

Abstract
Aims/hypothesis
Type 2 diabetes is a growing global public health challenge. Investigating quantitative traits, including fasting glucose, fasting insulin and HbA1c, that serve as early markers of type 2 diabetes progression may lead to a deeper understanding of the genetic aetiology of type 2 diabetes development. Previous genome-wide association studies (GWAS) have identified over 500 loci associated with type 2 diabetes, glycaemic traits and insulin-related traits. However, most of these findings were based only on populations of European ancestry. To address this research gap, we examined the genetic basis of fasting glucose, fasting insulin and HbA1c in participants of the diverse Population Architecture using Genomics and Epidemiology (PAGE) Study.
Methods
We conducted a GWAS of fasting glucose (n = 52,267), fasting insulin (n = 48,395) and HbA1c (n = 23,357) in participants without diabetes from the diverse PAGE Study (23% self-reported African American, 46% Hispanic/Latino, 40% European, 4% Asian, 3% Native Hawaiian, 0.8% Native American), performing transethnic and population-specific GWAS meta-analyses, followed by fine-mapping to identify and characterise novel loci and independent secondary signals in known loci.
Results
Four novel associations were identified (p < 5 × 10−9), including three loci associated with fasting insulin, and a novel, low-frequency African American-specific locus associated with fasting glucose. Additionally, seven secondary signals were identified, including novel independent secondary signals for fasting glucose at the known GCK locus and for fasting insulin at the known PPP1R3B locus in transethnic meta-analysis.
Conclusions/interpretation
Our findings provide new insights into the genetic architecture of glycaemic traits and highlight the continued importance of conducting genetic studies in diverse populations.
Keywords: Fine-mapping, Genome-wide association study, Glucose, Glycaemic traits, HbA1c, Insulin, Transethnic population
Introduction
Type 2 diabetes is a growing public health challenge, affecting approximately 14.6% of the US population [1] and expected to double in prevalence in the next two decades [2–4]. Investigating the genetic architecture of quantitative traits, including fasting glucose, fasting insulin and HbA1c, that serve as early markers of type 2 diabetes progression may lead to a deeper understanding of type 2 diabetes aetiology. For example, prior genome-wide association studies (GWAS) of glycaemic traits identified novel loci in genes and pathways related to glucose metabolism, circadian rhythm regulation, and cell proliferation and development [5, 6], as well as erythrocyte characteristics that can influence HbA1c [7].
Despite the success of prior glycaemic trait GWAS, which have identified nearly 600 loci [5, 6, 8–11], most of these findings were identified in populations primarily of European ancestry. Such limited ancestral diversity reduces our ability to map novel loci [12–18]. Additionally, locus characterisation and fine-mapping can be improved through multi-ethnic studies that increase sample size and leverage differences in linkage disequilibrium (LD) structure between diverse populations [19–22].
This study examined the genetic architecture of fasting glucose, fasting insulin and HbA1c in participants of the diverse Population Architecture using Genomics and Epidemiology (PAGE) Study [23]. We aimed to identify novel genetic loci and independent secondary association signals at previously identified regions and characterise these loci through transethnic fine-mapping.
Methods
Ethics statements
Approval by the Institutional Review Boards was obtained for each participating cohort. Informed consent was obtained from all participants, and the study was conducted in accordance with the principles of the Declaration of Helsinki.
Study population
This study included adults without diabetes who self-identified as African American (AA), Hispanic/Latino (HA), Asian (ASN), Native Hawaiian (HI), Native American (NAm), European (EA) or other race/ethnicity, enrolled in the Atherosclerosis Risk in Communities (ARIC) study, the Ichan Mount Sinai School of Medicine’s BioMe Biobank (BioMe), the Coronary Artery Risk Development in Young Adults Study (CARDIA), the Multiethnic Cohort (MEC) Study, the Hispanic Community Health Study/Study of Latinos (HCHS/SOL) and the Women’s Health Initiative (WHI) (see electronic supplementary material [ESM] Methods for details). These studies were part of the PAGE Study consortium, an NIH-funded effort to characterise the genetic architecture of complex traits among historically underrepresented populations through large-scale genetic epidemiology research [23].
In this paper, we stratified populations based on self-identified race/ethnicity due to historical reasons (e.g. genotyping datasets and study recruitment) and in recognition of the shared lived experiences of people based on self-identified grouping. To address confounding by population stratification, we included ancestral principal components in our models. We conducted two main analyses: transethnic analyses in the entire population; and analyses stratified by self-identified race/ethnicity. Participants who self-identified as ‘other race/ethnicity’ were included in all transethnic analyses but because of lack of power due to small sample sizes, no population-specific analyses for this group are presented.
Trait measurement
Fasting glucose and fasting insulin concentrations (fasting > 8 h) were measured using standard assays at baseline visits; for all cohorts except HCHS/SOL, HbA1c was measured at a subsequent visit. Glycaemic trait measurements among individuals with type 2 diabetes reflect their current glycaemic control, which is influenced by their access and adherence to medical treatment; therefore, individuals were excluded from analysis if they reported a previous diabetes diagnosis or fasting glucose concentrations consistent with diabetes (≥ 7.0 mmol/l). Because HbA1c was not measured at the same time point as fasting glucose and fasting insulin in most cohorts and was only added as a diagnostic criterion for diabetes in 2009 [24], after the majority of data were collected, individuals with HbA1c ≥ 48.0 mmol/mol (6.5%) were not excluded from the study population. However, for HbA1c analyses, individuals with extreme HbA1c values (HbA1c ≥ 65.0 mmol/mol [8.1%]) were excluded. Individuals with BMI >70 kg/m2 were also excluded for all traits.
Contributing samples were genotyped using multiple platforms (ESM Methods, ESM Table 1). A total of 53,426 samples were genotyped on the MEGA array, which was specifically designed to increase variant coverage across multiple ethnic groups [25, 26]. Additionally, 28,477 participants with fasting glucose measurements, 12,296 participants with HbA1c measurements and 26,965 participants with fasting insulin measurements from ARIC, BioMe, CARDIA, MEC and WHI were previously genotyped using either Illumina or Affymetrix arrays within each individual study/stratum. All studies used standard quality control filters (ESM Table 1). Ancestral principal component analysis was conducted to evaluate and adjust for population substructure, as previously described in Wojcik et al [26].
Statistical analyses
Fasting glucose concentrations, natural-log-transformed fasting insulin concentrations, and HbA1c measurements were each adjusted for age at trait measurement, sex, age × sex interaction, BMI (kg/m2), smoking status, self-reported race/ethnicity and study centre (see ESM Methods for details of covariate measurements), after which residuals were computed and inverse-normally transformed within each genetic dataset (e.g. population-specific for ARIC or substudy for WHI). In sensitivity analyses, models were estimated excluding BMI. Association analyses for each dataset were performed using SUGEN version 8.10 (https://github.com/dragontaoran/SUGEN), which implemented a generalised estimating equation method that accounts for relatedness, while adjusting for ten ancestral principal components [27]. Subsequently, fixed-effects models with inverse variance weighting were used to pool dataset-specific variant effect estimates and their SEs across populations as well as within populations using METAL version 2011-03-25 (http://csg.sph.umich.edu/abecasis/Metal/download/), after applying genomic control correction [28]. Variants with an effective n < 30 or an imputation R2 < 0.4 within a given dataset were excluded from meta-analysis. To account for testing of multiple traits across multiple ancestries, we defined novel loci as those in which the lead variant reached a genome-wide significance threshold of p < 5.0 × 10−9, as done previously [26], and were located more than 500 KB from any previously established loci for the given glycaemic trait.
