

Abstract
The molybdenum/tungsten—bis-pyranopterin guanine dinucleotide family of formate dehydrogenases (FDHs) plays roles in several metabolic pathways ranging from carbon fixation to energy harvesting because of their reaction with a wide variety of redox partners. Indeed, this metabolic plasticity results from the diverse structures, cofactor content, and substrates used by partner subunits interacting with the catalytic hub. Here, we unveiled two noncanonical FDHs in Bacillus subtilis, which are organized into two-subunit complexes with unique features, ForCE1 and ForCE2. We show that the formate oxidoreductase catalytic subunit interacts with an unprecedented partner subunit, formate oxidoreductase essential subunit, and that its amino acid sequence within the active site deviates from the consensus residues typically associated with FDH activity, as a histidine residue is naturally substituted with a glutamine. The formate oxidoreductase essential subunit mediates the utilization of menaquinone as an electron acceptor as shown by the formate:menadione oxidoreductase activity of both enzymes, their copurification with menaquinone, and the distinctive detection of a protein-bound neutral menasemiquinone radical by multifrequency electron paramagnetic resonance (EPR) experiments on the purified enzymes. Moreover, EPR characterization of both FDHs reveals the presence of several [Fe-S] clusters with distinct relaxation properties and a weakly anisotropic Mo(V) EPR signature, consistent with the characteristic molybdenum/bis-pyranopterin guanine dinucleotide cofactor of this enzyme family. Altogether, this work enlarges our knowledge of the FDH family by identifying a noncanonical FDH, which differs in terms of architecture, amino acid conservation around the molybdenum cofactor, and reactivity.
Keywords: bioenergetics, electron paramagnetic resonance (EPR), metalloenzyme, quinone, bacterial metabolism
Abbreviations: BV, benzyl viologen; ECD, electrochemical detection; EPR, electron paramagnetic resonance; FDHs, formate dehydrogenases; ForC, formate oxidoreductase catalytic; ForE, formate oxidoreductase essential; Mo/W-bis-PGD, molybdenum/tungsten–bis-pyranopterin guanine dinucleotide; MSK, menasemiquinone
Formate dehydrogenases (FDHs) catalyze the oxidation of formate into CO2 but have also been shown to catalyze the reverse reaction in methanogenic and/or acetogenic microorganisms, namely the reduction of CO2 into formate (1). Metal-dependent FDHs (hereafter named FDHs) are exclusively found in prokaryotes, and 3D-structure–based phylogeny demonstrates that such enzymes were already present in the last universal common ancestor (2). In addition, paleogeochemistry suggests that in ancient times, compounds such as CO2 and CH4 were very abundant and that iron, molybdenum, and tungsten were available and in soluble forms (3, 4). Indeed, FDHs harbor molybdenum or tungsten at the active site as well as one to five iron–sulfur cluster(s) (1, 5, 6, 7, 8). The molybdenum or tungsten atom is coordinated by the dithiolene sulfurs of two organic pyranopterin guanine dinucleotides (Mo/W-bis-PGD) (1, 9), a cysteine or a selenocysteine and a sulfido group, all being essential for the activity (10, 11, 12). In addition, an histidine and an arginine residue located in proximity to the Mo atom (13) are strictly conserved in all FDHs described so far (Fig. 1A) and required for activity (14). These residues have been proposed to be involved in correct orientation or stabilization of the formate in the active site (15, 16, 17), but their role in activity is still debated. They could also act as a switch to obstruct alternatively the substrate and the product tunnels during catalysis, as suggested by the 3D structure of the FDHs from Rhodobacter capsulatus and Desulfovibrio vulgaris Hildenborough and formyl-methanofuran dehydrogenase from Methanothermobacter wolfeii (18, 19, 20). Altogether, there is still a need for clarifying the exact role of these conserved residues surrounding the catalytic site in FDH activity.
Figure 1.

Bacillus subtilis genome encodes putative noncanonical FDHs.A, alignment of the amino acids involved in the active site of characterized FDH enzymes and the two putative FDH proteins from B. subtilis (red asterisks). Consensus sequence (blue box) shows which amino acids are conserved with a 50% threshold. FDHs have been organized into three classes according to the sequence and organization of their active site (Fig. S7): Class I: C/UH RGQ or C/UQ RGH, class II: C/UH RGE, and class III: C/UH RGH. Overview of the full sequence alignment is shown in Figure S1B. B, genomic organization of yjgCD (ForCE1) and yrhED (ForCE2) in B. subtilis. C, structural model of YjgC generated with I-TASSER (89) using FdsA from R. capsulatus (PDB ID: 6TGA). FeS clusters are named according to their proximity to the Mo cofactor in the 3D structural model. FDHs, formate dehydrogenases.
From a physiological point of view, FDHs are involved in a wide range of anaerobic metabolic pathways, such as energy harvesting and carbon fixation (6, 9). This is due to the interaction of the catalytic subunit that harbors the Mo/W-bis-PGD cofactor with partner subunits with the greatest diversity among the Mo/W-bis-PGD enzyme superfamily (9). In methanogenic archaea, FDH is part of a complex that facilitates a two-step reduction of CO2 to formate and then to formyl-methanofuran (20). In bacteria, the membrane-bound FDH complex couples formate oxidation to quinols production in anaerobic respiratory chains (21), and the soluble FDH from Cupriavidus necator and R. capsulatus couples formate oxidation to NADH reduction that feed the reductive pentose phosphate cycle (22, 23). To summarize, FDHs are a group of heterogenous enzymes that display different subunit composition from the single monomeric subunit FdhF from Escherichia coli to the six-subunit complex formyl-methanofuran dehydrogenase (13, 20), and partner subunits hold a variety of redox cofactors ([Fe-S] clusters, hemes, F420, FMN) that allow reaction with a wide variety of physiological redox partners, such as cytochromes, ferredoxins, NAD, and quinones (1, 5, 6, 7, 8). Altogether, identification and characterization of FDHs with variations in the amino acids surrounding the catalytic site or new subunit composition could improve our understanding of catalysis and expand the metabolic pathway repertory in which FDHs are participating.
In the archetypal gram-positive bacterium, Bacillus subtilis, an FDH activity has been detected in cells grown aerobically in the rich medium (24). This is intriguing as FDHs are usually part of anaerobic metabolic pathways, and B. subtilis is not able to produce formate (25). These observations prompted us to search for the enzyme(s) involved in this activity. Combining bioinformatics with biochemistry, enzymology, and electron paramagnetic resonance (EPR) spectroscopy, we have identified and characterized two atypical FDH enzymes in B. subtilis and members of a new subfamily of FDHs. Notably, formate oxidation activity is measured, whereas the conserved His residue in the active site is naturally substituted by a Gln. Moreover, both FDHs display a new architecture in which their catalytic subunit is associated with an unprecedented partner subunit that allows reactivity with menaquinone.
Results
The YjgC and YrhE sequences from B. subtilis encode putative metal-dependent FDHs
Scrutiny of the B. subtilis 168 genome indicates that it encodes for yjgC and yrhE genes, whose ∼110-kDa predicted proteins are paralogous with 61% of identity. Both putative oxidoreductases contain a C-terminal domain typical for Mo/W-bis-PGD enzymes and characterized by motifs allowing the coordination of one Mo/W-bis-PGD cofactor and one [4Fe-4S] cluster (Fig. S1A and Table S1). Furthermore, both sequences contain an N-terminal extension with binding motifs for three [4Fe-4S] clusters and one [2Fe-2S] cluster as determined by similarities (Fig. S1A). In particular, this region shares homologies with the N-terminal domain of the NuoG subunit of the NADH:ubiquinone oxidoreductase (i.e., complex I) and the catalytic subunit of NAD-dependent [NiFe]-hydrogenases (19, 26, 27). Such domain organization is also shared with other FDHs, notably FdsA from C. necator and R. capsulatus (Figs. S1B and S2A). A close-up alignment of the active site residues with previously characterized FDH sequences and YjgC and YrhE sequences is shown in Figure 1A. Whereas the typical metal Cys ligand and the strictly conserved Arg residue are present, the highly conserved His residue is substituted by a Gln in YjgC and YrhE. Phylogenetic analysis identified a large group of YjgC/YrhE-related sequences with the similar substitution in several bacterial and archaeal phyla, which are distinct from FdsA-related sequences (Fig. S2A). The presence of the Gln next to this Cys essential residue in the primary sequence questions the activity of these enzymes as FDHs. However, for the evidence that B. subtilis possesses at least one FDH is the existence of a putative fdhD gene encoding a sulfurtransferase crucial for FDH activity (12). In the B. subtilis genome, the fdhD gene is distant from the yjgC and yrhE loci (53′ versus 18′ and 39′, respectively). Interestingly, the yjgC gene is predicted to be organized in operon with yjgD as yjgC and yjgD are coexpressed with a positive correlation of 0.98 (28, 29). The same holds true for the gene yrhE predicted to be in operon with yhrD as both are adjacent and coexpressed with a positive correlation of 0.99 (Fig. 1B). The yjgD and yrhD genes encode 21-kDa and 17-kDa polypeptides, with weak sequence overall identity (20%), and contain a DUF1641 domain of 38 amino acids of unknown function (PFAM PF07849). While YjgC/YrhE are related to FdsA, as illustrated by the YjgC structural model based on the 3D structure of R. capsulatus FdsA (Fig. 1C) and the phylogenic analysis (Figs. 1A and S1), YjgD and YrhD are distinct from FdsD or any of the other partner subunits previously characterized for an FDH, the complex I and [Ni-Fe] hydrogenases (6). In addition, YjgD and YrhD are predicted to be cofactor-less and have a distinct sequence from the FdsC/FdhD sulfurtransferase or any specific chaperone involved in Mo-bis-PGD maturation (30, 31). Altogether, these in silico analyses suggest that YjgC and YrhE are the enzymes involved in the FDH activity previously detected in B. subtilis (24) and question the role of YjgD and YrhD.
