
Fig. 2: Detection of single amino acid substitutions in single peptides.
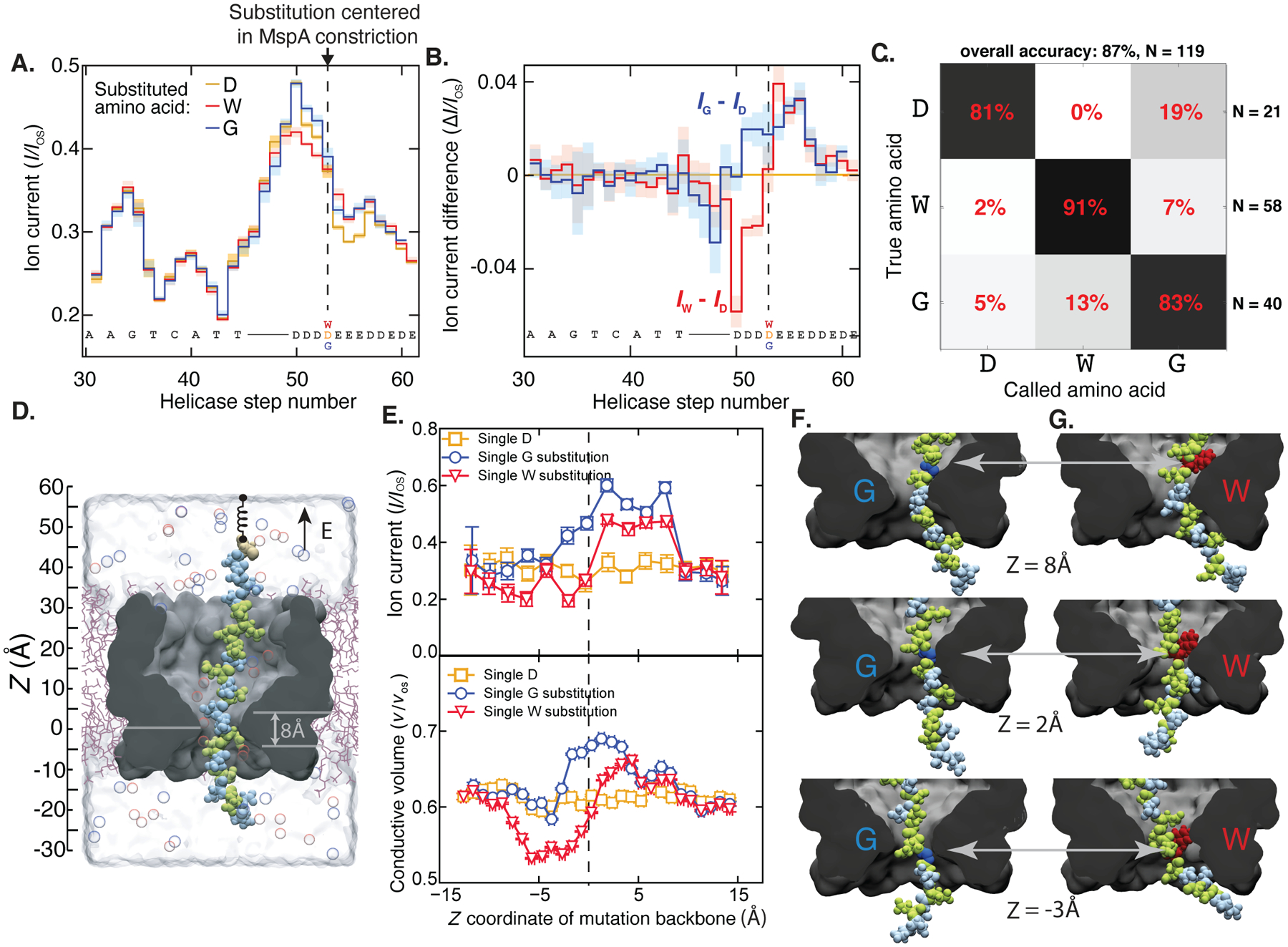
(A) Consensus ion current sequences for each of the three measured variants (D, gold; W, red; G, blue), which differ significantly at the site of the amino acid substitution. (B) Difference in ion current between the W (red) and G (blue) variants and the D variant. Error bars are standard deviations. (C) Confusion matrix showing error modes of a blind classifier in identifying variants of reads, demonstrating an 87% single-read accuracy. (D) All-atom model where a reduced-length MspA pore (grey) confines a polypeptide chain (Glu: green, Asp: light blue; Cys: beige). The top end of the peptide is anchored using a harmonic spring potential, representing the action of the helicase at the rim of a full-length MspA. Water and ions are shown as semitransparent surface and spheres, respectively. (E) Top: Ionic current in MspA constriction versus z coordinate of the mutated residue backbone from MD simulations. Bottom: Fraction of nanopore construction volume available for ion transport. Vertical and horizontal error bars denote standard errors and standard deviations, respectively. (G,H) Representative molecular configurations observed in MD simulations of peptide variants. Glycine and tryptophane residues are shown in dark blue and red, respectively. Significant peptide/pore surface interactions are observed.