Fine-mapping
To identify independent secondary signals, stepwise conditional analyses were performed for the transethnic meta-analysis results, conditioning on the most significant variants (known and novel) identified in our GWAS and applying genomic control correction. After conditioning on the top genome-wide significant (p < 5 × 10−9) variant, variants identified within a 1 MB region of the variant with a p value < 5.0 × 10−8 were considered significant, independent signals. These conditional analyses were repeated, adding in the conditional lead variants until no variant had a conditional p value less than the locus-specific significance (p < 5.0 × 10−8). To determine whether identified secondary signals at known loci were independent from known secondary signals, we also conditioned on known variants reported in the literature.
We subsequently performed fine-mapping of novel primary analysis loci and independent secondary loci using FINEMAP version 1.4_x86_64 (http://www.christianbenner.com) [29]. All variants within ±1 MB of each novel primary and independent secondary variants were included for fine-mapping, restricting to variants with a stratum specific effective n > 30 and imputation R2 > 0.4. If variants demonstrated population-specific significance, a population-specific LD matrix was constructed; for all other variants with genome-wide significance in the transethnic meta-analysis, a combined ancestry LD matrix was constructed by computing population-specific LD matrices and subsequently weighting by population sample size. We then computed the posterior probabilities of k causal variants at each reported locus and constructed a 95% credible set (CS). LocusZoom plots [30] of the CS top variants were generated to visualise the signals identified at each locus.
Replication
Replication of novel loci was performed under a common analysis plan; variant proxies in high LD (D′ and r2 > 0.9 in the population of interest) were used if the variant of interest was not genotyped or well-imputed in the following four multi-ethnic studies: Jackson Heart Study (JHS); Cameron County Hispanic Cohort (CCHC); Reasons for Geographical And Racial Differences in Stroke (REGARDS) Study; and Multi-Ethnic Study of Atherosclerosis (MESA). Additionally, published summary statistics from the China Health and Nutrition Survey (CHNS) cohort [31] and an analysis of individuals of EA ancestry from Lagou et al and the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) [32] were also included for replication analyses (ESM Methods). We used the R package MetaSubtract version 1.60 (https://cran.r-project.org/web/packages/MetaSubtract/) [33] to remove overlapping EA ARIC cohort results from the Lagou et al summary statistics before their inclusion in replication (ESM Methods). A maximum of n = 8459, n = 92,432, n = 3406 and n = 6476 AA, EA, HA and ASN participants, respectively, were identified for replication of fasting glucose, fasting insulin and HbA1c novel variants. Replication data were not available for HI and NAm populations. Significance was determined using Bonferroni correction (0.05/number of significant novel independent signals). All replication results were meta-analysed in transethnic and population-specific analyses, using METAL [28].
Functional annotation
Finally, to characterise the putative functionality of variants, we performed bioinformatic follow-up for all novel primary and independent secondary variants, as well as the top variants identified in each fine-mapping CS. We used the UCSC Genome Browser Islet Regulome tracks [34–36], which include data on chromatin classes, cytokine-induced regulatory elements and enhancer hubs in both adult human islets and pancreatic progenitors. Additionally, we created a custom UCSC Genome Browser analysis hub of important regions (e.g. enhancer and repressor activities, DNase I hypersensitive sites [DHS] and transcribed regions) in the pancreas and insulin-responsive tissues, including skeletal muscle, liver and adipose tissue, using GTEx [37] and Roadmap Epigenome Project [38] data.
Results
Study overview
After exclusions, a total of 52,267, 23,357 and 48,395 participants were available for fasting glucose, HbA1c and fasting insulin GWAS, respectively (ESM Table 2), of which collectively over half were either self-reported AA or HA (maximum 23% AA, 46% HA, 40% EA, 4% ASN, 3% HI, 0.8% NAm). The mean age of participants was 54.5 years and they were overweight (mean ± SD BMI 28.0 ± 5.7), with a greater representation of female participants (72%). Glycaemic trait distributions were similar across studies and self-reported race/ethnic groups, with mean ± SD fasting glucose levels ranging from 4.5 ± 0.5 mmol/l to 5.5 ± 0.6 mmol/l, HbA1c levels ranging from 34.0 ± 3.5 (5.3%) mmol/mol to 38.6 ± 3.2 (5.7%) mmol/mol and fasting insulin levels ranging from 32.3 ± 19.7 pmol/l to 80.9 ± 59.0 pmol/l.
Identification of significant loci
In the transethnic meta-analysis, we identified a total of 13, 13 and 11 genome-wide significant (p < 5.0 × 10−9) loci for fasting glucose, HbA1c and fasting insulin, respectively (Fig. 1 and ESM Table 3, ESM Fig. 1). Several loci and, in some cases, several top variants were shared across glycaemic traits: G6PC2 for fasting glucose and HbA1c (shared top variant: rs560887); GCKR for fasting glucose and fasting insulin (shared top variant: rs1260326); SLC2A2 for fasting glucose and HbA1c (shared top variant: rs1879442); and GCK for fasting glucose and HbA1c. Effect estimates for significant variants were generally consistent across populations (Fig. 2 and ESM Fig. 1), although statistical significance varied, often in accordance with minor allele frequency (MAF) and/or sample size.
Fig. 1.
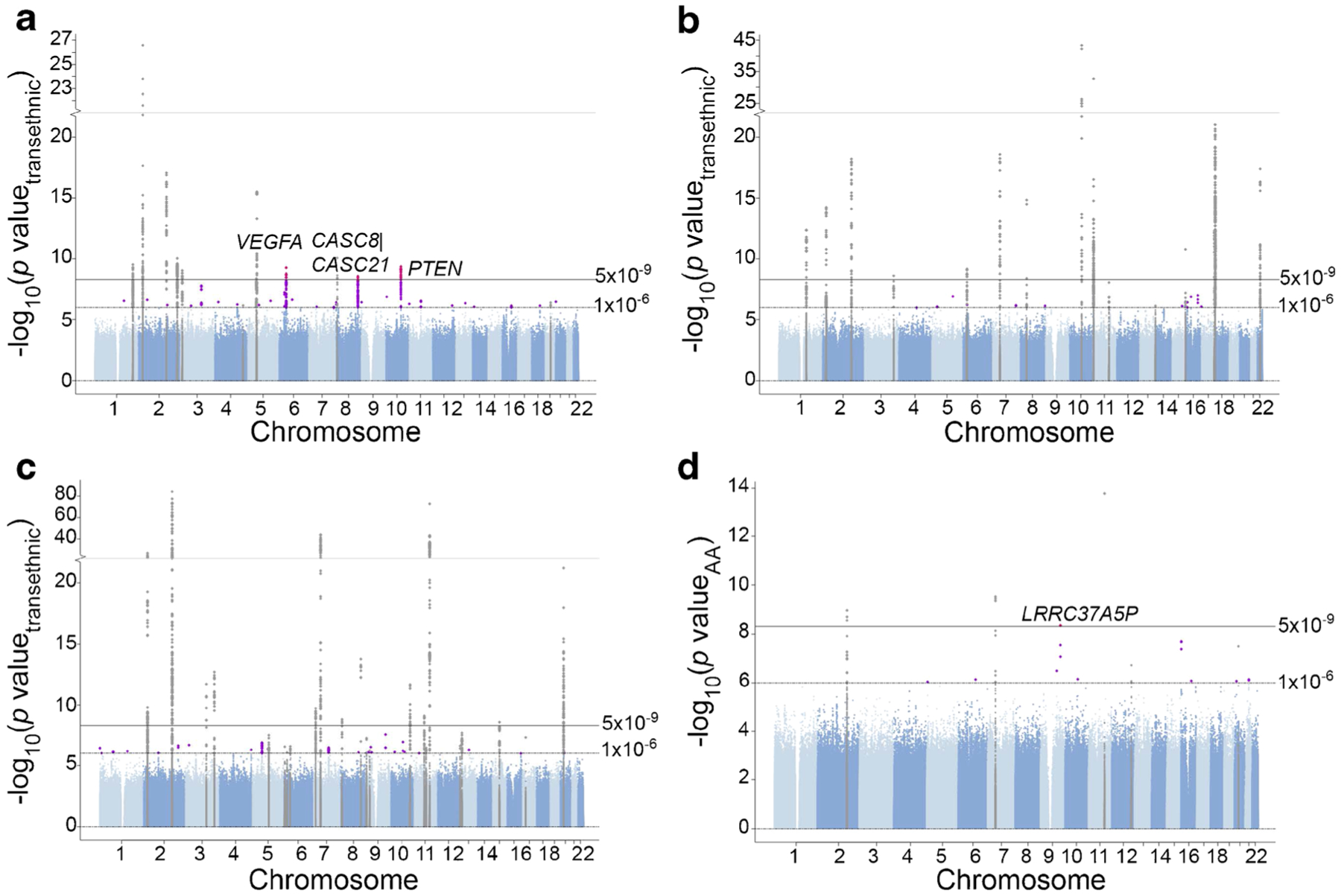
Manhattan plots for glycaemic trait association analyses in PAGE, adjusting for BMI. (a) Fasting insulin transethnic meta-analysis results. (b) HbA1c transethnic meta-analysis results. (c) Fasting glucose transethnic meta-analysis results. (d) Fasting glucose AA-specific meta-analysis results. Known loci are shown in grey; novel loci with p value < 1 × 10−6 are shown in purple; novel loci with p value < 5 × 10−9 are shown in pink
Fig. 2.
