

SUMMARY
The gut microbiota benefits the host by limiting enteric pathogen expansion (colonization resistance), partially via the production of inhibitory metabolites. Propionate, a short-chain fatty acid produced by microbiota members, is proposed to mediate colonization resistance against Salmonella enterica serovar Typhimurium (S. Tm). Here, we show that S. Tm overcomes the inhibitory effects of propionate by using it as a carbon source for anaerobic respiration. We determine that propionate metabolism provides an inflammation-dependent colonization advantage to S. Tm during infection. Such benefit is abolished in the intestinal lumen of Salmonella-infected germ-free mice. Interestingly, S. Tm propionate-mediated intestinal expansion is restored when germ-free mice are monocolonized with Bacteroides thetaiotaomicron (B. theta), a prominent propionate producer in the gut, but not when mice are monocolonized with a propionate-production-deficient B. theta strain. Taken together, our results reveal a strategy used by S. Tm to mitigate colonization resistance by metabolizing microbiota-derived propionate
Graphical Abstract
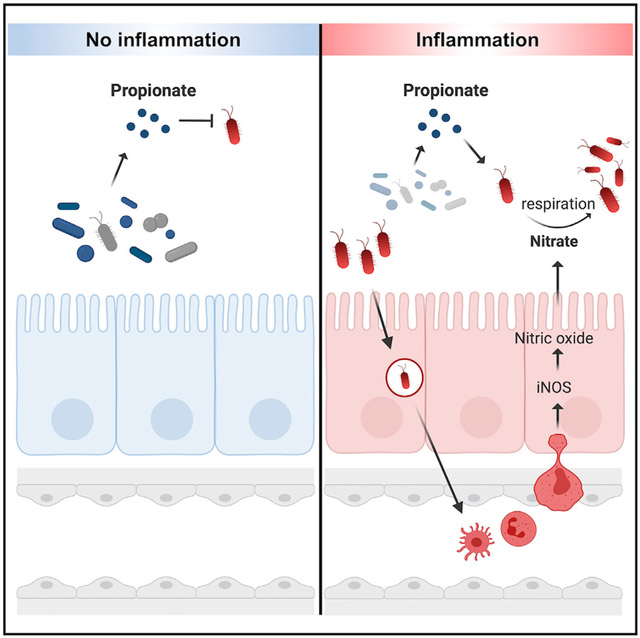
In brief
Propionate, a short-chain fatty acid produced by the gut microbiota, is proposed to mediate colonization resistance against Salmonella enterica serovar Typhimurium (S. Tm). Here, Shelton et al. show that nitrate-dependent propionate metabolism fuels pathogen expansion in the inflamed gut, allowing S. Tm to overcome propionate’s inhibitory effects.
INTRODUCTION
The intestines are occupied by a complex microbial community, the gut microbiota, mainly composed of obligate anaerobic bacteria. By residing in the gut, the microbiota contribute to host health through nutrient production (Sonnenburg et al., 2005; Bäckhed et al., 2004), immune education (Lathrop et al., 2011; reviewed by Zheng et al., 2020), and protection against enteric pathogens (colonization resistance) (van der Waaij et al., 1971; Bohnhoff et al., 1964). Colonization resistance is accomplished through diverse mechanisms (Sassone-Corsi and Raffatellu, 2015). For instance, the gut microbiota can indirectly inhibit pathogen expansion by activating the host’s immune response or by enhancing the intestinal mucosal barrier (Petersson et al., 2011; Ivanov et al., 2009; Benson et al., 2009). On the other hand, direct antagonism of enteric pathogens by the gut microbiota is achieved through niche competition or the production of inhibitory molecules (Deriu et al., 2013; Sorbara and Pamer, 2019).
A proposed microbiota-derived metabolite that mediates colonization resistance is propionate (Jacobson et al., 2018; Horswill et al., 2001). Propionate, an abundant short-chain fatty acid (Byrne and Dankert, 1979), is generated by the fermentation of sugars by anaerobic bacteria, specifically members of the Bacteroides genus (den Besten et al., 2013). As a predicted component of colonization resistance, propionate has previously been studied for its specific role in inhibiting Salmonella enterica serovar Typhimurium (S. Tm). The complete mechanism by which propionate exerts its toxic effect on S. Tm remains unknown. Initially, propionate was shown to inhibit S. Tm by generating toxic by-products produced during propionate catabolism (Horswill et al., 2001). Propionate was recently shown to acidify the intracellular space of S. Tm and significantly reduce S. Tm’s growth rate (Jacobson et al., 2018). However, the mechanisms employed by enteric pathogens to overpower propionate-mediated colonization resistance, a key step for successful gut colonization, remain largely unknown.
S. Tm has evolved several mechanisms to overcome colonization resistance. Upon infection, S. Tm invades the intestinal epithelium and activates the host’s innate immune system and inflammatory response (Galän and Curtiss, 1989). As a result, the host produces reactive nitrogen species, specifically nitric oxide, to inhibit the growth of the pathogen (Alam et al., 2002). Host-generated nitric oxide can react with other compounds in the intestinal lumen to generate nitrate (Szabó et al., 2007; Rivera-Chávez and Bäumler, 2015). Interestingly, S. Tm can take advantage of the nitrate generated by the host immune response by using it as an alternative electron acceptor to fuel anaerobic respiration (Barrett and Riggs, 1982). By performing anaerobic respiration, S. Tm can outgrow the resident microbiota whose metabolism relies on fermentation (Lopez et al., 2012; McLaughlin et al., 2019). In addition to the energetic benefits of anaerobic respiration compared with fermentation, S. Tm’s metabolic adaptation in the inflamed gut enables the pathogen to access new nutrient niches and metabolize novel carbon sources (Thiennimitr et al., 2011; Faber et al., 2017).
The carbon sources utilized by S. Tm during anaerobic respiration remain largely uncharacterized. Interestingly, is it possible that S. Tm may use propionate as a carbon source during infection, as this pathogen possesses the machinery necessary for propionate catabolism (Horswill and Escalante-Semerena, 1997; Horswill and Escalante-Semerena, 1999). Specifically, the prpBCDE operon encodes the enzymes required to convert propionate into pyruvate through the 2-methylcitrate cycle (Horswill and Escalante-Semerena, 1999). Furthermore, genes in the prp operon are nonfunctional in extraintestinal serovars of S. Tm, raising the possibility that propionate metabolism is required for successful S. Tm colonization in the inflamed intestinal lumen (Nuccio and Bäumler, 2014). In this study, we show that the ability of S. Tm to metabolize propionate relies on nitrate-dependent anaerobic respiration. We then use conventional and germ-free mouse models of S. Tm gastroenteritis to demonstrate that S. Tm uses inflammation-dependent anaerobic respiration to overcome propionate-mediated colonization resistance.
RESULTS
Propionate supports S. Tm growth during anaerobic respiration in vitro
Previous investigations into the function of the prp operon have focused on the use of propionate as a carbon source under aerobic conditions (Horswill et al., 2001; Rondon et al., 1995) (Figures 1A and S1A). However, as an enteric pathogen, S. Tm encounters propionate in the intestinal lumen, which is mainly anaerobic (Friedman et al., 2018). Therefore, we sought to investigate the ability of S. Tm to metabolize propionate under conditions relevant to intestinal disease. We first determined if propionate could support S. Tm growth in vitro if low oxygen levels (0.8%) or alternative electron acceptors (i.e., DMSO, TMAO, fumarate, tetrathionate, or nitrate) generated during S. Tm-induced inflammation were available (Rivera-Chávez and Bäumler, 2015; Nuccio and Bäumler, 2014; Winter et al., 2013) (Figure 1B). Interestingly, we observed that propionate significantly increased S. Tm growth when nitrate was added to the medium (Figure 1B). Propionate increased S. Tm growth slightly when oxygen was present, but to a much lesser extent than nitrate, suggesting that oxygen is not the preferred electron acceptor used by S. Tm for propionate catabolism. The other alternative electron acceptors tested (i.e., DMSO, TMAO, fumarate, and tetrathionate) did not support S. Tm growth with propionate (Figure 1B). We next tested if a range of propionate concentrations (5–200 mM), physiologically relevant for mice and humans (Byrne and Dankert, 1979; den Besten et al., 2013), supported S. Tm growth in the presence or absence of nitrate. S. Tm was able to grow in increasing concentrations of propionate up to 50 mM when nitrate was present in the medium (Figure 1C), suggesting that S. Tm may be able to metabolize high levels of propionate during infection. Notably, S. Tm could not ferment propionate as no growth is observed in the absence of nitrate (Figure 1C).
Figure 1. Propionate fuels S. Tm growth in the presence of nitrate in vitro.

(A) Simplified model of propionate catabolism in S. Tm. Genes in the prpBCDE operon (red) metabolize propionate into pyruvate. prpE, propionyl-CoA synthase; prpC, methylcitrate synthase; prpD, methylcitrate dehydratase; prpB, 2-methylisocitrate lyase.
