

Abstract
Idiopathic pulmonary fibrosis (IPF), a devastating, fibroproliferative, chronic lung disorder, is associated with expansion of fibroblasts/myofibroblasts, which leads to excessive production and deposition of extracellular matrix. IPF is typically clinically identified as end-stage lung disease, after fibrotic processes are well-established and advanced. Fibroblasts have been shown to be critically important in the development and progression of IPF. We hypothesize that differential chromatin access can drive genetic differences in IPF fibroblasts relative to healthy fibroblasts. To this end, we performed assay of transposase-accessible chromatin sequencing to identify differentially accessible regions within the genomes of fibroblasts from healthy and IPF lungs. Multiple motifs were identified to be enriched in IPF fibroblasts compared with healthy fibroblasts, including binding motifs for TWIST1 and FOXA1. RNA sequencing identified 93 genes that could be annotated to differentially accessible regions. Pathway analysis of the annotated genes identified cellular adhesion, cytoskeletal anchoring, and cell differentiation as important biological processes. In addition, single nucleotide polymorphism analysis showed that linkage disequilibrium blocks of IPF risk single nucleotide polymorphisms with IPF-accessible regions that have been identified to be located in genes that are important in IPF, including MUC5B, TERT, and TOLLIP. Validation studies in isolated lung tissue confirmed increased expression for TWIST1 and FOXA1 in addition to revealing SHANK2 and CSPR2 as novel targets. Thus, modulation of differential chromatin access may be an important mechanism in the pathogenesis of lung fibrosis.
Keywords: idiopathic pulmonary fibrosis, fibroblasts, RNA sequencing, ATAC sequencing, epigenetics
The most common type of restrictive lung disease is idiopathic pulmonary fibrosis (IPF), a lethal lung disease defined by chronic, progressive, and irreversible interstitial fibrosis. In established IPF, hazy opacities, reduced inspiratory lung volumes, and chest radiographs showing bilateral reticular infiltrates predominately in the lower lobes indicate the presence of interstitial lung disease (1). Several genetic predispositions for IPF have previously been identified. These include genes associated with telomerase maintenance (telomerase RNA component [TERC], telomerase reverse transcriptase [TERT]) (1), along with others such as Toll-interacting protein (TOLLIP), mucin-5B (MUC5B), and MAM domain–containing glycosylphosphatidylinositol anchor protein 2 (MDGA2) (2). The prognosis of IPF with conventional treatment is poor, and minimal treatment exists. If left untreated, the median survival rate for IPF is 3–5 years. For long-term survival, the only strategy is lung transplantation. However, donor lung supply is limited, and lung transplant outcomes are still inferior to those of other organ transplants. Therefore, identification of nontransplant therapies continues to be essential.
IPF is often detected as an end-stage diffuse fibrotic disease. IPF progresses from foci of fibrotic lesions in the lower lobes, to diffuse lung disease, with intermittent fibrotic lesions visible in the upper lobes. The upper lobes often still contain some normal-appearing tissue (1, 3). Distinct differences in expression profiles between upper and lower lobes has recently been shown (4). Key effector cells in fibrotic foci are activated fibroblasts, myofibroblasts. Whereas under physiological conditions, myofibroblasts are pivotal for wound repair, aberrant activation drives these cells to become hyperproliferative with increased extracellular matrix production and apoptotic resistance (5, 6). These cellular characteristics contribute to the destruction of lung architecture and are important in driving the active fibrotic lesions observed in IPF.
The cellular characteristics of fibroblasts in IPF have been shown to differ from normal fibroblasts (7). These studies have been performed from cells isolated from highly fibrotic lung regions, yet the genetic signature from fibroblasts in areas of active fibrotic lesions is not fully known. The upper lobes of explanted IPF lungs contain normal-appearing tissue with intermittent fibrotic foci, suggesting active fibrotic lesions in these foci. Gene expression profiles of myofibroblasts isolated from the upper lobes in patients with IPF may offer early disease markers and help us delve into the mechanisms that promote aberrant tissue repair in IPF.
Our understanding of the cellular and molecular pathways associated with fibroblasts in active IPF lesions remains limited. We have used assay for transposase-accessible chromatin sequencing (ATAC-Seq) on upper lobe cultured fibroblasts from healthy controls and IPF patients to identify differentially accessible regions (DARs) and transcription factor binding sites within the genome of these cells. Additionally, we have used RNA sequencing (RNA-Seq) to identify differentially expressed genes (DEGs) that could reflect early disease in IPF. Our hypothesis is that fibroblasts isolated from the upper lobes from IPF patients will present with distinct transcriptomic and epigenetic signatures compared with healthy fibroblasts. These upper lobe signatures may provide insight in the progression of fibrosis in IPF.
Methods
Human Samples
Human-explanted lung samples from IPF patients were deidentified and obtained from the transplantation programs at Memorial Hermann Hospital and The Houston Methodist Hospital. The collection of material was approved by the following institutional review board approvals (HSC-MS-15–1049 and HSC-MS-08–0354). IPF lung tissue was collected at the time of lung explantation and processed on site within 60 minutes. Normal human lung tissues were obtained from the International Institute for the Advancement of Medicine; they were collected from lungs that were unused for transplantation because of reasons unrelated to obvious acute or chronic pulmonary disease. Basic demographic and clinical data from control- and IPF-derived tissues are included in Table E1 in the data supplement.
Cell Culture
Primary human lung fibroblasts were isolated directly from the upper half of the upper lobe close to apical regions from human lungs (explant IPF lungs or normal healthy lungs) using the explant method. This area of the lung in IPF patients has reduced fibrosis compared with lower regions of the lung as demonstrated histologically and following Ashcroft scores (Figure E1). Fibroblasts were cultured in Dulbecco’s modified Eagle medium containing 10% FBS (Sigma-Aldrich), penicillin/streptomycin (Sigma-Aldrich), plasmocin (Invivogen), and amphotericin B (Sigma-Aldrich). Passage 3 and 4 were used for all subsequent analysis.
Histology and Morphometry
Formalin-fixed paraffin embedded lung samples were cut at 5 μm thickness and stained for Masson’s trichome. Ashcroft scores were performed by investigators blinded to group status, as previously described (8).
RNA Isolation
RNA was isolated using Trizol and the Qiagen RNeasy kit (Qiagen) according to the manufacturer’s instructions. RNA was stored at –80°C.
Preprocessing and Differential Analysis of RNA Sequencing Data
The raw reads obtained from paired-end RNA sequencing were mapped to human reference genome hg38 using Hisat2 (9) with default parameters. Gene expression level was calculated as fragments per Kb per million reads with Cufflinks v2.2.1 (10) and count with htseq-count (11); the annotation gtf file is gencode v28 from GENCODE (12). R edgeR (13) was used to obtain the DEGs between IPF patients and health controls with false discovery rate (FDR) cutoff of 0.01 and fold change cutoff of 1.5.