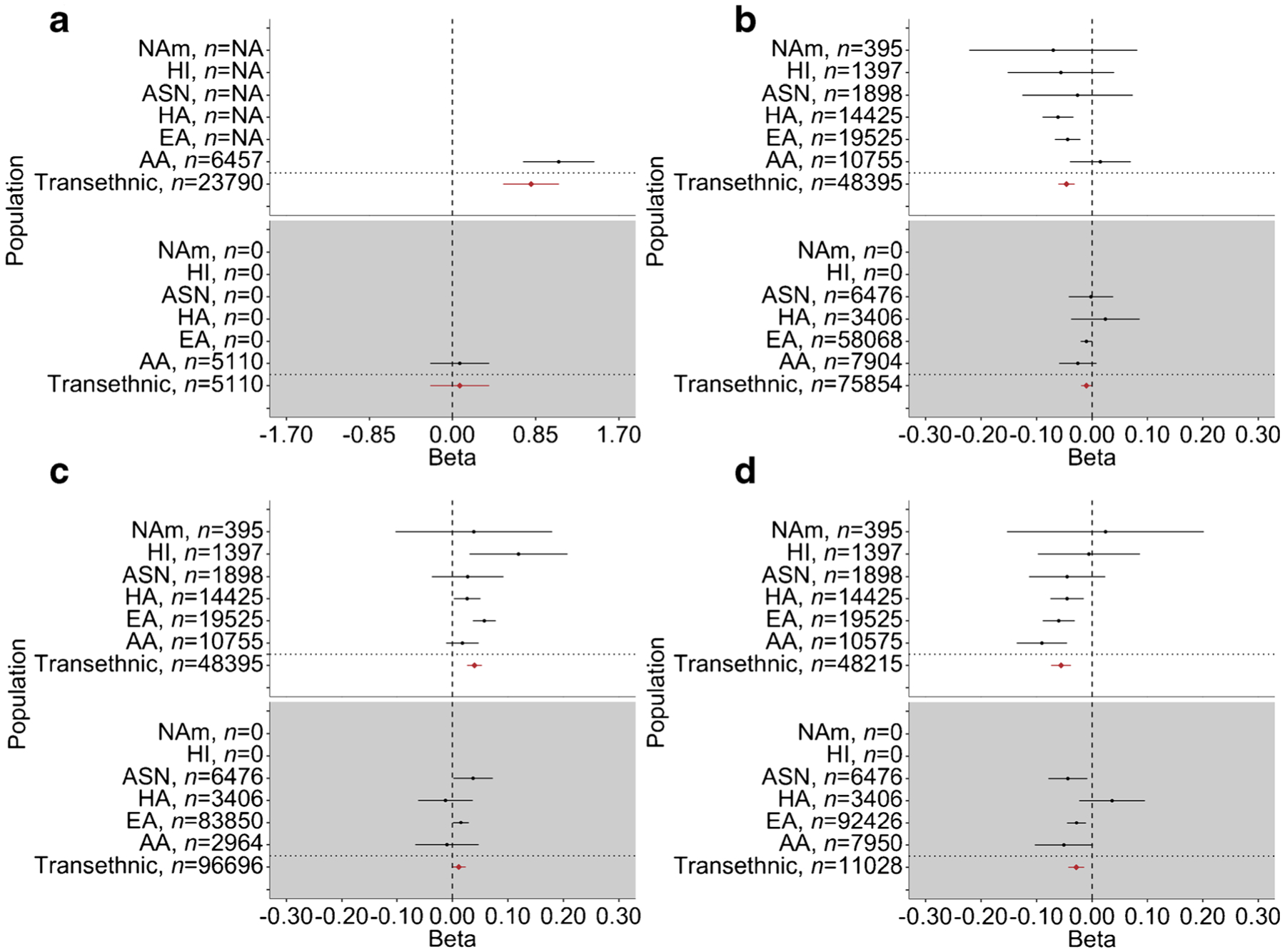
Forest plots of primary GWAS and replication transethnic and population-specific meta-analysis effect estimates and 95% CIs for the four novel variants identified in the PAGE Study. (a) Fasting glucose variant rs571025315 at LRRC37A5P locus, which was genome-wide significant (p < 5 × 10−9) only in AA-specific meta-analysis. Effective n < 30 for all other populations in the primary analysis, indicated by sample size n = NA in the primary analysis panel. (b) Fasting insulin variant rs9472142 at VEGFA locus. (c) Fasting insulin variant rs35131928 at CASC8/CASC21 locus; EA REGARDS replication data used proxy variant rs10956361 in lieu of rs35131928 (D′ = 1 and r2 = 1 with rs35131928 in EA PAGE data). (d) Fasting insulin variant rs10887773 at PTEN locus. PAGE Study GWAS results for transethnic and population-specific meta-analyses are shown against a white background; transethnic and population-specific meta-analyses of replication results are shown against a grey background. Replication data sources, by population, are as follows: AA, JHS, REGARDS; EA, REGARDS, MESA, MAGIC; HA, MESA, CCHC; and ASN, MESA, CHNS
Three of the 34 significant loci identified in transethnic GWAS were novel (± 500 KB from a known variant) at time of analysis (January 2020) and were associated with fasting insulin: the VEGFA (also known as MVCD1, VEGF or VPF) locus on chromosome 6 (lead variant rs9472142, p = 5.56 × 10−10); the CASC8/CASC21 (also known as CARLO1, CARLo-1, LINC00860, CARLO2, CARLo-2 or LINC01244) locus on chromosome 8 (lead variant rs35131928, p = 2.70 × 10−9); and the PTEN (also known as 10q23del, BZS, CWS1, DEC, GLM2, MHAM, MMAC1, PTEN1, PTENbeta or TEP1) locus on chromosome 10 (lead variant rs10887773, p = 4.55 × 10−10) (Table 1, Figs 1, 2). Wide variation in MAF was observed across populations for lead variants at these three novel loci, particularly for rs9472142 at the VEGFA locus (MAF range 0.12–0.36) and rs10887773 at the PTEN locus (MAF range 0.10–0.37). Effect estimates were generally directionally consistent across populations (Fig. 2). We also identified a fourth novel locus associated with fasting glucose in the population-specific meta-analysis of self-identified African Americans: the LRRC37A5P (also known as C9orf29) locus on chromosome 9 (lead variant rs571025325, pAA = 4.58 × 10−9) (Table 1, Figs 1, 2), with a MAF of 0.0037.