(B) NCE minimal medium containing 40 mM of an alternative electron acceptor alone or 40 mM alternative electron acceptor + 10 mM propionate was inoculated with S. Tm and grown anaerobically for 24 h. Alternatively, S. Tm was grown in a hypoxic chamber under 0.8% oxygen in NCE minimal medium with or without 10 mM propionate. Fold change calculated by comparing growth on oxygen or alternative electron acceptor + 10 mM propionate with growth with oxygen or alternative electron acceptor alone.
(C) NCE minimal medium containing increasing propionate concentration with or without 40 mM nitrate was inoculated with S. Tm. The OD600 of S. Tm was measured after 24 h of anaerobic growth.
(D–F) Relative transcription of prpR (D), prpB (E), and prpC (F) in NCE minimal medium supplemented with propionate, nitrate, or both propionate and nitrate was determined by qRT-PCR. Transcription of target genes was normalized to gyrB rRNA.
(G) NCE minimal medium containing propionate or propionate and nitrate was inoculated with WT S. Tm, ΔprpC, or a complemented strain of ΔprpC (−ΔprpC_pprpC). The medium was supplemented with 200 μM isopropyl β-D-1-thiogalactopyranoside to induce expression of prpC in the complemented mutant strain. The OD600 of each strain was measured after 24 h of anaerobic growth. Each dot represents one biological replicate (average of triplicate technical replicate per biological replicate). Bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not statistically significant. See also Figure S1.
To determine whether anaerobic respiration affects expression of the prpBCDE operon, we measured changes in S. Tm gene transcription when propionate, nitrate, or propionate and nitrate were available. Propionate alone induced expression of prpR, the transcriptional activator of the prpBCDE operon (Figure 1D). However, addition of nitrate (nitrate + propionate medium) was necessary to increase expression of the propionate utilization genes prpB and prpC (Figures 1E and 1F), supporting the specific role of nitrate in propionate catabolism. We next confirmed that the prpBCDE operon was necessary for S. Tm growth in the presence of propionate and nitrate. Deletion of the prpBCDE operon or prpC in S. Tm blunted the pathogen’s growth on propionate under anaerobic respiration conditions (Figures 1G, S1B, and S1C). However, no defects in growth were observed when mutants were given glucose or glycerol as a carbon source (Figures S1D and S1E). Growth on propionate could be restored in ΔprpC by reintroducing the prpC gene by plasmid complementation (Figure 1G). These experiments reveal that nitrate respiration supports propionate catabolism in vitro through the prpBCDE operon.
Nitrate respiration allows S. Tm to overcome the inhibitory effects of propionate under low pH
The ability of S. Tm to use propionate as a carbon source under anaerobic respiration conditions may be confounded by the acidifying effects of this short-chain fatty acid (Jacobson et al., 2018; Ricke, 2003), particularly in the low pH of the intestines (ranging from pH 7.4 to 5.7) (Sorbara et al., 2019). Thus, we determined how changes in pH impact the growth of S. Tm with propionate and nitrate in vitro. Notably, S. Tm grew significantly more at pH 7.0, 6.5, and 6.0 when propionate and nitrate were present than with propionate alone (Figures 2A-2C). Wildtype (WT) S. Tm also grew significantly better than ΔprpC, which failed to grow when given propionate and nitrate at all pH values tested (Figures 2A-2C). The growth of WT S. Tm at pH 6.0 was reduced compared with growth at pH 6.5 and 7.0 (Figure 2C). However, decreased growth is observed when S. Tm is grown with glycerol and nitrate at pH 6.0, suggesting that reduced growth at pH 6.0 is not specific to propionate metabolism (Figures S2A and S2B).
Figure 2. Nitrate prevents inhibitory effects of propionate on S. Tm in vitro.

(A–C) NCE minimal medium was adjusted to pH 7.0 (A), pH 6.5 (B), and pH 6.0 (C), respectively. The medium was supplemented with 10 mM propionate or 10 mM propionate + 40 mM nitrate and inoculated with WT S. Tm or ΔprpC. The culture was grown anaerobically for 14 h, and samples were taken every 2 h to plate for CFUs. n = 4.
(D–F) Cultures grown in (A–C) were used to calculate the generation time of WT S. Tm or ΔprpC at pH 7.0 (D), pH 6.5 (E), and pH 6.0 (F). Each dot represents one biological replicate (average of triplicate technical replicate per biological replicate). Bars represent mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S2.
Previous research revealed that propionate-mediated colonization resistance by decreasing the growth rate of S. Tm (Jacobson et al., 2018). To address if nitrate prevented this effect of propionate, the generation time of S. Tm at pH 7.0, 6.5, and 6.0 was calculated when S. Tm was grown in the presence of propionate and nitrate or propionate alone. The addition of nitrate significantly decreased the generation time of WT S. Tm at all pH values tested (Figures 2D-2F). Furthermore, the generation time of WT S. Tm was markedly shorter than ΔprpC at each pH tested when propionate and nitrate were present (Figures 2D-2F). Although nitrate led to a significant reduction in generation time for ΔprpC at pH 6.5, no difference in generation time was observed at pH 7.0 and 6.0 when both nitrate and propionate were available (Figures 2D-2F). These data show that nitrate can mitigate the inhibitory effects of propionate on S. Tm growth by promoting propionate metabolism through the prpBCDE operon.
Propionate metabolism provides a growth advantage to S. Tm in the inflamed gut
After discovering that S. Tm can metabolize propionate under anaerobic respiration conditions in vitro, we next investigated whether propionate catabolism provided S. Tm with a colonization advantage in vivo. We infected C57BL/6 mice, pretreated with streptomycin, with an equal mixture of WT S. Tm and ΔprpC. Four days after infection, the bacterial load of each strain was determined by plating the colon contents on selective medium, and the ratio of S. Tm WT and ΔprpC (competitive index) was calculated. To determine the role of propionate catabolism in S. Tm colonization of systemic sites, we also assessed the competitive index of S. Tm WT versus ΔprpC mutant in the spleen and liver sampled from infected mice. Interestingly, we observed a significant competitive advantage for WT S. Tm over ΔprpC in the colon contents, but not in the liver or spleen (Figure 3A). These experiments reveal that propionate metabolism benefits S. Tm during infection specifically in the gastrointestinal tract.
Figure 3. Propionate utilization confers an advantage to S. Tm in an inflammation-dependent mechanism.

(A) Streptomycin-pretreated C57BL/6 mice were inoculated with an equal mixture of WT S. Tm and ΔprpC. The competitive index in the colon content and homogenized samples from the liver or spleen was determined 4 days after infection.
(B) Streptomycin-pretreated C57BL/6 mice were inoculated with an equal mixture of the indicated S. Tm strains. The competitive index in the colonic content was determined 4 days after infection.
(C) Combined histopathology score of pathological lesions in the cecum of mice from (B) (n = 5).
(D) Representative images of hematoxylin and eosin-stained cecal tissue of mice from (C). Scale bars, 200 μm.
(E) Propionate concentration in the cecal content was determined by liquid chromatography/mass spectrometry (LC/MS) 2 days after infection.
(F) Streptomycin-pretreated C57BL/6 mice were inoculated with an equal mixture of WT S. Tm and ΔprpC. Mice received propionate supplementation (20 mM) in the drinking water for the duration of the experiment. The competitive index in the colonic content was determined 4 days after infection. Each dot represents data from one animal (biological replicate). Bars represent mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001. See also Figure S3.
Next, we investigated whether inflammation was required for propionate metabolism to confer an advantage to S. Tm. C57BL/6 mice, pretreated with streptomycin, developed significant intestinal inflammation characterized by edema, epithelial damage, infiltration of inflammatory cells in the submucosa and exudate in the intestinal lumen 4 days after S. Tm infection (Figures 3C and 3D). The competitive advantage for WT S. Tm over ΔprpC in the colon contents increased over time (Figures S3A and S3B), suggesting that propionate utilization may be dependent on inflammation induction by the pathogen. S. Tm uses two type III secretion systems (T3SS) to invade the intestinal epithelium and perform intracellular replication (Galán and Curtiss, 1989). A mutant strain (ΔinvA ΔspiB) of S. Tm is defective in both T3SS and does not cause inflammation in a mouse model (Coombes et al., 2005; Raffatellu et al., 2009). Thus, we constructed a ΔprpC mutant in the S. Tm inflammation-deficient background (ΔinvA ΔspiB ΔprpC) and then infected streptomycin-pretreated C57BL/6 mice with an equal mixture of ΔinvA ΔspiB and ΔinvA ΔspiB ΔprpC. The competitive index was determined 4 days after infection. In contrast to the competitive advantage observed for WT over ΔprpC, no competitive advantage was observed for ΔinvA ΔspiB over ΔinvA ΔspiB ΔprpC, revealing that inflammation is required for propionate metabolism to be advantageous to S. Tm (Figures 3B and S3C). Histopathology analysis confirmed that infection with S. Tm ΔinvA ΔspiB did not induce intestinal inflammation (Figures 3C and 3D). The concentration of propionate was measured in the feces of mice infected with WT or ΔinvA ΔspiB and no significant differences were observed (Figure 3E), indicating that the lack of an advantage of ΔinvA ΔspiB over ΔinvA ΔspiB ΔprpC was not due to decreased levels of propionate in ΔinvA ΔspiB-infected mice.