ATAC-Seq Data Analysis
ATAC-Seq was performed on three individual IPF fibroblasts and three individual healthy controls. All libraries were sequenced on the Illumina HiSeq 2500 with 80 bp single-end reads. Illumina adapters were first trimmed by Trim galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and then reads were aligned to hg38 with bowtie2 (14) using default parameters. The low-quality reads (MAPQ < 10, PCR duplicates) were excluded from the further analysis. All mapped reads were offset by +4 bp for the + strand and –5 bp for the – strand (15). For each sample, peaks were called using MACS2 (16) with following parameters: “-q 0.05 -B –nomodel –shift 37 –extsize 73 –broad –keep-dup all.” Encode blacklist regions and regions in mitochondrial and chromosome Y were excluded from called peaks. The significant change of chromatin accessibility between the two groups was accessed using edgeR (13) on the basis of the counts of peak regions, with FDR (false discovery rate) cutoff of 0.05 and fold change cutoff of 1.5. HOMER (17) was applied for peak annotation and motif analysis. MEME-ChIP was also applied for motif analysis (18). A schematic overview of the data processing is given in Figure E2.
Data Sharing
All analyzed ATAC-Seq and RNA-Seq data will be made available and deposited in an online database.
Validation qPCR and PCR Array
Total RNA was isolated from frozen lung tissue. Individual primers for validation quantitative PCR (qPCR) are described in Table E2 and were run using SYBR Green.
For qPCR experiments that consisted of two groups, an unpaired two-tailed Student’s t test with a Welch correction was performed. The Grubbs' test was used to detect outliers. GraphPad Prism 7.0 or higher was used to analyze the data.
Results
Single Nucleotide Polymorphisms in Differentially Associated Regions
Healthy and IPF fibroblasts were isolated from the upper half of the upper lobe of healthy controls and IPF patients. ATAC using sequencing was performed to assess chromatin accessibility differences between healthy and IPF fibroblasts. Quality control of ATAC-Seq libraries showed similar genomic distribution of merged peak regions in IPF and healthy fibroblasts (Figure E3). The successful detection of accessible regions is also supported by the observation of strong enrichment of reads around transcription start sites (Figure E4). As ATAC-Seq reads are enriched at transcription start sites (19), this method allows for transcription factor binding prediction. DNA binding-site motif analysis shows DARs that become either more or less accessible in IPF fibroblasts compared with healthy fibroblasts. Twenty-seven unique genome-wide association study SNPs were known to be associated with IPF from the EBI database (https://www.ebi.ac.uk/gwas/efotraits/EFO_0000768); linkage disequilibrium (LD) blocks of those SNPs were obtained from SNiPA database (https://snipa.helmholtz-muenchen.de/snipa3/) using the 1000 Genome Project Phase 3 data of European population with the cutoff of r2 < 0.8. We then identified 20 LD blocks out of 27 SNPs (Table E3). Except the LD block of rs7725218, all the 19 LD blocks contained IPF peaks, and LD block of rs11191865 contained more IPF DARs. Our results demonstrated the presence of SNPs previously reported in IPF that included TERT, TOLLIP, MUC5B, GOSR2, and TYH1, among others (2, 20, 21). These results demonstrate that these SNPs are conserved in our IPF samples and validate the IPF origin of our fibroblasts. The genomic distribution of peak regions in our ATAC-Seq samples in healthy and IPF fibroblasts is shown in Figure E5.
DEGs in Healthy and IPF Fibroblasts
To identify gene expression changes, RNA-Seq was performed for both healthy and IPF fibroblasts to find DEGs in healthy versus IPF fibroblasts. We used a cutoff FDR <0.05 and fold change >1.5. Our volcano plot and heat map show a total of 858 DEGs between healthy and IPF fibroblasts, with 350 genes that were upregulated and 508 downregulated in IPF versus healthy fibroblasts (Figure 1A), and the resulting heat map (Figure 1B) reveals clear differences between IPF and controlling derived fibroblasts. The top 40 DEGs are listed in Table 1. Importantly, using both ATAC-Seq and RNA-Seq samples we performed a principal component analysis plot including all regions or genes demonstrating a clear separation between our control and IPF samples (Figure E6).
Figure 1.
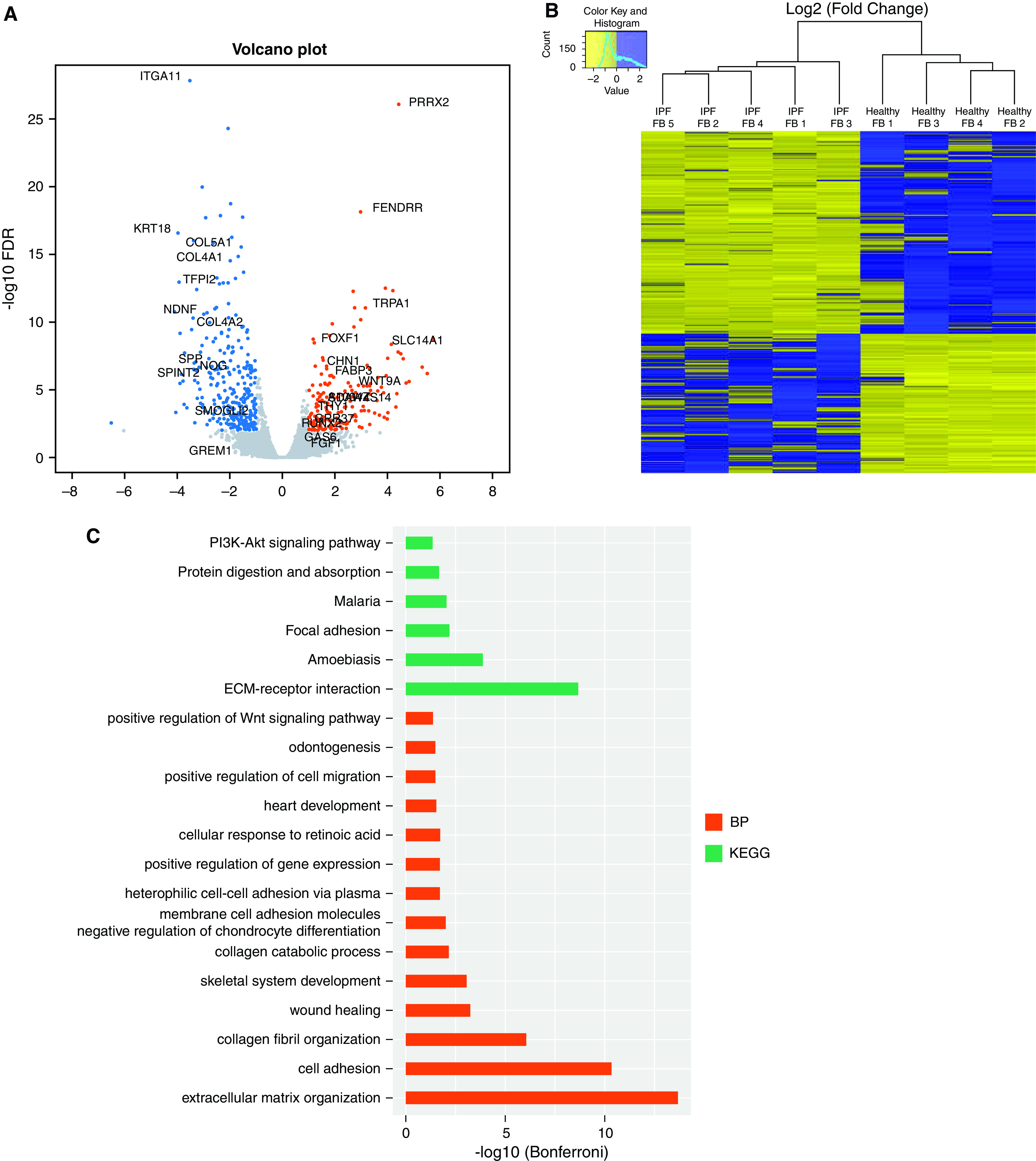
RNA sequencing analysis of healthy and idiopathic pulmonary fibrosis (IPF) fibroblasts (FBs). (A) Volcano plot and (B) heatmap of differentially expressed genes (DEGs) in healthy and IPF FBs by RNA sequencing. Cutoff FDR was <0.05 and fold change >1.5. (C) Annotation of differentially expressed genes in healthy and IPF FBs using DAVID. BP = biological process; DAVID = database for annotation, visualization and integrated discovery; ECM = extracellular matrix; FDR = false discovery rate; KEGG = Kyoto Encyclopedia of Genes and Genomes.