Table 1.
Novel genome-wide-significant (p<5 × 10−9) loci discovered in genome-wide association study of fasting glucose, fasting insulin and HbA1c via transethnic and population-specific meta-analysis
| Trait | Lead variant | Chr:Position | Ref allele | Alt allele | Locus | Effect (SE) of alternative allele | MAF | p value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | EA | HA | ASN | HI | NAm | Transethnic | AA | EA | HA | ASN | HI | NAm | |||||||
| Transethnic meta-analysis | |||||||||||||||||||
| Fasting insulin | rs9472142 | 6:43818942 | C | T | VEGFA | −0.046 (0.007) | 0.36 | 0.29 | 0.24 | 0.12 | 0.25 | 0.25 | 5.56×10−10 | 2.78×10−2 | 1.48×10−4 | 1.26×10−5 | 6.04×10−1 | 2.48×10−1 | 3.64×10−1 |
| Fasting insulin | rs35131928 | 8:128350707 | C | CA | CASC8/CASC21 | 0.040 (0.007) | 0.34 | 0.40 | 0.46 | 0.49 | 0.33 | 0.42 | 2.70×10−9 | 2.25×10−1 | 3.21×10−8 | 2.78×10−2 | 4.02×10−1 | 8.02×10−3 | 5.91×10−1 |
| Fasting insulin | rsl 0887773 | 10:89765945 | G | T | PTEN | −0.056 (0.009) | 0.10 | 0.14 | 0.20 | 0.37 | 0.28 | 0.19 | 4.55×10−10 | 8.51×10−5 | 3.69×10−5 | 2.95×10−3 | 1.97×10−1 | 9.01×10−1 | 7.89×10−1 |
| AA-specific meta-analysis | |||||||||||||||||||
| Fasting glucose | rs571025325 | 9:114379301 | G | A | LRRC37A5P | 1.10 (0.19) | 0.0037 | 1.98×10−6 | 1.05×10−3 | 0 | 3.50×10−5 | 0 | 2.84×10−8 | 4.58×10−9 | NAa | NAa | NAa | NAa | NAa |
NA, effective n < 30 and population-specific meta-analyses were not computed
Replication of lead variants at four novel loci
Replication of lead variants or proxy variants at the four potentially novel loci was performed through transethnic meta-analysis of independent AA (n range 1311–4986), ASN (n range 667–5809), EA (n range 1054–97,348) and HA (n range 1189–2217) cohorts, with EA fasting insulin results from published summary statistics from Lagou et al contributing the largest sample size. Lead variants for all three novel fasting insulin loci showed directionally consistent effects, although considerable effect attenuation was observed. The PTEN lead variant was significant at the Bonferroni-corrected significance level of p = 0.0125 (α = 0.05/4 signals) in independent transethnic meta-analysis and the other two fasting insulin loci showed suggestive significance, particularly CASC8/CASC21 (p = 0.0174) (Fig. 2 and ESM Table 4). The fourth locus (fasting glucose, LRRC37A5P), which was observed only in AA-specific meta-analysis, did not show evidence of replication (p = 0.62), although only 41 of the 5110 replication dataset participants were expected to carry at least one copy of the minor allele (ESM Table 4). Furthermore, in Chen et al’s [39] recently published glycaemic traits GWAS, our VEGFA, PTEN and CASC8/CASC21 lead variants showed significance in transethnic (VEGFA and PTEN), EA-specific (VEGFA, PTEN) and East Asian-specific (PTEN, CASC8/CASC21) meta-analyses; however, these results are not an independent replication as they contain overlapping data from the ARIC, BioMe, WHI, HCHS/SOL and several replication cohorts used here (ESM Table 4).
Secondary analyses at known glycaemic trait loci
Through stepwise conditional analysis, we identified seven significant secondary signals at known glycaemic trait loci, including two previously unreported fasting glucose (GCK [also known as FGQTL3, GK, GLK, HHF3, HK4, HKIV, HXKP, LGLK, MODY2 or PNDM1], rs55908146) and fasting insulin (PPP1R3B [also known as GL, PPP1R4 or PTG], rs330941) secondary signals that remained significant after conditioning upon known variants (Table 2 and ESM Table 5). Wide variation in MAF was observed across populations for both novel independent secondary signals rs330941 (MAF range 0.22–0.49) and rs55908146 (MAF range 0.15–0.32) (Table 2).
Table 2.
Significant (p<5 × 10−8) previously unreported secondary signals at known fasting insulin and fasting glucose loci
| Trait | Secondary Varianta | Chr:Position | Ref allele | Alt allele | Locus | Effect (SE) of alt allele in conditional analysis | Primary analysis transethnic p valueb | Conditional analysis transethnic p valuec | Primary conditioning variant(s)d | LD D’e | LD r2f | MAF | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transethnic | AA | EA | HA | ASN | HI | NAm | ||||||||||||
| Fasting insulin | rs330941 | 8:9018657 | C | T | PPP1R3B | 0.045 (0.007) | 4.79×10−7 | 2.37×10−10 | rs4841132 | 0.18 | 0.005 | 0.43 | 0.29 | 0.39 | 0.49 | 0.22 | 0.27 | 0.43 |
| Fasting glucose | rs55908146 | 7:44180226 | G | A | GCK/MYL7 | 0.043 (0.008) | 8.03×10−6 | 1.75×10−8 | rs2908286, rs2908290 | 0.04, 0.24 | 0.001, 0.014 | 0.28 | 0.15 | 0.32 | 0.30 | 0.26 | 0.25 | 0.30 |
Lead variant from conditional analysis reaching locus-specific significance
p value of the secondary variant in the primary GWAS analysis, not adjusted for the primary variant
p value of the secondary variant, adjusted for the primary variant(s)
Lead variant(s) from the primary analysis and previous stepwise conditional analysis
LD D′ between primary variant(s) and secondary variant in PAGE Study data (transethnic LD generated from unrelated subset of AA, HA, ASN, HI and NAm participants)
LD r2 between primary variant(s) and secondary variant in PAGE Study data (transethnic LD generated from unrelated subset of AA, HA, ASN, HI and NAm participants)
Fine-mapping
To identify the most likely causal variant(s) for the four putatively novel loci and two novel independent secondary signals, we subsequently utilised FINEMAP to estimate the number of causal variants per locus and generate a 95% CS for each causal variant. For three of the four novel loci (LRRC37A5P, CASC8/CASC21, PTEN) we estimated one causal variant at each locus (k = 1) (Table 3); at these loci, the top variants in our GWAS analyses (rs571025325, rs35131928, rs10887773) were identified as the variants most likely to be causal, although with varying posterior probabilities of being the top causal variant (range 0.06–0.79) (ESM Tables 6, 7, 8). The broad range of posterior probabilities by locus reflects the size of the LD block. For the fourth novel locus (VEGFA), the highest posterior probability was observed for k = 2 causal variants, with our top GWAS variant rs9472142 identified as the top variant in CSVEGFA1 (Table 3 and ESM Table 9); the top variant in CSVEGFA2 (rs6910726) was just under the significance threshold in our stepwise conditional analysis, with p = 4.20 × 10−6 (ESM Table 5).
Table 3.