In our experiments, we noticed that streptomycin treatment caused decreased propionate levels in the intestinal lumen of mice when compared with untreated controls (data not shown). Antibiotic-induced lower propionate levels could interfere with S. Tm growth in intestinal lumen, since propionate’s ability to inhibit S. Tm growth is concentration dependent (Jacobson et al., 2018) (Figure 1C). Thus, we restored propionate levels in our antibiotic treatment mouse model by providing 20 mM propionate in the drinking water for the duration of the experiment (Figure 3F). The competitive advantage for WT over ΔprpC was still present when mice received propionate supplementation, suggesting that streptomycin effects on propionate intestinal levels did not play a role in our phenotype. In addition, to circumvent the effects of antibiotics in the gut microbiota and propionate levels, we performed our in vivo competition experiments between WT and ΔprpC S. Tm in CBA/J mice, since this mouse strain develops S. Tm gastroenteritis in the absence of antibiotic treatment at 14 days post-infection (Figure S3D). Importantly, CBA/J mice infected with S. Tm have higher intestinal propionate levels when compared with antibiotic-treated C57BL/6 mice (Figure S3E). WT S. Tm was able to outcompete a ΔprpC mutant at day 13 post-infection in the cecum content of CBA/J (Figure S3F), where pathogen-induced gastroenteritis is established (Figure S3D). The phenotype was less striking in the colon (Figure S3F), probably because S. Tm-induced inflammation is restricted to the cecum in this mouse model (Figure S3D). Our results demonstrate that propionate catabolism continues to provide a growth advantage to S. Tm in the intestinal lumen during inflammation in the presence of higher propionate concentrations.
Nitrate respiration is required for S. Tm to benefit from propionate metabolism in vivo
We observed in vitro that nitrate is the preferred alternative electron acceptor to support S. Tm growth on propionate (Figure 1). To perform anaerobic respiration using nitrate as an alternative electron acceptor, S. Tm relies on three nitrate reductases, nar-GHI, narZYV, and napABC (Lopez et al., 2015). Indeed, a ΔnapA ΔnarZ ΔnarG mutant strain of S. Tm is unable to perform nitrate-dependent anaerobic respiration (Lopez et al., 2012, 2015). To investigate if nitrate respiration is required for propionate metabolism to be advantageous in vivo, we infected streptomycin-pretreated C57BL/6 mice with an equal mixture of S. Tm ΔnapA ΔnarZ ΔnarG and S. Tm ΔnapA ΔnarZ ΔnarG ΔprpC and measured the competitive index 4 days after infection. In contrast to the competitive advantage observed for WT over ΔprpC, no advantage was observed for ΔnapA ΔnarZ ΔnarG over ΔnapA ΔnarZ ΔnarG ΔprpC (Figures 4A and S4A), suggesting that nitrate respiration is required for propionate metabolism to benefit S. Tm in vivo.
Figure 4. Inflammation generated nitrate required for propionate catabolism by S. Tm.

(A) Streptomycin-pretreated C57BL/6 mice were inoculated with an equal mixture of the indicated S. Tm strains. The competitive index in the colon content was determined 4 days after infection.
(B) Streptomycin-pretreated C57BL/6 mice were inoculated with WT S. Tm, ΔinvA ΔspiB, or mock treated. mRNA levels of Nos2 were measured in the cecal mucosa 4 days post-infection and normalized to β-actin mRNA levels.
(C–E) Streptomycin-pretreated C57BL/6 WT mice and Nos2-deficient mice were inoculated with an equal mixture of the WT S. Tm and ΔprpC mutant. One group was treated with aminoguanidine (AG) as indicated. (C) The competitive index in the cecal content was determined 4 days post-infection. (D) Nitrate concentration in the cecal and colonic mucus layer was determined by a modified Griess assay. (E) Combined histopathology score of pathological lesions in the cecum of mice from (C) (n = 5). (F) Propionate concentration in the cecal content of mice from (C) was determined by liquid chromatography/mass spectrometry (LC/MS) 2 days after infection.
(F and G) Streptomycin-pretreated C57BL/6 mice were inoculated with an equal mixture of WT S. Tm and ΔprpC in the indicated S. Tm strain backgrounds. The competitive index in the cecal content (F) and nitrate concentration in the cecal and colonic mucous layer (G) were determined 4 days post-infection. Each dot represents data from one animal (biological replicate). Bars represent mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S4.
As an alternative approach, we investigated if propionate metabolism was advantageous to S. Tm if the availability of inflammation-derived nitrate was decreased. During Salmonella infection, inflammatory monocytes and intestinal epithelial cells upregulate inducible nitric oxide synthase (iNOS) encoded by Nos2, leading to the production of nitric oxide (Lopez et al., 2012; McLaughlin et al., 2019), in a T3SS-dependent manner (Figures 4B and S4B). Nitric oxide reacts with superoxide radicals to form peroxynitrate, which can then decompose into nitrate (Winter et al., 2013). Therefore, we repeated the competitive infection assays in mice treated with the iNOS inhibitor aminoguanidine (Winter et al., 2013), and in iNOS (Nos2)-deficient mice. In mock-treated WT mice, we observed a competitive advantage for WT S. Tm over ΔprpC (Figures 4C and S4C). However, this advantage was abrogated both in mice treated with aminoguanidine and in Nos2-deficient mice (Figures 4C and S4C). Nitrate measurements from colonic and cecal mucosa revealed a significant decrease in nitrate levels in mice treated with aminoguanidine and in Nos2-deficient mice (Figure 4D). Despite differences in nitrate levels, inflammation was similar between the three treatment groups (Figure 4E). Propionate levels were similar in the feces of aminoguanidine-treated mice and were elevated in the feces of Nos2−/− mice compared with mock-treated WT mice (Figure 4F), confirming that lack of a competitive advantage of WT S. Tm over ΔprpC in aminoguanidine-treated or in Nos2−/− mice was not due to decreased propionate or differences in inflammation between experimental groups. Together, these data reveal that induction of host-derived nitrate supports propionate utilization by S. Tm during pathogen-induced gastroenteritis.
Previous research demonstrates that expression of the effector protein SopE by S. Tm is required to induce significant nitrate production by the host (Lopez et al., 2012, 2015). Therefore, it is possible that the nitrate-dependent propionate catabolism only happens during infection with S. Tm strains that express the SopE effector (SopE+). To test this hypothesis, we repeated our in vivo competition experiments between a WT and ΔprpC mutant using WT S. Tm SL1344 (SopE+), ΔsopE S. Tm SL1344 (SopE−) and WT S. Tm IR715 (SopE−) strain backgrounds. Indeed, the advantage of the WT S. Tm over a ΔprpC mutant was present in mice infected with the S. Tm SL1344 background, but it was absent when mice were infected with ΔsopE S. Tm SL1344 and WT S. Tm IR715 strains (Figure 4G). Interestingly, in vivo infection with ΔsopE S. Tm SL1344 and S. Tm IR715 induced significantly lower nitrate levels in the intestinal mucosa in comparison with infection with S. Tm SL1344 (Figure 4H). Our experiments demonstrate that SopE-dependent nitrate production is required to support propionate catabolism by S. Tm during intestinal inflammation.
Microbiota-derived propionate provides a growth advantage to S. Tm in the presence of nitrate
While we predict that the advantage of WT S. Tm over ΔprpC (Figure 3) is due to the ability of WT S. Tm to overcome propionate-mediated colonization resistance, it is possible that other microbiota-derived metabolites may contribute to this phenotype. Therefore, we sought to examine the advantage of WT versus ΔprpC using an in vitro approach in which propionate levels can be controlled. As a source of propionate, we used Bacteroides thetaiotaomicron, a representative of the Bacteroides genus and a predominant propionate producer in the gut (Reichardt et al., 2014). As a negative control, we also cultured B. theta BT1686-89, an isogenic propionate-production-deficient B. theta mutant strain (Kovatcheva-Datchary et al., 2015). B. theta BT1686-89 was grown anaerobically in mucin broth for 4 days to adjust for a growth defect (Figures S5A and S5B), while WT B. theta was grown for 2 days (Figures S5B). Growth of WT B. theta led to an accumulation of propionate in the medium while B. theta BT1686-89 supernatant contained significantly less propionate (Figure 5A). Then, to determine whether microbiota-derived propionate provides a growth advantage to S. Tm, we cultured WT S. Tm and S. Tm ΔprpC in supernatant from WT B. theta or B. theta BT1686-89, and B. theta supernatants were either left untreated or supplemented with nitrate (Figures 5B and 5C). In the absence of nitrate, no differences in growth were observed between WT S. Tm or ΔprpC cultured in supernatant from either WT B. theta or B. theta BT1686-89 (Figure 5A). However, addition of nitrate enabled WT S. Tm to grow to significantly higher levels than ΔprpC in supernatant from WT B. theta, but not in supernatant from B. theta BT1686-89 (Figures 5B and 5C). Restoring propionate levels in supernatant of B. theta BT1686-89 (Figure 5A) rescued WT S. Tm’s ability to grow significantly more than ΔprpC in the presence of nitrate (Figures 5B and 5C). These results indicate that the growth advantage of WT S. Tm over ΔprpC is specific to propionate and not due to other B. theta-derived metabolites. Moreover, these results demonstrate that microbe-derived propionate can fuel S. Tm growth in the presence of nitrate in a prpBCDE-dependent manner.
