. Chem. 2020, 7, 895 10.3389/fchem.2019.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.; Xiao C.; Glass L. M.; Sun J. MolTrans: Molecular Interaction Transformer for drug–target interaction prediction. Bioinformatics 2021, 37, 830–836. 10.1093/bioinformatics/btaa880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Tan X.; Wang D.; Zhong F.; Liu X.; Yang T.; Luo X.; Chen K.; Jiang H.; Zheng M. TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020, 36, 4406–4414. 10.1093/bioinformatics/btaa524. [DOI] [PubMed] [Google Scholar]
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser Ł.; Polosukhin I. In Attention is All you Need, NeurIPS Proceedings, Advances in Neural Information Processing Systems 30 (NIPS 2017), 2017; pp 5998–6008.
- Devlin J.; Chang M.-W.; Lee K.; Toutanova K. In BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding, Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, 2019.
- Brown T.; Mann B.; Ryder N.; Subbiah M.; Kaplan J. D.; Dhariwal P.; Neelakantan A.; Shyam P.; Sastry G.; Askell A.. et al. In Language Models are Few-Shot Learners, NeurIPS Proceedings, Advances in Neural Information Processing Systems 33 (NeurIPS 2020), 2020.
- Keskar N. S.; McCann B.; Varshney L. R.; Xiong C.; Socher R.. Ctrl: A Conditional Transformer Language Model For Controllable Generation; GitHub, 2019.
- Öztürk H.; Özgür A.; Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.; Keum J.; Nam H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129 10.1371/journal.pcbi.1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen M.; Zhang Z.; Niu S.; Sha H.; Yang R.; Yun Y.; Lu H. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409. 10.1021/acs.jproteome.6b00618. [DOI] [PubMed] [Google Scholar]
- Rives A.; Meier J.; Sercu T.; Goyal S.; Lin Z.; Liu J.; Guo D.; Ott M.; Zitnick C. L.; Ma J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. bioRxiv 2020, 622803 10.1101/622803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnaggar A.; Heinzinger M.; Dallago C.; Rihawi G.; Wang Y.; Jones L.; Gibbs T.; Feher T.; Angerer C.; Bhowmik D.; et al. ProtTrans: Towards Cracking the Language of Life’s Code Through Self-Supervised Deep Learning and High Performance Computing. bioRxiv 2020, 2020.07.12.199554 10.1101/2020.07.12.199554. [DOI] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W. R.; Bridgland A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Shraga A.; Olshvang E.; Davidzohn N.; Khoshkenar P.; Germain N.; Shurrush K.; Carvalho S.; Avram L.; Albeck S.; Unger T.; et al. Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chem. Biol. 2019, 26, 98.e5–108.e5. 10.1016/j.chembiol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Tebben A. J.; Ruzanov M.; Gao M.; Xie D.; Kiefer S. E.; Yan C.; Newitt J. A.; Zhang L.; Kim K.; Lu H.; et al. Crystal structures of apo and inhibitor-bound TGF[beta]R2 kinase domain: insights into TGF[beta]R isoform selectivity. Acta Crystallogr., Sect. D: Struct. Biol. 2016, 72, 658–674. 10.1107/S2059798316003624. [DOI] [PubMed] [Google Scholar]
- Mi H.; Thomas P.. PANTHER Pathway: An Ontology-Based Pathway Database Coupled with Data Analysis Tools. Protein Networks and Pathway Analysis. Methods in Molecular Biology; Methods in Molecular Biology; Humana Press, 2009; Vol. 563, pp 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M.; Ovchinnikov S.; Steinegger M. ColabFold - Making protein folding accessible to all. bioRxiv 2021, 2021.08.15.456425 10.1101/2021.08.15.456425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S.; Karanicolas J. Correction to When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode?. J. Med. Chem. 2017, 60, 5940. 10.1021/acs.jmedchem.7b00868. [DOI] [PubMed] [Google Scholar]
- Tang J.; Szwajda A.; Shakyawar S.; Xu T.; Hintsanen P.; Wennerberg K.; Aittokallio T. Modeling: Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. 10.1021/ci400709d. [DOI] [PubMed] [Google Scholar]
- Tang J.; Szwajda A.; Shakyawar S.; Xu T.; Hintsanen P.; Wennerberg K.; Aittokallio T. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. 10.1021/ci400709d. [DOI] [PubMed] [Google Scholar]
- Salvat R. S.; Parker A. S.; Choi Y.; Bailey-Kellogg C.; Griswold K. E. Mapping the Pareto Optimal Design Space for a Functionally Deimmunized Biotherapeutic Candidate. PLoS Comput. Biol. 2015, 11, e1003988 10.1371/journal.pcbi.1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S.; Shi S.; Ueda H. R.. SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery; GitHub, 2019.
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. Epik: a software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Dhanjal J. K.; Bhargava P.; Kaul A.; Wang J.; Zhang H.; Kaul S. C.; Wadhwa R.; Sundar D. Withanone and Withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 2022, 1–13. 10.1080/07391102.2020.1775704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K. J., et al. , Scalable algorithms for molecular dynamics simulations on commodity clusters. SC ‘06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery: Tampa, Florida, 2006, pp. 43−43.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.