Fine-mapping posterior probabilities of k causal variants at novel primary GWAS and independent secondary signal loci
| Trait | GWAS index variant | Locus | Posterior probability (no. of causal variants is k) | ||||
|---|---|---|---|---|---|---|---|
| k=1 | k=2 | k=3 | k=4 | k=5 | |||
| Primary GWAS analysis loci | |||||||
| Fasting glucose | rs571025325 | LRRC37A5P | 0.62 | 0.38 | 4.74×10−7 | 0 | 0 |
| Fasting insulin | rs9472142 | VEGFA | 0.37 | 0.48 | 0.15 | 0.01 | 0 |
| Fasting insulin | rs35131928 | CASC8/CASC21 | 0.72 | 0.28 | 0.0002 | 0 | 0 |
| Fasting insulin | rs10887773 | PTEN | 0.78 | 0.22 | 0 | 0 | 0 |
| Conditional analysis loci | |||||||
| Fasting insulin | rs330941 | PPP1R3B | 0.05 | 0.77 | 0.19 | 0 | 0 |
| Fasting glucose | rs55908146 | GCK/MYL7 | 0 | 0.04 | 0.37 | 0.54 | 0.06 |
For the two novel independent secondary signals, the highest posterior probabilities were estimated for k = 2 (PPP1R3B) and k = 4 (GCK) causal variants (Table 3). Because we did not perform any LD pruning, we identified CSs containing many variants in high LD with each other, and therefore low individual posterior probabilities of being the top causal variant in each CS. For example, at the PPP1R3B locus, for the variants in CSPPP1R3B1, the posterior probabilities of being the top causal variant range between 0.11 and 0.26 (ESM Table 10). The top three variants in CSPPP1R3B2, including the most significant variant from our conditional analysis, rs330941, are in high LD with each other but not the CSPPP1R3B1 variants, and posterior probabilities for these three variants range from 0.24 to 0.37 (ESM Table 10). The novel GCK secondary signal rs55908146 was among the top five variants in CSGCK3, all of which had a probability of being the top variant in CSGCK3 of about 0.10, additionally suggesting an LD block (ESM Table 11). LocusZoom plots of the loci with more than one CS showed that the CSs have little shared LD (ESM Fig. 2).
Functional annotation
We performed bioinformatic follow-up of the novel primary loci and known loci with independent secondary signals using the UCSC Genome Browser Islet Regulome tracks [34–36] and a custom UCSC analysis hub of important regions (e.g. enhancer and repressor activities, DHS and transcribed regions) in the pancreas and insulin-responsive tissues including skeletal muscle, liver and adipose tissue. However, functional annotation of the top variants in the fine-mapping CSs for each loci did not indicate a clear potential mechanism through which variants may act; gene expression in the GTEx dataset [40] showed ubiquitous levels of expression across tissues for most of the loci, and human pancreatic islet chromatin state data showed chromatin state markers of expression in the general regions of many of the loci (data not shown).
Discussion
Examining the genetic architecture of glycaemic traits in a diverse study, we identified three novel (at time of analysis, January 2020) fasting insulin loci shared across populations and a fourth low-frequency fasting glucose locus specific to self-identified AAs. Additionally, we identified two previously unreported independent secondary signals in the PPP1R3B and GCK loci associated with fasting insulin and fasting glucose, respectively. These results emphasise the continued need for more GWAS in diverse populations to assess the genetic heterogeneity of complex diseases.
While this paper was under review, Chen et al and the MAGIC consortium published a large-scale transancestry analysis of glycaemic traits, aggregating GWAS data from up to 281,416 individuals without diabetes [39]. They identified the novel fasting insulin-associated PTEN locus identified here (r2 = D′ = 1 between our identified variant rs10887773 and Chen et al’s variant rs12769346), as well as a fasting insulin variant in the VEGFA locus. However, after conditioning on Chen et al’s top variant (rs998584), our identified VEGFA top variant remained genome-wide significant (p < 5 × 10−9). Additionally, there was low LD between the VEGFA variants (r2PAGE rs9472142 and MAGIC rs998584 = 0.03, D′PAGE rs9472142 and MAGIC rs998584 = 0.35); we note that rs9472152, which was contained within both of our VEGFA fine-mapping 95% CSs, is located near rs998584, with r2rs9472125 and MAGIC rs998584 = 0.01 and D′rs9472125 and MAGIC rs998584 = 0.61 between the two variants, as calculated from the PAGE combined ancestry LD. The independent fasting insulin and fasting glucose secondary signals we identified in the PPP1R3B and GCK loci were not among the variants identified at these loci by Chen et al.
Although there was overlap in the cohorts in our PAGE data and in Chen et al, including ARIC, BioMe, WHI and HCHS/SOL, in the PAGE Study much of our contributing genetic data from these cohorts were newly genotyped on the MEGA array, which was specifically designed to increase variant coverage across multiple ancestry groups [25, 26]. Additionally, the distribution of ancestry groups varied across the two analyses: PAGE data had a higher percentage of non-EA participants (% non-EA range 60.0% [fasting insulin] to 62.4% [fasting glucose]) than Chen et al, in which approximately 30% of participants were non-EA. While the PAGE Study’s statistical power is diminished by a smaller sample size, due to the increased ancestral diversity and finer genotyping on the MEGA array, we identified two loci not identified by Chen et al and one that was reported by Chen et al [39]. Both approaches provide complementary information on the genetic architecture of glycaemic traits in diverse populations.
The three novel fasting insulin loci identified via transethnic meta-analysis (VEGFA, CASC8/CASC21 and PTEN) and the novel fasting glucose AA-specific locus (LRRC37A5P) harbour genes with biologically plausible roles in insulin signalling and beta cell function. VEGFA has been associated with type 2 diabetes [41], waist/hip ratio [42, 43] and erythrocyte traits [44, 45]. Novel variant rs9472142, in CSVEGFA1, is in high LD (r2EA = 0.97) with an identified VEGFA type 2 diabetes variant (rs9472138), supporting an early role of this signal prior to type 2 diabetes onset [22]. Mouse models have also demonstrated that VEGFA signalling is necessary for pancreas specification and differentiation and plays important roles in pancreatic islet blood vessel maintenance and blood flow [46]. CASC8/CASC21 are cancer susceptibility genes and have not been previously associated with insulin or type 2 diabetes, although the CASC8 locus has been associated with BMI-adjusted waist/hip ratio in individuals of African ancestry [47]. The low probability for any single variant identified in fine-mapping CS1 for CASC8/CASC21 indicates an LD block or haplotype for this locus. PTEN is involved in the negative regulation of insulin signalling [48] and has been associated with type 2 diabetes [41, 49]. A low probability for any single variant in fine-mapping CSPTEN1 also indicates a likely LD block or haplotype for this locus. Although several variants in our final novel locus, LRRC37A5P, have previously shown suggestive significance (p < 1.0 × 10−6) in association with diastolic BP in a transethnic meta-analysis of the metabolic syndrome [50], this locus has not previously been associated with fasting glucose. The pseudogene LRRC37A5P is next to the PTGR1 gene encoding an enzyme involved in the inactivation of chemotactic factor, leukotriene B4, which is associated with insulin resistance and obesity [51, 52].
Fine-mapping of known fasting insulin and fasting glucose PPP1R3B and GCK loci containing novel independent secondary signals yielded results consistent with our stepwise conditional analyses. Multiple CSs, including those containing our identified secondary signals, were predicted for each locus. PPP1R3B contributes to insulin signalling through an insulin–Akt–protein phosphatase 1 regulatory subunit 3G (PPP1R3G)–protein phosphatase 1 regulatory subunit 3B (PPP1R3B) regulatory axis, in which PPP1R3B binds to dephosphorylated glycogen synthase (GS), thus relaying insulin signals for hepatic glycogen synthesis [53]. Rare PPP1R3B missense variants may increase the risk of type 2 diabetes, possibly through altered GS function and altered lipid metabolism [54]. GCK encodes the enzyme glucokinase, which acts to maintain glucose homeostasis and has been previously associated with fasting glucose and type 2 diabetes [5, 11, 14, 55–58]. Specific GCK mutations also cause Mendelian disease phenotypes including MODY2 and permanent neonatal diabetes mellitus (PNDM) [59–61]. Continuing to identify the spectrum of natural variation across populations of genes that alter risk for glycaemic traits and type 2 diabetes will enable improvements in risk prediction models for diverse populations.