Figure 5. Bacteroides-produced propionate is a carbon source for S. Tm if nitrate is present.

(A–C) Mucin broth was inoculated with WT B. thetaiotaomicron (WT) or B. thetaiotaomicron BT1686-89 (BT1686-89). B. theta BT1686-89 was cultured anaerobically for 4 days and WT B. theta was cultured for 2 days. (A) Propionate concentration in the digested mucin broth from WT or BT1686-89 B. theta culture (supplemented or not with propionate) was determined by liquid chromatography/mass spectrometry (LC/MS). (B) Filter-sterilized WT B. theta or B. theta BT1686-89-digested mucin broth was inoculated with an equal mixture of WT S. Tm and ΔprpC. Nitrate and propionate (40 and 10 mM, respectively) were added where indicated. Cultures were plated after 16 h of anaerobic growth. (C) Competitive index was calculated from CFU counts in (A). Nitrate and propionate were added as indicated. Each dot represents one biological replicate (average of triplicate technical replicate per biological replicate). Bars represent mean ± SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S5.
Metabolism of Bacteroides-produced propionate supports S. Tm growth in vivo
To determine whether Bacteroides-produced propionate promotes S. Tm intestinal colonization in vivo, we investigated the role of S. Tm propionate utilization during intestinal infection in a germ-free mouse model. Germ-free mice were left germ-free or were colonized with WT B. theta or B. theta BT1686-89 (Figure 6A). In addition, a subset of mice colonized with B. theta BT1686-89 were treated with propionate in their drinking water (Figure 6A). Measurement of propionate in the feces of mice 7 days after colonization revealed that mice given WT B. theta had significantly higher levels of propionate than mice colonized with B. theta BT1686-89 (Figure 6B). Supplementation with propionate in the drinking water of mice colonized with B. theta BT1686-89 increased the amount of propionate in the feces (Figure 6B). Colonization of the three groups with their respective strain of Bacteroides was confirmed after 7 days (Figure 6C). Next, we infected each group with an equal mixture of WT S. Tm and ΔprpC. WT S. Tm was able to outcompete the ΔprpC mutant in mice colonized with WT B. theta 3 days after infection (Figures 6D and 6E). The WT S. Tm competitive advantage was abrogated in mice colonized with B. theta BT1686-89 but restored if B. theta BT1686-89 mice are given propionate in the drinking water (Figures 6D and 6E). All experimental groups had equal levels of intestinal inflammation, revealing that differences in propionate-dependent intestinal colonization by S. Tm in mice colonized with WT B. theta or B. theta BT1686-89 were not due to altered host immune responses (Figure 6F). Collectively, these experiments show that S. Tm can overcome the inhibitory effects of Bacteroides-derived propionate in the inflamed gut.
Figure 6. S. Tm utilization of microbiota-derived propionate provides an advantage during infection.

(A) Schematic representation of the experiment and groups used. Germ-free mice were colonized with either WT B. theta (WT) or B. theta BT1686-89 for 7 days before infection with an equal mixture of WT S. Tm and a ΔprpC mutant.
(B) Propionate concentration in the feces was measured after 7 days of colonization with different strains of Bacteroides and propionate supplementation.
(C) Before infection, fecal samples were collected from monocolonized mice and plated on blood agar to confirm equal colonization between different Bacteroides strains.
(D) Monocolonized mice were infected with an equal mixture of the WT S. Tm and ΔprpC mutant. The abundance of each strain in the colon content was determined by selective plating 3 days post-infection.
(E) Competitive index was calculated from CFU counts in (D).
(F) Combined histopathology score of pathological lesions in the cecum of mice from (D). Each dot represents data from one animal (biological replicate). Bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DISCUSSION
For decades, the antimicrobial properties of propionate have been leveraged to limit Salmonellae infection in agricultural animals (Ricke, 2003; Iba and Berchieri, 1995). However, in this study, we report findings that S. Tm can overcome the inhibitory effects of propionate if inflammation-derived nitrate is available. Our data show that S. Tm upregulates machinery used to perform propionate catabolism if nitrate is present. This leads to an advantage in vivo as strains of S. Tm that can perform propionate metabolism have an advantage over strains that cannot catabolize propionate. Collectively, we describe a previously unexplored mechanism by which S. Tm contends with a component of colonization resistance by performing anaerobic respiration in the inflamed gut.
A recent report determined that propionate production by gut microbiota members inhibited S. Tm colonization in vivo (Jacobson et al., 2018). For the study, the researchers utilized a mouse model of chronic S. Tm infection, characterized by lower bacterial burden and mild intestinal inflammation (Jacobson et al., 2018). In contrast, we investigated the interaction between propionate and S. Tm in a mouse model of S. Tm-induced gastroenteritis that results in severe inflammation (Barthel et al., 2003) (Figures 3C and 3D). S. Tm causes inflammation by invading the intestinal epithelium and triggering an immune response (Galán and Curtiss, 1989). Consequently, innate immune cells release reactive oxygen and nitrogen species that react to generate alternative electron acceptors, including tetrathionate and nitrate (McLaughlin et al., 2019; Winter et al., 2013; Winter et al., 2010). The presence of alternative electron acceptors provides S. Tm with the opportunity to perform anaerobic respiration and outgrow the obligate anaerobic microbiota (Lopez et al., 2012; McLaughlin et al., 2019, Winter et al., 2010). Furthermore, the inflamed gut is a unique niche in which S. Tm alters its metabolism and begins to utilize carbon sources that require respiration (Nuccio and Bäumler, 2014). Some of these carbon sources have been identified, including 1,2-propanediol, ethanolamine, and fructose-asparagine (Thiennimitr et al., 2011; Faber et al., 2017; Ali et al., 2014). However, many remain unknown (Nuccio and Bäumler, 2014). Here, we show that S. Tm can metabolize propionate via anaerobic respiration, and this provides S. Tm with an advantage in vivo. Interestingly, propionate utilization was specific to the availability of nitrate in vitro and in vivo, supporting the idea that nitrate is involved in the regulation of the prpBCDE operon. By utilizing a different model of S. Tm infection, we show a new mechanism by which S. Tm mitigates the effects of propionate.
Multiple mechanisms have been proposed by which propionate inhibits Salmonella and other enteric pathogens. Initial studies hypothesized that propionate reduced S. Tm growth through the generation and accumulation of toxic intermediates (Horswill et al., 2001). Subsequent research focused on the ability of propionate to diffuse into the cytoplasm of the bacteria and decrease intracellular pH (Jacobson et al., 2018; den Besten et al., 2013). This is predicted to impede the ability of S. Tm to colonize its host (Jacobson et al., 2018). Additional modes of inhibition include repression of S. Tm invasion through destabilization of hilD, a regulator of Salmonella Pathogenicity Island 1 (Hung et al., 2013). However, past studies that examined the inhibitory effects of propionate did not consider the presence of inflammation-derived alternative electron acceptors. Indeed, we showed that the addition of nitrate to cultures containing propionate increased the growth rate of S. Tm despite acidic conditions. Therefore, we propose that inflammation-derived nitrate provides S. Tm with the ability to overcome propionate-induced toxicity.
By leveraging a monocolonized gnotobiotic mice model, we determined that the advantage observed for WT S. Tm over a prpC-deficient mutant is specific to microbiota-derived propionate. The 2-methylcitrate cycle is fueled by 1,2-propanediol and propionate; both metabolites are produced by members of the microbiota (Faber et al., 2017). A previous study identified that 1,2-propanediol fuels S. Tm growth during infection (Faber et al., 2017). In this work, we showed that the advantage observed for WT S. Tm over a prpC-deficient mutant is specific to propionate and not 1,2-propanediol using germ-free mice colonized with the specific strains of B. theta. Future work should examine how S. Tm integrates microbiota-derived 1,2-propanediol and propionate to grow in vivo.
In conclusion, this study shows that intestinal inflammation enables S. Tm to overcome propionate-mediated colonization resistance. Together, our findings provide the paradigm-shifting perspective that, during infection, microbiota-derived propionate may aid in intestinal pathogen colonization. Therefore, in addition to the role of propionate in promoting colonization resistance, we propose that, during infection, microbiota-derived propionate may also support S. Tm expansion in the intestinal tract. The inhibitory and beneficial effects of propionate during pathogen colonization may have significant implications on the use of microbiota-derived propionate as an antimicrobial treatment during S. Tm gastroenteritis and will be an important area of future research.
Limitations of the study
Previous work (Hung et al., 2013) demonstrated that propionate metabolism impacts invasion of S. Tm. Therefore, the competitive advantage of WT S. Tm over a ΔprpC mutant observed in our studies could also be a consequence of differences in the invasiveness of each strain. Future work will need to evaluate the impact of inflammation-dependent propionate catabolism in regulation of S. Tm invasion. Furthermore, our study shows that nitrogen regulates the expression of the prpBCDE operon; however, a complete understanding of the mechanism by which inflammation-derived nitrogen regulates propionate catabolism is still needed. In this work, we examined the role of propionate utilization by S. Tm in a mouse model of gastroenteritis. Although mouse models provide excellent tools to dissect host-microbe biology, differences between the gut microbiota of humans and mice prevent direct translation of our findings to S. Tm infection in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mariana X. Byndloss (mariana.x.byndloss@vumc.org).
Materials availability
All unique reagents generated in this study are available from the lead contact without restriction.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry
All animal experiments were approved by the Institution of Animal Care and Use Committee at Vanderbilt University Medical Center. Female C57BL/6J, CBA/J mice and Nos2-deficient (on the C57BL/6J background), aged 6 weeks, were obtained from The Jackson Laboratory. Mice were housed in individually ventilated cages with ad libitum access to chow and water. Germ-free (G.F.) Swiss Webster mice were initially purchased from Taconic Farms and maintained by the investigators at Vanderbilt University Medical Center. Experiments in this study were performed with 6-week-old male and female G.F. mice.
Animals were randomly assigned to treatment groups before experimentation. At the end of the experiment, mice were humanely euthanized using carbon dioxide inhalation. Animals that had to be euthanized for humane reasons before reaching the predetermined time point were excluded from the analysis.
Bacterial strains
S. Tm strains (Key Resources Table) were routinely grown aerobically at 37°C in L.B. Broth (10 g/L tryptone, 5 g/L yeast extract, 10 g/L sodium chloride) or on L.B. agar plates (10 g/L tryptone, 5 g/L yeast extract, 10 g/L sodium chloride, 15 g/L agar). When appropriate, L.B. agar plates and broth were supplemented with 100 μg/mL streptomycin (Strep), 30 μg/mL chloramphenicol (Cm), 100 μg/mL carbenicillin (Carb), 50 μg/mL kanamycin (Kan), or 50 μg/mL nalidixic acid (NaI). B. thetaiotaomicron strains were cultured in an anaerobic chamber (85% nitrogen, 10% hydrogen, 5% carbon dioxide, Coy Lab Products). B. thetaiotaomicron strains (Table S1) were routinely cultured on blood agar plates (37 g/L brain heart infusion medium, 15 g/L agar, 50 mL sheep blood).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| S. Typhimurium, SL1344 StrepR | Hoiseth and Stocker, 1981 | SL1344 |
| S. Typhimurium, SL1344 ΔprpC | This study | WJ207 |
| S. Typhimurium, SL1344 ΔprpC phoN::KanR | This study | WJ216 |
| S. Typhimurium, SL1344 ΔprpBCDE | This study | WJ195 |
| S. Typhimurium, SL1344 ΔprpBCDE phoN::KanR | This study | WJ177 |
| S. Typhimurium, SL1344 ΔinvA | This study | WJ227 |
| S. Typhimurium, SL1344 ΔinvAΔspiB | This study | WJ231 |
| S. Typhimurium, SL1344 ΔinvA ΔspiB ΔprpC | This study | CS019 |
| S. Typhimurium, SL1344 ΔinvA ΔspiB ΔprpC phoN::KanR | This study | CS031 |
| S. Typhimurium, SL1344 ΔnapA ΔnarZ narG::pCAL | Lopez et al., 2012 | CAL55 |
| S. Typhimurium, SL1344 ΔnapA ΔnarZ narG::pCAL5 phoN:: CmR | This study | CS048 |
|
S. Typhimurium, SL1344 ΔnapA ΔnarZ narG::pCAL5 ΔprpC |
This study | CS045 |
|
S. Typhimurium, SL1344 ΔnapA ΔnarZ narG::pCAL5 ΔprpC phoN:: KanR |
This study | CS046 |
| S. Typhimurium, SL1344 ΔcyxA | This study | WJ378 |
| S. Typhimurium, SL1344 ΔcyxA ΔprpC | This study | WJ380 |
| S. Typhimurium, SL1344 ΔsopE | This study | WJ377 |
| S. Typhimurium, SL1344 ΔsopE ΔprpC | This study | WJ379 |
| S. Typhimurium ATCC14028 NalR | Stojiljkovic et al., 1995 | IR715 |
| S. Typhimurium, IR715 ΔprpC | This study | NS017 |
| S. Typhimurium, IR715 ΔprpC::KanR | This study | NS039 |
| E. coli, S17-1 λpir, zxx::RP4 2-(TetR::Mu) (KanR::Tn7) λpir recA1 thi pro hsdR (r−m+) | (Simon et al., 1983) | S17-1 λpir |
| B. thetaiotaomicron, VPI 5482 Δtdk | Koropatkin et al., 2008 | VPI5482 |
| B. thetaiotaomicron, Δtdk Δ1686-1689 | Kovatcheva-Datchary et al., 2015 | BT1686-89 |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth, Miller (Luria Bertani) | Becton Dickinson | Cat# 244520 |
| LB Agar, Miller (Luria Berani | Becton Dickinson | Cat# 244620 |
| Platinum PCR Supermix High Fidelity | Invitrogen | 12532016 |
| PCR Supermix | Invitrogen | 10572014 |
| Bacto Brain Heart Infusion | Becton Dickinson | Cat# 237500 |
| Sheep Blood | Fischer BioReagents | Cat# R54008 |
| Sodium Nitrate | Sigma-Aldrich | Cat# S5506 |
| Potassium tetrathionate | Sigma-Aldrich | Cat# P2926 |
| Trimethylamine N-oxide | Sigma-Aldrich | Cat# T0514 |
| Sodium Fumarate | Alfa Aesar | Cat# 1127622 |
| Sodium Propionate | Alfa Aesar | Cat# 1744036 |
| Glycerol | Fisher BioReagents | Cat# G33-4 |
| D (+)- Glucose | Acros Organics | Cat# 410950010 |
| Isopropyl β-D-1-thiogalactopyranoside | Fischer BioReagents | Cat# BP1755 |
| iQ SYBR Green Supermix | Bio-Rad | 170-880 |
| Mucin from porcine stomach, Type II | Sigma-Aldrich | Cat# M2378 |
| Aminoguanidine HCl | Sigma-Aldrich | Cat# 396494 |
| Vanadium (III) Chloride | Acros Organics | Cat# 1970000250 |
| Sulfanilamide | Fischer BioReagents | Cat# 04525-100 |
| N- (1-naphthyl)ethylenediamine dichloride | Sigma-Aldrich | Cat# 222488 |
| Tri Reagent | Molecular Research | Cat# TR118 |
| Critical commercial assays | ||
| Gibson Assembly | NEB | Cat# E2611L |
| SurePrep TrueTotal R.N.A. Purification Kit | Fisher BioReagents | Cat# BP2800 |
| iScript gDNA Clear cDNA synthesis kit | Bio-Rad | Cat# 1725035 |
| Zymoclean Gel DNA Recovery Kit | Zymo Research | D4001 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Cat# 000664 |
| Mouse: CBA | Jackson Laboratory | Cat# 000656 |
| Germ-free Swiss Webster | Byndloss Lab | N/A |
| Oligonucleotides | ||
| Primers used in this study, see Table S1 | This study | N/A |
| Recombinant DNA | ||
| Plasmid: pRDH10, ori(R6K) mobRP4 sacRB CmR TetR | Kingsley et al., 1999 | pRDH10 |
| Plasmid: pKD46, CarbR PBAD-gam-beta-exo oriR101 repA101ts | Datsenko and Wanner, 2000 | N/A |
| Plasmid: pKD13, KanR FRT KmR FRT PS1 PS4 oriR6Kγ | Datsenko and Wanner, 2000 | N/A |
| Plasmid: pCP20, CarbR CmR c/857 λPRflp oripSC101ts | Datsenko and Wanner, 2000 | N/A |
| Plasmid: Up- and downstream region of prpC (SL1344) in pRDH10 | This study | pWJ65 |
| Plasmid: Up- and downstream region of prpBCDE (SL1344) in pRDH10 | This study | pWJ50 |
| Software and algorithms | ||
| GraphPad Prism 8 | GrapPad Software | N/A |
METHOD DETAILS
Construction of bacterial strains and plasmids
S. Tm mutants were generated from either SL1344 or IR715 parent strain. Primer sequences for mutant construction can be found in Table S1. To construct a ΔprpC mutant in the SL1344 parent strain, upstream and downstream regions of approximately 0.5 kb in length were amplified by PCR and then purified. The pRDH10 suicide vector was digested with SalI, purified, and assembled with the prpC PCR fragments to form pRDH10:ΔprpC. pRDH10:ΔprpC was then transformed into E. coli S17-1 λpir. Conjugation was then performed at 30°C, and exconjugants in which the suicide plasmid had integrated into the chromosome of S. Tm were recovered on L.B. agar plates containing streptomycin and chloramphenicol. Subsequent sucrose selection was performed on sucrose plates (5% sucrose, 10 g/L tryptone, 5 g/L yeast extract, 10 g/L sodium chloride, 15 g/L agar) to select for a second crossover events. PCR was performed to detect events that lead to the unmarked deletion of prpC. The ΔprpBCDE mutant strain was constructed as described above, but with primers designed for regions upstream of prpB and downstream of prpE in order to create pRDH10:ΔprpBCDE. To delete prpC from the IR715 parent strain, lambda red recombination was used. The KanR cassette was amplified from pKD13 using the F3 and R3 prpC-deletion primers with homology to upstream and downstream regions of prpC. The resulting PCR product was integrated into the prpC region in a wildtype strain containing the plasmid pKD46, followed by the selection of ΔprpC:Kan mutants. The KanR cassette was removed using the plasmid PCP20 (Datsenko and Wanner, 2000). To generate ΔinvA ΔspiB, invA was deleted from SL1344 using the lambda red recombination method as described above (Datsenko and Wanner, 2000). The KanR cassette was amplified from pKD13 using the primers with homology to upstream and downstream of invA. The resulting PCR product was integrated into the invA region in a wildtype strain containing the plasmid pKD46, followed by the selection of ΔinvA:Kan mutants. The KanR cassette was removed using the plasmid PCP20 (Datsenko and Wanner, 2000). The double mutant ΔinvA ΔspiB was then constructed by deleting spiB from ΔinvA using lambda red recombination as detailed previously. To generate ΔinvA ΔspiB ΔprpC, prpC was deleted from ΔinvA ΔspiB using the lambda red recombination method as described above. To generate both ΔsopE and ΔsopE ΔprpC, sopE was deleted from WT SL1344 and SL1344 ΔprpC using lamba red recombination as described above. To construct a ΔnapA ΔnarG ΔnarZ ΔprpC mutant, conjugation was performed with ΔnapA ΔnarG ΔnarZ (Lopez et al., 2012) and S17-1 λpir transformed with pRDH10:ΔprpC. Exconjugants were recovered on L.B. agar plates containing streptomycin, carbenicillin, and chloramphenicol, and then sucrose selection was performed for second crossover events. PCR was performed to detect events that lead to the unmarked deletion of prpC. To introduce a selectable marker, the phoN:KanR or phoN:CmR mutation was transduced by phage P22 HT int-105 (Schmieger, 1972) into strains as indicated in Table S1. To complement the prpC deletion, the prpC gene was amplified and then combined with the BamHI-digested pUHE21-2lacIq plasmid using Gibson Assembly. The resulting plasmid was transformed into E. coli DH5α, and selected for on L.B. agar plates containing carbenicillin. Plasmid ligation was confirmed by PCR, and pUHE21-2lacIq:prpC was then transformed into ΔprpC. B. thetaiotaomicron containing a deletion in genes BT1686-89 (B. theta BT1686-89) was constructed previously (Kovatcheva-Datchary et al., 2015), and both wildtype B. thetaiotaomicron VPI-5482 Δtdk (B. theta) (Koropatkin et al., 2008) and B. theta BT1686-89 strains were provided to the investigators by Dr. Eric Martens.
In vitro growth assays
Non-Carbon E Salts (NCE media) containing 3.94 g/L monopotassium phosphate, 5.9 g/L dipotassium phosphate, 4.68 g/L ammonium sodium hydrogen phosphate tetrahydrate, 2.46 g/L magnesium sulfate heptahydrate, 1 mM magnesium sulfate, 0.1% casamino acids, 1% vitamin and mineral supplements (A.T.C.C.) was supplemented with 40 mM of an electron acceptor (dimethyl sulfoxide (DMSO), trimethylamine N-oxide (TMAO), fumarate, potassium tetrathionate, or sodium nitrate) or a combination of 10 mM propionate and an electron acceptor. To test the growth of S. Tm under hypoxic conditions, NCE media with or without 10 mM propionate was placed in a hypoxic chamber set to contain 0.8% oxygen. For anaerobic growth experiments, media was placed in the anaerobic chamber 48 h prior to inoculation. Overnight aerobic cultures of S. Tm strains were harvested, washed in PBS, and resuspended in NCE media. Wildtype S. Tm was then added to anaerobic or hypoxic media containing different electron acceptors (plus or minus propionate) at a final concentration of 1 × 104 CFU/mL. Growth was determined after 24 h by spreading serial ten-fold dilutions on LB agar plates.
To measure anaerobic growth of wildtype S. Tm, ΔprpC, and ΔprpBCDE with propionate and/or nitrate, overnight cultures of each strain were diluted into anaerobic NCE media containing 40 mM glycerol and sodium nitrate. Strains were incubated for four hours, harvested, washed in PBS, and resuspended in NCE media. In a 96 well plate, strains were added to NCE media containing propionate, nitrate, or a combination of both at a final OD600 = 0.001. OD600 was measured after 24 h using the Epoch 2 plate reader (BioTek). Similarly, growth of wildtype S. Tm, ΔprpC, and ΔDprpBCDE in NCE media containing 40 mM glycerol and nitrate or 5 mM glucose was determined as described above with the exception that no anaerobic back-dilution was done.
To confirm growth of a complemented ΔprpC strain, overnight cultures of wildtype S. Tm + pUHE21-2lacIq, ΔprpC + pUHE21-2lacIq, and ΔprpC + pUHE21-2lacIq:prpC were harvested, washed in PBS, and resuspended in NCE media. Strains were then added to NCE media containing 200 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 10 mM propionate or 200 μM IPTG, 10 mM propionate, and 40 mM nitrate at a final O.D. = 0.001. OD600 was measured after 24 h using the Epoch 2 plate reader (BioTek). In vitro growth assays were performed in triplicate with different colonies.
In vitro gene expression
Overnight cultures of wildtype S. Tm were harvested and 1 × 109 CFU was added to 5 mL of NCE media or NCE media supplemented with either 10 mM propionate, 40 mM nitrate, or a combination of both. Cultures were incubated for 4 h prior to R.N.A. extraction (performed according to instructions for SurePrep TrueTotal RNA Purification Kit). R.N.A. (500 ng) was reverse transcribed using an iScript gDNA Clear cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using S.Y.B.R. green (SsoAdvanced; Bio-Rad) for prpR, prpB, and prpC. The expression of target genes was normalized to that of gyrB. Primers are listed in Table S2.
Growth and generation time of S. Tm at decreasing pH
NCE media was adjusted to pH 7.0, pH 6.5, and pH 6.0 using 10 M hydrochloric acid (HCl). 10 mL of media from each pH was supplemented with 10 mM propionate or 10 mM propionate +40 mM nitrate and placed in the anaerobic chamber. Overnight cultures of wildtype S. Tm and ΔprpC were washed in PBS and resuspended in NCE media. Cultures were adjusted to an O.D. = 0.1 and then diluted 1:100 into pH-adjusted media. Samples were incubated at 37°C, and aliquots were removed every two hours for 14 h. Aliquots were then serially diluted and plated onto LB agar to determine bacterial numbers. Generation time (G) was calculated according to (Todar, 2006) and using the following formula: where T2 equals the time at the end of exponential phase, and T1 is the time at the beginning of the exponential phase. B2 corresponds to the bacterial number at T2, and B1 equals the bacterial number at T1.
Growth of Bacteroides thetaiotaomicron strains in mucin broth
Porcine mucin was dissolved in 1x NCE salts at a final concentration of 0.5% (w/v). Mucin broth was inoculated with a fresh colony of B. theta BT1686-89 and incubated under anaerobic conditions for 96 h at 37°C or inoculated with wildtype B. theta and incubated for 48 h at 37°C. To measure the growth of B. theta BT1686-89, aliquots were removed from cultures, and ten-fold serial dilutions were plated on blood agar plates to calculate bacterial numbers. Digested mucin broth from each strain was filter-sterilized (0.5 μm pore size) before propionate measurements or competitive growth assays.
Competitive growth assays
Competition assays were performed in either digested mucin broth or NCE Media. Propionate (10 mM) or nitrate (40 mM) were added as indicated. A 1:1 ratio of two overnight bacterial strains at a final concentration of 1 × 104 CFU/mL were added to the media and incubated anaerobically for 18 h. Bacterial numbers were determined by plating serial dilutions on selective LB Agar plates. In vitro competition assays were performed in triplicate with different colonies.
Animal experiments
Streptomycin-treated mouse model:
Groups of 6 to 7-week-old C57BL/6 mice were treated with 5 g/L streptomycin in the drinking water for 48 h which was then removed 24 h before infection with S. Tm. For competitive infections, mice were orally inoculated with a 1:1 mixture of 1 × 109 CFU of each strain. Fecal samples were collected two days after infection for propionate measurement. Four days after infection, samples for histopathology, cecal tissue for RNA extraction, colonic luminal content for bacterial plating, and cecal content for propionate measurements were collected. In some experiments, mice were given 1 g/Laminoguanidine hydrochloride in their drinking water immediately after infection with S. Tm.
CBA mouse model:
Groups of 6 – 7-week-old female CBA mice were orally inoculated with a 1:1 mixture of 1 × 109 CFU of S. Tm and ΔprpC (both SL1344 and IR715 backgrounds). Fecal samples were collected at days 1, 3, 5, 7, 9, and 11 after infection for bacterial plating and propionate measurements. Thirteen days after infection, samples for histopathology, colonic luminal content and cecal content for bacterial plating, and propionate measurements were collected.
Germ-Free Swiss Webster mice:
Groups of 6-week-old germ-free mice were colonized with approximately 1 × 108 CFU of wildtype B. theta or B. theta BT1686-89. Four days after colonization a subset of mice colonized with B. theta BT1686-89 were given 20 mM propionate in the drinking water. After 7 days, fecal samples were collected from mono-colonized mice to measure B. theta colonization and propionate levels. Mono-colonized and germ-free mice were then infected with an equal mixture of 1 × 107 CFU of wildtype S. Tm and ΔprpC. Three days after infection with S. Tm samples were collected as described above.
Nitrate measurements
Intestinal nitrate measurements were performed as described previously (Byndloss et al., 2017). Briefly, mice were euthanized, and the intestine was removed and divided along its sagittal plane. The mucus layer was gently scraped from the tissue and homogenized in 200 μL PBS and then placed on ice. Samples were centrifuged at 5,000 × g for 10 min at 4°C to remove the remaining solid particles. The supernatant was then filter sterilized (0.2-μm pore size). Measurement of intestinal nitrate followed an adaptation of the Griess assay. In this assay, nitrate was first reduced to nitrite by combining 50 μL of each sample with 50 μL of Griess reagent 1 containing vanadium(III) chloride (0.5 M HCl, 0.2 mM VCl3, 1% sulfanilamide), and then the mixture was incubated at room temperature for 10 min. Next, 50 μL of Griess reagent 2 (0.1% [1-naphthyl]ethylenediamine dichloride) was added to each sample. Absorbance at 540 nm was measured immediately after the addition of Griess reagent 2 to detect any nitrite present in the samples. The samples were then incubated for 8 h at room temperature (to allow for reduction of nitrate to nitrite), and the absorbance at 540 nm was measured again. The initial absorbance (prior to reducing nitrate to nitrite) was subtracted from the absorbance after 8 h to determine nitrate concentrations in the mucus layer.
Quantification of Nos2 expression by qRT-PCR
Cecal tissue was homogenized using a FastPrep-24 and RNA extracted using the TRI reagent method. RNA (1 μg) was reverse transcribed using an iScript gDNA Clear cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed with SYBR green (SsoAdvanced; Bio-Rad) for Nos2 (Primers listed in Table S2). The expression of Nos2 was normalized to the housekeeping gene Act2b, encoding β-actin.
Propionate measurements
Extraction and normalization.
Fecal matter was weighed, diluted to a final density of 0.125 g/mL in MeOH/H2O (1:5) and homogenized with a cordless Pellet Pestle tissue grinder equipped with disposable polypropylene mixers (Fisher). Insoluble debris was removed by centrifugation (12,000 × g, 30 min, 5°C); the supernatants were transferred to clean Eppendorf tubes and stored at −20°C until the day of analysis.
Propionate Analysis.
Propionate was derivatized with the reagent dansylhydrazine and the carboxyl activating agent 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and measured as its corresponding dansylhydrazone derivative (Tan et al., 2014). Briefly, fecal extracts (10 μL) were spiked with a stable isotope-labeled internal standard propionate-d5 (1 nmol) and derivatized in H2O/DMSO (2:1) containing 50 mM sodium phosphate buffer (pH = 4), 12.5 mg/mL dansylhydrazine, and 12.5 mg/mL EDC. Due to the limited stability of EDC in water, stock solutions of EDC should be made up in ice-cold water and used immediately. After two hours at room temperature, dansylated derivatives were extracted once with ethyl acetate (750 μL). The organic (top) layer was transferred to a clean Eppendorf tube, dried under a gentle stream of nitrogen gas, and reconstituted in 150 μL of acetonitrile/water (1:1) prior to analysis. Calibration standards for unlabeled propionate were prepared in water, derivatized, and extracted in the same manner. LC-MS/MS analysis was performed using a Thermo TSQ Quantum mass spectrometer interfaced to a Thermo HTC PAL refrigerated autosampler and a Thermo Surveyor HPLC pump. A Waters XTerra MS analytical column (2.1 mm × 100 mm, 3.5 μm) was used for all chromatographic separations. Mobile phases were made up of 0.2% acetic acid and 15 mM ammonium acetate in (A) H2O/CH3CN (9:1) and in (B) CH3CN/CH3OH/H2O (90:5:5). Gradient conditions were as follows: 0 to 1 min, B = 0%; 1 to 8 min, B = 0% to 100%; 8 to 10 min, B = 100%; 10 to 10.5 min, B = 100% to 0%; 10.5 to 15 min, B = 0%. The flow rate was maintained at 300 μL/min; a software-controlled divert valve was used to transfer eluent from 0 to 2.0 min of each chromatographic run to waste. The total chromatographic run time was 15 min. The autosampler tray temperature and the column compartment temperature were maintained at 5°C and 50°C respectively. The sample injection volume was 10 μL. The autosampler injection valve and the sample injection needle were flushed and washed sequentially with mobile phase B (two cycles) and mobile phase A (two cycles) between each injection. The mass spectrometer was operated in positive ion mode. Quantitation was based on single reaction monitoring detection of the following dansylated analogues: propionate, m/z 322 ↑ 235, C.E. 15; propionate-d5: m/z 327 ↑ 235, C.E. 15. The following optimized source parameters were used for the detection of analytes and internal standards. N2 sheath gas 40 psi; N2 auxiliary gas 5 psi; spray voltage 4 kV; capillary temperature 300°C; tube lens voltage 120 V; declustering voltage 20 V. Data acquisition and quantitative spectral analysis were done using Thermo-Finnigan Xcalibur version 2.0.7 SP1 and Thermo-Finnigan LCQuan version 2.7, respectively. Calibration curves were constructed by plotting peak area ratios (analyte/internal standard) against analyte concentrations for a series of nine calibration standards, ranging from 0.01 to 100 nmol propionate. A weighting factor of 1/C2 was applied in the linear least-squares regression analysis to maintain homogeneity of variance across the concentration range (%RE ≤ 15% at C > LLOQ).
Histopathology scoring
Formalin fixed cecal tissue sections were stained with hematoxylin and eosin, and a veterinary pathologist performed a blinded evaluation using criteria shown in Table S3 as described previously (Spees et al., 2013). Representative images were taken using a Leica DM750 microscope and a Leica ICC50W camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical data analysis was performed using Graphpad PRISM. Fold changes of ratios (bacterial competitive index and mRNA levels), and bacterial numbers were transformed logarithmically prior to statistical analysis. An unpaired Student’s t test was used on the transformed data to determine whether differences between groups were statistically significant (p < 0.05). When more than two treatments were used, statistically significant differences between groups were determined by one-way ANOVA followed by Tukey’s HSD test (between >2 groups). Significance of differences in histopathology was determined by a one-tailed non-parametric test (Mann-Whitney).
Supplementary Material
Highlights.
Nitrate-dependent anaerobic respiration enables propionate metabolism by S. Tm
Propionate metabolism supports S. Tm intestinal expansion in the inflamed gut
Propionate utilization by S. Tm is dependent on the SPI-1 effector SopE
Bacteroides-derived propionate fuels S. Tm intestinal growth in gnotobiotic mice
ACKNOWLEDGMENTS
We thank Dr. Eric Martens for kindly providing Bacteroides thetaiotaomicron and Bacteroides thetaiotaomicron BT1686-89. C.D.S. was supported by the Dorothy Beryl and Theodore Roe Austin Pathology Research Fund and NIH T32 Training Grant T32AI112541. W.Y. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) by the Ministry of Education 2020R1A6A3A03037326. N.G.S. was supported by NIH T32 Training Grant T32ES007028-46 and GT15104 from the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program. N.J.F. was supported by NIH T32 Training Grant T32DK007673. Work in M.X.B.’s laboratory was supported by V Scholar grant V2020-013 from The V Foundation for Cancer Research, Vanderbilt Digestive Disease Pilot and Feasibility grant P30 058404, A.C.S. Institutional Research grant IRG-19-139-59, VICC GI SPORE grant P50CA236733, United States-Israel Binational Science Foundation grant 2019136, and Vanderbilt Institute for Clinical and Translational Research grant VR53102.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110180.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- Alam MS, Akaike T, Okamoto S, Kubota T, Yoshitake J, Sawa T, Miyamoto Y, Tamura F, and Maeda H (2002). Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun 70, 3130–3142. 10.1128/iai.70.6.3130-3142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MM, Newsom DL, González JF, Sabag-Daigle A, Stahl C, Steidley B, Dubena J, Dyszel JL, Smith JN, Dieye Y, et al. (2014). Fructose-asparagine is a primary nutrient during growth of Salmonella in the inflamed intestine. PLoS Pathog. 10, e1004209. 10.1371/journal.ppat.1004209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, and Gordon JI (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U S A 101, 15718–15723. 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EL, and Riggs DL (1982). Evidence of a second nitrate reductase activity that is distinct from the respiratory enzyme in Salmonella typhimurium. J. Bacteriol 150, 563–571. 10.1128/jb.150.2.563-571.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Rüssmann H, and Hardt WD (2003). Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun 71, 2839–2858. 10.1128/iai.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Pifer R, Behrendt CL, Hooper LV, and Yarovinsky F (2009). Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6, 187–196. 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnhoff M, Miller CP, and Martin WR (1964). Resistance of the mouse’s intestinal tract to experimental Salmonella infection. I. Factors which interfere with the initiation of infection by oral inoculation. J. Exp. Med 120, 805–816. 10.1084/jem.120.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, et al. (2017). Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BM, and Dankert J (1979). Volatile fatty acids and aerobic flora in the gastrointestinal tract of mice under various conditions. Infect. Immun 23, 559–563. 10.1128/iai.23.3.559-563.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK, Coburn BA, Potter AA, Gomis S, Mirakhur K, Li Y, and Finlay BB (2005). Analysis of the contribution of Salmonella pathogenicity islands 1 and 2 to enteric disease progression using a novel bovine ileal loop model and a murine model of infectious enterocolitis. Infect. Immun 73, 7161–7169. 10.1128/iai.73.11.7161-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, and Bakker BM (2013). The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res 54, 2325–2340. 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriu E, Liu JZ, Pezeshki M, Edwards RA, Ochoa RJ, Contreras H, Libby SJ, Fang FC, and Raffatellu M (2013). Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14, 26–37. 10.1016/j.chom.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F, Thiennimitr P, Spiga L, Byndloss MX, Litvak Y, Lawhon S, Andrews-Polymenis HL, Winter SE, and Bäumler AJ (2017). Respiration of microbiota-derived 1,2-propanediol drives Salmonella expansion during colitis. PLoS Pathog. 13, e1006129. 10.1371/journal.ppat.1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman ES, Bittinger K, Esipova TV, Hou L, Chau L, Jiang J, Mesaros C, Lund PJ, Liang X, FitzGerald GA, et al. (2018). Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. U S A 115, 4170–4175. 10.1073/pnas.1718635115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán JE, and Curtiss R 3rd. (1989). Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U S A 86, 6383–6387. 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiseth SK, and Stocker BA (1981). Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239. 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- Horswill AR, Dudding AR, and Escalante-Semerena JC (2001). Studies of propionate toxicity in Salmonella enterica identify 2-methylcitrate as a potent inhibitor of cell growth. J. Biol. Chem 276, 19094–19101. 10.1074/jbc.M100244200. [DOI] [PubMed] [Google Scholar]
- Horswill AR, and Escalante-Semerena JC (1997). Propionate catabolism in Salmonella typhimurium LT2: two divergently transcribed units comprise the prp locus at 8.5 centisomes, prpR encodes a member of the sigma-54 family of activators, and the prpBCDE genes constitute an operon. J. Bacteriol 179, 928–940. 10.1128/jb.179.3.928-940.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill AR, and Escalante-Semerena JC (1999). Salmonella typhimurium LT2 catabolizes propionate via the 2-methylcitric acid cycle. J. Bacteriol 181, 5615–5623. 10.1128/jb.181.18.5615-5623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CC, Garner CD, Slauch JM, Dwyer ZW, Lawhon SD, Frye JG, McClelland M, Ahmer BM, and Altier C (2013). The intestinal fatty acid propionate inhibits Salmonella invasion through the post-translational control of HilD. Mol. Microbiol 87, 1045–1060. 10.1111/mmi.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba AM, and Berchieri A Jr. (1995). Studies on the use of a formic acid-propionic acid mixture (Bio-add) to control experimental Salmonella infection in broiler chickens. Avian Pathol. 24, 303–311. 10.1080/03079459508419071. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A, Lam L, Rajendram M, Tamburini F, Honeycutt J, Pham T, Van Treuren W, Pruss K, Stabler SR, Lugo K, et al. (2018). A gut commensal-produced metabolite mediates colonization resistance to Salmonella infection. Cell Host Microbe 24, 296–307.e297. 10.1016/j.chom.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley RA, Reissbrodt R, Rabsch W, Ketley JM, Tsolis RM, Everest P, Dougan G, Bäumler AJ, Roberts M, and Williams PH (1999). Ferrioxamine-mediated iron(III) utilization by Salmonella enterica. Appl. Environ. Microbiol 65, 1610–1618. 10.1128/aem.65.4.1610-1618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin NM, Martens EC, Gordon JI, and Smith TJ (2008). Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115. 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, Hallen A, Martens E, Björck I, and Bäckhed F (2015). Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab. 22, 971–982. 10.1016/j.cmet.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, and Hsieh CS (2011). Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254. 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Rivera-Chávez F, Byndloss MX, and Bäumler AJ (2015). The periplasmic nitrate reductase NapABC supports luminal growth of Salmonella enterica serovar Typhimurium during colitis. Infect. Immun 83, 3470–3478. 10.1128/iai.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Winter SE, Rivera-Chávez F, Xavier MN, Poon V, Nuccio SP, Tsolis RM, and Bäumler AJ (2012). Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. mBio 3. 10.1128/mBio.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PA, Bettke JA, Tam JW, Leeds J, Bliska JB, Butler BP, and van der Velden AWM (2019). Inflammatory monocytes provide a niche for Salmonella expansion in the lumen of the inflamed intestine. PLoS Pathog. 15, e1007847. 10.1371/journal.ppat.1007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio SP, and Bäumler AJ (2014). Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. mBio 5, e00929–e00914. 10.1128/mBio.00929-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO, Roos S, Holm L, and Phillipson M (2011). Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol 300, G327–G333. 10.1152/ajpgi.00422.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, et al. (2009). Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5, 476–486. 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, and Louis P (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. Isme j 8, 1323–1335. 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke SC (2003). Perspectives on the use of organic acids and short chain fatty acids as antimicrobials. Poult. Sci 82, 632–639. 10.1093/ps/82.4.632. [DOI] [PubMed] [Google Scholar]
- Rivera-Chávez F, and Bäumler AJ (2015). The pyromaniac inside you: Salmonella metabolism in the host gut. Annu. Rev. Microbiol 69, 31–48. 10.1146/annurev-micro-091014-104108. [DOI] [PubMed] [Google Scholar]
- Rondon MR, Horswill AR, and Escalante-Semerena JC (1995). DNA polymerase I function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by Salmonella typhimurium LT2. J. Bacteriol 177, 7119–7124. 10.1128/jb.177.24.7119-7124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi M, and Raffatellu M (2015). No vacancy: how beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol 194, 4081–4087. 10.4049/jimmunol.1403169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieger H (1972). Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet 119, 75–88. 10.1007/bf00270447. [DOI] [PubMed] [Google Scholar]
- Simon R, Priefer U, and Pühler A (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology1, 784–791. [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan foraging in vivo by an intestineadapted bacterial symbiont. Science 307, 1955–1959. 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- Sorbara MT, Dubin K, Littmann ER, Moody TU, Fontana E, Seok R, Leiner IM, Taur Y, Peled JU, van den Brink MRM, et al. (2019). Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J. Exp. Med 216, 84–98. 10.1084/jem.20181639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbara MT, and Pamer EG (2019). Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol. 12, 1–9. 10.1038/s41385-018-0053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spees AM, Wangdi T, Lopez CA, Kingsbury DD, Xavier MN, Winter SE, Tsolis RM, and Bäumler AJ (2013). Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. mBio 4. 10.1128/mBio.00430-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic I, Bäumler AJ, and Heffron F (1995). Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J. Bacteriol 177, 1357–1366. 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H, and Radi R (2007). Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov 6, 662–680. 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tan B, Lu Z, Dong S, Zhao G, and Kuo MS (2014). Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal. Biochem 465, 134–147. 10.1016/j.ab.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, and Bäumler AJ (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U S A 108, 17480–17485. 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todar K (2006). Todar’s Online Textbook of Bacteriology (University of Wisconsin-Department of Bacteriology; ). [Google Scholar]
- van der Waaij D, Berghuis-de Vries JM, and Lekkerkerk L.-v. (1971). Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J. Hyg. (Lond) 69, 405–411. 10.1017/s0022172400021653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Liwinski T, and Elinav E (2020). Interaction between microbiota and immunity in health and disease. Cell Res 30, 492–506. 10.1038/s41422-020-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.