

Abstract
Metabolic reprogramming of cancer cells creates a unique tumor microenvironment (TME) characterized by the limited availability of nutrients, which subsequently affects the metabolism, differentiation, and function of tumor-infiltrating T lymphocytes (TILs). TILs can also be inhibited by tumor-derived metabolic waste products and low oxygen. Therefore, a thorough understanding of how such unique metabolites influence mammalian T cell differentiation and function can inform novel anti-cancer therapeutic approaches. Here, we highlight the importance of these metabolites in modulating various T cell subsets within the TME, dissecting how these changes might alter clinical outcomes. We explore potential TME metabolic determinants that might constitute candidate targets for cancer immunotherapies, ideally leading to future strategies for reprogramming tumor metabolism to potentiate anti-cancer T cell functions.
Keywords: Metabolic reprogramming, Metabolites, T cells, Tumor microenvironment, Tumor immunotherapy
Abnormally accumulated metabolites in the TME are a hallmark of cancers
The metabolism of tumor cells differs from healthy or normal mammalian cells because of the acquisition and maintenance of malignant properties (1–3). Malignant cells undergo metabolic reprogramming to fulfill the energy needs for tumor growth and progression in the tumor microenvironment (TME) (4). Therefore, a better understanding of the molecular processes of cancer metabolism can facilitate the development of novel and effective candidate anticancer therapies targeting such cellular processes. This notion is strongly supported by recent studies in preclinical models and cancer patients. For instance, glycolysis is considered an attractive target for cancer therapy because increased glucose uptake has been observed in human and mouse tumors (5). Furthermore, inhibition of lactate dehydrogenase A (LDH-A) -- converting pyruvate to lactate in the glycolysis pathway in mammalian cells-- has suppressed tumor growth in mouse tumor models of non-small cell lung cancer (NSCLC), suggesting that metabolic reprogramming might be an effective therapeutic strategy for treating certain cancers (6).
Accordingly, metabolite concentrations can be measured to assess the metabolic activity and dysregulated metabolism of tumors (7). For instance, increased concentrations of metabolic intermediates in glucose, amino acid, and tricarboxylic acid cycle (TCA) metabolism have been documented in breast tumor tissues from transgenic mouse models overexpressing a panel of oncogenes, relative to controls (8). Furthermore, the metabolic signatures comprising groups of metabolites in mammary tissues have been associated with clinical outcomes and may have putative prognostic value for breast cancer patients (8). In addition, the metabolic states of cancer cells in the TME can significantly influence the functions of various immune cells, including T cells (9). Moreover, abnormally accumulated metabolites in the TME, such as lactate, are also harmful for immune cells and can reduce their anti-tumor efficacy (10). Indeed, the types of nutrients used by T cells and key metabolic processes such as glycolysis, and lipid or mitochondrial metabolism can selectively direct CD4+ T cell differentiation programs to engage unique functional properties in mice (11). We and others argue that the precise evaluation of specific metabolite concentrations in cancer patients through metabolomics can be utilized for clinical diagnosis; but also, be potentially applied to develop targeted candidate therapies. In addition, understating how specific metabolites in the suppressive TME might regulate T cell fates and functions to achieve anti-cancer immunity is vital for developing precise approaches to achieving immunity in various malignancies and for the success and responsiveness of immunotherapies.
In this article, we provide an overview of the current landscape of alterations of major metabolites associated with glucose and lipid metabolism in the TME, including lactate, citrate, fatty acid (FA), and cholesterol, which have been linked to anti-tumor immunity in recent studies (Box 1 and Figure 1). In addition, we highlight the importance of certain metabolites in the TME as candidate therapeutic targets that might improve anti-tumor immunity by reprogramming metabolic pathways in tumor and/or immune cells.
Box 1. Key metabolites in glucose and lipid metabolism.
Glucose is utilized by cells to generate adenosine triphosphate (ATP) in the majority of eukaryotic systems, where glucose is being broken down via a series of enzymatic reactions to release stored energy, known as glycolysis (120). Under normal aerobic conditions, glycolysis produces pyruvate, which is imported into mitochondria and subsequently converted into acetyl-CoA, entering the TCA cycle, also known as the Krebs cycle. Under anaerobic conditions, pyruvate is converted to lactate by lactate dehydrogenase (LDH) in the cytosol (11). Throughout these two metabolic processes, citrate and lactate are the major metabolites harboring important functions for host energy metabolism (Figure 1). Lactate, a hydroxycarboxylic acid, is the end product of anaerobic glycolysis, existing as two stereoisomers, L-lactate and D-lactate, and L-lactate is predominant physiological enantiomer (121). Citrate (citric acid) is the first molecule that can be produced from lactic acid fermentation from pyruvate when oxygen is lacking during the metabolic process of TCA cycle (122, 123). In addition, extracellular citrate can be transported to cells through a plasma membrane-specific variant of the mitochondrial citrate transporter (pmCiC) and affected cell metabolism through citrate-dependent metabolic pathways (61).
Lipid metabolism provides energy as well as the structural and functional building blocks for lipids in cells, such as cell membrane lipids (124). Fatty acid (FA) and cholesterol are key metabolites in lipid metabolism (Figure 1). Indeed, FA synthesis and oxidation (β-oxidation) are two important pathways in lipid metabolism (124). According to the presence or absence of one or more double bonds in the backbone, FAs can be classified into saturated FAs (having no double bond), monounsaturated FAs (MUFAs, having one double bond), and polyunsaturated FAs (PUFAs, having more double bonds)(45, 125). The major storage form of FAs is in triglycerides (TAG), and large amounts are also esterified to cholesterol or phospholipids. FAs are mainly derived from the hydrolysis of fats or synthesis from two carbon units (acetyl- CoA) in the liver, adipose tissue, and lactating breast (126). When FAs are de novo synthesized or taken up from the extracellular microenvironment, they can be converted into TAG and stored in lipid droplets (LDs) as an energy source(127). Cholesterol is either absorbed through diet or synthesized de novo by cells(127). Cholesterol is transported into cells as two bounding forms in clusters of lipids and proteins, high-density lipoprotein (HDL) and low-density lipoprotein (LDL) (127). Indeed, cholesterol, cholesterol metabolites, and esters, are also major components of the plasma membrane and other cellular organelles, maintaining cellular homeostasis (128). Furthermore, cholesterol can be a precursor for steroid hormones or be enriched in lipid rafts thus acting as an important molecule for intracellular signal transduction (128).
Figure 1. Source and production of key metabolites.
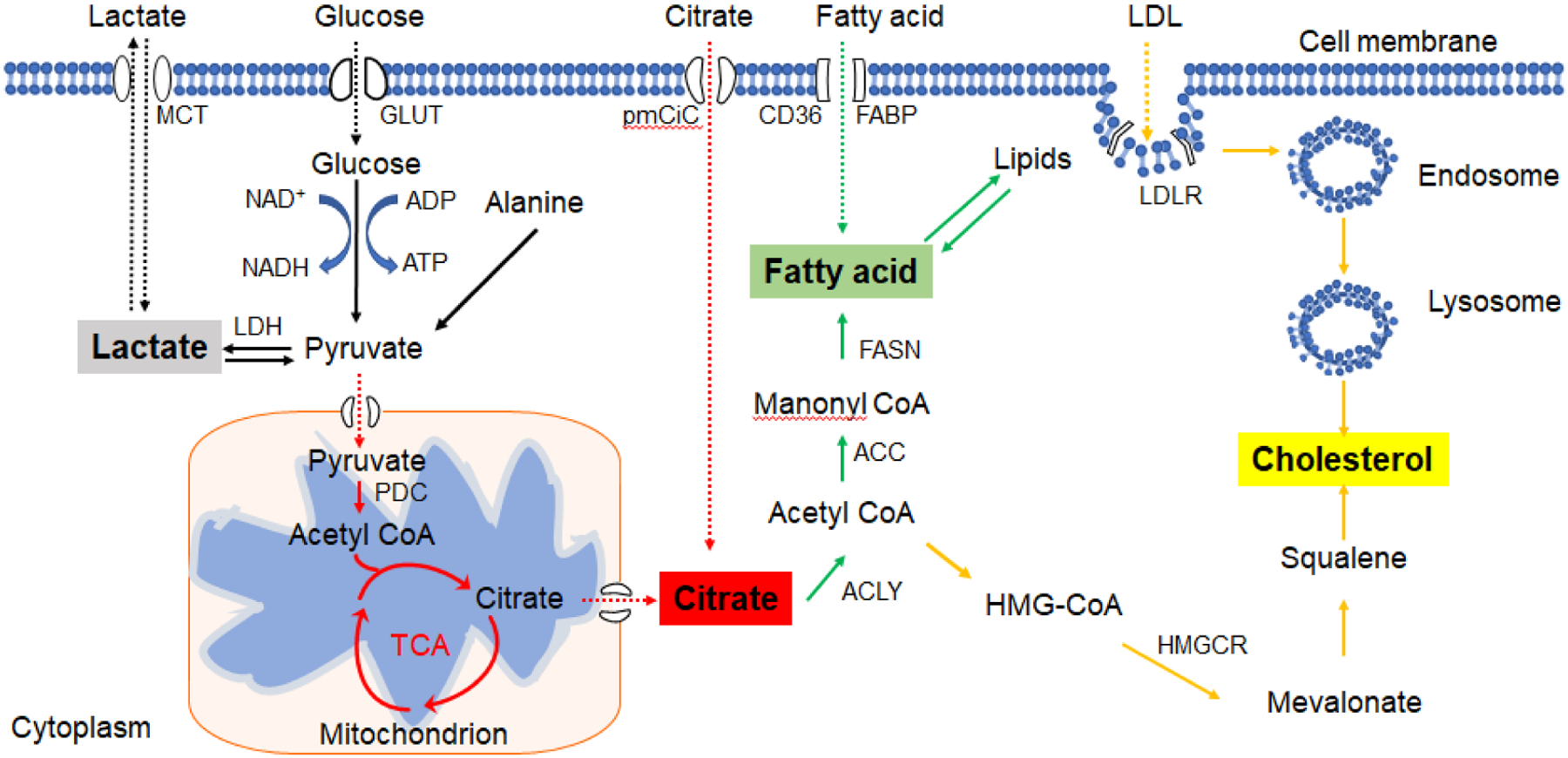
Glucose is hydrolyzed via a series of enzymatic reactions to produce pyruvate, which then is imported into the mitochondria and consequently converted into acetyl-CoA to enter the TCA. Under anaerobic conditions, pyruvate is converted to lactate by LDH. Citrate is formed during TCA cycle. FAs are mainly derived from the hydrolysis of fats or synthesis from two carbon units (acetyl- or malonyl-CoA). FAs can also be up-taken from the extracellular microenvironment. Cholesterol is transported to the lysosomes through the endocytic pathway; it is then hydrolyzed to free cholesterol molecules. Cholesterol synthesis starts from the mevalonate pathway and is eventually yielded after multistep enzymatic reactions. MCT: monocarboxylate transporters; GLUT: glucose transporter; ATP: adenosine triphosphate; ADP: adenosine diphosphate; NAD+: nicotinamide adenine dinucleotide; NADH: nicotinamide adenine dinucleotide (NAD) + hydrogen (H); LDH: lactate dehydrogenase; PDC: pyruvate dehydrogenase complex; TCA: tricarboxylic acid cycle; FAs: Fatty acids. FABP: fatty acid-binding protein; FASN: fatty acid synthase; ACC: Acetyl CoA carboxylase; ACLY: ATP citrate lyase; LDL: low-density lipoprotein; HMGCR: 3-Hydroxy-3-Methylglutaryl-CoA Reductase
Metabolites can regulate T cell differentiation and development
Many metabolic pathways occur in a cell to keep the cell living, growing, and dividing. In humans, important metabolic pathways, including glycolysis, the citric acid cycle, oxidative phosphorylation, the pentose phosphate pathway, fatty acid β-oxidation, gluconeogenesis, and others, produce a large number of metabolites with unique biological functions (12). Furthermore, metabolites derived from these metabolic processes are also directly involved in T cell activation and development (11).