Strengths of this study include the large study size and representation of multiple ancestrally, ethnically and racially diverse populations, including HA and AA populations, which shoulder a large burden of hyperglycaemia and type 2 diabetes in the USA and historically have been understudied in genetic epidemiology research. However, because the greatest proportion of participants were from HA, AA and EA populations, this study was limited in its ability to detect associations specific to East Asian, South Asian, HI and NAm populations. Additionally, our transethnic fine-mapping approach utilised a combined ancestry LD matrix that was constructed by computing population-specific LD matrices and subsequently weighting by population sample size. This weighted LD matrix approach is limited by the fact that it ‘averages’ LD patterns across populations, thus potentially missing ancestry-specific LD differences. Nevertheless, we applied this approach because it accounts for potentially more than two causal variants at a given loci. Developing computationally scalable fine-mapping methods that leverage ancestry-specific LD patterns while accounting for more than two causal variants is an area of active research.
Furthermore, only the fasting insulin association at the PTEN locus replicated in a transethnic meta-analysis of several multi-ethnic studies, although both the VEGFA and CASC8/CASC21 loci showed suggestive significance. Our inability to replicate several identified loci likely reflects the increasing limitations of replication in large-scale ‘mega-biobank’ studies, since meta-analysis of multiple small independent replication studies, as performed here, may be underpowered [62]. Furthermore, replicating rare variants like the AA-specific LRRC37A5P variant is a known challenge, especially since rare variants tend to be population-specific [63]. To further interrogate rare loci identified in populations thus far underrepresented in GWAS, there must be a continued effort to increase the ancestral diversity of the populations studied in GWAS and all biomedical research.
In summary, this study of glycaemic traits in the diverse PAGE Study identified three novel fasting insulin loci: one AA-specific rare fasting glucose locus; and two novel independent secondary signals at known fasting glucose and fasting insulin loci. These findings reinforce the need to conduct genetic association studies in participants of diverse backgrounds to yield new insights into the genetics of glycaemic traits.
Supplementary Material
Research in context.
What is already known about this subject?
Previous genome-wide association studies (GWAS) have identified over 500 loci associated with type 2 diabetes, and glycaemic and insulin-related traits
Most of these findings were generated in populations of European ancestry
What is the key question?
Can novel primary loci and independent secondary signals associated with fasting glucose, fasting insulin and HbA1c be identified in transethnic and population-specific meta-analyses in the diverse Population Architecture using Genomics and Epidemiology (PAGE) Study?
What are the new findings?
We identified three novel fasting insulin loci in transethnic meta-analysis, and a novel low-frequency African American-specific locus
We also identified two novel independent secondary signals in known fasting glucose and fasting insulin loci
How might this impact on clinical practice in the foreseeable future?
These findings provide new insights into the genetic architecture of glycaemic traits and highlight the importance of conducting genetic studies in diverse populations
Acknowledgements
The PAGE consortium thanks the staff and participants of all PAGE studies for their important contributions. The complete list of PAGE members can be found at http://www.pagestudy.org/page-investigators/. We thank the staff and participants of the ARIC study for their important contributions. More detail about the ARIC study may be found at: https://sites.cscc.unc.edu/aric/. We thank the WHI investigators and staff for their dedication and the study participants for making the programme possible. Full listing of WHI investigators can be found at https://www.whi.org/researchers/DocumentsWriteaPaper/WHIInvestigatorShortList.pdf. We also thank the investigators, the staff and the participants of MESA for their valuable contributions. A full list of participating MESA institutions and investigators can be found at http://www.mesa-nhlbi.org and https://www.mesa-nhlbi.org/aboutMESAPersonnel.aspx, respectively. We also thank the staff and participants of CARDIA for their important contributions. More details about CARDIA may be found at: https://www.cardia.dopm.uab.edu/. Representatives of the National Institute of Neurological Disorders and Stroke were involved in the review of the manuscript but were not directly involved in the collection, management, analysis or interpretation of data. The authors thank the other investigators, the staff, and the participants of the REGARDS study for their valuable contributions. A full list of participating REGARDS investigators and institutions can be found at: https://www.uab.edu/soph/regardsstudy/. The authors wish to thank the staff and participants of the JHS. We also thank the National Institute for Nutrition and Health, China Center for Disease Control and Prevention, Beijing Municipal Center for Disease Control and Prevention, and the Chinese National Human Genome Center at Shanghai.
Funding
The PAGE Study is funded by the National Human Genome Research Institute with co-funding from the National Institute on Minority Health and Health Disparities. Assistance with data management, data integration, data dissemination, genotype imputation, ancestry deconvolution, population genetics, analysis pipelines and general study coordination was provided by the PAGE Coordinating Center (NIHU01HG007419). Genotyping services were provided by the Center for Inherited Disease Research, which is fully funded through a federal contract from the National Institutes of Health (NIH) to The Johns Hopkins University, contract number HHSN268201200008I. Genotype data quality control and quality assurance services were provided by the Genetic Analysis Center in the Biostatistics Department of the University of Washington, through support provided by the Center for Inherited Disease Research contract. PAGE data and materials included in this report were funded through the following studies and organisations:
The ARIC study is funded in whole or in part by federal funds from the National Heart, Lung and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract nos HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I), R01HL087641, R01HL059367 and R01HL086694, National Human Genome Research Institute contract U01HG004402, and National Institutes of Health contract HHSN268200625226C. Infrastructure was partly supported by grant no. UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
The Mount Sinai BioMe Biobank is supported by The Andrea and Charles Bronfman Philanthropies.
The CARDIA Study is supported by contracts HHSN268201800003I, HHSN268201800004I, HHSN268201800005I, HHSN268201800006I and HHSN268201800007I from the National Heart, Lung and Blood Institute (NHLBI). CARDIA is also partially supported by the Intramural Research Program of the National Institute on Aging (NIA) and an intra-agency agreement between NIA and NHLBI (AG0005). GWAS genotyping and data analyses were funded in part by grants U01-HG004729 and R01-HL093029 from the National Institutes of Health to M. Fornage.
The MEC characterisation of epidemiological architecture is funded through the NHGRI PAGE programme (U01HG004802 and its NHGRI ARRA supplement). The MEC study is funded by the National Cancer Institute (R37CA54281, R01CA63, P01CA33619, U01CA136792 and U01CA98758).
The WHI programme is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, and US Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005.
The HCHS/SOL is a collaborative study supported by contracts from the National Heart, Lung and Blood Institute (NHLBI) to the University of North Carolina (HHSN268201300001I / N01-HC-65233), University of Miami (HHSN268201300004I / N01-HC-65234), Albert Einstein College of Medicine (HHSN268201300002I / N01-HC 65235), University of Illinois at Chicago (HHSN268201300003I / N01-HC-65236 Northwestern University), and San Diego State University (HHSN268201300005I / N01-HC-65237). The following Institutes/Centres/Offices have contributed to the HCHS/SOL through a transfer of funds to the NHLBI: National Institute on Minority Health and Health Disparities; National Institute on Deafness and Other Communication Disorders; National Institute of Dental and Craniofacial Research; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute of Neurological Disorders and Stroke; and NIH Institution-Office of Dietary Supplements. The Genetic Analysis Center at the University of Washington was supported by NHLBI and NIDCR contracts (HHSN268201300005C AM03 and MOD03).