The YjgCD and YrhED are protein complexes with formate-oxidizing activity
To test the activity of YjgC and YrhE as FDHs and the importance of YjgD and YrhD, the corresponding proteins were purified. First, the yjgCD and yrhED operons were expressed in B. subtilis under the control of an inducible promoter and a 6-histidine tag was added at the N terminus of the YjgC and YrhE proteins. Affinity purification of these proteins showed that YjgC and YrhE copurified with YjgD and YrhD, respectively, as identified by SDS-PAGE and MS analysis (Fig. 2A, Tables S2 and S3). MS data are available via ProteomeXchange with identifier PXD028742. These results suggest that proteins YjgD and YrhD interact physically with YjgC and YrhE, respectively. A full steady-state enzyme kinetic analysis of formate oxidation by YjgCD and YrhED using benzyl viologen (BV) as an artificial electron acceptor is shown in Figure 2B. Using the Michaelis–Menten plot, a kcat of 96 s−1 was measured for the YjgCD heterodimer (MW = 132 kDa) and a kcat of 56 s−1 for the YhrED heterodimer (MW = 125 kDa) (Table 1). Both values are in the same range as the ones determined for FDH of R. capsulatus (36 s−1) and C. necator (201 s−1) (22, 26). Apparent KMformate of 5 mM for YjgCD and 1 mM for YrhED are larger than the values of 0.281 mM and 0.31 mM determined for FDH of R. capsulatus and C. necator, respectively. As observed for other FDHs (22, 32, 33), both enzyme preparations exhibit a basic pH optimum, pH 10 for the YjgCD complex and pH 8.5 for YrhED (Fig. 2C). In conclusion, the YjgCD and YrhED are protein complexes with formate-oxidizing activity with kinetic parameters comparable with other FDHs despite the fact that the amino acid sequence within the active site deviates from the consensus residues typically associated with FDH activity. Henceforth, the YjgC subunit was renamed ForC for formate oxidoreductase catalytic subunit and the YjgD subunit ForE for formate oxidoreductase essential subunit for reasons that will be presented at the end of the Results section. As B. subtilis encodes two FDHs, YjgCD was renamed ForCE1 and YrhED was renamed ForCE2.
Figure 2.

YjgCD (ForCE1) and YrhED (ForCE2) are formate-oxidizing enzymes.A, purified proteins are analyzed on Coomassie Brilliant Blue–stained 15% SDS-polyacrylamide gel. Molecular standards are indicated in kilodalton. Lane 1: YjgC N-terminus 6his-tagged copurifies with YjgD; lane 2: YrhE N-terminus 6his-tagged copurifies with YrhD; lane 3: YjgC C-terminus 8his-tagged was purified without YjgD. B, Michaelis–Menten plot for the formate:benzyl viologen oxidoreductase reaction of YjgCD (ForCE1) at pH 10 and YrhED (ForCE2) at pH 8.6. C, pH dependence of kcat (s−1) for YjgCD (ForCE1) and YrhED (ForCE2).
Table 1.
Enzymatic catalytic constants for formate oxidation by YjgCD, YjgC, and YrhED using either benzyl viologen (top) or menadione (bottom) as electron acceptors
| FDH | KMformate (mM) | kcat (s−1) | Vmax (U) | pH |
|---|---|---|---|---|
| YjgCD/ForCE1 | 5.1 ± 0.9 | 96.1 ± 4 | 43.7 ± 1.9 | 10 |
| YrhED/ForCE2 | 1.1 ± 0.1 | 55.8 ± 1.6 | 26.6 ± 0.8 | 8.6 |
| YjgC/ForC1 | ND | 2.13 ± 0.05 | 1.16 ± 0.03 | 10 |
| KMmenadione (mM) | kcat (s−1) | Vmax (U) | pH | |
|---|---|---|---|---|
| YjgCD/ForCE1 | 0.025 ± 0.003 | 16.5 ± 0.6 | 7.5 ± 0.3 | 9 |
| YrhED/ForCE2 | 0.023 ± 0.005 | 26.4 ± 1.8 | 12.6 ± 0.9 | 9 |
| YjgC/ForC1 | ND | 0 | 0 | 9 |
Abbreviation: ND, not determined.
EPR analysis of ForCE1 and ForCE2
To characterize the cofactor content of the two enzymes, we next examined the EPR properties of purified ForCE1 and ForCE2 enzymes, either in their air-oxidized state or after incubation with a large excess of sodium formate or sodium dithionite. When measured in the 50- to 70-K temperature range, the air-oxidized enzymes mainly display two essentially isotropic and partially overlapping EPR signals originating from two slow-relaxing S = ½ species with g-values centered at 2.004 and 1.993. Their relative intensities are different in the two enzymes (Fig. 3A, traces 1 and 2). In a first step, the g = 1.993 signal that exhibits a partially resolved structure in both enzymes was investigated in detail. Optimally detected in the air-oxidized ForCE2 sample, this signal was further characterized by performing echo-detected field-swept experiments at W-band (∼94 GHz) or continuous wave EPR measurements at Q-band (∼34 GHz) (Fig. 3B, inset and trace 2). At both frequencies, the g = 1.993 signal is well isolated and displays a rhombic shape well reproduced by considering a single species with g-values g1,2,3 = 1.9971, 1.9933, and 1.9890. Whereas this model is unable to account for the resolved structure on the X-band EPR spectrum, the addition of a strongly coupled proton with nearly isotropic 1H hyperfine coupling tensor having principal values A1,2,3 = 13, 13, and 11 MHz allows reproducing the observed splittings (Fig. 3B, trace 1). Eventually, the multifrequency EPR spectra are well simulated using the same set of g- and A-tensors as given above (Fig. 3B). The signal-giving species displays g-values fully consistent with those of a 4 d1 Mo(V) ion with bis-pterin coordination (34). Notably, the corresponding g-tensor anisotropy (g1 – g3 ∼ 0.008) is unusually low and most closely resembles that of the slow-type Mo(V) signal Desulfovibrio alaskensis NCIMB 13491 FDH (g1 – g3 ∼ 0.012) (35). Moreover, the Mo(V) g-values in ForCE2 belong clearly to the correlation found for the variations of the g-tensor of Mo(V) species with a six-sulfur coordination (9, 34, 36) (Fig. S3). Finally, the magnitude of the proton hyperfine coupling tensor is comparable with that resolved on the Mo(V) EPR signal detected in C. necator FDH and assigned to a molybdenum-SH group (26). Therefore, the g = 1.993 signal similarly detected in ForCE1 and ForCE2 is unambiguously assigned to a Mo(V) species generated at their active site.
Figure 3.
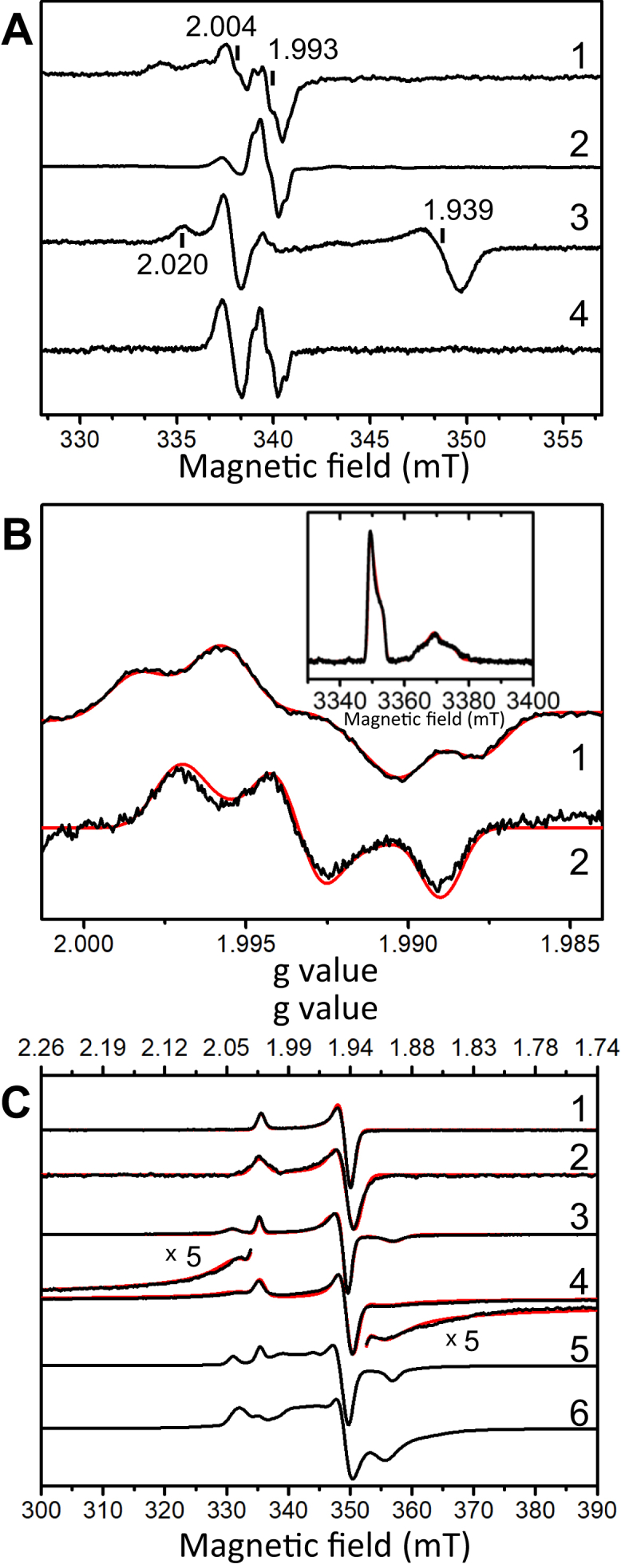
EPR characterization of the cofactors in ForCE1 and ForCE2.A, air-oxidized (1 and 2) and formate-reduced (3 and 4) ForCE1 (1 and 3) and ForCE2 (2 and 4) collected at 50 K (1) or 70 K (2–4). Other experimental conditions were as follows: microwave power, 4 mW, field modulation amplitude, 0.4 mT (1 and 2) or 0.2 mT (3 and 4) at 100 kHz, and microwave frequency, ∼9.48 GHz. Vertical lines indicate remarkable g values given in the figure and discussed in the main text. B, continuous wave Mo(V) EPR spectra in air-oxidized ForCE2 samples measured at X band (9.47864 GHz) (1) and Q band (34.1187 GHz) (2) frequencies. Other experimental conditions were as follows: temperature, 70 K (1) and 50 K (2), microwave power, 1 mW (1) and 0.1 mW (2), and field modulation amplitude, 0.1 mT (1) and 0.5 mT (2) at 100 kHz. The inset shows the echo-detected field-swept EPR spectrum measured at 50 K at W-band (94.0014 GHz) frequency. Other experimental conditions are as follows: microwave pulse lengths, 20 ns and 40 ns for π/2 and π pulses, respectively. C, EPR spectra of dithionite-reduced ForCE1 (1, 3, and 5) and ForCE2 (2, 4, and 6) measured at 100 K (1 and 2), 50 K (3 and 4), or 15 K (5 and 6). Other experimental conditions were as follows: microwave power, 1 mW (1) or 10 mW (2–6), modulation amplitude 1 mT (1, 2, 4–6) or 0.4 mT (3) at 100 kHz, microwave frequency, ∼ 9.48 GHz. Spectral simulations are displayed as red traces superimposed to the experimental spectra and have been performed using the parameters given in the main text and supporting information (Table S8).
Interestingly, incubation of the enzymes with a large excess of sodium formate leads to an increase of the g = 2.004 signal relative to that of the Mo(V) species (Fig. 3A, traces 3 and 4). In addition, an axial signal with g-values 2.020 and 1.939 is detected in the ForCE1 spectrum measured at 50 K (Fig. 3A, trace 3). These observations are explained by partial reduction of the enzymes by sodium formate and are consistent with the fact that the latter is the physiological substrate. The same axial signal is observed on the EPR spectrum of the dithionite-reduced samples of ForCE1 and ForCE2 measured at 100 K, whereas the Mo(V) signal and the g = 2.004 species are no longer detected, consistent with further reduction of the latter and of the Mo(V) into the EPR-silent Mo(IV) state in these samples (Fig. 3C, traces 1 and 2). The slow relaxation properties of this axial signal and its g-values are typical for S = ½ [2Fe-2S]1+ clusters (37) and very similar to those detected in related FDHs from Clostridium thermoaceticum, Methylosinus trichosporium, or C. necator (26, 38, 39, 40). Upon lowering the temperature to 50 K, new signals are detected peaking at g = 2.047 and 1.897 in ForCE1 (Fig. 3C, trace 3), and g = 2.030 and 1.912 in ForCE2 (Fig. 3C, trace 4). These are tentatively assigned to the g1 and g3 values of an additional slow relaxing S = ½ [4Fe-4S]1+ cluster. The corresponding intermediate g2 value is estimated by numerical simulations (shown as red solid lines in Fig. 3C, traces 3 and 4), yielding to g1,2,3 = 2.047, 1.948, 1.897 and 2.030, 1.940, 1.912 for this cluster in ForCE1 and ForCE2, respectively. Further lowering the temperature to 15 K leads to the appearance of additional overlapping signals peaking at 1.99 and 1.96, which most likely arise from an additional fast-relaxing [4Fe-4S]1+ center (Fig. 3C, traces 5 and 6). Being the sole EPR active center detected at 100 K, the [2Fe-2S] cluster was used as an internal reference for spin quantitation measurements. Double integration of the EPR spectrum of a dithionite-reduced ForCE1 sample buffered at pH 7.5 and measured at 15 K in nonsaturating conditions (i.e., 0.1 mW) indicated that the detected signals contribute to about 4.7 ±0.4 spins/ForCE1, assuming one [2Fe-2S] center per enzyme.
Overall, the present EPR analysis allows identifying the signature of several metal centers in ForCE1 and ForCE2, including that of their Mo-bis-PGD cofactor, a [2Fe-2S] cluster and at least two other [4Fe-4S] centers with distinct relaxation properties. Importantly, the detected species have quite similar EPR signatures in both enzymes in agreement with the high identity percentage between ForC1 and ForC2 subunits.
A menasemiquinone radical stabilized within ForCE1 and ForCE2
An unexpected outcome of the EPR analysis presented above is the detection of an intense isotropic radical signal at g = 2.004 in ForCE1 and ForCE2 after incubation with sodium formate (Fig. 3A, traces 3 and 4). The radical peak-to-peak linewidth (∼0.88 mT) is similar to that typically observed for protein-bound isoprenoid quinones (41, 42). To further identify its origin, its redox properties were investigated using EPR-monitored redox titration experiments performed on purified ForCE1 buffered at pH 6.0 and 7.5. The EPR amplitude of the radical measured against ambient redox potential changes according to a bell-shaped curve as expected for quinone species undergoing two successive one-electron reduction steps between the fully oxidized quinone and the fully reduced quinol state with an EPR-active semiquinone intermediate (Fig. 4A) (42, 43, 44, 45, 46). When the data were fitted using theoretical curves based on Equation 1, midpoint potential values (E1, E2) = (−42 mV, −90 mV) and (−136, −175 mV) were obtained at pH 6 and 7.5, yielding two-electron midpoint potential values Em of −66 mV and −156 mV, respectively. Such values are consistent with those of low-potential menaquinones (Em, pH 7.5 MK/MKH2 = −100 ± 10 mV) (47) such as MK-7, the major quinone present in B. subtilis membranes (48). The −60 mV/pH unit dependence of Em in the investigated pH range indicates a 2H+/2e− reaction as measured for semiquinone radicals stabilized in exchangeable Q sites (43, 44, 45). In addition, the similar −60 mV/pH unit dependence of both E1 and E2 indicates that the neutral menasemiquinone (MSK) form (i.e., MSKH•) is stabilized in this pH range. The maximum concentration of the radical was estimated to be 0.5 MSK/ForCE1 from double integration of its EPR spectrum measured under nonsaturating conditions and using the [2Fe-2S] cluster as internal standard, assuming one [2Fe-2S]/enzyme.
Figure 4.

ForCE1 and ForCE2 bind MK-7 as an electron sink.A, normalized redox titration curves of the MSK radical in ForCE1 buffered at pH = 6 (black) or 7.5 (red). Experimental points (filled squares or filled circles) have been fitted to the theoretical curve as described in the Experimental procedures section. Experimental conditions were as follows: temperature, 50 K, microwave power, 10 mW, and modulation amplitude, 0.5 mT (pH 7.5) or 1 mT (pH 6) at 100 kHz. B, g-scale representation of the cw EPR spectra (black traces) and their simulation (red traces) of the MSK. radical in ForCE1 buffered at pH = 6 and measured at X band (9.4812 GHz) (1), Q band (34.094 GHz) (2), and W band (94.244 GHz) (3). Spectra were simulated using the same rhombic g-tensor with principal values g1,2,3 = 2.0054, 2.0051, and 2.0023 and the linewidths given in supporting information (Table S8). Experimental conditions were as follows: temperature, 50 K, microwave power, 4 mW (1), 0.1 mW (2), or 0.05 mW (3), and field modulation amplitude at 100 kHz, 0.4 mT (1 and 3) or 0.5 mT (2). C, ForCE1 and ForCE2, but not ForC1, bind large quantities of MK-7. HPLC-ECD analysis of the MK-7 standard (15 pmol) and of lipid extracts corresponding to ∼40 pmol of purified proteins. D, Michaelis–Menten plot for the formate:menadione oxidoreduction reaction of ForCE1 at pH 9. E, Michaelis–Menten plot for the formate:menadione oxidoreduction reaction of ForCE2 at pH 9. F, pH dependence of kcat (s−1) for ForCE1.
The chemical nature of the radical was further confirmed by performing EPR experiments at higher microwave frequencies, as large unresolved hyperfine couplings typically obscure the g anisotropy of radicals measured at standard X-band frequencies (9–10 GHz) (49). Whereas the g tensor anisotropy of the radical in ForCE1 is only partially resolved at the Q band (∼34 GHz), its g3 component is clearly resolved at the W band (∼94 GHz), the overall line shape remaining predominantly axial (Fig. 4B). Such features are again expected for protein-bound isoprenoid semiquinones (42, 50). In addition, EPR spectra recorded at X, Q, and W bands are well simulated using the same rhombic g-tensor with principal values g1,2,3 = 2.0054, 2.0051, 2.0023. These are very close to those measured for other protein-bound MSK such as in the QD site of E. coli nitrate reductase (42, 46) (Fig. S4) or in the QA site of Zn-substituted reaction center from Blastochloris viridis (41). Therefore, the EPR signal of the radical detected in ForCE1 can be assigned to a neutral MSK radical bound to the enzyme based on its EPR properties evaluated at several microwave frequencies as well as on its redox characteristics. This assignment also holds for the radical species detected in ForCE2, which exhibits similar g-values, as concluded from the comparative analysis of the W band EPR spectra of the radical measured in ForCE1 and in ForCE2 (Fig. S4).
In addition, our data reveal that the detected MSK is highly stabilized in ForCE1 in the pH range from 6.0 to 7.5. The magnitude of the MSK stability constant is virtually independent of pH (KS, pH = 6.0 ∼ 7, KS, pH = 7.5 ∼ 5) and much larger than that expected for the unbound species, that is, KS, free ∼ 10−15. Hence, the calculated occupancy level Rocc of the ForCE1 Q-site that corresponds to the fraction of the site occupied by MK, MSK, or MKH2 (irrespective of their protonation state) is close to unity at both pH values (Rocc ∼ 0.8 at pH = 6.0 and ∼ 0.9 at pH 7.5).
We confirmed the nature of the MK bound in purified ForCE1 and ForCE2 by analyzing lipid extracts with HPLC coupled to electrochemical detection and MS (HPLC-ECD-MS) (Fig. 4C). Lipid extracts from ForCE1 and ForCE2 show a predominant peak at 9.8 min, which coincides with the MK-7 standard. MS confirmed the identity of MK-7 (Fig. S6A). A minor peak of MK-8 was also detected (Fig. 4C), as supported by MS analysis (Fig. S6B). While the ForCE1-MK-7 ratio inferred by HPLC-ECD analysis shows a stoichiometric value of 1:0.87, the ForCE2-MK-7 ratio is above 1:1 stoichiometry (1:1.63) (Table S5). Nevertheless, the ForCE1-MK-7 ratio is in agreement with the amount of one MK-7 per complex quantified by EPR analysis.
ForCE1 and ForCE2 reduce MK-7 analogs
Having demonstrated that ForCE1 and ForCE2 enzymes are able to stabilize an MSK radical, we hypothesized that MK-7 is the electron acceptor during formate oxidation in B. subtilis. The enzymatic activity of purified ForCE1 and ForCE2 was analyzed in the presence of formate and menadione, a structural analog of MK-7. Full steady-state enzyme kinetic analyses are shown in Figure 4, D and E. To model experimental data, we used a Michaelis–Menten plot and found similar apparent low KMmenadione values for both enzymes (25 μM for ForCE1 and 23 μM for ForCE2) and a kcat of 16.5 and 26.4 s−1 for ForCE1 and ForCE2, respectively (Table 1). The pH dependence of the formate:menadione oxidoreductase activity was determined and showed a pH optimum at ≈ 9 for both enzymes (Figs. 4F and S5). Altogether, our in vitro kinetic analyses show that ForCE1 and ForCE2 are able to couple formate oxidation to menadione reduction, and we propose that MK-7 is the physiological electron acceptor during formate oxidation.
ForE1 is an essential subunit for formate oxidoreductase activity of ForC1
To assess the importance of ForE1 or ForE2 in FDH activity, the yjgC (forC1) and yrhE (forC2) genes were expressed under the control of an inducible promoter in absence of yjgD (forE1) and yrhD (forE2), respectively. Despite many attempts, we were unable to purify the ForC2 subunit His-tagged in C-terminal when produced without ForE2. On the contrary, we were able to purify ForC1 His-tagged in C-terminal (Fig. 2A, Table S4). This protein was able to oxidize formate with BV as an artificial electron acceptor but at a residual kcat = 2 s−1 compared with 96 s−1 for the ForCE1 complex (Table 1). HPLC-ECD analysis of ForC1 showed that very low quantities of MK-7 are associated with ForC1, as compared with ForCE1 (Fig. 4C, Table S5), and ForC1 is devoid of formate:menadione oxidoreductase activity (Table 1). Thus, the ForE1 subunit is required for formate:menadione oxidoreductase activity of ForC1 and most likely for MK-7 binding. Altogether, these results demonstrate the essential role of ForE1 in ForCE1 activity.
Discussion
FDHs have been widely studied as they reversibly catalyze formate oxidation into CO2, CO2 reduction being of high biotechnological interest. In contrast to other members of the Mo/W-bis-PGD enzyme superfamily, FDHs exhibit a plethora of quaternary structure, subunit, and redox center composition and cellular localization allowing their integration in several anaerobic metabolic pathways as reviewed (5, 7, 9, 51). Here, we report on the identification, isolation, and characterization of two atypical FDHs, ForCE1 and ForCE2, in the aerobic soil bacterium, B. subtilis. In contrast with other characterized FDHs, ForCEs (i) have a unique subunit composition, (ii) display modification of the hitherto conserved amino acid residues surrounding the metal active site, and (iii) bind MK-7 while being cytosolic enzymes as shown by the homologous expression system reported here. Notably, ForCE1 and ForCE2 have each a Mo-bis-PGD cofactor and bear up to 5 [Fe-S] clusters as determined by EPR spectroscopy. In addition, both enzymes are able to perform formate:menadione oxidoreduction and have the ability to strongly bind MK-7 as indicated by HPLC-ECD-MS analysis, which leads to the detection of an isotropic radical signal by EPR spectroscopy, assigned to an MSK signal.
Multimeric FDHs work in concert with other subunits to oxidize formate or receive electrons from a donor to reduce CO2. In other terms, partner subunits allow FDH integration into a specific metabolic pathway. Gene(s) encoding partner subunit(s) are organized into operons with the gene encoding the catalytic subunit. In FDHs from B. subtilis, genes encoding catalytic and partner subunits are organized into forCE1 and forCE2 operons. Our results have shown that purified ForC1 produced in absence of ForE1 binds weakly to MK-7 and that it has lost formate:menadione oxidoreductase activity. Accordingly, we suggest that ForE is the partner subunit that allows coupling formate oxidation to quinone reduction. Such reaction has been demonstrated for the respiratory FDH complex exemplified in E. coli (21). However, the partner subunits FdnHI are very distinct from ForE at the sequence level. The Mo-bis-PGD containing the FdnG subunit is the formate oxidation site connected through a 5 [Fe-S] cluster electron wire (in FdnGH) to the quinone reduction site in the transmembrane cytochrome FdnI. In contrast, the ForC subunit harbors the Mo-bis-PGD cofactor formate oxidation site as well as 5 [Fe-S] clusters in the same polypeptide and the menaquinone reduction site hypothetically located at the ForCE interface. It must be noted that ForCE sequences are not predicted to contain any transmembrane segment, neither hemes, as confirmed by EPR analysis of ForCE1 and ForCE2. Therefore, we conclude that the way the formate:menaquinone oxidoreduction occurs into ForCEs is unprecedented.
The ForC catalytic subunit presents high sequence similarity with FdsA from R. capsulatus and C. necator (Fig. S1) and a ForC1 structural model using the former has been built (Fig. 1C). In this model, electrons are logically shuttled from the Mo-bis-PGD catalytic site along the [Fe-S] clusters (C1 to C4/C5) to the physiological electron acceptor. We hypothesize that MK-7 binds to the N-terminal region of ForC1 in such a way that the distance between the MK-7 and one of the two most N-terminus [Fe-S] clusters, namely the [4Fe-4S] (C4) or the [2Fe-2S] (C5) cluster (Fig. 1C), is compatible with physiological electron transfer (52). It is interesting to note that EPR-monitored redox titration experiments performed on purified ForCE1 buffered at pH 6.0 and 7.5 have provided values of Em (MK/MKH2) about 55 mV lower than that of the menaquinone pool. Such a shift can be explained by an ∼ 80-fold tighter binding of oxidized MK than MKH2 to the ForCE1 Q-site, which would be expected for a quinone reduction site in which binding of the substrate (menaquinone) is favored over that of the product (menaquinol). This situation is very similar to that described for the plastosemiquinone bound to the quinone reduction site QB in photosystem II (Δ = −50 mV) (53). In addition, similar values have been measured for the demethylmenasemiquinone stabilized at the quinol oxidation site of the membrane-bound nitrate reductase from E. coli (Δ = −30 or −80 mV) (54), and tighter binding of ubiquinol than ubiquinone has been discussed in the quinone reduction site of the bc1 complex from yeast (Δ ∼ +24 to +54 mV) (55) or from Rhodobacter sphaeroides (Δ ∼ +60 mV) (56).
Within the enzymatic complex, the ForE1 subunit could help stabilize menaquinone binding by providing a structural interface with ForC1 as in its absence, the amount of copurified MK-7 drops dramatically to 1.6%. Moreover, ForE1 may participate to the structural integrity of ForC1 as its formate:BV oxidoreductase activity dropped by 50-fold when produced and purified in the absence of ForE1. Using several techniques, we have shown that MK-7 copurified with ForCE1 in a molar ratio (1:0.87). As discussed above, ForCE enzymes are not predicted to contain any transmembrane helices and are purified without detergents. Nevertheless, they bind menadione with an apparent affinity (KMmenadione = 20 μM) in the same range as the membrane integral nitrate reductase A from E. coli and the peripheral type-II NADH:quinone oxidoreductase from Caldalkalibacillus thermarum (57, 58). Finally, ForCE1 is also able to bind MK-8 at a low but significant ratio, consistent with low MK-8 abundance in B. subtilis membrane (48). To recruit menaquinone, it is reasonable to assume that ForCE contacts the membrane. Even if ForCE1 and ForCE2 have been purified without detergents, they could loosely bind the cytoplasmic side of the membrane through amphipathic helices similarly to the peripheral NdhII from C. thermarum that couples NADH oxidation to menaquinone reduction (59). Evaluation of this hypothesis is the object of undergoing studies in our laboratory.
ForC1 and ForC2 from B. subtilis are distinct from other members of the FDH family notably because of the residues at vicinity of the Mo atom such as the natural substitution of the strictly conserved His residue adjacent to Cys or Secys (C/UH) by a Gln (C/UQ) (Figs. 1A and S2A). This change from a positively charged and polar amino acid (His) by a polar one (Gln) is intriguing and prompted us to examine the active site region by sequence alignment and examination of the 3D structures of FDHs (Fig. S7). This scrutinous analysis allowed us to pinpoint an His residue located beside the strictly conserved Arg active residue and whose function may be to preserve at least one positive charge in close proximity to the active site (Fig. S7A). Furthermore, this analysis sheds light into the systematic occurrence of at least one His residue close to the active site with the occurrence of a Gln residue (Class I: C/UH RGQ or C/UQ RGH as in ForC, Fig. S7, A–C), a Glu residue (Class II: C/UH RGE, Fig. S7, D and E), or an His residue (class III: C/UH RGH, Fig. S7, F and G). As mentioned before, the importance of these residues and their mechanistic role at the active site are still unknown, and this will be investigated in detail in forthcoming studies.
Phylogenetic analysis indicates that the ForC proteins of B. subtilis are the first characterized representatives of a new subfamily of FDHs with a CQ RGH motif. Indeed, using the ForC1 sequence as bait, 377 sequences with an N-terminal extension allowing the coordination of 4 [Fe-S] clusters were identified. These sequences belong to the previously mentioned classes I and III. Strikingly, the 176 sequences with the CQ RGH catalytic motif (Fig. S2A sequences shown in blue) cluster distinctly from the 201 sequences with the CH RGQ/H catalytic motif (Fig. S2A sequences shown in gray). The 176 sequences are mainly found in Firmicutes, but some representatives are found in other bacterial phyla (Proteobacteria, Actinobacteria, Deinococcus, Acidobacteria, and Planctomycetes) or in Archaea (Fig. S2B). Furthermore, the vast majority of sequences with the CQ RGH motif are synteny encoded with a gene encoding a protein with a DUF1641 domain, presumably a homolog of ForE.
While the forC1 and forC2 genes are paralogous and proteins have 61 % of identity, ForE1 and ForE2 share weak sequence identity (20%). However, both enzymatic complexes achieve the same reaction with comparable kinetic behavior and most likely interact loosely with the membrane to recruit menaquinone. A notable difference between both complexes resides in the way the corresponding operons are regulated. While genes encoding FDHs are typically regulated by anaerobiosis, fermentation, and formate induction (60, 61), forCE1 and forCE2 do not seem expressed under these metabolic conditions (24, 28) and are upregulated by different environmental and cellular cues. Notably, the forCE1 operon is part of the SigB-dependent general stress regulon and is upregulated in swarming conditions, in the presence of high salt or ethanol concentrations (28, 62, 63). On the other hand, the forCE2 transcription is upregulated after germination and in exponential growth and downregulated by the ResED two-component system that activates genes in oxygen-limited conditions (28, 64).
Altogether, our results highlight the originality of these noncanonical FDHs, expand our knowledge on the FDHs' building blocks, and pave the way for developing new biocatalysts for CO2 reduction.
Experimental procedures
Bacterial strains, media, and culture conditions
B. subtilis 168 (trpC2) strain and derivatives are described in Table S6. Genomic integration of reporter sequences was achieved using the pSPH1/pSPH2 plasmids (obtained by Dr Henrik Strahl von Schulten) by transformation at the amyE locus (65). Target integration was confirmed by amylase sensitivity. Chromosomic inactivation of target genes was obtained by transformation of gDNA from KO strains (source BGSC). Clones were verified by PCR. For routine growth, cells were propagated in LB medium (tryptone, 10 g/l, yeast extract, 5 g/l, NaCl, 5 g/l). When necessary, antibiotics were used at the following concentrations: spectinomycin (100 μg ml−1), kanamycin (10 μg ml−1), and erythromycin (1.25 μg ml−1).
For overproduction of recombinant proteins, B. subtilis 168 derivatives (4088 amyE::pSHP1-6hisyjgCD, 4233 amyE::pSHP2-yjgC8his, 4230 amyE::pSHP-6hisyrhED, 4192 amyE::pSHP-yrhE8his) were grown in 4.8 L of the LB medium with 2 μM sodium molybdate at 37 °C until A600 = 0.3; cells were then submitted to a salt shock (0.4 M NaCl) for 30 min, followed by induction with 0.8% xylose for 3 h (4088 and 4233 strains) and expression overnight (4230 and 4192 strains) at 37 °C. Harvested cells were washed once in 20 mM sodium phosphate, pH 7.5, and 50 mM Na2SO4, and cells were kept at −80 °C until use.
Plasmid construction
Genes were cloned into pSPH1 and pSPH2 to allow induction with the xylose promoter at the amyE locus. Plasmids were constructed as follows: the pET28HT-YjgCD: yjgCD fragment was amplified with primers 898-899 using B. subtilis gDNA as the template and inserted by ligation into a previously excised pET28dHT-gapR at NdeI-KpnI sites to add a 6-his tag sequence followed by a TEV cleavage site at the 5′ end of yjgC.; pSPH1-6hisyjgCD: 6his-yjgCD was amplified with primers 941 to 952 using pET28HT-YjgCD as the template and introduced by ligation into pSHP1 at AvrII-XhoI sites; pSPH1-6hisyrhED: yrhED was amplified with primers 967 to 968 using B. subtilis 168 gDNA as the template and introduced into pSPH1 at AvrII-EcoRI sites using Gibson Assembly Cloning kit NEB with the linker primer 966. Cloning of yrhE at this site adds a sequence encoding a 6-his tag followed by a TEV cleavage site at the 5′ end. pSPH2-yjgC8his: yjgC was amplified with primers 944 to 950 and introduced into pSPH2 at XhoI-SpeI sites. Cloning of yjgC at this site adds a sequence encoding an 8-his tag at the 3′ end; pSPH1-yrhE8his: pSPH2-yjgC8his was digested at XhoI-SfiI sites to release yjgC fragment. Then, a yrhE fragment was amplified with primers 988 to 1042 using B. subtilis gDNA as the template and introduced by ligation.
All plasmids were verified by sequencing, and primers are listed in Table S7.
Protein purification
All steps were performed at 4 °C with an ÄKTA FPLC system (GE Healthcare). Frozen cells, typically 10 to 15 g of wet weight, were thawed and suspended in 90 ml of 20 mM sodium phosphate, pH 7.5, and 50 mM Na2SO4 (buffer A). Cell suspension was treated with protease inhibitor cocktail (Roche), lysozyme (1 mg/ml during 30 min at 37 °C), DNase I, and 0.5 mM EDTA and disrupted by 1 to 2 passages through a French pressure cell at 1 bar. Cells debris were removed by centrifugation for 40 min at 100,000g. The supernatant was applied onto 15 ml Ni-NTA affinity column (GE Healthcare). The column was washed with buffer A supplemented with 50 mM imidazole, and the enzyme eluted in buffer A supplemented with 500 mM imidazole and 10 mM KNO3. The enzyme was washed in 50 mM MES, pH 6, 50 mM Na2SO4, 5% glycerol, and 10 mM KNO3 using a PD-10 desalting column (Pharmacia), concentrated with Amicon 100 kDa (Millipore) and frozen in liquid nitrogen.
Activity assays and kinetic analysis
FDH activity was determined using an AvaSpec-2048L spectrophotometer, inside an anaerobic chamber, with an atmosphere of 100% N2, at room temperature (RT), with stirring. For formate-BV oxidoreduction measurements, the reduction of BV was monitored at 600 nm (Ɛ600 nm (BV+) = 7.0945 mM−1 cm−1) with 1.75 mM BV in 25 mM CHES, pH 8.6 (6his-YrhED), or 17.5 mM BV in 25 mM CHES, pH 10 (YjgC-8his/6his-YjgCD), supplemented with 50 mM sodium formate at RT. One unit of activity is defined as the amount of FDH capable of oxidizing 1 μmol of formate per minute per milligram of the enzyme. Kinetic analysis was measured at substrate concentrations ranging from 0.5 μM to 150 mM sodium formate. For formate:menadione oxidoreduction measurements, the reduction of 60 μM menadione was monitored at 260 nm (Ɛ260 nm (menadione) = 17.2 mM−1 cm−1) in 25 mM CHES, pH 9, and 50 mM sodium formate at RT. One unit of activity is defined as the amount of FDH capable of reducing 1 μmol of menadione per minute per milligram of the enzyme. Kinetic analysis was measured at substrate concentrations ranging from 0.5 μM to 200 μM menadione. Kinetic parameters were calculated by direct fitting of the Michaelis–Menten equation: y = Vmax × S/(KM + S) using OriginPro. The influence of pH on FDH activity was assessed using a buffer mix containing 20 mM MES, CHAPS, CHES, and Hepes at RT. The pH was adjusted from 6.5 to 11 with HCl and NaOH.
Quinone extraction and analysis
Quinones were extracted from solutions of purified ForCE1, ForCE2, or ForC1 essentially as described (66). Briefly, 20 to 50 μl of purified proteins (corresponding to 0.1–0.25 mg protein) was transferred into glass tubes and completed up to 200 μl with water. 50 μl KCl 3 M and 3 ml methanol were added, and the tubes were vortexed for 1 min. 2 ml petroleum ether (40–60° boiling range) was added, and vortex was repeated for 1 min. The tubes were centrifuged at 700 rpm for 1 min at RT, the upper phase was collected, and the methanol phase was extracted again with 2 ml petroleum ether. Both petroleum ether phases were combined in 5 ml Eppendorf tubes and dried under a nitrogen flow. Dried lipid extracts were resuspended in 100 μl ethanol, and fractions corresponding to 5 and 20 μg protein were analyzed by HPLC coupled to ECD and MS as previously described (67). The probe temperature was 400 °C, the cone voltage was 80 V, and MS spectra were recorded between m/z 500 and 900 with a scan time of 0.5 s.
Commercial MK-7 (Sigma) was used to generate standard curves ranging from 2 to 100 pmoles. The concentration of the MK-7 solution was determined using an extinction coefficient Ɛ248 nm = 18,900 M−1 cm−1 (68). The standard curve for the ECD signal was used to quantify the peaks (MK-7 and MK-8) obtained for the protein samples. Technical duplicates (extraction and analysis) were performed for each protein preparation, and the quantifications of MKs represent the average of the four values obtained from two measurements (5 and 20 μg protein) for each duplicate.
Redox titrations
Preparation of EPR samples
Formate-reduced samples of ForCE1 were prepared in 50 mM MES buffer, pH 6.0, with 80 μM of protein as followed. The air-oxidized sample was incubated with 100 mM sodium formate in a glove box and frozen immediately. Subsequently, the sample was thawed in the glove box and further reduced with 13 mM sodium dithionite before freezing. Formate-reduced samples of ForCE2 were prepared in 20 mM Tris/propane, pH 6.5, and 5% glycerol containing 50 μM of protein as followed. The air-oxidized sample was incubated with 30 mM sodium formate in a glove box before freezing or incubated with 5 mM sodium dithionite in a glove box before freezing.
Redox titrations of the MSK radical in ForCE1 were carried out anaerobically at RT (about 25 °C) either in a glove box or in an airtight vessel flushed with oxygen-free argon. Redox potentials were measured with a combined Pt-Ag/AgCl/KCl (3M) microelectrode and are given in the text with respect to the standard hydrogen electrode. The following redox mediators were used at 10 μM final concentrations: phenazine methosulfate, phenazine ethosulfate, methylene blue, resorufin, indigo carmine, anthraquinone 2,6 disulfate, flavin mononucleotide, phenosafranine, neutral red, and methyl viologen. Reductive titrations were carried out by stepwise addition of an anaerobic sodium dithionite solution (100 mM or 1 mM). Samples were anaerobically transferred into calibrated EPR tubes that were rapidly frozen.
The normalized variation of the peak-to-peak amplitude of the MSK EPR signal was fitted to a theoretical curve corresponding to two successive one-electron redox processes:
| (1) |
Where E1 and E2 are the midpoint potentials of the MK/MSK and MSK/MKH2 couples, respectively. α = F/RT where R and F are the molar gas and Faraday constants, respectively, and T is the absolute temperature. The two-electron midpoint potential of the MK/MKH2 couple, which corresponds to the redox potential for which the amount of MSK is maximum, is Em = (E1 + E2)/2. The stability constant KS of MSK defined with respect to the comproportionation reaction is as follows:
| (2) |
The occupancy level Rocc was calculated as the ratio between the normalized MSK concentration per enzyme inferred from spin quantitation experiments (assuming one [2Fe-2S] cluster taken as internal reference per enzyme) and the normalized maximal MSK concentration (MSKmax) given by the following equation.
| (3) |
Differences in the binding constants of MK (KMK) and MKH2 (KMKH2) manifest as a shift in the Em from that of free MK/MKH2 given by the following equation:
| (4) |
The affinity of the site for MSK is a determinant of E1 and E2 but not of Em.
Multifrequency EPR spectroscopy
X and Q band EPR spectra were measured on a Bruker Elexsys E500 spectrometer. For X band measurements, the spectrometer was equipped with an ER4012ST rectangular cavity fitted to an ESR900 helium flow cryostat (Oxford Instruments). For Q band measurements, a standard resonator equipped with an CF 935 cryostat (Oxford Instruments) was used. W band experiments were performed on a Bruker Elexsys E680 spectrometer equipped with a 6 T superconducting magnet and a 2-kG high-resolution sweep coil. Spectra were recorded with the standard W-band resonator fitted with a Bruker cryogen-free system (Stinger). For the echo-detected field-swept experiment, the two-pulse echo intensity was measured as a function of magnetic field at fixed time interval τ = 200 ns between the two microwave pulses, using a shot repetition time of 500 μs.
A field correction was applied to the magnetic field by simulating the overlapping spectral contribution of Mn2+, assuming g(Mn2+) = 2.00101 ± 0.00005 and an isotropic hyperfine coupling constant a(Mn2+) = −(8.710 ± 0.003) mT (69).
Spectral simulations
Numerical simulations of EPR spectra were performed with the EasySpin package (release 5.0.12) using MATLAB (The MathWorks, Inc) (70). A field-independent (unresolved hyperfine couplings, H-strain) linewidth model was used to simulate the EPR signal of the MSK and the Mo(V) species, whereas those of the FeS centers were simulated using a field-dependent (g-strain) linewidth model. For H-strain, the full width at half maximum of Gaussian lines along the g-tensor principal axes was adjusted, whereas the corresponding g-strain distributions were considered uncorrelated. Parameters used for the simulations shown in this work are provided in Table S8.
MS analysis
Purified proteins were subjected to an SDS-PAGE, and the stacking gel band corresponding to total proteins was excised and submitted to in-gel trypsin digestion for proteomic analysis as described previously (71) and with the following adapted modifications. Spectra were processed by Proteome Discoverer software (ThermoFisher, version 2.4.1.15) using the Sequest HT algorithm with the Bacillus subtilis database (Taxonomy ID: 224308, version 2016-08-20, downloaded from the NCBI by Protein Center including 5573 entries). In this study, proteins were also filtered by a minimum number of Peptide Spectral Match of 10. The list of identified proteins is available in Tables S2–S4.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (72) with the dataset identifier PXD028742 and 10.6019/PXD028742.
Supporting information
This article contains supporting information (73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Romain Mercier for providing plasmids pSPH1/pSPH2, originally constructed by H. Strahl von Schulten. We also thank Matt Cabeen, Elodie Foulquier-Khadaroo, and Anne Galinier for providing protocols and advice for B. subtilis genetics and culture. We thank Anne Galinier and Frédéric Biaso for scientific discussions. We also thank Genevieve Shirley Dobihal and David Rudner for providing genomic DNA Bacillus subtilis 168 trpC2 ΔyjgD::ery, strain was originally constructed by Koo et al. (88). Marseille Protéomique (MaP) has been supported by IBiSA (Infrastructures en Biologie Santé et Agronomie) and Aix Marseille Université. We are grateful to the EPR facilities available at the French EPR network (RENARD, IR CNRS 3443) and the Aix-Marseille EPR center. This work was supported by the French National Research Agency (ANR MOLYERE project, Grant No. 16-CE29-0010-01), the CNRS, and the CNRS Energy unit (Cellule Energie) through the project MICROBIOCO2.
Author contributions
A. W., R. A.-C., and S. G. methodology; A. U., F. P., M. B., F. S., G. G., R. L., A. W., R. A.-C., and S. G. investigation; A. M., B. G., A. W., R. A.-C., and S. G. writing–review and editing; A. M., B. G., and S. G. resources; A. M., B. G., and S. G. funding acquisition.
Funding and additional information
A. U. and M. B. were supported by a French MESR fellowship and R. A.-C. by an ANR MOLYERE project post-doctoral fellowship.
Edited by Ruma Banerjee
Footnotes
Present address for Rodrigo Arias-Cartín: Stress Adaptation and Metabolism Unit, Department of Microbiology, Pasteur Institute, CNRS, Microbiologie Integrative et Moléculaire (UMR 2001), Paris, France.
Contributor Information
Rodrigo Arias-Cartín, Email: rodrigo.ariascartin@pasteur.fr.
Stéphane Grimaldi, Email: stephane.grimaldi@univ-amu.fr.
Anne Walburger, Email: walburger@imm.cnrs.fr.
Supporting information
References
- 1.Niks D., Hille R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Protein Sci. 2019;28:111–122. doi: 10.1002/pro.3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoepp-Cothenet B., van Lis R., Philippot P., Magalon A., Russell M.J., Nitschke W. The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2012;2:263. doi: 10.1038/srep00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anbar A.D. Elements and evolution. Science. 2008;322:1481–1483. doi: 10.1126/science.1163100. [DOI] [PubMed] [Google Scholar]
- 4.Catling D.C., Zahnle K.J. The Archean atmosphere. Sci. Adv. 2020;6:9. doi: 10.1126/sciadv.aax1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maia L.B., Moura I., Moura J.J.G. Molybdenum and tungsten-containing formate dehydrogenases: Aiming to inspire a catalyst for carbon dioxide utilization. Inorg. Chim. Acta. 2017;455:350–363. [Google Scholar]
- 6.Nielsen C.F., Lange L., Meyer A.S. Classification and enzyme kinetics of formate dehydrogenases for biomanufacturing via CO2 utilization. Biotechnol. Adv. 2019;37:107408. doi: 10.1016/j.biotechadv.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Magalon A., Ceccaldi P., Schoepp-Cothenet B. 2016. The prokaryotic Mo/W-bisPGD enzymes family, Molybdenum and Tungsten Enzymes; pp. 143–191. [Google Scholar]
- 8.Hartmann T., Schwanhold N., Leimkühler S. Assembly and catalysis of molybdenum or tungsten-containing formate dehydrogenases from bacteria. Biochim. Biophys. Acta. 2015;1854:1090–1100. doi: 10.1016/j.bbapap.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Grimaldi S., Schoepp-Cothenet B., Ceccaldi P., Guigliarelli B., Magalon A. The prokaryotic Mo/W-bisPGD enzymes family: A catalytic workhorse in bioenergetic. Biochim. Biophys. Acta. 2013;1827:1048–1085. doi: 10.1016/j.bbabio.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Arnoux P., Ruppelt C., Oudouhou F., Lavergne J., Siponen M.I., Toci R., Mendel R.R., Bittner F., Pignol D., Magalon A., Walburger A. Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nat. Commun. 2015;6:6148. doi: 10.1038/ncomms7148. [DOI] [PubMed] [Google Scholar]
- 11.Schrapers P., Hartmann T., Kositzki R., Dau H., Reschke S., Schulzke C., Leimkühler S., Haumann M. Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from Rhodobacter capsulatus. Inorg. Chem. 2015;54:3260–3271. doi: 10.1021/ic502880y. [DOI] [PubMed] [Google Scholar]
- 12.Thomé R., Gust A., Toci R., Mendel R., Bittner F., Magalon A., Walburger A. A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. J. Biol. Chem. 2012;287:4671–4678. doi: 10.1074/jbc.M111.327122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyington J.C., Gladyshev V.N., Khangulov S.V., Stadtman T.C., Sun P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science. 1997;275:1305–1308. doi: 10.1126/science.275.5304.1305. [DOI] [PubMed] [Google Scholar]
- 14.Maia L.B., Moura J.J.G., Moura I. Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2015;20:287–309. doi: 10.1007/s00775-014-1218-2. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann T., Schrapers P., Utesch T., Nimtz M., Rippers Y., Dau H., Mroginski M.A., Haumann M., Leimkühler S. The molybdenum active site of formate dehydrogenase is capable of catalyzing C–H bond cleavage and oxygen atom transfer reactions. Biochemistry. 2016;55:2381–2389. doi: 10.1021/acs.biochem.6b00002. [DOI] [PubMed] [Google Scholar]
- 16.Popov V.O., Lamzin V.S. NAD+-dependent formate dehydrogenase. Biochem. J. 1994;301:625–643. doi: 10.1042/bj3010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yilmazer B., Isupov M.N., De Rose S.A., Bulut H., Benninghoff J.C., Binay B., Littlechild J.A. Structural insights into the NAD+-dependent formate dehydrogenase mechanism revealed from the NADH complex and the formate NAD+ ternary complex of the Chaetomium thermophilum enzyme. J. Struct. Biol. 2020;212:107657. doi: 10.1016/j.jsb.2020.107657. [DOI] [PubMed] [Google Scholar]
- 18.Oliveira A.R., Mota C., Mourato C., Domingos R.M., Santos M.F.A., Gesto D., Guigliarelli B., Santos-Silva T., Romão M.J., Cardoso Pereira I.A. Toward the mechanistic understanding of enzymatic CO2 reduction. ACS Catal. 2020;10:3844–3856. [Google Scholar]
- 19.Radon C., Mittelstädt G., Duffus B.R., Bürger J., Hartmann T., Mielke T., Teutloff C., Leimkühler S., Wendler P. Cryo-EM structures reveal intricate Fe-S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat. Commun. 2020;11:1912. doi: 10.1038/s41467-020-15614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner T., Ermler U., Shima S. The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science. 2016;354:114–117. doi: 10.1126/science.aaf9284. [DOI] [PubMed] [Google Scholar]
- 21.Jormakka M., Törnroth S., Byrne B., Iwata S. Molecular basis of proton motive force generation: Structure of formate dehydrogenase-N. Science. 2002;295:1863–1868. doi: 10.1126/science.1068186. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann T., Leimkühler S. The oxygen-tolerant and NAD+-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 2013;280:6083–6096. doi: 10.1111/febs.12528. [DOI] [PubMed] [Google Scholar]
- 23.Oh J.I., Bowien B. Structural analysis of the fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. J. Biol. Chem. 1998;273:26349–26360. doi: 10.1074/jbc.273.41.26349. [DOI] [PubMed] [Google Scholar]
- 24.Glaser P., Danchin A., Kunst F., Zuber P., Nakano M.M. Identification and isolation of a gene required for nitrate assimilation and anaerobic growth of Bacillus subtilis. J. Bacteriol. 1995;177:1112–1115. doi: 10.1128/jb.177.4.1112-1115.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano M.M., Dailly Y.P., Zuber P., Clark D.P. Characterization of anaerobic fermentative growth of Bacillus subtilis: Identification of fermentation end products and genes required for growth. J. Bacteriol. 1997;179:6749–6755. doi: 10.1128/jb.179.21.6749-6755.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niks D., Duvvuru J., Escalona M., Hille R. Spectroscopic and kinetic properties of the molybdenum-containing, NAD+-dependent formate dehydrogenase from Ralstonia eutropha. J. Biol. Chem. 2016;291:1162–1174. doi: 10.1074/jbc.M115.688457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuchan K., Baymann F., Baffert C., Brugna M., Nitschke W. The dyad of the Y-junction- and a flavin module unites diverse redox enzymes. Biochim. Biophys. Acta Bioenerg. 2021;1862:148401. doi: 10.1016/j.bbabio.2021.148401. [DOI] [PubMed] [Google Scholar]
- 28.Nicolas P., Mäder U., Dervyn E., Rochat T., Leduc A., Pigeonneau N., Bidnenko E., Marchadier E., Hoebeke M., Aymerich S., Becher D., Bisicchia P., Botella E., Delumeau O., Doherty G., et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 29.Zhu B., Stülke J. SubtiWiki in 2018: From genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 2018;46:D743–D748. doi: 10.1093/nar/gkx908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Böhmer N., Hartmann T., Leimkühler S. The chaperone FdsC for Rhodobacter capsulatus formate dehydrogenase binds the bis-molybdopterin guanine dinucleotide cofactor. FEBS Lett. 2014;588:531–537. doi: 10.1016/j.febslet.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 31.Magalon A., Mendel R.R. Biosynthesis and insertion of the molybdenum cofactor. EcoSal Plus. 2015;6:2. doi: 10.1128/ecosalplus.ESP-0006-2013. [DOI] [PubMed] [Google Scholar]
- 32.Axley M.J., Grahame D.A., Stadtman T.C. Escherichia coli formate-hydrogen lyase. Purification and properties of the selenium-dependent formate dehydrogenase component. J. Biol. Chem. 1990;265:18213–18218. [PubMed] [Google Scholar]
- 33.Schuchmann K., Müller V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science. 2013;342:1382–1385. doi: 10.1126/science.1244758. [DOI] [PubMed] [Google Scholar]
- 34.Grimaldi S., Biaso F., Burlat B., Guigliarelli B. 2016. Electron paramagnetic resonance studies of molybdenum enzymes, Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations; pp. 68–120. [Google Scholar]
- 35.Brondino C.D., Passeggi M.C.G., Caldeira J., Almendra M.J., Feio M.J., Moura J.J.G., Moura I. Incorporation of either molybdenum or tungsten into formate dehydrogenase from Desulfovibrio alaskensis NCIMB 13491; EPR assignment of the proximal iron-sulfur cluster to the pterin cofactor in formate dehydrogenases from sulfate-reducing bacteria. J. Biol. Inorg. Chem. 2004;9:145–151. doi: 10.1007/s00775-003-0506-z. [DOI] [PubMed] [Google Scholar]
- 36.Biaso F., Burlat B., Guigliarelli B. DFT investigation of the molybdenum cofactor in periplasmic nitrate reductases: Structure of the Mo(V) EPR-active species. Inorg. Chem. 2012;51:3409–3419. doi: 10.1021/ic201533p. [DOI] [PubMed] [Google Scholar]
- 37.Dermoun Z., De Luca G., Asso M., Bertrand P., Guerlesquin F., Guigliarelli B. The NADP-reducing hydrogenase from Desulfovibrio fructosovorans: Functional interaction between the C-terminal region of HndA and the N-terminal region of HndD subunits. Biochim. Biophys. Acta. 2002;1556:217–225. doi: 10.1016/s0005-2728(02)00364-x. [DOI] [PubMed] [Google Scholar]
- 38.Deaton J.C., Solomon E.I., Watt G.D., Wetherbee P.J., Durfor C.N. Electron paramagnetic resonance studies of the tungsten-containing formate dehydrogenase from Clostridium thermoaceticum. Biochem. Biophys. Res. Commun. 1987;149:424–430. doi: 10.1016/0006-291x(87)90384-6. [DOI] [PubMed] [Google Scholar]
- 39.Jollie D.R., Lipscomb J.D. Formate dehydrogenase from Methylosinus trichosporium OB3b. Purification and spectroscopic characterization of the cofactors. J. Biol. Chem. 1991;266:21853–21863. [PubMed] [Google Scholar]
- 40.Young T., Niks D., Hakopian S., Tam T.K., Yu X., Hille R., Blaha G.M. Crystallographic and kinetic analyses of the FdsBG subcomplex of the cytosolic formate dehydrogenase FdsABG from Cupriavidus necator. J. Biol. Chem. 2020;295:6570–6585. doi: 10.1074/jbc.RA120.013264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardiner A.T., Zech S.G., MacMillan F., Käss H., Bittl R., Schlodder E., Lendzian F., Lubitz W. Electron paramagnetic resonance studies of zinc-substituted reaction centers from Rhodopseudomonas viridis. Biochemistry. 1999;38:11773–11787. doi: 10.1021/bi990661c. [DOI] [PubMed] [Google Scholar]
- 42.Grimaldi S., Lanciano P., Bertrand P., Blasco F., Guigliarelli B. Evidence for an EPR-detectable semiquinone intermediate stabilized in the membrane-bound subunit NarI of nitrate reductase A (NarGHI) from Escherichia coli. Biochemistry. 2005;44:1300–1308. doi: 10.1021/bi048009r. [DOI] [PubMed] [Google Scholar]
- 43.Hastings S.F., Kaysser T.M., Jiang F., Salerno J.C., Gennis R.B., Ingledew W.J. Identification of a stable semiquinone intermediate in the purified and membrane bound ubiquinol oxidase-cytochrome bd from Escherichia coli. Eur. J. Biochem. 1998;255:317–323. doi: 10.1046/j.1432-1327.1998.2550317.x. [DOI] [PubMed] [Google Scholar]
- 44.Ingledew W.J., Ohnishi T., Salerno J.C. Studies on a stabilisation of ubisemiquinone by Escherichia coli quinol oxidase, cytochrome bo. Eur. J. Biochem. 1995;227:903–908. doi: 10.1111/j.1432-1033.1995.tb20217.x. [DOI] [PubMed] [Google Scholar]
- 45.Sato-Watanabe M., Itoh S., Mogi T., Matsuura K., Miyoshi H., Anraku Y. Stabilization of a semiquinone radical at the high-affinity quinone-binding site (QH) of the Escherichia coli bo-type ubiquinol oxidase. FEBS Lett. 1995;374:265–269. doi: 10.1016/0014-5793(95)01125-x. [DOI] [PubMed] [Google Scholar]
- 46.Lanciano P., Magalon A., Bertrand P., Guigliarelli B., Grimaldi S. High-stability semiquinone intermediate in nitrate reductase A (NarGHI) from Escherichia coli is located in a quinol oxidation site close to heme bD. Biochemistry. 2007;46:5323–5329. doi: 10.1021/bi700074y. [DOI] [PubMed] [Google Scholar]
- 47.Liebl U., Pezennec S., Riedel A., Kellner E., Nitschke W. The Rieske FeS center from the gram-positive bacterium PS3 and its interaction with the menaquinone pool studied by EPR. J. Biol. Chem. 1992;267:14068–14072. [PubMed] [Google Scholar]
- 48.Sato T., Yamada Y., Ohtani Y., Mitsui N., Murasawa H., Araki S. Production of menaquinone (vitamin K2)-7 by Bacillus subtilis. J. Biosci. Bioeng. 2001;91:16–20. doi: 10.1263/jbb.91.16. [DOI] [PubMed] [Google Scholar]
- 49.Un S., Dorlet P., Rutherford A.W. A high-field EPR tour of radicals in photosystems I and II. Appl. Magn. Reson. 2001;21:341–361. [Google Scholar]
- 50.Grimaldi S., Ostermann T., Weiden N., Mogi T., Miyoshi H., Ludwig B., Michel H., Prisner T.F., MacMillan F. Asymmetric binding of the high-affinity Q(H)(∗)(-) ubisemiquinone in quinol oxidase (bo3) from Escherichia coli studied by multifrequency electron paramagnetic resonance spectroscopy. Biochemistry. 2003;42:5632–5639. doi: 10.1021/bi034010z. [DOI] [PubMed] [Google Scholar]
- 51.Hille R., Hall J., Basu P. The mononuclear molybdenum enzymes. Chem. Rev. 2014;114:3963–4038. doi: 10.1021/cr400443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Page C.C., Moser C.C., Chen X., Dutton P.L. Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 53.De Causmaecker S., Douglass J.S., Fantuzzi A., Nitschke W., Rutherford A.W. Energetics of the exchangeable quinone, QB, in Photosystem II. Proc. Natl. Acad. Sci. U. S. A. 2019;116:19458–19463. doi: 10.1073/pnas.1910675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rendon J., Pilet E., Fahs Z., Seduk F., Sylvi L., Hajj Chehade M., Pierrel F., Guigliarelli B., Magalon A., Grimaldi S. Demethylmenaquinol is a substrate of Escherichia coli nitrate reductase A (NarGHI) and forms a stable semiquinone intermediate at the NarGHI quinol oxidation site. Biochim. Biophys. Acta. 2015;1847:739–747. doi: 10.1016/j.bbabio.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Covian R., Zwicker K., Rotsaert F.A., Trumpower B.L. Asymmetric and redox-specific binding of quinone and quinol at center N of the dimeric yeast cytochrome bc1 complex. Consequences for semiquinone stabilization. J. Biol. Chem. 2007;282:24198–24208. doi: 10.1074/jbc.M700662200. [DOI] [PubMed] [Google Scholar]
- 56.Robertson D.E., Prince R.C., Bowyer J.R., Matsuura K., Dutton P.L., Ohnishi T. Thermodynamic properties of the semiquinone and its binding site in the ubiquinol-cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J. Biol. Chem. 1984;259:1758–1763. [PubMed] [Google Scholar]
- 57.Giordani R., Buc J., Cornish-Bowden A., Cárdenas M.L. Kinetics of membrane-bound nitrate reductase A from Escherichia coli with analogues of physiological electron donors. Eur. J. Biochem. 1997;250:567–577. doi: 10.1111/j.1432-1033.1997.0567a.x. [DOI] [PubMed] [Google Scholar]
- 58.Blaza J.N., Bridges H.R., Aragão D., Dunn E.A., Heikal A., Cook G.M., Nakatani Y., Hirst J. The mechanism of catalysis by type-II NADH:quinone oxidoreductases. Sci. Rep. 2017;7:40165. doi: 10.1038/srep40165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heikal A., Nakatani Y., Dunn E., Weimar M.R., Day C.L., Baker E.N., Lott J.S., Sazanov L.A., Cook G.M. Structure of the bacterial type II NADH dehydrogenase: A monotopic membrane protein with an essential role in energy generation. Mol. Microbiol. 2014;91:950–964. doi: 10.1111/mmi.12507. [DOI] [PubMed] [Google Scholar]
- 60.Oh J.I., Bowien B. Dual control by regulatory gene fdsR of the fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. Mol. Microbiol. 1999;34:365–376. doi: 10.1046/j.1365-2958.1999.01613.x. [DOI] [PubMed] [Google Scholar]
- 61.Pinske C., Sawers R.G. Anaerobic formate and hydrogen metabolism. EcoSal Plus. 2016;7:1. doi: 10.1128/ecosalplus.ESP-0011-2016. [DOI] [PubMed] [Google Scholar]
- 62.Höper D., Völker U., Hecker M. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J. Bacteriol. 2005;187:2810–2826. doi: 10.1128/JB.187.8.2810-2826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petersohn A., Brigulla M., Haas S., Hoheisel J.D., Völker U., Hecker M. Global analysis of the general stress response of Bacillus subtilis. J. Bacteriol. 2001;183:5617–5631. doi: 10.1128/JB.183.19.5617-5631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye R.W., Tao W., Bedzyk L., Young T., Chen M., Li L. Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions. J. Bacteriol. 2000;182:4458–4465. doi: 10.1128/jb.182.16.4458-4465.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dubnau D., Davidoff-Abelson R. Fate of transforming DNA following uptake by competent Bacillus subtilis. I. Formation and properties of the donor-recipient complex. J. Mol. Biol. 1971;56:209–221. doi: 10.1016/0022-2836(71)90460-8. [DOI] [PubMed] [Google Scholar]
- 66.Szyttenholm J., Chaspoul F., Bauzan M., Ducluzeau A.-L., Chehade M.H., Pierrel F., Denis Y., Nitschke W., Schoepp-Cothenet B. The controversy on the ancestral arsenite oxidizing enzyme; deducing evolutionary histories with phylogeny and thermodynamics. Biochim. Biophys. Acta. 2020;1861:148252. doi: 10.1016/j.bbabio.2020.148252. [DOI] [PubMed] [Google Scholar]
- 67.Pelosi L., Vo C.-D.-T., Abby S.S., Loiseau L., Rascalou B., Hajj Chehade M., Faivre B., Goussé M., Chenal C., Touati N., Binet L., Cornu D., Fyfe C.D., Fontecave M., Barras F., et al. Ubiquinone biosynthesis over the entire O2 range: Characterization of a conserved O2-independent pathway. mBio. 2019;10 doi: 10.1128/mBio.01319-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMillan D.G.G., Marritt S.J., Butt J.N., Jeuken L.J.C. Menaquinone-7 is specific cofactor in tetraheme quinol dehydrogenase CymA. J. Biol. Chem. 2012;287:14215–14225. doi: 10.1074/jbc.M112.348813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burghaus O., Plato M., Rohrer M., Moebius K., MacMillan F., Lubitz W. 3-mm High-field EPR on semiquinone radical anions Q.cntdot.- related to photosynthesis and on the primary donor P.cntdot.+ and acceptor QA.cntdot.- in reaction centers of Rhodobacter sphaeroides R-26. J. Phys. Chem. 1993;97:7639–7647. [Google Scholar]
- 70.Stoll S., Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 71.Santin Y.G., Doan T., Lebrun R., Espinosa L., Journet L., Cascales E. In vivo TssA proximity labelling during type VI secretion biogenesis reveals TagA as a protein that stops and holds the sheath. Nat. Microbiol. 2018;3:1304–1313. doi: 10.1038/s41564-018-0234-3. [DOI] [PubMed] [Google Scholar]
- 72.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gumerov V.M., Zhulin I.B. TREND: A platform for exploring protein function in prokaryotes based on phylogenetic, domain architecture and gene neighborhood analyses. Nucleic Acids Res. 2020;48:W72–W76. doi: 10.1093/nar/gkaa243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Letunic I., Bork P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.George G.N., Hilton J., Temple C., Prince R.C., Rajagopalan K.V. Structure of the molybdenum site of dimethyl sulfoxide reductase. J. Am. Chem. Soc. 1999;121:1256–1266. [Google Scholar]
- 76.Jepson B.J.N., Anderson L.J., Rubio L.M., Taylor C.J., Butler C.S., Flores E., Herrero A., Butt J.N., Richardson D.J. Tuning a nitrate reductase for function. J. Biol. Chem. 2004;279:32212–32218. doi: 10.1074/jbc.M402669200. [DOI] [PubMed] [Google Scholar]
- 77.Barber M.J., May H.D., Ferry J.G. Inactivation of formate dehydrogenase from Methanobacterium formicicum by cyanide. Biochemistry. 1986;25:8150–8155. [Google Scholar]
- 78.Jepson B.J.N., Mohan S., Clarke T.A., Gates A.J., Cole J.A., Butler C.S., Butt J.N., Hemmings A.M., Richardson D.J. Spectropotentiometric and structural analysis of the periplasmic nitrate reductase from Escherichia coli. J. Biol. Chem. 2007;282:6425–6437. doi: 10.1074/jbc.M607353200. [DOI] [PubMed] [Google Scholar]
- 79.Najmudin S., González P.J., Trincão J., Coelho C., Mukhopadhyay A., Cerqueira N.M.F.S.A., Romão C.C., Moura I., Moura J.J.G., Brondino C.D., Romão M.J. Periplasmic nitrate reductase revisited: A sulfur atom completes the sixth coordination of the catalytic molybdenum. J. Biol. Inorg. Chem. 2008;13:737–753. doi: 10.1007/s00775-008-0359-6. [DOI] [PubMed] [Google Scholar]
- 80.Gangeswaran R., Lowe D.J., Eady R.R. Purification and characterization of the assimilatory nitrate reductase of Azotobacter vinelandii. Biochem. J. 1993;289:335–342. doi: 10.1042/bj2890335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Butler C.S., Charnock J.M., Bennett B., Sears H.J., Reilly A.J., Ferguson S.J., Garner C.D., Lowe D.J., Thomson A.J., Berks B.C., Richardson D.J. Models for molybdenum coordination during the catalytic cycle of periplasmic nitrate reductase from Paracoccus denitrificans derived from EPR and EXAFS spectroscopy. Biochemistry. 1999;38:9000–9012. doi: 10.1021/bi990402n. [DOI] [PubMed] [Google Scholar]
- 82.Dementin S., Arnoux P., Frangioni B., Grosse S., Léger C., Burlat B., Guigliarelli B., Sabaty M., Pignol D. Access to the active site of periplasmic nitrate reductase: Insights from site-directed mutagenesis and zinc inhibition studies. Biochemistry. 2007;46:9713–9721. doi: 10.1021/bi700928m. [DOI] [PubMed] [Google Scholar]
- 83.Arnoux P., Sabaty M., Alric J., Frangioni B., Guigliarelli B., Adriano J.-M., Pignol D. Structural and redox plasticity in the heterodimeric periplasmic nitrate reductase. Nat. Struct. Biol. 2003;10:928–934. doi: 10.1038/nsb994. [DOI] [PubMed] [Google Scholar]
- 84.Simpson P.J.L., McKinzie A.A., Codd R. Resolution of two native monomeric 90kDa nitrate reductase active proteins from Shewanella gelidimarina and the sequence of two napA genes. Biochem. Biophys. Res. Commun. 2010;398:13–18. doi: 10.1016/j.bbrc.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 85.Butler C.S., Charnock J.M., Garner C.D., Thomson A.J., Ferguson S.J., Berks B.C., Richardson D.J. Thiocyanate binding to the molybdenum centre of the periplasmic nitrate reductase from Paracoccus pantotrophus. Biochem. J. 2000;352:859–864. [PMC free article] [PubMed] [Google Scholar]
- 86.Rivas M.G., González P.J., Brondino C.D., Moura J.J.G., Moura I. EPR characterization of the molybdenum(V) forms of formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774 upon formate reduction. J. Inorg. Biochem. 2007;101:1617–1622. doi: 10.1016/j.jinorgbio.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 87.Prisner T., Lyubenova S., Atabay Y., MacMillan F., Kröger A., Klimmek O. Multifrequency cw-EPR investigation of the catalytic molybdenum cofactor of polysulfide reductase from Wolinella succinogenes. J. Biol. Inorg. Chem. 2003;8:419–426. doi: 10.1007/s00775-002-0432-5. [DOI] [PubMed] [Google Scholar]
- 88.Koo B.-M., Kritikos G., Farelli J.D., Todor H., Tong K., Kimsey H., Wapinski I., Galardini M., Cabal A., Peters J.M., Hachmann A.-B., Rudner D.Z., Allen K.N., Typas A., Gross C.A. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 2017;4:291–305. doi: 10.1016/j.cels.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang J., Zhang Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (72) with the dataset identifier PXD028742 and 10.6019/PXD028742.