Metabolites implicated in T cell activation and proliferation
Recent studies have reported that citrate -- exported from mitochondria -- is required for CD4+ helper T (Th1) cell proliferation and epigenetic remodeling, as evidenced by the induction of histone acetylation and gene expression changes involved in T cell activation (13). Indeed, exported citrate generates acetyl-CoA and induces histone H3K9 and H3K27 acetylation in selected loci such as Ifng to promote specific gene expression, reprogramming Th1 cell activation (13, 14). In addition, exposure to increasing concentrations of Na L-lactate has suppressed the proliferation of murine and human effector T (Teff) cells independent of acidity and toxicity, which can be rescued through LDH inhibition (15).
In addition to glucose metabolites, low concentrations of FAs can positively influence the proliferative capacity of T cells, while high concentrations of FAs can directly induce T cell apoptosis in a dose-dependent manner (16–19). Mechnistically, FA-induced T cell apoptosis involves activation of caspase 3, induction of mitochondrial dysfunction (depolarization and ROS production), and lipid accumulation in FA-treated lymphocytes (16–20). Furthermore, the degree of saturation and length of FAs are also factors regulating T cell apoptosis. For instance, short-chain FAs (SCFAs) are not toxic to T cells even at high concentrations (17), while longer and more unsaturated FAs are toxic at low concentrations (17, 20). However, low and non-toxic concentrations of FAs may suppress T cell activation to varying degrees depending on the stimulus used for T cell activation; this was evidenced from palmitic acid (PA) studies showing its suppression on concanavalin A-induced T cell proliferation but not on T cells with anti-CD3 antibody stimulation in vitro (16, 18, 21). High-FA diets in rats can suppress splenic T cell proliferation, due to the presence of oils rich in saturated FAs, monounsaturated FAs, n-6 polyunsaturated fatty acids (PUFA), or n-3 PUFA in diets (22, 23). Cholesterol also can alter T cell proliferation, and dietary-induced hypercholesterolemia can enhance T cell receptor (TCR)-stimulated T cell proliferation and increase TCR signaling strength in CD4+ T cells, as evidenced from the increased expression of transcription factor Nur77 (24). Moreover, hypercholesterolemia has led to enhanced CD3 internalization and increased expression of T cell activation markers such as CD69; this has increased the proliferation of stimulated T cells, suggesting that T cells can be activated by cholesterol (24).
Metabolites can affect T cell subset differentiation
Metabolites not only affect T cell proliferation but can also alter CD4+ T cell subset differentiation. The presence of sodium lactate in Th subset polarizing conditions can induce a significant up-regulation of IL-17 and Rorc in all Th subsets, while Th1 and Th2 signature cytokines remain unmodified upon treatment with sodium lactate in vitro, suggesting that sodium lactate can promote Th17 differentiation (25). These findings were supported by studies from another report showing that Na L-lactate increased Th17 polarization in mouse splenocytes in vitro (15). Furthermore, addition of Na L-lactate to CD4+CD25− conventional T cells (Tconvs) cultured under Treg polarizing conditions (e.g. rhTGF-β1 and IL-2) led to heightened polarization of induced Treg (iTreg) among mouse splenocytes in vitro. However, in this work, Na L-lactate addition did not influence T cell metabolism or reactive oxygen species (ROS)-dependent stress signaling, suggesting that Na L-lactate could selectively drive Treg developmental processes (15). In addition, citrate can modulate the Th17/Treg cell balance in vivo (26). Specifically, citrate treatment inhibited the viability of Th17 cells and decreased the production of Th17-related cytokines, while elevating the viability of Treg cells and increasing the production of Treg-related cytokines in a chronic rat model of renal failure (26). These results suggested that citrate could harbor anti-inflammatory properties by regulating the ratio of Th17/Treg cells (26).
Dietary-induced hypercholesterolemia increases CD4+RORγt+IL-17A+ cell populations and promotes CD4+FoxP3+ Treg cell differentiation in the liver of C57BL/6J mice (27). Moreover, hypercholesterolemia elevates FoxP3 expression and peripheral Treg cell populations but does not affect proliferation of activated T cells, suggesting that there is an increase in peripheral Treg development (24). However, hypercholesterolemia also hampers intrahepatic Treg differentiation resulting in increased proatherogenic Th1 cell differentiation and Th1/Th17 ratios, suggesting that hypercholesterolemia can rebalance differentiated intrahepatic T cell subsets (27). Moreover, prolonged hypercholesterolemia can impair Treg but not effector T cell accumulation in atherosclerotic lesions, while reversal of hypercholesterolemia can prevent the loss of Treg cells in atherosclerotic lesions (28). This may indicate that hypercholesterolemia-mediated regulation of Treg cell differentiation may occur in an organ-dependent fashion.
From another angle, CD4+ Teff cells exhibit high levels of glycolysis, while iTreg cells rely more on FAs as their preferred energy source (29). In the absence of FAs or inhibition of FA oxidation (FAO), iTreg differentiation is markedly reduced in vitro (30). A recent study demonstrated that Th17 cell development requires de novo FA synthesis mediated by acetyl-CoA carboxylase 1 (ACC1) and the glycolytic-lipogenic pathway, while Treg cell development depends on exogenous FAs; this suggests that Treg and Th17 cells use distinct FA sources for their energy metabolism (31). Moreover, with the fermentation of dietary components, bacteria produce SCFAs, including acetate, propionate, and butyrate (32). This is relevant as bacterially derived SCFA concentrations can control the size of the gut colonic Treg pool (32). Indeed, supplementation of propionate can increase colonic Treg numbers in germ-free mice and thus, control chronic inflammation (32). Dietary supplement of SCFAs, especially butyrate and propionate, contribute to inducing Treg cells in the gut, and butyrate has been shown to mitigate mouse experimental colitis (mediated by the model of adoptively transferred CD4+CD45RBhigh T cells in Rag1−/− mice), suggesting that SCFAs can regulate differentiation of Treg cells in vivo (33, 34). Mechanistic studies further showed that butyrate treatment induced the epigenetic regulation of Foxp3 expression and promoted Treg cell polarization, as evidenced from enhanced histone H3 acetylation at the Foxp3 locus, relative to controls (33). In addition, FAs can also indirectly affect T cell activation, proliferation, and differentiation, through peroxisome proliferator-activated receptor (PPAR) signaling, which regulates lipid metabolism-associated pathways (35). Indeed, the majority of FAs can serve as ligands for PPAR activation in T cells (36, 37). Furthermore, FA-induced activation of CD4+ T cells also depends on mTOR signaling-mediated induction of PPARγ, as evidenced from the increased gene expression associated with FA uptake in CD4+ T cells controlled by PPARγ,(38). These studies highlight the importance of FAs in regulating CD4+ T cell differentiation.
CD8+ T cells require distinct FA metabolism to regulate memory differentiation and subset specialization (39, 40). Memory CD8+ T cells (TM) have elevated FAO compared with effector CD8+ T cells in vivo (39). CD8+ TM cells promote FA and triglyceride (TG) synthesis using extracellular-derived glucose, and then perform “cell-intrinsic lipolysis” to hydrolyze these lysosomally stored TGs using lysosomal acid lipase (LAL) in mice (39). Tissue-resident memory (TRM ) CD8+ T cells derived from skin have increased FA-binding proteins 4 and 5 associated with FA uptake and FAO compared with central memory CD8+ T cells (CD8+ TCM) or effector CD8+ T cells in OT-1 TCR transgenic mice after inoculation with recombinant vaccinia virus carrying ovalbumin (OVA) (40). In addition, recent studies showed that Tc9 cells (IL-9-secreting CD8+ T cell subset), harbored significantly lower amounts of intracellular cholesterol than type 1 CD8+ T cells (Tc1) (41). Manipulating cholesterol amounts by adding cholesterol or by using β-cyclodextrin (β-CD) to reduce cellular cholesterol content can uniquely control IL-9 protein expression and Tc9 differentiation in polarizing Tc9 cells in vitro, but not in other T cell subsets (41). These findings collectively suggest that FAs and cholesterol can regulate CD8+ T cell development.
Metabolites can control T cell functions
Lactate has been reported to suppress the motility of activated T cells in vitro (25). Sodium lactate restrains chemokine CXCR-CXCL10-induced glycolysis in CD4+ T cells, resulting in suppressed migration of CD4+ T cells. Furthermore, lactic acid inhibits the cytotoxic activity of CD8+ cytotoxic T lymphocytes (CTLs) (25). In addition, Na L-lactate reduces Teff proliferation but does not affect Treg suppressive activity or proliferation (15). Furthermore, high concentrations of lactic acid in the TME of patients suffering different malignancies impair IL-2 and IFN-γ production, disturb T cell metabolism, and reduce cytotoxic CTL activity (42).
Citrate has been shown to regulate memory T cell function during systemic bacterial infections in mice (43). During infection, citrate is converted to acetyl-CoA by ATP citrate lyase (ACLY) and expands the cellular acetyl-coenzyme pool, mediated by increased acetate in memory CD8+ T cells, resulting in enhanced acetylation of enzyme GAPDH, catalyzing glycolysis, and thus facilitating prompt memory CD8+ T cell recall responses in mice (43). Glucose-derived citrate was abundant in activated effector memory (Tem) CD8+ T cells generating acetyl-CoA by ACLY and subsequent histone acetylation; indeed, prevention of acetyl-CoA formation via citrate and blockage of histone acetyl-transferase activity can reduce IFN-γ secretion produced by activated Tem CD8+ T cells (44).
Both extracellular and intracellular FA contents can influence the functions of CD4+ and CD8+ T cell subsets (45). FAs can modulate cytokine secretion upon addition to T cells, including TNF, IL-2, IL-6, IL-8, IL-1β, IL-10, and IFN-γ (46–48). Addition of polyunsaturated FAs results in long-term activation of PKC and IL-2 synthesis in T cells treated by dioctanoylglycerol and ionomycin (48). Saturated FAs can also induce human T cell cytokine production even without T cell activation in vitro (46). In addition, IL-2 production and IL-2 receptor alpha-chain (CD25) expression are increased in lymphocytes from mice or rats receiving FA-rich diets (22, 49).
Increased cholesterol amounts in whole cells and plasma membranes are exhibited in activated mouse CD8+ T cells stimulated with anti-CD3/CD28 antibodies in vitro (50). Inhibiting cholesterol esterification by suppressing Acetyl-Coenzyme A acetyltransferase 1 (ACAT1) increases the plasma membrane cholesterol concentration in CD8+ T cells, resulting in enhanced TCR clustering and signaling, and immunological synapse formation (50). Reducing cholesterol esterification augments cytolytic granule and cytokine production, as well as CD8+ T cell cytotoxicity. In contrast, suppression of cholesterol biosynthesis with the HMG-CoA reductase inhibitor lovastatin, or of cholesterol transport with inhibitor U18666A, significantly decreases granule and CD8+ T cell cytokine production in vitro (50). Cholesterol treatment not only inhibits granzyme B, IFN-γ and TNF production in CD8+ T cells, but also decreases the proliferation and migratory ability of CD8+ T cells in mice (51). In addition, decreasing dietary cholesterol in ApoE−/− mice limits the generation of inflammatory phenotypes with decreased intracellular IL-10 and IL-12 production, mediated by CD4+ and CD8+ T cells (52). Decreasing cholesterol using β-CD can also promote Tc9 cell persistence and enhance antitumor immunity in vivo in MC38 tumor models (41).
Abnormal production of metabolites in the TME
Metabolic reprogramming in cancers has been investigated for over 90 years since Otto Warburg first described the glycolytic characteristics of cancer cells (53). The Warburg Effect provides an overall benefit that supports a TME conducive to cancer cell proliferation (54). The metabolites related to glucose and lipid metabolism are produced and distributed abnormally in the TME, also contributing to tumor development and progression (Figure 2) (55).
Figure 2. Effects of key metabolites on effective anti-tumor immunity in the suppressive tumor microenvironment.
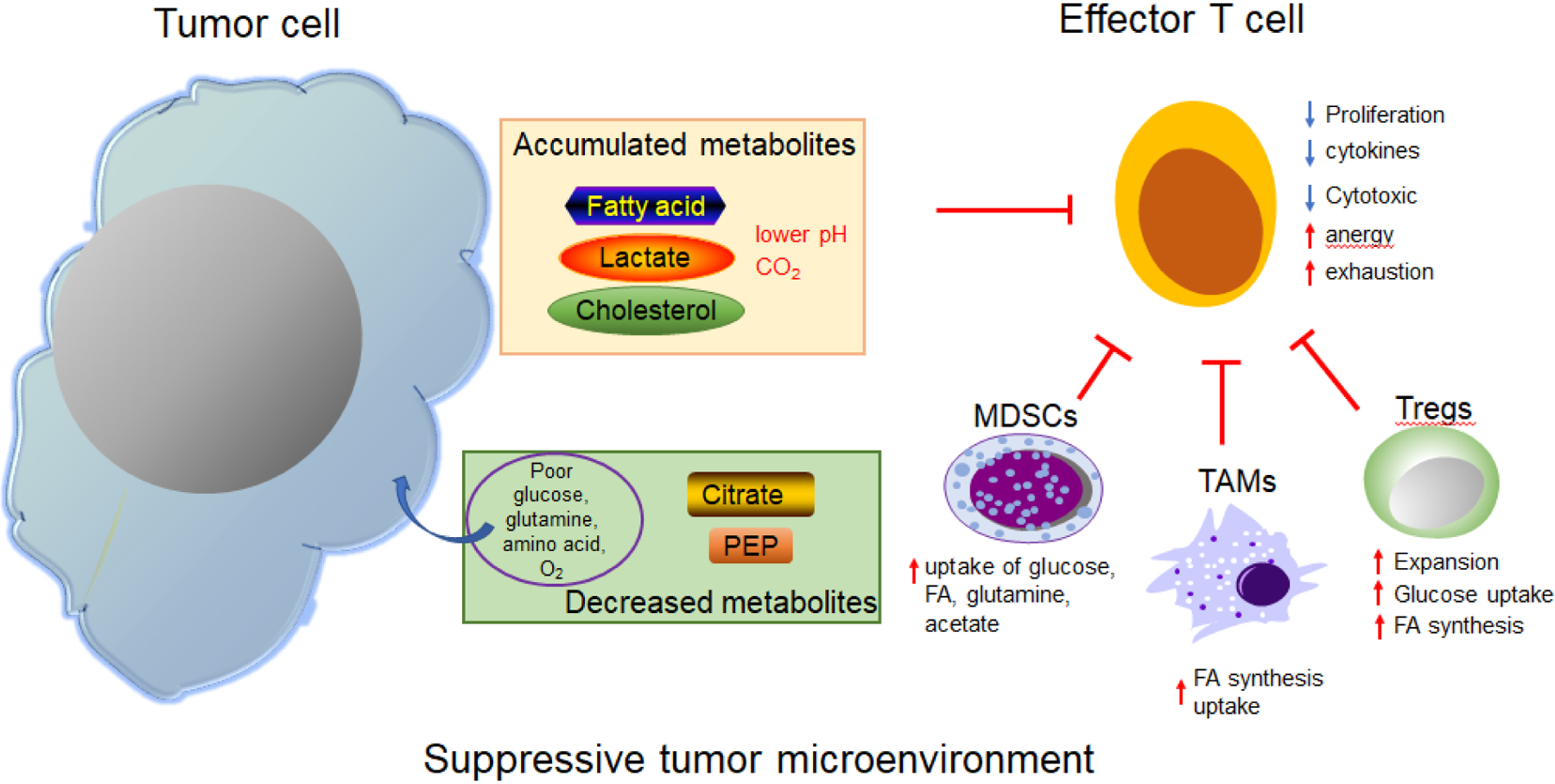
Tumor cells uptake large amounts of nutrients (glucose, glutamine, amino acids, etc.) from the TME for their energy demands, resulting in a nutrient-poor environment. Cancer cells have active aerobic glycolysis to produce lactic acid and contribute to an acidic environment. Accumulated lactic acid, fatty acid (FA), and cholesterol significantly suppress the proliferation, cytokine production, and cytotoxic activity of effector T cells; they also lead to the development of anergy or exhaustion in tumor-infiltrating CD8+ cells. Decreased amounts of some metabolites, such as citrate and PEP, also affect effector T cell functions in anti-tumor immunity. Tumor-infiltrating Tregs are activated and expanded in this specific TME, where they preferentially capture and utilize glucose to fuel glycolysis; meanwhile, they synthesize and accumulate FAs, which contribute to the suppression of antitumor immunity. In addition, tumor associated macrophages (TAM) exhibit increased FA synthesis, uptake, and oxidation. Myeloid derived suppressor cells (MDSC) also experience active glucose uptake, as well as the uptake of FA, glutamine, and acetate. PEP: phosphoenolpyruvate; Tregs: regulatory T cells;
Accumulation of metabolites in the TME
Tumor cells consume more glucose than that of normal cells, and rely on glycolysis for ATP production even in the presence of oxygen (53). Hypoxia in the TME also increases the gene expression of glucose transporters and glycolytic enzymes such as LDH (56). These factors result in production of large amounts of lactic acid and other metabolites through glycolysis in the TME of many cancer cell types (57). In addition to production from tumor cells, tumor-infiltrating innate and adaptive immune cells produce lactate in the TME (10). Furthermore, significantly elevated concentrations of serum lactate have been detected in patients with various cancers, including breast cancer, gastrointestinal cancer, urogenital cancer, lung cancer, sarcoma, melanoma, myeloid malignancies, lymphoid malignancies, and other types of cancers (42).
Unlike lactate, citrate concentrations are decreased in cancer cells (58, 59). The amounts of citrate are significantly decreased in the blood and urine of patients with multiple tumors, including metastatic prostate, lung, pancreas, colorectal, bladder, esophagus cancer, and neurological tumors (60). The decreased extracellular citrate is linked to an increase in metabolic demand of cancer cells (60). In addition, extracellular citrate can be up-taken and provided to cancer cells via a plasma membrane-specific variant of the mitochondrial citrate transporter (pmCiC), which affects cancer cell metabolism and cancer development in vivo in murine xenografts of human pancreatic cancer (61).
Lipid abundance is now recognized as an established hallmark of many tumors including melanoma, pancreatic cancer, and others (62, 63). In a mouse model of pancreatic ductal adenocarcinoma (PDA) and in human tumor specimens, the specific long-chain fatty acids (LCFAs) accumulated in the TME with PDA progression (62). In the B16 or MC38 implanted mouse tumor models, the tumor interstitial fluid (TIF) contained higher concentration of many species of FFAs, as well as acyl-carnitines, ceramides, and esterified cholesterol than those in serum from the same animal (63). In B16 cancer models, tumor tissues have greater amounts of free poly-unsaturated FAs and phosphatidylethanolamines enriched with poly-unsaturated fatty acyl than normal skin or spleen, as well as metastatic lung tissues compared with normal lung (64). In cancer cells, FAs are utilized for biosynthesis of membrane lipids and signaling molecules, including phospholipids (PLs) and other lipids (sterols, sphingolipids, and lyso-PLs) (65). Activation of de novo FA biosynthesis in cancer increases relative amounts of saturated and monounsaturated species but decreases polyunsaturated forms of membrane lipids, which results in protection of cancer cells from ROS-induced damages (66, 67). Deregulated FA synthesis can also contribute to the regulation of signaling pathways involved in cancer cell proliferation and survival (68). The abundance and saturation of cellular FAs determine the activity of signaling proteins in cancer cells, such as WNT proteins, SRC and RAS oncoproteins, which are frequently modified by esterification of a palmitoleoyl chain or thioesterification with saturated palmitoyl residues (68, 69).
Cholesterol amounts are also high in cancer cells. Cholesterol metabolism is activated in the TME associated with cancer progression, as evidenced from enhanced cholesterol biosynthesis, increased exogenous cholesterol uptake by LDLR, elevated cholesterol esterification by ACAT1, and increased oxysterol production (70). Enriched cholesterol has been shown in tumor tissues in mouse models with various tumor types, including B16 melanoma with subcutaneous and lung metastasis, LL2, and MC38 tumors (51). Furthermore, HMG-CoA reductase and LDL receptor activities are up-regulated in proliferating cancer cells, resulting in the rise of cholesterol content and consumption (71). In addition to cancer cells, cholesterol amounts are high in TILs from breast cancer and melanoma tumor-bearing mice (72).
Metabolite amounts are associated with clinical outcomes
Metabolic abnormalities in cancers are linked to cancer progression (73). Lactate accumulation in the TME is negatively associated with patient survival but highly related to the incidence of metastasis and therapeutic resistance (74). Elevated serum lactate concentrations in patients with malignant tumors are positively correlated with tumor burden, including in breast, gastrointestinal, urogenital, and lung cancers, as well as in sarcoma, lymphoid and myeloid malignancies, and melanoma (42). Excretion of high concentrations of lactate and expression of high amounts of LDHA in solid tumors are associated with poor prognosis and survival in human melanoma (75). Elevated LDHA and LDHA-associated lactic acid production and acidification inhibit the function and survival of T and NK cells with decreased expression of IFN-γ and granzyme B and increased cell apoptosis, contributing to tumor immune escape (75). Decreased extracellular citrate has been detected in blood and/or urine in patients with metastatic prostate, bladder, colorectal, lung, pancreas, and esophagus cancers, as well as neurological tumors, linked to the high metabolic demand of cancer cells (60). Reduced citrate in cancer cells is considered as an indicator of cancer aggressiveness in human prostate cancer (58).
FA uptake and biomass storage are increased in malignant human prostate tissue (76), while FAO is downregulated in multiple tumor types, including breast, gastric, and kidney cancer, colorectal carcinomas, and in lung, pancreatic, bladder, and stomach adenocarcinomas, as well as in esophageal and renal cell carcinomas (77). FAO activation may decrease cancer cell proliferation and be associated with improved clinical outcomes in cancer patients, including improved prognosis, and favorable responses to neoadjuvant chemotherapy or aromatase inhibition (77). Dysregulation of cholesterol homeostasis is also an important contributing factor to cancer development (78). Cholesterol synthesis is increased in sarcoma, acute myeloid leukemia, and melanoma, which is correlated with decreased patient survival (78). Furthermore, epidemiologic studies suggest a positive association between elevated serum cholesterol and high risk for developing certain cancer types, especially prostate cancer (79–81).
Effect of tumor-derived metabolites on T cells in the TME
The demand for nutrient resources and nutrient competition between cells can influence tumor growth, as well as immune cell survival and function (9). Furthermore, increasing evidence suggests that the abnormal accumulation of metabolites in the TME can directly control immune cell function and anti-tumor activity, especially in TILs (Figure 2).
Metabolites control T cell proliferation and function in the TME
Tumor-derived lactic acid can significantly suppress the proliferation and cytokine production of human CTLs and lead to a significant decrease in their cytotoxic activity (42). Lactic acid produced by tumor cells inhibits IFN-γ production by cancer-specific CTLs in melanoma spheroid cocultures (82). Tumor lactic acidosis has impaired TCR-triggered phosphorylation of JNK, C-Jun and p38, leading to the inhibition of CTLs with complete blockage of IFN-γ production and partial impairment of lytic granule exocytosis (83). In addition, acidification in the melanoma TME has led to reduced expression of granzyme B and decreased STAT5 and ERK activation in human and mouse CD8+ T cells (84). Succinate, an important intermediate in the TCA cycle, has been reported to be elevated in patients with lung cancer (85). Of note, cancer-derived succinate can promote macrophage polarization by activating succinate receptor SUCNR1, contributing to cancer metastasis (85). Succinate can also be converted into fumarate through succinate dehydrogenase (SDH) to regulate the activation, proliferation, and function of human CD4+ and CD8+ T cells (86). Lastly, SDH can inhibit Th1 proliferation and histone acetylation with reduced IFN-γ production (13).
Adapting to hypoglycemia and hypoxia conditions in the TME of a mouse melanoma model, CD8+ TILs have been shown to display increased transcript expression of PPAR-α and its downstream molecules involved in FA uptake and transport, TG synthesis and catabolism, as well as peroxisomal and mitochondrial FA catabolism, resulting in high FA uptake and enhanced oxidation of endogenous and exogenous FAs (87). Promoting FA catabolism has also improved CD8+ TILs anti-tumor functions in a mouse melanoma model (87). Specific deletion of ACC1 to decrease long-chain FA (LCFA) synthesis in T cells resulted in impaired CD8+ T cell peripheral persistence and homeostatic proliferation in naïve mice, and decreased antigen-specific CD8+ T cell accumulation and survival in OVA-specific TCR transgenic mice, relative to controls (88). Furthermore, providing exogenous FAs rescued ACC1-deficient CD8+ T cell growth and survival in those models (88). In the pancreatic TME, CD8+ TILs increased the uptake and accumulation of LCFAs in high-grade lesions, which impaired mitochondrial function and triggered major transcriptional reprogramming of pathways to subsequently reduce FA catabolism (62). Mechanistic studies further showed that intrapancreatic CD8+ T cells specifically down-regulated the very-long-chain acyl-CoA dehydrogenase (VLCAD), which decreased the metabolization of excess specific LCFA lipids in CD8+ T cells as well as the cytotoxic effect of the latter (62). Furthermore, enriched FAs in the TME have induced human and mouse CD8+ T cell ferroptosis with inhibited proliferation and production of cytotoxic effector molecules IFN-γ and TNF-α through increased FA scavenger receptor CD36 (64). In addition, intra-tumoral Treg cells from patients with breast cancer and melanoma, or melanoma mouse models, have also expressed high amounts of CD36, which has been associated with driving Treg metabolic reprogramming and mitochondrial fitness, as well as T reg accumulation in the TME (89).
Recent studies showed that tumor-specific Tc9 cells were superior effector cells compared with Tc1 for anti-tumor immunity (90). Moreover, cholesterol modulation could affect the anti-tumor activity of tumor-specific Tc9 cells (41). Indeed, reduced cholesterol increased the numbers of IFN-γ and granzyme B-producing Tc9 cells while addition of cholesterol decreased their numbers in the MC38-gp100 tumor-bearing mice (41). In addition, the cholesterol metabolite 27 hydroxycholesterol was shown to attract polymorphonuclear neutrophils and γδ T cells to the TME, but deplete CTLs in breast cancer tumor-bearing mice, facilitating breast cancer metastasis (91).
In addition to glucose and lipid metabolism, amino acids and glutamine are important energy sources for both tumor cells and immune cells, which also produce many metabolites regulating T cell differentiation and function in the TME (92). Recent studies have shown that methionine and S-adenosylmethionine (SAM) play important roles in regulating development and function of CD4+ and CD8+ T cells (93). Tumor cells compete with CD8+ T cells for methionine usage through high expression of methionine transporter SLC43A2, resulting in impaired anti-tumor function of CD8+ TILs in colon cancer patients and tumor-bearing mice as well (93). Mechanistically, decreased SAM provided less methyl for H3K79 methylation in T cells, resulting in decreased STAT5 expression and impaired function of CD8+ T cells (93). Furthermore, methionine and SAM could drive CD8+ T cell exhaustion, resulting in CD8+ T cell dysfunction and promotion of tumor growth in hepatocellular carcinoma mouse models (94). Alpha-ketoglutarate (aKG), a glutamine metabolite, can also regulate CD4+ T cell subset differentiation (95, 96). Decreased amounts of aKG causing limited extracellular glutamine have been shown to induce naïve CD4+ T cell differentiation into Treg cells (95). By contrast, treatment with cell-permeable aKG ex vivo inhibited Treg differentiation but promoted Th1 cell differentiation (96). Adoptive transfer of T cells treated with aKG maintained an inflammatory profile and delayed tumor growth in fibrosarcoma mouse models (96).
Metabolites can not only directly alter effector T cell function in the TME, but can also regulate signaling molecules or other types of immune cells, which in turn further alter effector T cells. Acidification of the TME mediated by tumor-derived lactic acid causes increased arginase I expression in macrophages and reduces the concentration of arginine in the TME, which inhibits T cell activation, proliferation, and effector functions in tumor-bearing mice (97). Tumor-infiltrating Treg cells in mouse melanoma models display intracellular lipid accumulation and an increased rate of FA synthesis. Meanwhile, tumor-infiltrating Treg cells preferentially capture and utilize glucose to fuel glycolysis (98). Both glycolytic and oxidative metabolism contribute to expansion of Treg cells in tumors, which can suppress effector T cell-mediated anti-tumor immunity (Figure 2) (98).
Metabolites control T cell fates in the TME
The abnormal accumulation of metabolites in the TME can also control the fates of T cells, such as induction of T cell exhaustion and anergy (84, 99). Recent studies have shown that tumor-derived lactate inhibits the expression of adhesion kinase family-interacting protein of 200KD (FIP200) in T cells by reducing cytosol NAD+, and increasing the inhibitory effects of adenylate-uridylate-rich elements within the 3’ untranslated region of Fip200mRNA, resulting in T cell autophagy deficiency, apoptosis, and poor anti-tumor immunity in ovarian cancer patients and tumor-bearing mice (99). Furthermore, tumor released lactic acid leads to high concentrations of acid and a low pH in the extracellular medium, and has been reported to lead to CD8+ TIL anergy in human cancer patients and in mouse melanoma models (84). In addition, long-term exposure to high cholesterol in the TME has resulted in high cholesterol content and an exhausted phenotype in CD8+ TILs in mouse melanoma models (51). Indeed, accumulated cholesterol disrupts lipid metabolism and increases endoplasmic reticulum (ER) stress in CD8+ TILs (51). Tc9 cells with decreased cholesterol displayed reduced apoptosis and better antitumor activity than cholesterol-sufficient Tc9 cells, while cholesterol-treated Tc9 cells exhibited poor survival and impaired antitumor activity in vivo in MC38-gp100 tumor models (41). Recent studies have demonstrated that FAs in the TME can induce human and mouse CD8+ T cell ferroptosis in a CD36-dependent manner (64). Reducing CD36 expression prevented CD8+T cell ferroptosis and enhanced CD8+ T cell cytotoxic cytokine production and anti-tumor function (64). These studies collectively underscore the importance of TME metabolites in directing the fates and functions of TILs in antitumor immunity.
Metabolite manipulation is a novel strategy for tumor immunotherapy
Given that tumor-derived metabolites can affect both tumor cell growth and TIL survival, proliferation, and function, development of novel strategies to reprogram metabolism and manipulate metabolites in the TME could provide new advances in cancer therapy. Indeed, many preclinical studies targeting metabolites for potential enhancement of anti-tumor efficacy have been explored and yielded impressive results (Table 1).
Table 1.
Metabolite-based anti-tumor immunotherapy
| Target metabolites | Treatments | Effects on T cells | Molecular mechanisms | References |
|---|---|---|---|---|
| Acidity or Lactate | Sodium bicarbonate | Promotes T cell persistence | Neutralizes TME acid | (101) |
| Esomeprazole (a proton pump inhibitor) | Increases tumor infiltration of CD8+ T cells, as well as fractions and absolute numbers of IFNγ+ CD44+CD8+ TILs; Enhance the ability of CD8+ T cells to recognize OVA257–264 peptide. | Increases TME pH | (84) | |
| Ketogenic diet or knockdown of LDH-A with shRNAs | Increases frequencies of CD4+ and CD8+ T cells; while reducing Treg and MDSC frequencies | Decreases lactate production by glycolytic tumors | (102) | |
| Conditional deletion of LDH-A in K-Ras: LDH-Aflfl-CreERTME mice | Increases numbers of CD3+ T cells and decrease the ratio of Foxp3+ to CD3+ T cells; Induces IL17- and IFNγ-producing CD8+ T (Tc17 and Tc1) cells. | Amplifies skewing of macrophages towards an M1-like phenotype; Increases T cell numbers in the TME by blocking PD-L1 expression. | (103) | |
| Citrate | Citrate | Promotes T cell infiltration in tumors; Enhances pro-inflammatory cytokine secretion. | Acts on the IGF1R-AKT-PTEN-eIF2α pathway and suppresses both glycolysis and TCA. | (104) |
| Phosphoenolpyruvate (PEP) | phosphoenolpyruvate carboxykinase 1 (PCK1)-overexpression | Sustains TCR-mediated Ca2+-NFAT signaling and effector functions by repressing sarco/ER Ca2+ATPase (SERCA) activity; Reprograms the metabolism in tumor-specific CD4+ and CD8+ T cells. | Increases PEP production through overexpression of PCK1; Regulates Ca2+-NFAT signaling in Th1 cells by inhibiting SERCA activity. | (108) |
| Fenofibrate (a PPAR-α agonist) | Enhances polyfunctionality of CD8+ T cells. | Increases FA uptake, catabolism, and PD1 expression in CD8+ TILs; | (87) | |
| Bezafibrate (an agonist of PGC-1α/PPAR complex) | Increases or maintains the number of functional CTLs by activating mitochondrial and cellular metabolism. | Promotes differentiation of naive T to effector T cells, upregulates FAO, and inhibits apoptosis of effector T cells; Increase numbers of functional effector T cells and improve effectiveness of PD-1 blockade. | (110) | |
| Fatty acid | Etomoxir | Reverses T cell proliferation and IFN-γ production; Diminishes the accumulation of CD4+FoxP3+ Tregs; Increases numbers of adoptively transferred OT-1 T cells (CD45.1+) infiltrating into tumors and numbers of IFN-γ-producing cells. | Inhibits FAO in tumor-infiltrating myeloid-derived suppressor cells (MDSC); Blocks immune inhibitory pathways and functions in tumor MDSC and decreases their production of inhibitory cytokines. | (111) |
| Genetic ablation of CD36 in CD8+ T cells | Increases cytotoxic cytokine production and enhances anti-tumor activity. | Decreases CD36-mediated fatty acid uptake by T cells. | (64) | |
| Cholesterol | Avasimibe | Enhances the production of cytokines and cytolytic granules, and increases killing and proliferation activity of CD8+ T cells. | Inhibits activity of ACAT1 and upregulates plasma membrane cholesterol in CD8+ T cells; Enhances TCR clustering and signaling and promotes formation of the immunological synapse. | (50) |
| Simvastatin or HMGCR shRNA | Reduces cholesterol content in CD8+ T cells; Decreases PD-1 and 2B4 expression and enhances CD8+ T cell antitumor activity. | Reduces cholesterol contents in T cells and/or the TME; Decrease tumor-infiltrating CD8+ T cell exhaustion. | (51) | |
| β-cyclodextrin (β-CD); simvastatin or lovastatin; knockdown of HMGCR and Abca1 with shRNAs | Decreases cholesterol content in Tc9 cells; Up-regulates IL-9 expression and production in Tc9 cells. | Cholesterol or its derivatives inhibit IL-9 expression through liver X receptor (LXR) Sumoylation-NF-κB signaling pathways; Reduces cholesterol in Tc9 cells; enhances Tc9 cell differentiation, persistence, and antitumor activity in an IL-9-dependent manner. | (41) | |
| Phospholipids | Methyl arachidonyl fluorophosphon ate (MAFP) and arachidonyl trifluoromethyl ketone (ATK) | Decreases the lipid droplet (LD) formation in CD4+ and CD8+ T cells and reverses T cell senescence and impaired functions induced by Treg cells. | Regulates lipid metabolism in CD4+ and CD8+ T cells and reverses the lipid accumulation in CD4+ and CD8+ T cells. | (72) |
Solid tumors are more acidic in comparison to normal tissues as a consequence of high glycolysis and poor cell perfusion, as well as accumulated acidic metabolites including lactate (100). Decreasing tumor acidity might help rescue effector T cell function for anti-tumor immunity (101). Using melanoma tumor models, studies have demonstrated that treatment of melanoma tumor-bearing mice with bicarbonate in drinking water can increase the acidic pH of tumors and neutralize the tumor acidity, resulting in inhibition of Yumm 1.1 tumor growth and enhanced therapeutic efficacy mediated by anti-PD-1 or anti-CTLA-4antibodies (inmmune checkpoint blockade (ICB)) in vivo (101). In addition, the combination of bicarbonate therapy and adoptive cell therapy with melanoma-specific Pmel TCR transgenic T cells can improve the long-term survival rates of B16 melanoma-bearing mice (101). Pan02 pancreatic cancer cell-derived lactate can inhibit NK cell cytolytic function and increase myeloid derived suppressor cell (MDSC) numbers in the TME (102). Ketogenic diets in Pan02-bearing mice lowered glucose concentrations and lactate production from glycolytic Pan02 tumors, resulting in decreased MDSC frequency, improved antitumor immune responses, and reduced tumor burden (102). Targeting key enzymes associated with metabolite production may also be an important alternative strategy to reprogram metabolism for cancer treatment. Recent studies have shown that high LDH-A expression and lactate production by macrophages in the TME are major inducers of T cell immunosuppression in lung cancer (103). Furthermore, myeloid-specific deletion of LDH-A in mice bearing K-Ras-driven tumors led to skewing of macrophages toward the so-called M1-like inflammatory phenotype, increasing T cell numbers and effector function by blocking lactate-driven PD-L1 expression on tumors (103). These studies suggest that indirectly targeting lactate by regulating LDH-A expression in macrophages might be an effective strategy to blunt tumoral immune escape (103), which certainly warrants further investigation.
Citrate could represent another possible therapeutic target (58, 59, 61). Citrate has inhibited tumor growth in diverse tumor models through suppression of glycolysis, the TCA cycle, and IGF-1R signaling, but promoted excessive lipid biosynthesis in tumor cells (104, 105). Oral administration of citrate has had antitumor effects in cancer patients with peritoneal mesothelioma and multiple endocrine neoplasia, possibly due to suppressed glycolysis (106, 107). Citrate treatment has also promoted T cell infiltration into tumors in lung cancer and Her2/Neu breast tumor-bearing mice (104). Another glycolytic metabolite, phosphoenolpyruvate (PEP), can serve as a metabolic checkpoint for controlling T cell-mediated anti-tumor responses (108, 109). Overexpression of phosphoenolpyruvate carboxykinase 1 (PCK1) metabolically reprogrammed tumor-specific CD4+ and CD8+ T cells to increase PEP production and enhance T cell anti-tumor responses, resulting in suppressed melanoma growth and prolonged survival of B16 melanoma-bearing mice (108). Exogenous addition of PEP to purified CD8+ TILs from B16 melanoma tumor-bearing mice during short-term in vitro cultures resulted in a significant increase in the frequency of CD8+ TILs producing IFN-γ and TNFα (109). Combination therapies of this with ICB antibodies (anti-PD1, anti-CTLA4, and anti-TIM3) also increased enolase activity in tumor-specific CD8+ TILs, resulting in enhanced CD8+ T cell anti-tumor activity and decreased tumor growth in the B16 mouse melanoma model (109).
Low O2 and glucose in the TME can promote FA catabolism to preserve CD8+ T cell functions (87). Treatment of fenofibrate, a PPAR-α agonist, increased FA catabolism in CD8+ T cells, which improved CD8+ T cell anti-tumor efficacy in a B16 melanoma BrafV600E tumor model (87). Bezafibrate, an agonist of PGC-1α/ PPAR complexes, has increased FA oxidation in CD8+ T cells, and the combination therapy with anti–PD-L1 antibody treatment has enhanced the survival and proliferation of tumor-reactive CTLs, leading in turn to enhanced antitumor immunity in MC38 colon cancer tumor-bearing mice (110). Etomoxir, a specific inhibitor of CPT1, has inhibited FA oxidation and impaired the functions of tumor-infiltrating MDSCs, with decreased FA uptake, ATP production, and immunosuppressive activity in 3LL lung cancer and MCA-38 colon cancer mouse models (111). Combinations of etomoxir and adoptive T cell therapy (ACT) with OT-1 T cells to treat OVA-expressing 3LL tumors, have also led to increased tumor infiltration by IFN-γ-producing cells (111). Many FA synthesis inhibitors have been recently developed for cancer treatment in clinical trials (68). CD36, the FA transporter, was recently reported as an important target for treating cancers (76). Silencing CD36 by specific shRNA in human prostate cancer PC3 cells reduced FA uptake and impaired cancer aggressiveness. Furthermore, utilizing patient-derived xenografts (PDXs) in mouse models of prostate cancer, blockade of CD36 with anti-CD36 monoclonal antibody (mAb) reduced tumor growth (76). Blockage of CD36 in CD8+ T cells has also enhanced anti-tumor efficacy mediated by CD8+ T cells treated with ICB in B16 melanoma models (64).
The TME can harbor high cholesterol amounts, which can induce TIL exhaustion (51). Reduced cholesterol content in tumor cells in the TME through shRNA targeting cholesterol synthesis enzyme 3-Hydroxy-3-Methylglutaryl-CoA reductase (HMGCR) or using cholesterol synthesis inhibitors, has significantly reduced PD-1 and 2B4 expression and lowered cholesterol content in CD8+ TILs in B16 melanoma tumor-bearing mice (51). Meanwhile, reducing cholesterol in T cells has enhanced CD8+ T cell-mediated antitumor activity, while cholesterol-treated CD8+ T cells exhibited impaired anti-tumor activity due to decreased IFN-γ and granzyme B production and cell proliferation, but increased cell apoptosis, relative to controls (51). In addition, inhibiting cholesterol esterification in CD8+ T cells through suppression of the cholesterol esterification enzyme ACAT1 with the specific inhibitor avasimibe, has led to enhanced cell proliferation and anti-tumor effects of CD8+ T cells in the B16F10 melanoma model (50). Mechanistically, ACAT1 inhibitor avasimibe treatment increased plasma membrane cholesterol amounts in CD8+ T cells, and augmented TCR clustering and signaling, as well as promoted immunological synapse formation, relative to controls (50). Treatments with β-CD to reduce cellular cholesterol content or with the HMGCR inhibitor simvastatin to inhibit endogenous cholesterol biosynthesis in Tc9 cells has maintained high IL-9 production and long-term survival of Tc9 cells to better perform anti-tumor activity in MC38 tumor-bearing mice (41).
Recent studies have demonstrated that increased senescent T cells exist in various types of cancer patients (112). Both tumor cells and tumor-associated Treg cells can induce senescence in effector T cells, which is another important dysfunctional state for T cells in the TME (112–116). In addition, increased amounts of cholesterol and FAs, as well as lipid droplets (LDs), have been identified in senescent T cells and in TILs from both melanoma and breast cancer patients, as well as from B16 and E0771 tumor-bearing mice (72). Mechanistic studies have further showed that elevated group IVA phospholipase A2 (cPLA2α) is responsible for the altered lipid metabolism and senescence induction in T cells mediated by Treg and tumor cells; reprogramming lipid metabolism with cPLA2α specific inhibitors, methyl arachidonyl fluorophosphonate (MAFP), and arachidonyl trifluoromethyl ketone (ATK), can prevent T cell senescence, resulting in enhanced anti-tumor immunity and immunotherapy efficacy in vivo in adoptive transfer T cell therapy of melanoma and breast cancer mouse models (72). These studies further support the concept that reprogramming of lipid metabolism is a novel and promising strategy for reconstituting effector T cell functions for tumor immunotherapy.
Concluding Remarks
It is well-recognized that metabolic dysfunction is a key hallmark of cancer. Metabolic reprogramming of cancer cells results in the accumulation of metabolites, which affect both cancer cells and tumor-infiltrating immune components. The tumor microenvironment metabolites form both glucose and lipid metabolism play a crucial role in controlling the differentiation, function and fate of TILs, thereby directly modifying clinical outcomes in cancer patients (10). Therefore, reprogramming the tumor cell metabolism in the TME, especially targeting key metabolites such as citrate, lactate, FA and cholesterol, has become a novel and promising strategy for tumor treatment (72).
Reprogramming metabolism in the TME and rebalance of nutrient utilization between cancer cells and T cells is indeed a key determinant for effective tumor immunity and immunotherapy (117). Although great progress has been made in this specific area of cancer research, there are still profound challenges to overcome in targeting cancer metabolism (see outstanding questions). First, accumulation of different types of metabolic products in the TME has potent effects suppressing effector T cells. However, the cell origins producing the individual metabolites and reasons for differential pathological effects with different cancer types remain unclear. Second, how these metabolites molecularly and uniquely regulate the function and fate of different types of T cells is still unknown and needs to be urgently investigated. Cancer cells and effector T cells have similar metabolic requirements and compete for nutrient resources within the TME (9). Therefore, another challenging issue is that targeting tumor-derived metabolites for cancer therapy may also inhibit immune cells including effector T cells in the TME. In addition, tumor cells have metabolic flexibility and heterogeneity, which may limit the efficacy of targeting one metabolic pathway for tumor therapy (118, 119). Therefore, more efforts are needed to conduct future studies to comprehensively understand the molecular and metabolic processes of tumor cells and tumor-infiltrating immune components during their interactions within the TME. A better understanding of these issues will be essential for development of precise strategies to selectively target different metabolites for tumor treatment in individual tumor types. In addition, new approaches that combine existing immunotherapies with strategies reprogramming metabolism in the TME to enhance infiltration, effector responses and longevity of tumor-specific T cells, are greatly needed.
Outstanding Questions.
Can we precisely characterize the metabolic profiles of tumor cells and tumor-infiltrating T cells in any individual cancer patient?
Can we identify the unique metabolites that induce T cell dysfunction in individual cancer types? Can we identify the specific and key enzymes that control these important metabolites in the tumor microenvironment?
Can we utilize dietary intervention to metabolically control T cell functions for enhanced anti-tumor immunity?
Can we develop novel strategies combining current immunotherapies with specific approaches to reprogram metabolism in T cells to enhance T cell effector functions for putative tumor treatments?
Highlights.
Metabolic dysfunction is a key hallmark of cancer, which results in the accumulation of metabolites in the tumor microenvironment (TME), affecting both cancer cells and immune components.
Abnormal production of metabolites in the TME is associated with negative clinical outcomes and disease progression of cancer patients.
TME metabolites derived from glucose and lipid metabolism can direct the differentiation, function, and fate of tumor-infiltrating T cells, thereby affecting anti-tumor immunity and immunotherapy.
Reprogramming of tumor metabolism in the TME, especially targeting key metabolites has emerged as a novel and promising strategy for the treatment of certain tumors.
Acknowledgments
Because of space limitations, the authors apologize that they cannot cite all relevant references in this research area. This work was partially supported by grants from the Melanoma Research Alliance (to G. P) and the NIH (R01CA184379, R01CA242188, R01CA237149, and R21AG067441 to G. P).
Glossary
- Lipid droplets
organelles composed of a neutral lipid core consisting mainly of triacylglycerols and cholesteryl esters surrounded by a phospholipid monolayer.
- Lipid rafts
lipid domains composed of sphingolipids and cholesterol in the outer exoplasmic leaflet; for modulating the membrane distribution of receptor and signaling molecules facilitating the assembly of active signaling platforms.
- Th1 cell
T helper type 1 cells-- one of the subsets of CD4+ T cells and involved in cell-mediated immune responses.
- Th2 cell
subset of CD4+ T cells, secreting IL-4, IL-5, IL-10, and IL-13; involved in humoral or antibody-mediated immune responses.
- Th17 cell
subset of CD4+ T cells characterized by IL-17 production; differentiation is regulated by transcription factors RORγt and RORα. They play important roles in autoimmune disorders and inflammation.
- Treg cell
Regulatory T cells, one subpopulation of CD4+ T cells with a suppression function to regulate and maintain immune homeostasis.
- FoxP3
member of the forkhead transcription factor family that mainly controls Treg differentiation.
- Tc9 cell
subset of CD8+ cytotoxic T cells which produce IL-9.
- Type 1 CD8+ T cell (Tc1)
subpopulations of cytotoxic CD8+ T cells, which can produce IFN-γ to perform effector immune responses.
- H3K9 and H3K27 acetylation
histone H3 acetylation at the lysine 9 residue (H3K9Ac) and lysine 27 residue (H3K27Ac), are histone markers associated with active transcription.
- Rorc
retinoic acid-receptor related orphan receptor C, encodes protein RORγt, an important nuclear receptor regulating Th17 cell development.
- Conventional T cell
expresses an αβ T cell receptor; present in peripheral blood, lymph nodes, and tissues; contains two major subsets, CD4+ Th cells and CD8+ cytotoxic cells basing on the respective expression of co-receptor CD4 or CD8.
- Peroxisome proliferator-activated receptors (PPARs)
panel of nuclear receptor proteins that function as transcription factors regulating gene expression for cell differentiation, development, metabolism.
- CD8+ memory T (Tm) cell
long-lived subset of CD8+ T cells, which are antigen specific and can elicit strong immune responses when encountering the same antigen.
- Tissue-resident memory (TRM) CD8+ T cells
memory lymphocyte population that resides in nonlymphoid tissues, such as the mucosal tissues and skin.
- Central memory CD8+ T (CD8+ Tcm) cell
subset of memory CD8+ T cells with high expression of CD62L and CCR7; have a centralized location within secondary lymphoid organs and superior proliferative abilities for handling systemic infections.
- Effector memory CD8+ T (CD8+ Tcm) cell
subset of memory CD8+ T cells with low expression of CD62L and CCR7; have cytotoxic ability and localize to inflamed tissues to provide protection against infection.
- Cell-intrinsic lipolysis
process mediated by lysosomal acid lipase, hydrolyzing TAG, DAG, and CE to generate free FA and cholesterol in lysosomes.
- OT-1 TCR transgenic mice
contain transgenic inserts for mouse Tcra-V2 and Tcrb-V5 genes; T cells from these mice recognize ovalbumin residues 257–264 in the context of H2Kb for studying the role of peptides in positive selection and the response of CD8+ T cells to antigens.
- Immunological synapse
special cell-cell communication between an antigen-presenting cell or target cell and e.g. a lymphocyte, with subsequent cell activation.
- ApoE
apolipoprotein E protein is a major cholesterol carrier for lipid transport in lipid metabolism, neurobiology, and neurodegenerative diseases. Deficient mice are used e.g. in atherosclerosis models.
- Lytic granule exocytosis
process in which special organelles containing a series of proteins are secreted from cells and mediate target cell destruction after activation of CTLs and NK cells.
- Ferroptosis
type of programmed cell death; accompanied by a great amount of iron and lipid peroxidation during cell death.
- ER stress
protective stress response of cells; occurs when the capacity of the ER to fold proteins becomes saturated.
- Melanoma-specific Pmel TCR transgenic T cells
T cells expressing transgenic TCR specific for the epitope of pmel-17 corresponding to B16 melanoma antigen gp100.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interests.
References
- 1.DeBerardinis RJ, Chandel NS. 2016. Fundamentals of cancer metabolism. Sci Adv 2: e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlova NN, Thompson CB. 2016. The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23: 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–74 [DOI] [PubMed] [Google Scholar]
- 4.Romero-Garcia S, Lopez-Gonzalez JS, Baez-Viveros JL, Aguilar-Cazares D, Prado-Garcia H. 2011. Tumor cell metabolism: an integral view. Cancer Biol Ther 12: 939–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG. 2011. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10: 671–84 [DOI] [PubMed] [Google Scholar]
- 6.Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, Wang X, Lorkiewicz PK, Schatzman S, Bousamra M 2nd, Lane AN, Higashi RM, Fan TW, Pandolfi PP, Sukhatme VP, Seth P. 2014. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 19: 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, Scalbert A. 2013. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res 41: D801–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai C, Arceo J, Arnold J, Sreekumar A, Dovichi NJ, Li J, Littlepage LE. 2018. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab 6: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL. 2015. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162: 1229–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. 2016. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front Immunol 7: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almeida L, Lochner M, Berod L, Sparwasser T. 2016. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol 28: 514–24 [DOI] [PubMed] [Google Scholar]
- 12.Ganeshan K, Chawla A. 2014. Metabolic regulation of immune responses. Annu Rev Immunol 32: 609–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, Kluger Y, Maher LJ 3rd, Rabinowitz J, Craft J, Flavell RA. 2019. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571: 403–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. 2016. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354: 481–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, Akimova T, Liu, Bhatti TR, Han R, Laskin BL, Baur JA, Blair IA, Wallace DC, Hancock WW, Beier UH. 2017. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25: 1282–93 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zurier RB, Rossetti RG, Seiler CM, Laposata M. 1999. Human peripheral blood T lymphocyte proliferation after activation of the T cell receptor: effects of unsaturated fatty acids. Prostaglandins Leukot Essent Fatty Acids 60: 371–5 [DOI] [PubMed] [Google Scholar]
- 17.Lima TM, Kanunfre CC, Pompeia C, Verlengia R, Curi R. 2002. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol In Vitro 16: 741–7 [DOI] [PubMed] [Google Scholar]
- 18.Gorjao R, Cury-Boaventura MF, de Lima TM, Curi R. 2007. Regulation of human lymphocyte proliferation by fatty acids. Cell Biochem Funct 25: 305–15 [DOI] [PubMed] [Google Scholar]
- 19.Takahashi HK, Cambiaghi TD, Luchessi AD, Hirabara SM, Vinolo MA, Newsholme P, Curi R. 2012. Activation of survival and apoptotic signaling pathways in lymphocytes exposed to palmitic acid. J Cell Physiol 227: 339–50 [DOI] [PubMed] [Google Scholar]
- 20.Cury-Boaventura MF, Gorjao R, de Lima TM, Newsholme P, Curi R. 2006. Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci 78: 1448–56 [DOI] [PubMed] [Google Scholar]
- 21.Ioan-Facsinay A, Kwekkeboom JC, Westhoff S, Giera M, Rombouts Y, van Harmelen V, Huizinga TW, Deelder A, Kloppenburg M, Toes RE. 2013. Adipocyte-derived lipids modulate CD4+ T-cell function. Eur J Immunol 43: 1578–87 [DOI] [PubMed] [Google Scholar]
- 22.Mito N, Kitada C, Hosoda T, Sato K. 2002. Effect of diet-induced obesity on ovalbumin-specific immune response in a murine asthma model. Metabolism 51: 1241–6 [DOI] [PubMed] [Google Scholar]
- 23.Yaqoob P, Newsholme EA, Calder PC. 1994. The effect of dietary lipid manipulation on rat lymphocyte subsets and proliferation. Immunology 82: 603–10 [PMC free article] [PubMed] [Google Scholar]
- 24.Mailer RKW, Gistera A, Polyzos KA, Ketelhuth DFJ, Hansson GK. 2017. Hypercholesterolemia Enhances T Cell Receptor Signaling and Increases the Regulatory T Cell Population. Sci Rep 7: 15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg FM, Mauro C. 2015. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol 13: e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ou Y, Li S, Zhu X, Gui B, Yao G, Ma L, Zhu D, Fu R, Ge H, Wang L, Jia L, Tian L, Duan Z. 2016. Citrate Attenuates Adenine-Induced Chronic Renal Failure in Rats by Modulating the Th17/Treg Cell Balance. Inflammation 39: 79–86 [DOI] [PubMed] [Google Scholar]
- 27.Mailer RKW, Gistera A, Polyzos KA, Ketelhuth DFJ, Hansson GK. 2017. Hypercholesterolemia Induces Differentiation of Regulatory T Cells in the Liver. Circ Res 120: 1740–53 [DOI] [PubMed] [Google Scholar]
- 28.Maganto-Garcia E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. 2011. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 124: 185–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. 2015. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125: 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. 2011. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186: 3299–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, Sparwasser T. 2014. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20: 1327–33 [DOI] [PubMed] [Google Scholar]
- 32.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. 2013. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341: 569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H. 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446–50 [DOI] [PubMed] [Google Scholar]
- 34.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonoda J, Pei L, Evans RM. 2008. Nuclear receptors: decoding metabolic disease. FEBS Lett 582: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi JM, Bothwell ALM. 2012. The Nuclear Receptor PPARs as Important Regulators of T-Cell Functions and Autoimmune Diseases. Molecules and Cells 33: 217–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Echeverria F, Ortiz M, Valenzuela R, Videla LA. 2016. Long-chain polyunsaturated fatty acids regulation of PPARs, signaling: Relationship to tissue development and aging. Prostaglandins Leukot Essent Fatty Acids 114: 28–34 [DOI] [PubMed] [Google Scholar]
- 38.Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. 2016. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun 7: 13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Sullivan D, van der Windt GJW, Huang SCC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, Hsu FF, Birnbaum MJ, Pearce EJ, Pearce EL. 2014. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 41: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan YD, Tian T, Park CO, Lofftus SY, Mei SL, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS. 2017. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543: 252-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, Wang A, Yang M, Qian J, Dong C, Yi Q. 2018. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J Exp Med 215: 1555–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M. 2007. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109: 3812–9 [DOI] [PubMed] [Google Scholar]
- 43.Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, Krzyzaniak MA, King CG, Burgener AV, Fischer M, Develioglu L, Belle R, Recher M, Bonilla WV, Macpherson AJ, Hapfelmeier S, Jones RG, Hess C. 2016. Memory CD8(+) T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 44: 1312–24 [DOI] [PubMed] [Google Scholar]
- 44.Bantug GR, Fischer M, Grahlert J, Balmer ML, Unterstab G, Develioglu L, Steiner R, Zhang L, Costa ASH, Gubser PM, Burgener AV, Sauder U, Loliger J, Belle R, Dimeloe S, Lotscher J, Jauch A, Recher M, Honger G, Hall MN, Romero P, Frezza C, Hess C. 2018. Mitochondria-Endoplasmic Reticulum Contact Sites Function as Immunometabolic Hubs that Orchestrate the Rapid Recall Response of Memory CD8(+) T Cells. Immunity 48: 542–55 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. 2017. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front Immunol 8: 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stentz FB, Kitabchi AE. 2006. Palmitic acid-induced activation of human T-lymphocytes and aortic endothelial cells with production of insulin receptors, reactive oxygen species, cytokines, and lipid peroxidation. Biochem Biophys Res Commun 346: 721–6 [DOI] [PubMed] [Google Scholar]
- 47.Fernanda Cury-Boaventura M, Cristine Kanunfre C, Gorjao R, Martins de Lima T, Curi R. 2006. Mechanisms involved in Jurkat cell death induced by oleic and linoleic acids. Clin Nutr 25: 1004–14 [DOI] [PubMed] [Google Scholar]
- 48.Szamel M, Rehermann B, Krebs B, Kurrle R, Resch K. 1989. Activation signals in human lymphocytes. Incorporation of polyunsaturated fatty acids into plasma membrane phospholipids regulates IL-2 synthesis via sustained activation of protein kinase C. J Immunol 143: 2806–13 [PubMed] [Google Scholar]
- 49.Moussa M, Le Boucher J, Garcia J, Tkaczuk J, Ragab J, Dutot G, Ohayon E, Ghisolfi J, Thouvenot JP. 2000. In vivo effects of olive oil-based lipid emulsion on lymphocyte activation in rats. Clin Nutr 19: 49–54 [DOI] [PubMed] [Google Scholar]
- 50.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CC, Chang TY, Zhang T, Zhou P, Song BL, Liu W, Sun SC, Liu X, Li BL, Xu C. 2016. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531: 651–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma XZ, Bi EG, Lu Y, Su P, Huang CJ, Liu LT, Wang Q, Yang MJ, Kalady MF, Qian JF, Zhang AJ, Gupte AA, Hamilton DJ, Zheng CY, Yi Q. 2019. ( )Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metabolism 30: 143-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chyu KY, Lio WM, Dimayuga PC, Zhou J, Zhao X, Yano J, Trinidad P, Honjo T, Cercek B, Shah PK. 2014. Cholesterol lowering modulates T cell function in vivo and in vitro. PLoS One 9: e92095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warburg O 1961. [On the facultative anaerobiosis of cancer cells and its use in chemotherapy]. Munch Med Wochenschr 103: 2504–6 [PubMed] [Google Scholar]
- 54.Liberti MV, Locasale JW. 2016. The Warburg Effect: How Does it Benefit Cancer Cells? (vol 41, pg 211, 2016). Trends in Biochemical Sciences 41: 287- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Shi Z, Xu X, Yu Z, Mi J. 2019. The influence of microenvironment on tumor immunotherapy. FEBS J 286: 4160–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. 2019. Hypoxia-Modified Cancer Cell Metabolism. Front Cell Dev Biol 7: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walenta S, Mueller-Klieser WF. 2004. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol 14: 267–74 [DOI] [PubMed] [Google Scholar]
- 58.Icard P, Lincet H. 2016. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target. Drug Resist Updat 29: 47–53 [DOI] [PubMed] [Google Scholar]
- 59.Huang L, Wang C, Xu H, Peng G. 2020. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim Biophys Acta Rev Cancer 1873: 188332. [DOI] [PubMed] [Google Scholar]
- 60.Mycielska ME, Milenkovic VM, Wetzel CH, Rummele P, Geissler EK. 2015. Extracellular Citrate in Health and Disease. Curr Mol Med 15: 884–91 [DOI] [PubMed] [Google Scholar]
- 61.Mycielska ME, Dettmer K, Rummele P, Schmidt K, Prehn C, Milenkovic VM, Jagla W, Madej GM, Lantow M, Schladt M, Cecil A, Koehl GE, Eggenhofer E, Wachsmuth CJ, Ganapathy V, Schlitt HJ, Kunzelmann K, Ziegler C, Wetzel CH, Gaumann A, Lang SA, Adamski J, Oefner PJ, Geissler EK. 2018. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res 78: 2513–23 [DOI] [PubMed] [Google Scholar]
- 62.Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, Nava Lauson CB, Tiberti S, Raimondi A, Jones MA, Reyzer M, Bates BM, Spraggins JM, Patterson NH, McLean JA, Rai K, Tacchetti C, Tucci S, Wargo JA, Rodighiero S, Clise-Dwyer K, Sherrod SD, Kim M, Navin NE, Caprioli RM, Greenberg PD, Draetta G, Nezi L. 2020. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med 217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, Farsakoglu Y, Varanasi SK, Low JS, Tang W, Wang H, McDonald B, Tripple V, Downes M, Evans RM, Abumrad NA, Merghoub T, Wolchok JD, Shokhirev MN, Ho PC, Witztum JL, Emu B, Cui G, Kaech SM. 2021. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54: 1561–77 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, Yi Q. 2021. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr., 2013. Cellular fatty acid metabolism and cancer. Cell Metab 18: 153–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hilvo M, Denkert C, Lehtinen L, Muller B, Brockmoller S, Seppanen-Laakso T, Budczies J, Bucher E, Yetukuri L, Castillo S, Berg E, Nygren H, Sysi-Aho M, Griffin JL, Fiehn O, Loibl S, Richter-Ehrenstein C, Radke C, Hyotylainen T, Kallioniemi O, Iljin K, Oresic M. 2011. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res 71: 3236–45 [DOI] [PubMed] [Google Scholar]
- 67.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. 2010. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 70: 8117–26 [DOI] [PubMed] [Google Scholar]
- 68.Rohrig F, Schulze A. 2016. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16: 732–49 [DOI] [PubMed] [Google Scholar]
- 69.Levental I, Grzybek M, Simons K. 2010. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry 49: 6305–16 [DOI] [PubMed] [Google Scholar]
- 70.Huang B, Song B-l, Xu C. 2020. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nature Metabolism 2: 132–41 [DOI] [PubMed] [Google Scholar]
- 71.Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, Pezzi V, Sirianni R. 2018. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front Endocrinol (Lausanne) 9: 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A, Huang L, Zhao Y, Lin W, Hsueh EC, Shen L, Shao Q, Hoft DF, Ford DA, Peng G. 2021. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med 13 [DOI] [PubMed] [Google Scholar]
- 73.Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. 2017. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14: 113. [DOI] [PubMed] [Google Scholar]
- 74.Parks SK, Mueller-Klieser W, Pouysségur J. 2020. Lactate and Acidity in the Cancer Microenvironment. 4: 141–58 [Google Scholar]
- 75.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, Schreml S, Haferkamp S, Kohl E, Karrer S, Berneburg M, Herr W, Mueller-Klieser W, Renner K, Kreutz M. 2016. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24: 657–71 [DOI] [PubMed] [Google Scholar]
- 76.Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, Porter LH, Niranjan B, Whitby ST, Lo J, Huang C, Schittenhelm RB, Anderson KE, Furic L, Wijayaratne PR, Matzaris M, Montgomery MK, Papargiris M, Norden S, Febbraio M, Risbridger GP, Frydenberg M, Nomura DK, Taylor RA. 2019. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Science Translational Medicine 11 [DOI] [PubMed] [Google Scholar]
- 77.Aiderus A, Black MA, Dunbier AK. 2018. Fatty acid oxidation is associated with proliferation and prognosis in breast and other cancers. Bmc Cancer 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuzu OF, Noory MA, Robertson GP. 2016. The Role of Cholesterol in Cancer. Cancer Res 76: 2063–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pelton K, Freeman MR, Solomon KR. 2012. Cholesterol and prostate cancer. Curr Opin Pharmacol 12: 751–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS. 2012. Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years’ follow up. BMC Cancer 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allott EH, Howard LE, Cooperberg MR, Kane CJ, Aronson WJ, Terris MK, Amling CL, Freedland SJ. 2014. Serum lipid profile and risk of prostate cancer recurrence: Results from the SEARCH database. Cancer Epidemiol Biomarkers Prev 23: 2349–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feder-Mengus C, Ghosh S, Weber WP, Wyler S, Zajac P, Terracciano L, Oertli D, Heberer M, Martin I, Spagnoli GC, Reschner A. 2007. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three-dimensional architectures by antigen-specific cytotoxic T lymphocytes. Br J Cancer 96: 1072–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. 2012. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer 131: 633–40 [DOI] [PubMed] [Google Scholar]
- 84.Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, Santinami M, Borghi M, Fais S, Bellone M, Rivoltini L. 2012. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 72: 2746–56 [DOI] [PubMed] [Google Scholar]
- 85.Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, Yeh CC, Peng YJ, Kuo YY, Wen HT, Lin HC, Hsiao CW, Wu KK, Kung HJ, Hsu YJ, Kuo CC. 2020. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell 77: 213–27 e5 [DOI] [PubMed] [Google Scholar]
- 86.Nastasi C, Willerlev-Olsen A, Dalhoff K, Ford SL, Gadsboll AO, Buus TB, Gluud M, Danielsen M, Litman T, Bonefeld CM, Geisler C, Odum N, Woetmann A. 2021. Inhibition of succinate dehydrogenase activity impairs human T cell activation and function. Sci Rep 11: 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu XW, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ. 2017. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 32: 377-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. 2014. Regulator of Fatty Acid Metabolism, Acetyl Coenzyme A Carboxylase 1, Controls T Cell Immunity. Journal of Immunology 192: 3190–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, Mohmood SR, Fernandez-Garcia J, Tsai CH, Schulze I, Picard F, Meylan E, Silverstein R, Goldberg I, Fendt SM, Wolchok JD, Merghoub T, Jandus C, Zippelius A, Ho PC. 2020. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol 21: 298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu Y, Hong B, Li H, Zheng Y, Zhang M, Wang S, Qian J, Yi Q. 2014. Tumor-specific IL-9-producing CD8+ Tc9 cells are superior effector than type-I cytotoxic Tc1 cells for adoptive immunotherapy of cancers. Proc Natl Acad Sci U S A 111: 2265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, Sullivan PM, Kemper JK, Gunn MD, McDonnell DP, Nelson ER. 2017. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8: 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, Yi P, Tang L, Pan Q, Rao S, Liang J, Tang Y, Su M, Luo X, Yang Y, Shi Y, Wang H, Zhou Y, Liao Q. 2021. The cancer metabolic reprogramming and immune response. Mol Cancer 20: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawlowska A, Xia H, Li J, Liao P, Yu J, Vatan L, Szeliga W, Wei S, Grove S, Liu JR, McLean K, Cieslik M, Chinnaiyan AM, Zgodzinski W, Wallner G, Wertel I, Okla K, Kryczek I, Lyssiotis CA, Zou W. 2020. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585: 277–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hung MH, Lee JS, Ma C, Diggs LP, Heinrich S, Chang CW, Ma L, Forgues M, Budhu A, Chaisaingmongkol J, Ruchirawat M, Ruppin E, Greten TF, Wang XW. 2021. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun 12: 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, Yong C, Surh N, Marie JC, Huehn J, Zimmermann V, Kinet S, Dardalhon V, Taylor N. 2015. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8: ra97. [DOI] [PubMed] [Google Scholar]
- 96.Matias MI, Yong CS, Foroushani A, Goldsmith C, Mongellaz C, Sezgin E, Levental KR, Talebi A, Perrault J, Riviere A, Dehairs J, Delos O, Bertand-Michel J, Portais JC, Wong M, Marie JC, Kelekar A, Kinet S, Zimmermann VS, Levental I, Yvan-Charvet L, Swinnen JV, Muljo SA, Hernandez-Vargas H, Tardito S, Taylor N, Dardalhon V. 2021. Article Regulatory T cell differentiation is controlled by aKG-induced alterations in mitochondrial metabolism and lipid homeostasis. Cell Reports 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. 2013. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer 133: 1107–18 [DOI] [PubMed] [Google Scholar]
- 98.Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, Severa M, Rizzo F, Coccia EM, Bonacina F, Mitro N, Norata GD, Rossetti G, Ranzani V, Pagani M, Giorda E, Wei Y, Matarese G, Barnaba V, Piconese S. 2018. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U S A 115: E6546–E55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, Bian Z, Maj T, He M, Liu RJ, He Y, Rattan R, Munkarah A, Guan JL, Zou W. 2017. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci Immunol 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. 2011. Imaging pH and metastasis. NMR Biomed 24: 582–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, Gillies RJ. 2016. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res 76: 1381–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Husain Z, Huang Y, Seth P, Sukhatme VP. 2013. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol 191: 1486–95 [DOI] [PubMed] [Google Scholar]
- 103.Seth P, Csizmadia E, Hedblom A, Vuerich M, Xie H, Li M, Longhi MS, Wegiel B. 2017. Deletion of Lactate Dehydrogenase-A in Myeloid Cells Triggers Antitumor Immunity. Cancer Res 77: 3632–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ren JG, Seth P, Ye H, Guo K, Hanai JI, Husain Z, Sukhatme VP. 2017. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci Rep 7: 4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao Y, Liu X, Si F, Huang L, Gao A, Lin W, Hoft DF, Shao Q, Peng G. 2021. Citrate Promotes Excessive Lipid Biosynthesis and Senescence in Tumor Cells for Tumor Therapy. Adv Sci (Weinh): e2101553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bucay AH. 2011. Clinical report: A patient with primary peritoneal mesothelioma that has improved after taking citric acid orally. Clinics and Research in Hepatology and Gastroenterology 35: 241- [DOI] [PubMed] [Google Scholar]
- 107.Halabe Bucay A 2009. Hypothesis proved…citric acid (citrate) does improve cancer: a case of a patient suffering from medullary thyroid cancer. Med Hypotheses 73: 271. [DOI] [PubMed] [Google Scholar]
- 108.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu XJ, Amezquita R, Tsui YC, Cui GL, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM. 2015. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162: 1217–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gemta LF, Siska PJ, Nelson ME, Gao X, Liu X, Locasale JW, Yagita H, Slingluff CL Jr., Hoehn KL, Rathmell JC, Bullock TNJ. 2019. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci Immunol 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chowdhury PS, Chamoto K, Kumar A, Honjo T. 2018. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res 6: 1375–87 [DOI] [PubMed] [Google Scholar]
- 111.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC. 2015. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res 3: 1236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu X, Hoft DF, Peng G. 2020. Senescent T cells within suppressive tumor microenvironments: emerging target for tumor immunotherapy. J Clin Invest 130: 1073–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, Gao A, Huang L, Hsueh EC, Ford DA, Hoft DF, Peng G. 2019. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab 29: 103–23 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC, Hoft DF, Peng G. 2018. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun 9: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, Han B, Huang Y, Zhang Y, Varvares MA, Hoft DF, Peng G. 2014. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med 6: 1294–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, Varvares MA, Hoft DF, Peng G. 2012. Human regulatory T cells induce T-lymphocyte senescence. Blood 120: 2021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reina-Campos M, Moscat J, Diaz-Meco M. 2017. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol 48: 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kishton RJ, Rathmell JC. 2015. Novel therapeutic targets of tumor metabolism. Cancer J 21: 62–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim J, DeBerardinis RJ. 2019. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab 30: 434–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lunt SY, Vander Heiden MG. 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–64 [DOI] [PubMed] [Google Scholar]
- 121.Connor H, Woods HF, Ledingham JG. 1983. Comparison of the kinetics and utilisation of D(−)-and L(+)-sodium lactate in normal man. Ann Nutr Metab 27: 481–7 [DOI] [PubMed] [Google Scholar]
- 122.Gray LR, Tompkins SC, Taylor EB. 2014. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 71: 2577–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haddad A, Mohiuddin SS. 2019. Biochemistry, Citric Acid Cycle. In StatPearls. Treasure Island (FL) [PubMed] [Google Scholar]
- 124.Masoro EJ. 1977. Lipids and lipid metabolism. Annu Rev Physiol 39: 301–21 [DOI] [PubMed] [Google Scholar]
- 125.de Jong AJ, Kloppenburg M, Toes RE, Ioan-Facsinay A. 2014. Fatty acids, lipid mediators, and T-cell function. Front Immunol 5: 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Menendez JA, Lupu R. 2007. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7: 763–77 [DOI] [PubMed] [Google Scholar]
- 127.Brown MS, Goldstein JL. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232: 34–47 [DOI] [PubMed] [Google Scholar]
- 128.Ikonen E 2008. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9: 125–38 [DOI] [PubMed] [Google Scholar]