The JHS is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD).
The REGARDS project is supported by cooperative agreement U01 NS041588 co-funded by the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institute on Aging (NIA), National Institutes of Health, Department of Health and Human Services.
MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079 and UL1-TR-001420. Funding for SHARe genotyping was provided by NHLBI contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, CA, USA) and the Broad Institute of Harvard and MIT (Boston, MA, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The CCHC is supported by MD000170 P20 funded from the National Center on Minority Health and Health Disparities (NCMHD), the University of Texas Houston Health Sciences Center, Center for Clinical and Translational Science CCTS-CTSA award UL1 TR00371 from NCATS.
The CHNS receives research grant funding from the National Institute for Health (NIH), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) for R01 HD30880, National Institute on Aging (NIA) for R01 AG065357, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) for R01DK104371 and R01HL108427, the NIH Fogarty grant D43 TW009077 since 1989, and the China-Japan Friendship Hospital, Ministry of Health for support for CHNS 2009, Chinese National Human Genome Center at Shanghai since 2009, and Beijing Municipal Center for Disease Prevention and Control since 2011.
-
Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756.
CLA, HMH, CGD, SFD, MG and KEN are supported by R01HL142825. HMH is also funded by NHLBI training grants T32 HL007055 and T32 HL129982-03, and ADA grant no. 1-19-PDF-045. KEN is also supported by R01HG010297-01. LMR is supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant KL2T2002490 and by R01HG010297. JEB and HGP are supported by R01HL142302-01; HGP is also supported by U01HG007416. CK is supported by S10OD028685. RJFL is supported by R01HG010297 and R01HL151152. MRI is supported by R01HL136666. SB and TCM are supported by U01HG007419. LSP is supported by Veterans Administration (VA) awards CSP no. 2008, I01 CX001899, I01 CX001737 and HSR&D IIR 07-138, and by NIH awards R21 DK099716, R18 DK066204, R03 AI133172, R21 AI156161, U01 DK091958, U01 DK098246 and UL1 TR002378.
The study sponsors/funders were not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report.
Authors’ relationships and activities
HMH receives a stipend from the American Heart Association for serving as a statistical editor for the journal Circulation Research. SAB has a financial interest in Adaptive Biotechnologies. LSP has served on Scientific Advisory Boards for Janssen, and has or had research support from Merck, Pfizer, Eli Lilly, Novo Nordisk, Sanofi, PhaseBio, Roche, AbbVie, Vascular Pharmaceuticals, Janssen, Glaxo SmithKline and the Cystic Fibrosis Foundation. LSP is also a cofounder and Officer and Board member and stockholder for a company, Diasyst, Inc., which markets software aimed to help improve diabetes management. The remaining authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Abbreviations
- AA
African American
- ARIC
Atherosclerosis Risk in Communities
- ASN
Asian
- CARDIA
Coronary Artery Risk Development in Young Adults Study
- CCHC
Cameron County Hispanic Cohort
- CHNS
China Health and Nutrition Survey
- CS
Credible set
- DHS
DNase I hypersensitive sites
- EA
European
- GS
Glycogen synthase
- GWAS
Genome-wide association studies
- HA
Hispanic/Latino
- HCHS/SOL
Hispanic Community Health Study/Study of Latinos
- HI
Native Hawaiian
- JHS
Jackson Heart Study
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- MAGIC
Meta-Analyses of Glucose and Insulin-related traits Consortium
- MEC
Multiethnic Cohort
- MESA
Multi-Ethnic Study of Atherosclerosis
- NAm
Native American
- PAGE
Population Architecture using Genomics and Epidemiology
- PPP1R3B
Protein phosphatase 1 regulatory subunit 3B
- REGARDS
Reasons for Geographical and Racial Differences in Stroke
- WHI
Women’s Health Initiative
Footnotes
Supplementary Information The online version contains peer-reviewed but unedited supplementary material available at https://doi.org/10.1007/s00125-021-05635-9.
Data availability
Full summary statistics from each of the population-specific and transethnic results are available at NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics).
References
- 1.Cowie C, Casagrande S, Geiss L (2018) Prevalence and incidence of type 2 diabetes and prediabetes. In: Cowie C, Casagrande S, Menke A et al. (eds) Diabetes in America: 3rd edition, vol 17–1468. National Institutes of Health, Bethesda, MD [Google Scholar]
- 2.Tancredi M, Rosengren A, Svensson AM et al. (2015) Excess mortality among persons with type 2 diabetes. N Engl J Med 373(18):1720–1732. 10.1056/NEJMoa1504347 [DOI] [PubMed] [Google Scholar]
- 3.Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S (2017) Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag 20(1):6–12. 10.1089/pop.2015.0181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang ES, Basu A, O’Grady M, Capretta JC (2009) Projecting the future diabetes population size and related costs for the U.S. Diabetes Care 32(12):2225–2229. 10.2337/dc09-0459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dupuis J, Langenberg C, Prokopenko I et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42(2):105–116. 10.1038/ng.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning AK, Hivert MF, Scott RA et al. (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44(6):659–669. 10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler E, Leong A, Liu CT et al. (2017) Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med 14(9):e1002383. 10.1371/journal.pmed.1002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prasad RB, Groop L (2015) Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel) 6(1):87–123. 10.3390/genes6010087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott RA, Scott LJ, Magi R et al. (2017) An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 66(11): 2888–2902. 10.2337/db16-1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prokopenko I, Langenberg C, Florez JC et al. (2009) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41(1):77–81. 10.1038/ng.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott RA, Lagou V, Welch RP et al. (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 44(9): 991–1005. 10.1038/ng.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haiman CA, Fesinmeyer MD, Spencer KL et al. (2012) Consistent directions of effect for established type 2 diabetes risk variants across populations: the Population Architecture Using Genomics and Epidemiology (PAGE) Consortium. Diabetes 61(6):1642–1647. 10.2337/db11-1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fesinmeyer MD, Meigs JB, North KE et al. (2013) Genetic variants associated with fasting glucose and insulin concentrations in an ethnically diverse population: results from the Population Architecture Using Genomics and Epidemiology (PAGE) study. BMC Med Genet 14:98. 10.1186/1471-2350-14-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bien SA, Pankow JS, Haessler J et al. (2017) Transethnic insight into the genetics of glycaemic traits: fine-mapping results from the Population Architecture Using Genomics and Epidemiology (PAGE) consortium. Diabetologia 60(12):2384–2398. 10.1007/s00125-017-4405-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu CT, Raghavan S, Maruthur N et al. (2016) Trans-ethnic Meta-analysis and functional annotation illuminates the genetic architecture of fasting glucose and insulin. Am J Hum Genet 99(1):56–75. 10.1016/j.ajhg.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigma Type 2 Diabetes Consortium, Estrada K, Aukrust I et al. (2014) Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. Jama 311(22):2305–2314. 10.1001/jama.2014.6511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moltke I, Grarup N, Jorgensen ME et al. (2014) A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512(7513):190–193. 10.1038/nature13425 [DOI] [PubMed] [Google Scholar]
- 18.Manning A, Highland HM, Gasser J et al. (2017) A low-frequency inactivating AKT2 variant enriched in the Finnish population is associated with fasting insulin levels and type 2 diabetes risk. Diabetes 66(7):2019–2032. 10.2337/db16-1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaitlen N, Pasaniuc B, Gur T, Ziv E, Halperin E (2010) Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet 86(1):23–33. 10.1016/j.ajhg.2009.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ong RT, Wang X, Liu X, Teo YY (2012) Efficiency of trans-ethnic genome-wide meta-analysis and fine-mapping. Eur J Hum Genet 20(12):1300–1307. 10.1038/ejhg.2012.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teo YY, Ong RT, Sim X, Tai ES, Chia KS (2010) Identifying candidate causal variants via trans-population fine-mapping. Genet Epidemiol 34(7):653–664. 10.1002/gepi.20522 [DOI] [PubMed] [Google Scholar]