Table 1.
Differentially Expressed Genes in Idiopathic Pulmonary Fibrosis Fibroblasts Compared to Healthy Fibroblasts
| Gene Symbol | Log2 (Fold Change) | Gene Symbol | Log2 (Fold Change) |
|---|---|---|---|
| Top Downregulated Genes | Top Upregulated Genes | ||
| PDPN | −4.0920827 | ITM2A | 5.74460609 |
| CPXM1 | −4.0497139 | LCE1E | 5.51594931 |
| KRT18 | −3.963184 | SLITRK2 | 5.31882352 |
| TSPAN18 | −3.9318555 | LCE2A | 4.81649601 |
| RARRES2 | −3.8886874 | CYSLTR1 | 4.7029308 |
| PKD1P5 | −3.8386864 | TOX | 4.5905139 |
| TENM2 | −3.7764473 | PRRX2 | 4.42152492 |
| AGT | −3.7339043 | LRATD1 | 4.40165336 |
| KIF21B | −3.7171292 | DUXAP9 | 4.35409129 |
| CECR2 | −3.6379231 | RORB | 4.3229159 |
| ITGA11 | −3.5238605 | CADPS | 4.20133148 |
| GPRC5C | −3.4537471 | SLC14A1 | 4.15324368 |
| RSPO3 | −3.3910054 | ST8SIA1 | 4.02093675 |
| FRAS1 | −3.3745218 | FER1L6 | 4.00672099 |
| TMEM233 | −3.3691099 | NELFE | 3.98025726 |
| CDK18 | −3.3514001 | KPRP | 3.94831062 |
| NEFH | −3.3382606 | ADAMTS19 | 3.86817991 |
| IGFN1 | −3.3304446 | TPTEP1 | 3.76492248 |
| SDK2 | −3.288493 | RUNX3 | 3.71662771 |
| UNC5C | −3.2667218 | NTSR1 | 3.67969715 |
Interestingly, the top 40 differentially expressed protein-coding genes revealed many targets that have been previously linked with lung fibrosis. These include podoplanin (PDNP) (22), keratin type II cytoskeletal 8 (KRT8) (23), angiotensin (AGT) (24), integrin alpha 11 (ITGA11) (25), R-spondin 3 (RSPO3) (26), and cysteinyl leukotriene type 1 receptor (CYSLTR1) (27). The other genes were presumed to be novel genes associated with IPF. Subsequent pathways and functional annotation of the DEGs in IPF fibroblasts were next analyzed using database for annotation, visualization and integrated discovery (DAVID) (Figure 1C). These analyses demonstrated that cellular adhesion and extracellular matrix (ECM) organization/receptor interaction pathways are enriched in IPF fibroblasts compared with normal cells.
Differentially Associated Regions in Healthy and IPF Fibroblasts
A heatmap of DARs between healthy and IPF fibroblasts was determined by ATAC-Seq with both enriched and reduced open chromatin regions in healthy versus IPF fibroblasts (Figure 2A), with the cutoff of FDR <0.05 and fold change >1.5. A total of 1,487 DARs were found with 904 DARs more accessible and 583 DARs less accessible regions in IPF fibroblasts compared with healthy fibroblasts. The significantly enriched motifs among those DARs through motif analysis are shown in Figure 2B. This revealed the known IPF target TWIST1 and also revealed novel targets such as Hepatocyte nuclear factor 3-alpha (FOXA1), Zinc finger and BTB domain containing 18 (ZBTB18), Core-binding factor subunit beta (CBFB), and Forkhead box P1 (FOXP1). These transcription factors are involved in development, organ-specific cell differentiation, and hematopoiesis. Our results revealed reduced accessibility for novel targets including CCAAT/enhancer-binding protein alpha (CEBPA), Trans-acting T cell-specific transcription factor GATA-6 (GATA6), Distal-less homeobox 1 (DLX1), MDS1 and EVI1 complex locus (MECOM), TGFB induced factor homeobox 1 (TGIF1), and MEIS homeobox 1 (MEIS1) (Figure 2B). The functions of these transcription factors were varied, but they are involved in body weight regulation, craniofacial patterning, development, cellular differentiation and proliferation, signal transduction, and survival of inhibitory neurons in the brain. We then gathered information of target genes of the 11 TFs using the TF2DNA, MotifMap, and CHEA databases (28–30). The obtained target genes that intersected with DEGs between IPF and healthy fibroblasts are the potential true target genes employed in the IPF disease state. Finally, the overlap in TF functionality between increased and decreased DARs was reflected in the biomolecular interaction network of TFs and their target genes using Cytoscape (Figure 3) (31). To generate this figure, the TF2DNA, MotifMap, and CHEA databases was used to predict the potential binding target genes for these transcription factors on the basis of experimental and theoretical sources. The predicted target genes of TFs obtained from the three databases were then compared with DARs associated genes and DEGs; the overlapped genes between predicted target genes of three databases and DARs associated DEGs have the highest possibilities to be true targets of TFs in our fibroblasts cell line (Table E4).
Figure 2.
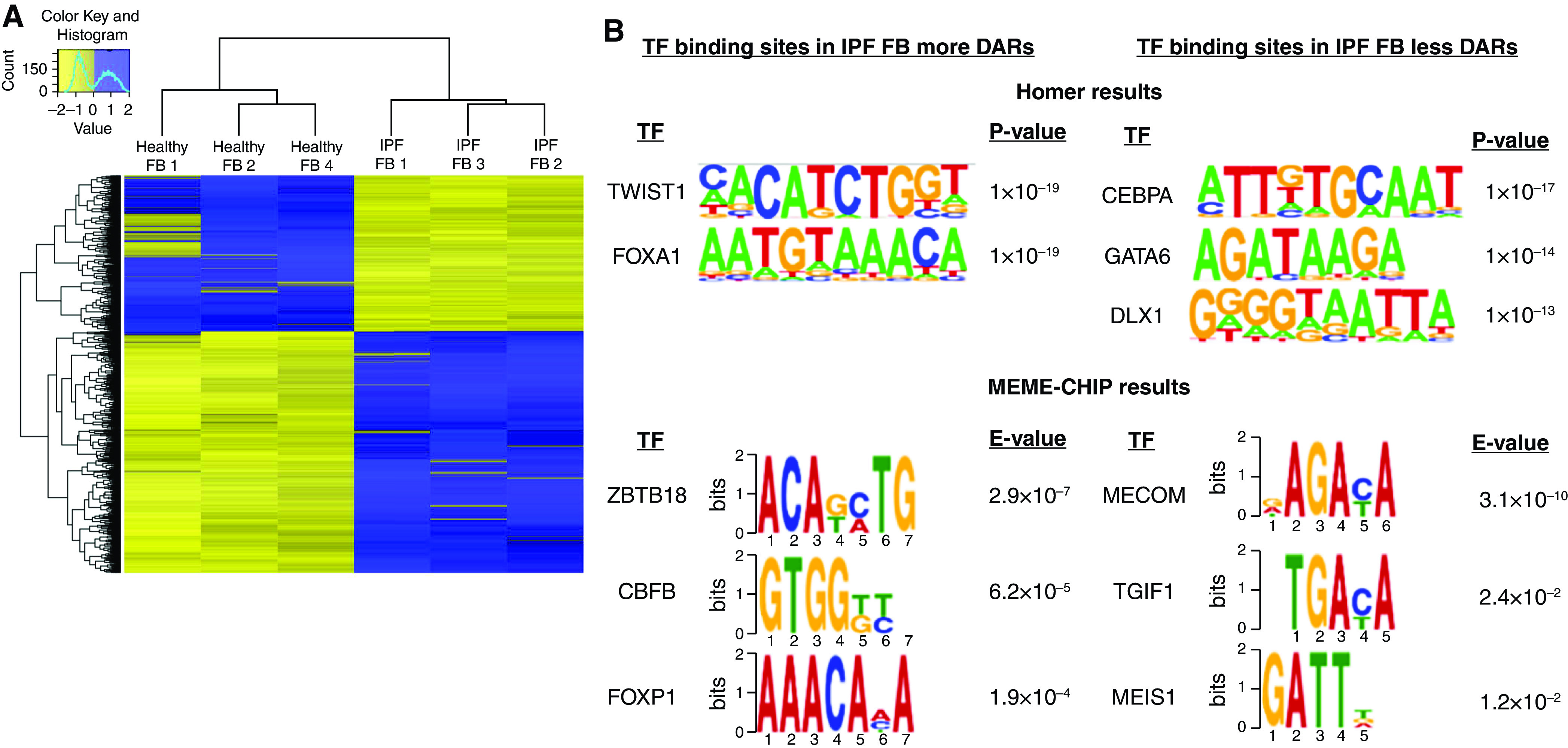
Assay for transposase-accessible chromatin sequencing analysis of healthy and IPF FBs. (A) Heatmap of differentially accessible regions (DARs) in healthy and IPF FBs by assay for transposase-accessible chromatin sequencing. Cutoff FDR was <0.05 and fold change >1.5. (B) Enriched motifs and related transcription factors (TFs) in differentially accessible regions in IPF fibroblasts using Homer and MEME-ChIP.
Figure 3.
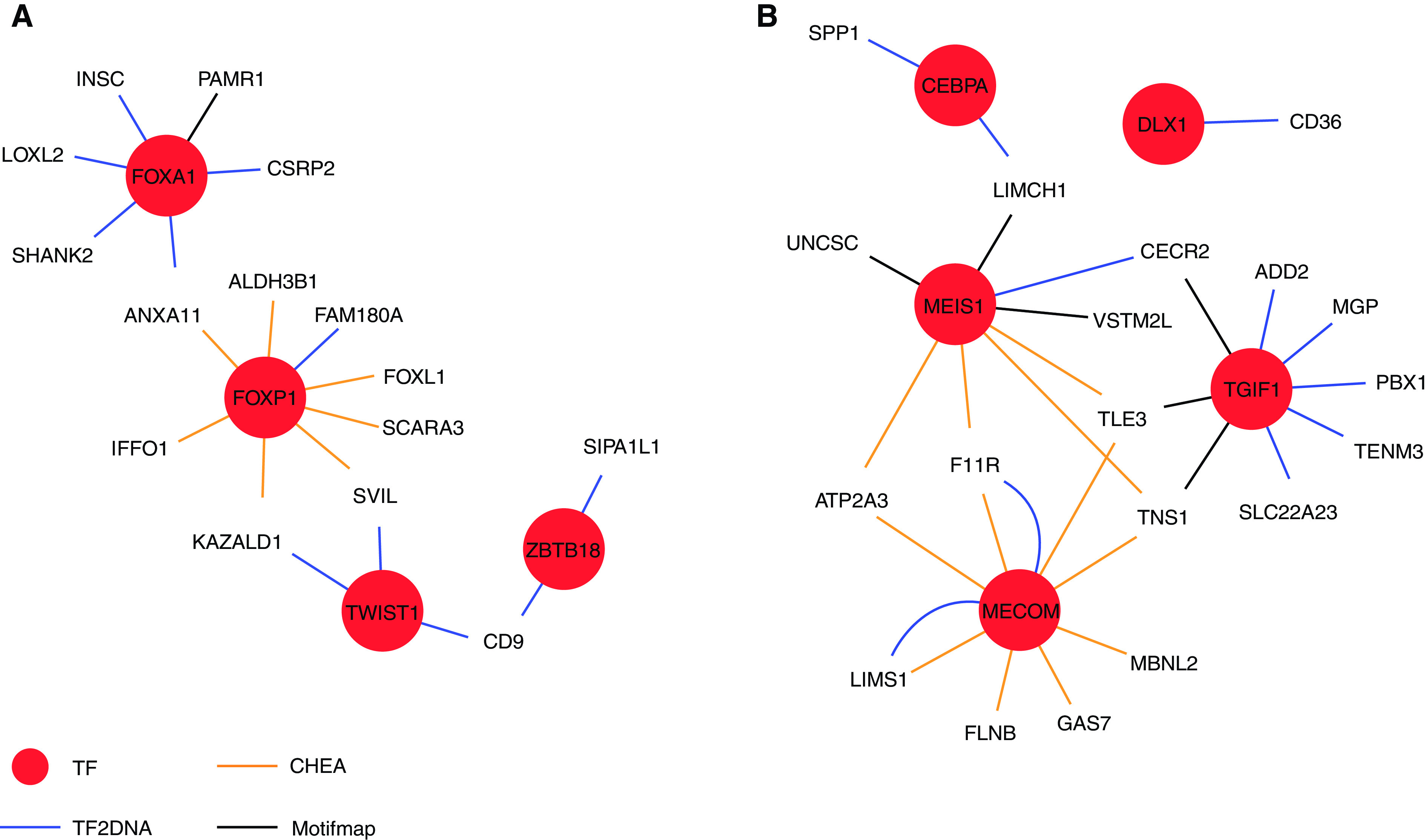
Networks of enriched TFs and their potential target genes. (A) TFs enriched in IPF FB more DARs; (B) TFs enriched in IPF FB less DARs.
DEGs Annotated to Differentially Associated Regions
Next, to understand the biological relevance of the DARs, we annotated the DEGs to the DARs and performed pathway analysis using DAVID. We identified 93 genes that could be annotated to DARs. The top 20 upregulated and downregulated differentially expressed annotated protein-coding genes are listed in Table 2. This analysis revealed targets associated with lung fibrosis that were not identified by DEG data alone, such as matrix gla protein (MGP) (32), interleukin-33 (IL33) (33), cluster of differentiation 36 (CD36) (34), secreted phosphoprotein 1 (SPP1) (35), and vascular cell adhesion molecule 1 (VCAM1) (36).
Table 2.
Differentially Expressed Genes Annotated to Differentially Accessible Chromatin Regions in Idiopathic Pulmonary Fibrosis Fibroblasts Compared to Healthy Fibroblasts
| Gene Symbol | Log2 (Fold Change) | Gene Symbol | Log2 (Fold Change) |
|---|---|---|---|
| Top Down-annotated Genes | Top Up-annotated Genes | ||
| PDPN | −4.092 | LRATD1 | 4.40165336 |
| CECR2 | −3.638 | KPRP | 3.94831062 |
| GPRC5C | −3.454 | WSCD1 | 3.51632614 |
| TMEM233 | −3.340 | INSC | 3.21226656 |
| UNC5C | −3.267 | WNT9A | 2.88724779 |
| CD36 | −3.237 | GCNT2 | 2.75540852 |
| MGP | −3.222 | HLF | 2.47426479 |
| ADD2 | −3.139 | SHANK2 | 2.28477953 |
| CYP7B1 | −2.854 | PKP1 | 2.24754 |
| IL33 | −2.797 | FABP3 | 1.96071504 |
| VSTM2L | −2.776 | GPNMB | 1.79752248 |
| F11R | −2.721 | CAPS | 1.69276489 |
| TENM3 | −2.634 | FOXL1 | 1.68227658 |
| KRT8 | −2.613 | VCAM1 | 1.64710903 |
| ROR2 | −2.489 | DENND3 | 1.6295145 |
| NMNAT2 | −2.457 | ADAMTS14 | 1.62248557 |
| GAS7 | −2.438 | SVIL | 1.61732069 |
| SPP1 | −2.413 | SCARA3 | 1.54750868 |
| MAN1C1 | −2.367 | FAM180A | 1.40541947 |
| ZBED6CL | −2.354 | FAR2 | 1.31814203 |
Next, we validated our results by qPCR our ATAC-Seq data using isolated lung tissue; these revealed increased expression levels for the TF: FOXA1 and TWIST1 (Figures 4A and 4B) consistent with our ATAC-Seq results. Interestingly, although our ATAC-Seq data identified increased DAR for FOXP1 and ZBTB18, no increased expression in isolated lung tissue was identified (Figures 4B and 4C). Regarding TFs that were identified to have reduced DAR, only GATA6 expression was reduced in isolated lung tissue (Figure 4E); no differences in CEBPA, MECOM, TGIF1, and MEIS1 were detected (Figures 4F–4I). Next, we sought to validate the predicted gene network identified in Figure 3. These studies revealed increased expression for the FOXP1 predicted targets cysteine and glycine rich protein 2 (CSRP2) and SH3 and multiple ankyrin repeat domains 2 (SHANK2) in lung tissue samples (Figures 5A and 5B) but not INSC spindle orientation adaptor protein (INSC) or lysyl oxidase like 1 (LOXL1) (Figures 5C and 5D). Similarly, no changes in supervillin (SVIL1) or kazal type serine peptidase inhibitor domain 1 (KAZALD1), downstream targets of TWIST1, were identified between IPF and control samples (Figures 5E and 5F). In addition to these targets, we also determined expression levels for the pro-fibrotic genes Col1a2, fibronectin (FN1), and periostin (POSTN). These profibrotic markers were elevated in IPF compared with control lung samples (Figures 5G–5I). Importantly, our validation studies were performed in a distinct set of control and IPF samples to enhance the significance of our results.
Figure 4.
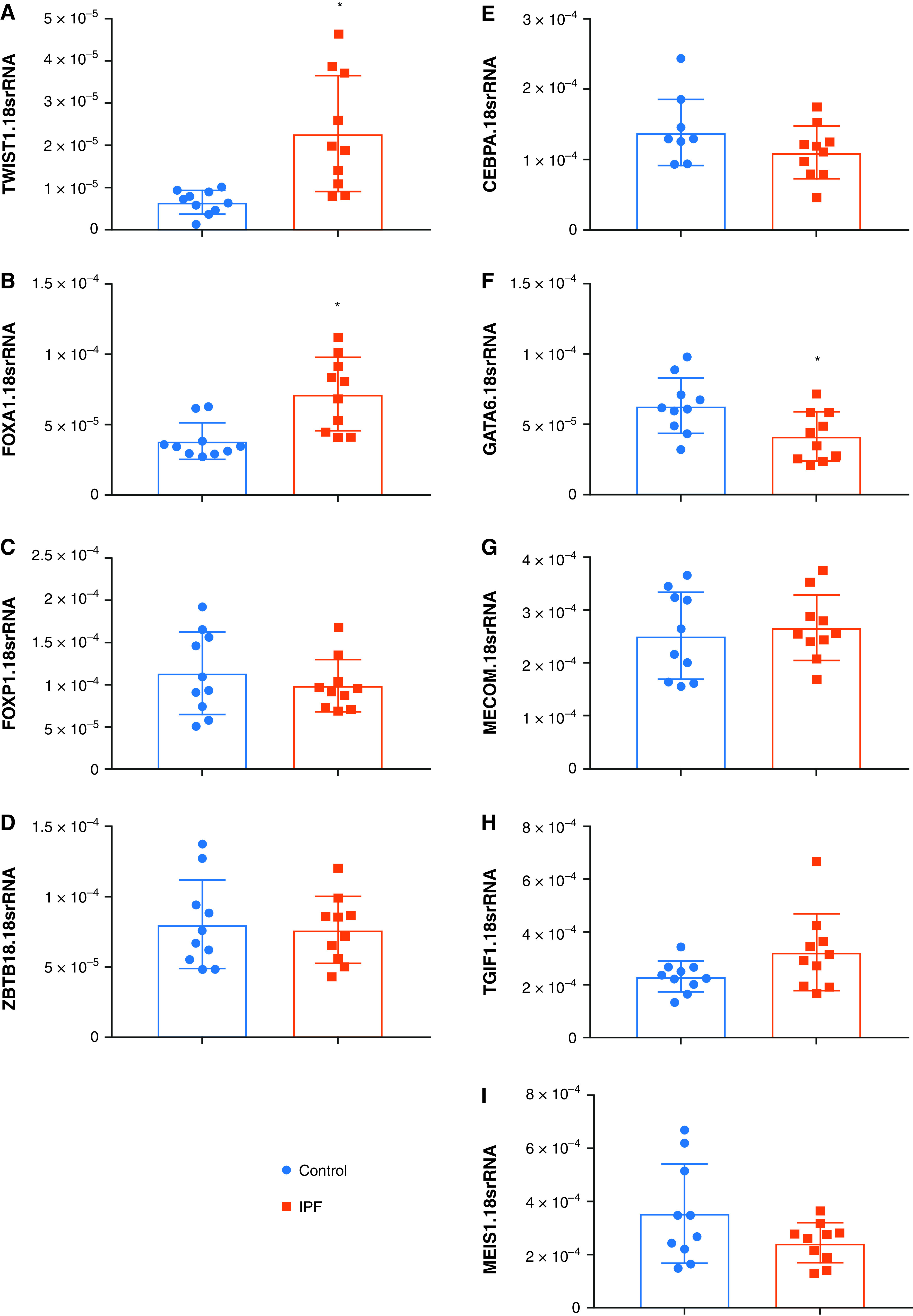
(A–I) Expression levels for the TFs: TWIST1 (A), FOXA1 (B), FOXP1 (C), ZBTB18 (D), CEBPA (E), GATA6 (F), MECOM (G), TGIF (H) and MEIS1 (I) from control or IPF lung tissue. Significance level *P < 0.05 refers to comparisons between Control and IPF.
Figure 5.
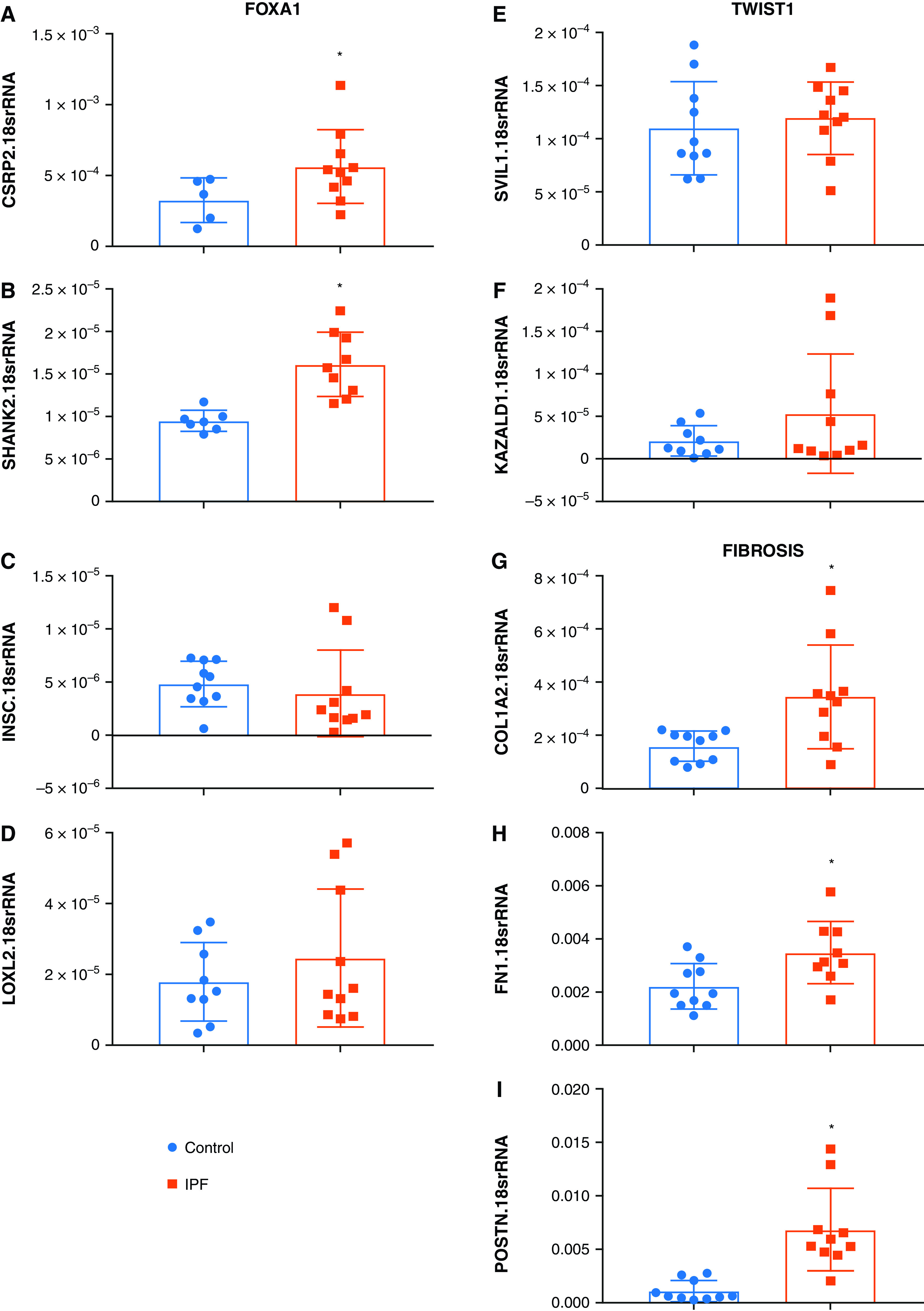
(A–I) Expression levels for predicted target genes for FOXA1: CSRP2 (A), SHANK2 (B), INSC (C), LOXL2 (D); the TWIST1 targets: SVIL1 (E), KAZALD1 (F), and the fibrotic markers: COL1A2 (G), FN1 (H), and POSTN (I) from control or IPF lung tissue. Significance level *P < 0.05 refers to comparisons between Control and IPF.
Discussion
The primary goal of this study was to identify alterations in chromatin accessibility in IPF fibroblasts from upper lobes where intermittent fibrosis is present. These upper lobe signatures may provide insight in the development of active fibrotic lesions. To this end we used fibroblasts isolated from upper lobes of explanted IPF lungs and control unused transplant lungs and compared gene expression profiles using RNA sequencing and chromatin accessibility using ATAC-Seq. The novelty of this work is that we used fibroblasts isolated from active fibrotic lesions, present in the upper lobes of IPF patients. Additionally, we also used ATAC-Seq alongside RNA-Seq to identify genetic differences following changes in chromatin accessibility. These findings were validated using isolated human lung tissue from control or IPF lungs.
Our data show that in early lesions of IPF, there are significant gene expression differences between IPF and healthy fibroblasts. A recent study indicated a global gene expression difference between the upper lobes of healthy controls and IPF patients, despite using normal-appearing tissue in the upper lobes of IPF patients (4). This study verifies the validity of using upper lobe–isolated fibroblasts to compare transcriptomic and epigenetic differences between healthy and IPF fibroblasts.
Multiple studies have performed genome wide transcriptomic profiling of human lung tissue of healthy controls and IPF patients (37–39). Human lung tissue contains various cell types of which the composition drastically changes in fibrotic disease, where myofibroblasts become a major representing cell type. We therefore chose to use lung tissue–derived fibroblasts for our study. A major advantage of our study is that we isolated fibroblasts from the same anatomic region within the explanted lungs, thereby limiting the heterogeneity of the samples. Primary cells isolated from human tissues are notoriously known for the sample heterogeneity. Nonetheless, despite using four to five individual donors per group, we observe nice clustering of the samples, which may be explained by the fact that fibroblasts were isolated from the same anatomical regions within the explanted lungs. The results were then validated in control and IPF lung samples.
Our fibroblasts were cultured to passage 3 or 4 before analysis was performed. Despite in vitro culture, we still see fibrosis-related pathways, including cell motility, ECM organization, and cell migration, being significantly affected in IPF fibroblasts compared with healthy controls. Furthermore, we observe various developmental processes altered in the IPF fibroblasts.
Previous reports have identified specific genes regulated by epigenetic changes in IPF samples (40, 41). Additionally, several groups identified widespread changes in methylation patterns in IPF lungs (42, 43). Nevertheless, knowledge regarding widespread epigenetic changes in specific cell types within IPF lungs remains limited. We therefore used ATAC-Seq to assess chromatin accessibility changes between healthy and IPF fibroblasts. A major advantage of ATAC-Seq compared with chromatin immunoprecipitation sequencing is that it does not require specific antibody pull-down, thereby preventing missing potential genetic targets regulated by the antibody-targeted proteins (44). ATAC-Seq revealed differentially accessible chromatin regions between healthy and IPF fibroblasts. As seen in our RNA-Seq results, using three individual donors per group for ATAC-Seq, we again observe nice clustering of the samples.
With both RNA- and ATAC-Seq data available, we next annotated our DEGs to our differentially accessible chromatin regions, followed by pathway analysis. Genes annotated to the less accessible regions in IPF fibroblasts allocate to various fibrotic-related pathways. These pathways include cytoskeleton anchoring, signal transduction, and cell differentiation. Surprisingly, genes annotated to the more accessible regions in IPF fibroblasts allocated to development, negative regulation of cell proliferation, and cell adhesion, suggesting that reduced chromatin accessibility may be important in the regulation of these pathways in IPF fibroblasts.
Accessible chromatin regions are enriched within transcription start sites (19), allowing transcription factor binding motif analysis. The binding motifs for both FOXA1 and TWIST1 were enriched in IPF fibroblasts compared with controls. Both FOXA1 and TWIST1 were confirmed in our validation experiments. The role of FOXA1 in lung fibrosis has not been fully identified; however, increased expression of this transcription factor has been identified in a rat model of acute lung injury that developed fibrotic injury (45). Elevated FOXA1 has also been detected in non–small-cell lung cancer where it modulated invasion, proliferation, and migration of highly invasive A549 cells (46). Intriguingly, two previously unreported targets in IPF associated with FOXA1, CSPR2 and SHANK2, were identified in our qPCR data. Although our studies link CSPR2 with FOXA1, TGF-β can also increase its expression (47). Furthermore, increased CSPR2 has been linked with promoting epithelial and mesenchymal cancer cell invasion through acting bundling necessary for invadopodia (48). SHANK2 has also been identified as a major tumor amplicon that regulates Hippo signaling (49), an important mechanism in mechanosignaling of fibroblasts in fibrosis (50). Taken together, these results point at a potential role of FOXA1 in lung fibrosis, given the similar changes in cell physiology of fibroblasts in IPF (5, 6). TWIST1 is a previously recognized mediator in the development of IPF, primarily through epithelial to mesenchymal transition (EMT) and T cell dysregulation (51); intriguingly, its downstream targets identified by out analysis were not validated by qPCR.
We also discovered several other binding motifs to be less accessible, including CEBPA, DLX1, TGIF1, and GATA6, although only GATA6 was confirmed by our qPCR experiments. In contrast to our studies, GATA-6 was found to be overexpressed in differentiated, quiescent myofibroblasts and induced ECM deposition through a TGF-beta dependent mechanism in fibrotic foci in IPF (52).
We also compared our results with other studies that examined in early disease changes in lung fibrosis. Herein, we compared our DEG with the 70 DEGs identified by Meltzer et al (53), where we uncovered seven genes (PDLIM4, PTGFRN, ANTXR1, TSHZ2, FNDC1, COL10A1, TSHZ2) that overlapped in both studies. Only three genes (POSTN, THBS2, THY1) overlapped between our dataset and the studies from McDonough et al (54) and two (COL3A1 and COL1A1) with those of Guillotin et al (55). Two enriched biological terms, cell adhesion and ECM organization, were also mentioned to be related to IPF in two studies (54, 55) that correlated with our results. Contrary to our initial studies using isolated fibroblasts for ATAC-Seq or RNA-Seq studies, from upper-lobe/apical regions of the lung, these experiments (53–55) were performed using frozen tissue specimens from explanted lungs undergoing lung transplantation for IPF (N = 10–11) or from controls (N = 6). Thus it is likely that they represent end-stage/established fibrosis, and like our validation experiments, expression changes specific to fibroblasts may be masked by expression levels of other cells.
A limitation of our study is that we did not compare fibrotic and nonfibrotic zones of the upper lobes. We also did not compare areas of severe versus diffuse fibrosis in IPF lungs. Nonetheless, we used primary human fibroblasts isolated from apical regions of both healthy control and IPF lungs. In this region of the lung, the extent of fibrosis is reduced compared with lower regions of the lung (Figure E1). Next, we validated our studies using tissue from the upper portion of the lung. Although our validation studies increased the N number of control and IPF samples, discoveries made from our isolated fibroblasts may have been masked in our qPCR validation experiments using the whole lung, where expression levels of other cells in addition to fibroblasts are represented.
Overall, our study shows that fibroblasts from the upper regions of IPF lungs may play a pivotal role in unraveling pathways in active fibrotic lesions. Differential chromatin access may be an important feature for future research into novel therapeutic targets against deleterious functions in IPF fibroblasts. Additionally, fibroblasts isolated from human lungs and subsequently cultured in vitro may provide an alternative for novel biomarker discovery.
Footnotes
Supported by National Institutes of Health grants 1UL1TR003167 (W.J.Z.), U01HL156059 (W.L.), R01HL138510 and R01HL157100 (H.K.-Q.), Cancer Prevention and Research Institute of Texas grants RP170668 (W.J.Z.) and RR160083 (W.L.), Welch Foundation grant AU-2000–20190330 (W.L.), Department of Defense grant W81XWH-19–1-0007 (H.K.-Q.), and American Heart Association grant 18IPA34170220 (H.K.-Q.).
Author Contributions: A.H. analyzed, interpreted the data, prepared figures, and drafted the manuscript. L.Z. performed the bioinformatic analysis, interpreted the data, and prepared the figures. T.C.J.M. interpreted the data and drafted the manuscript. T.C.J.M., F.X., and R.W. cultured the cells and isolated them for assay of transposase-accessible chromatin–seq (ATAC-Seq) and RNA-sequencing (RNA-seq) experiments. S.C. and W.B. performed the quantitative PCR (qPCR) studies and the Masson’s trichome staining. R.A.T., D.R., and S.S.S.K.J. identified provided tissue for cell isolation and provided clinical insight into the study. L.H., W.L., and W.J.Z. helped design the study and oversaw the ATAC-Seq, RNA-seq, and bioinformatic analyses. H.K.-Q. planned the study, interpreted and analyzed data, and drafted the manuscript. All authors read and approved the manuscript.
The Gene Expression Omnibus accession number for the ATAC-Seq data is GSE180242 and for RNA-seq is GSE180415.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0437OC on August 9, 2021
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med . 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 2. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med . 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunninghake GW, Lynch DA, Galvin JR, Gross BH, Müller N, Schwartz DA, et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest . 2003;124:1215–1223. doi: 10.1378/chest.124.4.1215. [DOI] [PubMed] [Google Scholar]
- 4. Luzina IG, Salcedo MV, Rojas-Peña ML, Wyman AE, Galvin JR, Sachdeva A, et al. Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis. Cell Immunol . 2018;325:1–13. doi: 10.1016/j.cellimm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000 Res . 2016;5:27158462. doi: 10.12688/f1000research.8190.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol . 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 7. Walsh SM, Worrell JC, Fabre A, Hinz B, Kane R, Keane MP. Novel differences in gene expression and functional capabilities of myofibroblast populations in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2018;315:L697–L710. doi: 10.1152/ajplung.00543.2017. [DOI] [PubMed] [Google Scholar]
- 8. Garcia-Morales LJ, Chen NY, Weng T, Luo F, Davies J, Philip K, et al. Altered hypoxic-adenosine axis and metabolism in group iii pulmonary hypertension. Am J Respir Cell Mol Biol . 2016;54:574–583. doi: 10.1165/rcmb.2015-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics . 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol . 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics . 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res . 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics . 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics . 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods . 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol . 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell . 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics . 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Höfer T, et al. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol . 2012;19:1185–1192. doi: 10.1038/nsmb.2419. [DOI] [PubMed] [Google Scholar]
- 20. Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med . 2017;5:869–880. doi: 10.1016/S2213-2600(17)30387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, et al. Pirfenidone Clinical Study Group A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet . 2008;45:654–656. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 22. Li C, Yu Y, Li W, Liu B, Jiao X, Song X, et al. Phycocyanin attenuates pulmonary fibrosis via the TLR2-MyD88-NF-κB signaling pathway. Sci Rep . 2017;7:5843. doi: 10.1038/s41598-017-06021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uhal BD, Kim JK, Li X, Molina-Molina M. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: autocrine mechanisms in myofibroblasts and macrophages. Curr Pharm Des . 2007;13:1247–1256. doi: 10.2174/138161207780618885. [DOI] [PubMed] [Google Scholar]
- 25. Bansal R, Nakagawa S, Yazdani S, van Baarlen J, Venkatesh A, Koh AP, et al. Integrin alpha 11 in the regulation of the myofibroblast phenotype: implications for fibrotic diseases. Exp Mol Med . 2017;49:e396. doi: 10.1038/emm.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang M, Haughey M, Wang NY, Blease K, Kapoun AM, Couto S, et al. Targeting the Wnt signaling pathway through R-spondin 3 identifies an anti-fibrosis treatment strategy for multiple organs. PLoS One . 2020;15:e0229445. doi: 10.1371/journal.pone.0229445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shimbori C, Shiota N, Okunishi H. Pranlukast, a cysteinyl leukotriene type 1 receptor antagonist, attenuates the progression but not the onset of silica-induced pulmonary fibrosis in mice. Int Arch Allergy Immunol . 2012;158:241–251. doi: 10.1159/000331439. [DOI] [PubMed] [Google Scholar]
- 28. Xie X, Rigor P, Baldi P. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics . 2009;25:167–174. doi: 10.1093/bioinformatics/btn605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics . 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pujato M, Kieken F, Skiles AA, Tapinos N, Fiser A. Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res . 2014;42:13500–13512. doi: 10.1093/nar/gku1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res . 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Brouwer B, Piscaer I, Von Der Thusen JH, Grutters JC, Schutgens RE, Wouters EF, et al. Should vitamin K be supplemented instead of antagonised in patients with idiopathic pulmonary fibrosis? Expert Rev Respir Med . 2018;12:169–175. doi: 10.1080/17476348.2018.1424544. [DOI] [PubMed] [Google Scholar]
- 33. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol . 2014;134:1422–1432.e11. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X, Chen Y, Lv L, Chen J. Silencing CD36 gene expression results in the inhibition of latent-TGF-beta1 activation and suppression of silica-induced lung fibrosis in the rat. Respir Res . 2009;10:36. doi: 10.1186/1465-9921-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J . 2019;54:1802441. doi: 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agassandian M, Tedrow JR, Sembrat J, Kass DJ, Zhang Y, Goncharova EA, et al. VCAM-1 is a TGF-β1 inducible gene upregulated in idiopathic pulmonary fibrosis. Cell Signal . 2015;27:2467–2473. doi: 10.1016/j.cellsig.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, et al. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One . 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Guardela BJ, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol . 2015;52:217–231. doi: 10.1165/rcmb.2013-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vukmirovic M, Herazo-Maya JD, Blackmon J, Skodric-Trifunovic V, Jovanovic D, Pavlovic S, et al. Identification and validation of differentially expressed transcripts by RNA-sequencing of formalin-fixed, paraffin-embedded (FFPE) lung tissue from patients with idiopathic pulmonary fibrosis. BMC Pulm Med . 2017;17:15. doi: 10.1186/s12890-016-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cisneros J, Hagood J, Checa M, Ortiz-Quintero B, Negreros M, Herrera I, et al. Hypermethylation-mediated silencing of p14(ARF) in fibroblasts from idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2012;303:L295–L303. doi: 10.1152/ajplung.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol . 2008;39:610–618. doi: 10.1165/rcmb.2007-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One . 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee JU, Son JH, Shim EY, Cheong HS, Shin SW, Shin HD, et al. Global DNA methylation pattern of fibroblasts in idiopathic pulmonary fibrosis. DNA Cell Biol . 2019;38:905–914. doi: 10.1089/dna.2018.4557. [DOI] [PubMed] [Google Scholar]
- 44. Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet . 2014;15:709–721. doi: 10.1038/nrg3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mu X, Wang H, Li H. Silencing of long noncoding RNA H19 alleviates pulmonary injury, inflammation, and fibrosis of acute respiratory distress syndrome through regulating the microRNA-423-5p/FOXA1 axis. Exp Lung Res . 2021;47:183–197. doi: 10.1080/01902148.2021.1887967. [DOI] [PubMed] [Google Scholar]
- 46. Li J, Zhang S, Zhu L, Ma S. Role of transcription factor FOXA1 in non-small cell lung cancer. Mol Med Rep . 2018;17:509–521. doi: 10.3892/mmr.2017.7885. [DOI] [PubMed] [Google Scholar]
- 47.Herrmann J, Borkham-Kamphorst E, Haas U, Van de Leur E, Fraga MF, Esteller M, et al. The expression of CSRP2 encoding the LIM domain protein CRP2 is mediated by TGF-beta in smooth muscle and hepatic stellate cells. Biochem Biophys Res Commun. 2006;345:1526–1535. doi: 10.1016/j.bbrc.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 48. Hoffmann C, Mao X, Dieterle M, Moreau F, Al Absi A, Steinmetz A, et al. CRP2, a new invadopodia actin bundling factor critically promotes breast cancer cell invasion and metastasis. Oncotarget . 2016;7:13688–13705. doi: 10.18632/oncotarget.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu L, Li P, Hao X, Lu Y, Liu M, Song W, et al. SHANK2 is a frequently amplified oncogene with evolutionarily conserved roles in regulating Hippo signaling. Protein Cell . 2021;12:174–193. doi: 10.1007/s13238-020-00742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–L357. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ning X, Zhang K, Wu Q, Liu M, Sun S. Emerging role of Twist1 in fibrotic diseases. J Cell Mol Med . 2018;22:1383–1391. doi: 10.1111/jcmm.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leppäranta O, Pulkkinen V, Koli K, Vähätalo R, Salmenkivi K, Kinnula VL, et al. Transcription factor GATA-6 is expressed in quiescent myofibroblasts in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol . 2010;42:626–632. doi: 10.1165/rcmb.2009-0021OC. [DOI] [PubMed] [Google Scholar]
- 53. Meltzer EB, Barry WT, D’Amico TA, Davis RD, Lin SS, Onaitis MW, et al. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: proof-of-principle. BMC Med Genomics . 2011;4:70. doi: 10.1186/1755-8794-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McDonough JE, Ahangari F, Li Q, Jain S, Verleden SE, Herazo-Maya J, et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight . 2019;4:e131597. doi: 10.1172/jci.insight.131597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guillotin D, Taylor AR, Platé M, Mercer PF, Edwards LM, Haggart R, et al. Transcriptome analysis of IPF fibroblastic foci identifies key pathways involved in fibrogenesis. Thorax . 2021;76:73–82. doi: 10.1136/thoraxjnl-2020-214902. [DOI] [PubMed] [Google Scholar]