- 22.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium et al. (2014) Genome-wide transancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46(3):234–244. 10.1038/ng.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matise TC, Ambite JL, Buyske S et al. (2011) The next PAGE in understanding complex traits: design for the analysis of population architecture using genetics and epidemiology (PAGE) study. Am J Epidemiol 174(7):849–859. 10.1093/aje/kwr160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International Expert Committee (2009) International expert committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 32(7):1327–1334. 10.2337/dc09-9033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bien SA, Wojcik GL, Zubair N et al. (2016) Strategies for enriching variant coverage in candidate disease loci on a multiethnic genotyping Array. PLoS One 11(12):e0167758. 10.1371/journal.pone.0167758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojcik GL, Graff M, Nishimura KK et al. (2019) Genetic analyses of diverse populations improves discovery for complex traits. Nature 570(7762):514–518. 10.1038/s41586-019-1310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin DY, Tao R, Kalsbeek WD et al. (2014) Genetic association analysis under complex survey sampling: the Hispanic Community Health Study/Study of Latinos. Am J Hum Genet 95(6):675–688. 10.1016/j.ajhg.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26(17):2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benner C, Spencer CC, Havulinna AS, Salomaa V, Ripatti S, Pirinen M (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32(10):1493–1501. 10.1093/bioinformatics/btw018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruim RJ, Welch RP, Sanna S et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26(18):2336–2337. 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spracklen CN, Shi J, Vadlamudi S et al. (2018) Identification and functional analysis of glycemic trait loci in the China Health and Nutrition Survey. PLoS Genet 14(4):e1007275. 10.1371/journal.pgen.1007275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagou V, Magi R, Hottenga JJ et al. (2021) Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun 12(1):24. 10.1038/s41467-020-19366-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nolte IM (2020) Metasubtract: an R-package to analytically produce leave-one-out meta-analysis GWAS summary statistics. Bioinformatics 36(16):4521–4522. 10.1093/bioinformatics/btaa570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miguel-Escalada I, Bonas-Guarch S, Cebola I et al. (2019) Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet 51(7):1137–1148. 10.1038/s41588-019-0457-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pasquali L, Gaulton KJ, Rodriguez-Segui SA et al. (2014) Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet 46(2):136–143. 10.1038/ng.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos-Rodriguez M, Raurell-Vila H, Colli ML et al. (2019) The impact of proinflammatory cytokines on the beta-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat Genet 51(11):1588–1595. 10.1038/s41588-019-0524-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carithers LJ, Moore HM (2015) The Genotype-Tissue Expression (GTEx) Project. Biopreserv Biobank 13(5):307–308. 10.1089/bio.2015.29031.hmm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature 518(7539):317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Spracklen CN, Marenne G et al. (2021) The trans-ancestral genomic architecture of glycemic traits. Nat Genet 53(6):840–860. 10.1038/s41588-021-00852-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.GTEx Consortium (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348(6235):648–660. 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vujkovic M, Keaton JM, Lynch JA et al. (2020) Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52(7):680–691. 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heid IM, Jackson AU, Randall JC et al. (2010) Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 42(11):949–960. 10.1038/ng.685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu Z, Guo Y, Shi H et al. (2020) Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J Allergy Clin Immunol 145(2):537–54 10.1016/j.jaci.2019.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vuckovic D, Bao EL, Akbari P et al. (2020) The polygenic and monogenic basis of blood traits and diseases. Cell 182(5):1214–1231 e1211. 10.1016/j.cell.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Astle WJ, Elding H, Jiang T et al. (2016) The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167(5):1415–1429 e1419. 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staels W, Heremans Y, Heimberg H, De Leu N (2019) VEGF-A and blood vessels: a beta cell perspective. Diabetologia 62(11): 1961–1968. 10.1007/s00125-019-4969-z [DOI] [PubMed] [Google Scholar]
- 47.Ng MCY, Graff M, Lu Y et al. (2017) Discovery and fine-mapping of adiposity loci using high density imputation of genome-wide association studies in individuals of African ancestry: African Ancestry Anthropometry Genetics Consortium. PLoS Genet 13(4):e1006719. 10.1371/journal.pgen.1006719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li YZ, Di Cristofano A, Woo M (2020) Metabolic role of PTEN in insulin signaling and resistance. Cold Spring Harb Perspect Med 10(8):a036137. 10.1101/cshperspect.a036137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spracklen CN, Horikoshi M, Kim YJ et al. (2020) Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582(7811):240–245. 10.1038/s41586-020-2263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willems EL, Wan JY, Norden-Krichmar TM, Edwards KL, Santorico SA (2020) Transethnic meta-analysis of metabolic syndrome in a multiethnic study. Genet Epidemiol 44(1):16–25. 10.1002/gepi.22267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ying W, Wollam J, Ofrecio JM et al. (2017) Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest 127(3):1019–1030. 10.1172/JCI90350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Esmaili S, George J (2015) Ltb4r1 inhibitor: a pivotal insulin sensitizer? Trends Endocrinol Metab 26(5):221–222. 10.1016/j.tem.2015.03.007 [DOI] [PubMed] [Google Scholar]
- 53.Li Q, Zhao Q, Zhang J et al. (2019) The protein phosphatase 1 complex is a direct target of AKT that links insulin signaling to hepatic glycogen deposition. Cell Rep 28(13):3406–3422 e3407. 10.1016/j.celrep.2019.08.066 [DOI] [PubMed] [Google Scholar]
- 54.Niazi RK, Sun J, Have CT et al. (2019) Increased frequency of rare missense PPP1R3B variants among Danish patients with type 2 diabetes. PLoS One 14(1):e0210114. 10.1371/journal.pone.0210114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose CS, Ek J, Urhammer SA et al. (2005) A −30G>A polymorphism of the beta-cell-specific glucokinase promoter associates with hyperglycemia in the general population of whites. Diabetes 54(10):3026–3031. 10.2337/diabetes.54.10.3026 [DOI] [PubMed] [Google Scholar]
- 56.Hwang JY, Sim X, Wu Y et al. (2015) Genome-wide association meta-analysis identifies novel variants associated with fasting plasma glucose in East Asians. Diabetes 64(1):291–298. 10.2337/db14-0563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horikoshi M, Mgi R, van de Bunt M et al. (2015) Discovery and fine-mapping of glycaemic and obesity-related trait loci using high-density imputation. PLoS Genet 11(7):e1005230. 10.1371/journal.pgen.1005230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki K, Akiyama M, Ishigaki K et al. (2019) Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat Genet 51(3):379–386. 10.1038/s41588-018-0332-4 [DOI] [PubMed] [Google Scholar]
- 59.Osbak KK, Colclough K, Saint-Martin C et al. (2009) Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 30(11):1512–1526. 10.1002/humu.21110 [DOI] [PubMed] [Google Scholar]
- 60.Bell GI, Polonsky KS (2001) Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 414(6865):788–791. 10.1038/414788a [DOI] [PubMed] [Google Scholar]
- 61.Kim SH (2015) Maturity-onset diabetes of the young: what do clinicians need to know? Diabetes Metab J 39(6):468–477. 10.4093/dmj.2015.39.6.468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huffman JE (2018) Examining the current standards for genetic discovery and replication in the era of mega-biobanks. Nat Commun 9(1):5054. 10.1038/s41467-018-07348-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bodmer W, Bonilla C (2008) Common and rare variants in multi-factorial susceptibility to common diseases. Nat Genet 40(6):695–701. 10.1038/ng.f.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full summary statistics from each of the population-specific and transethnic results are available at NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics).