

Abstract
Purpose of review
Here we summarize recent advancements in β cell replacement as a therapy for type 1 diabetes.
Recent findings
β cell replacement therapy has been proposed as a cure for type 1 diabetes with the introduction of the Edmonton protocol for cadaveric islet transplantation. To allow widespread use of this approach, efforts have focused on establishing an abundant source of insulin-producing β cells, protecting transplanted cells from ischemia-mediated death, immune rejection, and re-occurring autoimmunity. Recent developments addressing these issues include generation of insulin-producing cells from human pluripotent stem cells, different encapsulation strategies and prevention of ischemia upon transplant.
Summary
Despite significant advances in generating functional β cells from human pluripotent stem cells, several key challenges remain in regard to the survival of β cell grafts, protection from (auto-) immune destruction and implementation of additional safety mechanisms before a stem cell-based cell replacement therapy approach can be widely applied. Taking current findings into consideration, we outline a multilayered approach to design immune-privileged β cells from stem cells using state of the art genome editing technologies that if successfully incorporated could result in great benefit for diabetic patients and improve clinical results for cell replacement therapy.
Keywords: autoimmunity, β cells, cell therapy, diabetes, insulin, stem cells
INTRODUCTION
Type 1 diabetes (T1D) is an immune-mediated disease characterized by the specific destruction of pancreatic β cells resulting in insulin deficiency [1]. If left untreated T1D is life threatening and requires lifelong treatment with exogenous insulin via multiple daily injections or the automatic subcutaneous infusion of insulin by a pump. However, risk of acute hypoglycemia and long-term complications remain including retinopathy, nephropathy, neuropathy, and cardiovascular disease. Additionally, T1D incidence is increasing, especially in children less than 5 years of age [2]. Cell replacement therapy with insulin-producing β cells has been postulated as a cure for T1D and extensive research efforts have focused on this approach during the last two decades. In this review, we highlight recent advances in β cell replacement focusing on the generation of pancreatic cells from pluripotent stem cells, discuss approaches for their delivery and protection after transplantation and provide an opinion on how to best address remaining challenges for this technology.
CADAVERIC ISLET TRANSPLANTATION
One of the first successful attempts at β cell replacement for T1D occurred in the 2000s with the introduction of the Edmonton protocol, which infused cadaveric human donor islets, containing β cells, into the liver through the portal vein of long-standing T1D patients. Prevention of immune rejection included a regimen of nonsteroid immunosuppressive drugs (sirolimus, tacrolimus, and daclizumab) [3]. Importantly, the Edmonton protocol has achieved partial success in replacing b cells with the majority of T1D patients off exogenous insulin shortly after the procedure. Follow-up showed that exogenous insulin independence was only maintained in some patients after 15 months [4]. One of the major limitations was the need for immunosuppressive drugs, some of which impair β cell function and survival [5–7]. Additionally, systemic side-effects of immune suppressants are generally not deemed appropriate, especially for children and adolescents with T1D. Despite these limitations with the initial Edmonton protocol, intensive research has taken place primarily focused on improving outcomes for islet transplants. Recent data from the Collaborative Islet Transplant Registry indicate that insulin independence after islet transplantation has improved, lasting up to 3 years in 44% of patients [8]. The Edmonton protocol has demonstrated a proof-of-principle for β cell replacement therapy in established T1D patients and better glycemic control than exogenous insulin administration [9], sparking research into improving islet survival after transplantation and finding alternative cell sources for β cell replacement in diabetes. One attractive approach is the direct differentiation of stem cells into insulin-producing β cells.
STEM CELLS AS A SOURCE OF INSULINPRODUCING β CELLS
Human embryonic stem cells (hESCs) were first derived from the inner cell mass of fertilized blastocysts in 1998 by Thomson et al. [10]. Pluripotent hESCs divide rapidly and can, with appropriate signals provided, give rise to every cell type found in the human body. Although ethical concerns have surrounded hESCs, the advent of reprogramming human somatic cells into induced pluripotent stem cells [11], practically considered equivalent to hESCs, has largely removed these concerns. Thus, human pluripotent stem cells (hPSC) could potentially provide an unlimited supply of any desired cell type for replacement therapy. Extensive work to formulate step-wise direct differentiation protocols to guide hPSCs along key developmental steps resulted in the first generation of hormone-expressing endocrine cells in 2006 [12]. However, these differentiated endocrine cells were mostly poly-hormonal and lacked the ability to secrete insulin in response to glucose, the hallmark function of bona fide pancreatic β cells. Furthermore, differentiated cultures also contain various other pancreatic cell types, including pancreatic progenitors from different developmental stages. Strikingly, subsequent transplantation of such heterogenous pancreatic cells into immune-deficient mice resulted in the generation of glucose responsive β cells after several months [13]. Notably, grafts could maintain euglycemia in mice rendered diabetic by chemical ablation of endogenous β cells, illustrating the translational potential of direct differentiation of hPSC in vitro followed by in-vivo transplantation for cellular therapy. These experiments provided for the first-time formal evidence that hPSC can indeed give rise to functional β cells, albeit requiring in-vivo transplantation, and encouraged the research community to devise strategies to generate insulin-producing cells in vitro.
Recently, we and others have described the generation of glucose-responsive β-like cells, referred simply as β cells from this point forward, within a few weeks under cell culture conditions from hPSCs using large-scale suspension culture systems [14–16]. This finding has been subsequently reproduced by independent investigators [17–21]. It appears the key to successful generation of β cells in vitro is a highly efficient generation of competent pancreatic progenitor cells followed by strong induction signals for endocrine differentiation. In addition, the preservation of the generated β cell phenotype by identification and addition of several small molecules/growth factors to the later stages of culture seems to be equally important in the achieved success. Generated β cells exhibit several key features of bona fide β cells, including marker expression, ultrastructural features, and their ability to respond to changes in glucose levels by secreting insulin in a static assay [14]. Importantly, hPSC-derived β cells display functionality shortly after transplantation, a distinct benefit when compared with the longer time period required for pancreatic progenitor differentiation into functional β cells in vivo described earlier. However, upon close examination several aspects indicate that hPSC-derived β cells resemble functional, but immature neonatal β cells rather than mature β cells from adult donors, highlighting the continued need to further elucidate the key features that define and generate mature β cells. Despite these remaining questions, the successful demonstration of generating functional β cells has tremendously increased the existing excitement for using stem cell-derived pancreatic β cells for cell therapy in patients, both in commercial and academic settings. In addition, by generating large numbers of genetically identical β cells in the laboratory researchers can address remaining challenges for successful implementation of cell therapy in a reproducible manner. This ability is further enhanced by the rapidly evolving field of genome engineering of hPSC that now allows the establishment of novel human model systems that were previously restricted only to animal models [22]. During the last 4 years, when hPSC-derived β cells were first described [14–16], researchers used these cells to address questions such as (1) where to transplant and how to protect grafts from ischemia during the peri-transplant period, (2) how to shelter grafts from the patient‘s own immune system, both in allo- and autologous settings, and (3) how to provide additional safety mechanisms.
IMPROVING THE MICROENVIRONMENT
In normal physiological conditions, capillaries run through highly metabolically active pancreatic islets providing almost all cells in the islets with equal oxygen (averaging 5% oxygen tension) and nutrient levels [23]. Transplanted islets are generally infused through the portal vein and often results in an immediate 60% loss of transplanted β cells [24]. This is thought to be caused by ischemia occurring in cells located in the middle of the transplanted islet mass because of low oxygen concentration (approximately 1% O2) [25]. Ischemia is a major problem for transplantation technology as it accounts for a high level of cell loss. Recently, stem cell-derived β cell susceptibility to ischemia has been investigated. We showed that approximately half of all hPSC-derived islet clusters die within the first week upon subcutaneous transplantation in immune-deficient mice and to slightly lesser extent under the kidney capsule supporting the concept that greater vascularization of the transplant site increases graft survival [26■]. Interestingly, β cells are preferentially lost from grafts when compared with less differentiated pancreatic progenitor cells in line with the idea that greater differentiation status correlates with increased susceptibility of stress-induced cell death. We further showed that cells are killed by two independent mechanisms, nutrition deprivation and hypoxia that act synergistically in an in vivo setting. Supplementation of specific amino acids rescued nutrition deprivation. Generation of β cells under physiological oxygen tension of 5%, in contrast to common atmospheric 20% oxygen pressure, conferred hypoxia resistance. Combination of both approaches during transplantation significantly improves graft survival and provides a simple and effective means to reduce ischemia-induced cell death of hPSC-derived β cells.
However, even with this improved strategy β cells are preferentially lost from grafts and not all cells survive. Genetic lineage tracing approaches are required to determine their fate. Do β-like cells simply dedifferentiate into a more resilient progenitor cell state? Is transdifferentiation into other hormone expressing cell types occurring? What other mechanisms contribute to cell loss? Answering these critical questions will provide the field with the required information to further improve the immediate outcome of hPSC-derived cell transplants, especially for β cells.
ENCAPSULATION OF STEM CELL-DERIVED β CELLS
Another area of focus in the field has been the encapsulation of islets and β cells. Ideally, the device or membrane would block physical interactions with the patient’s own tissue and cells but allow exchange of nutrients and hormones. If successful, this technology could remove the need for lifelong immune suppression currently required for cadaveric islet grafts.
Currently, the first clinical trial using macroencapsulated hES-derived pancreatic progenitor cells is underway (NCT02239354) [27]. Progenitor cells are expected to further differentiate into functional β cells in patients over several weeks to months analogous to what has been observed using animal models [13]. However, while the research community awaits results from this first in human safety trial, modifications to the macroencapsulation device will likely be needed to enhance cell survival. An encapsulation strategy that allows blood vessels to vascularize the encapsulated cells may improve long-term survival and is under clinical investigation (NCT03162926).
Another concern with macroencapsulation devices is the potential for scar tissue formation because of a foreign body reaction. Despite potential technical roadblocks of macroencapsulation devices, trials in humans are tremendously important and will inform researchers of necessary refinements using this strategy. Indeed, different materials and compositions have been evaluated with respect to aptitude of foreign body reaction and their ability to allow diffusion of nutrients but impede immune cells [28,29]. Studies by us and others have also proposed alternative transplantation sites compared with subcutaneous tissue which is currently being used to allow ease of insertion and retrievability of grafts at defined time periods [30–33]. Additionally, the ability to retrieve the encapsulation device is important as there is the distinct potential of incompletely differentiated cells to give rise to a teratoma [34].
In sum, each study provides further insights into this attractive strategy, however none have yet achieved a major breakthrough. As an alternative to macroencapsulation devices, microencapsulation could provide protection and increase the surface area for nutrient absorption; however, this comes at the expense of retrievability. Alginate-based hydrogels have been extensively used to microencapsulate cell clusters, including islets, but strong immune-mediated reactions have been observed in mice, primates, and humans [35–40]. A recent combinatorial screening approach for chemical modification of alginates has largely overcome this hurdle and demonstrated only minimal foreign body reaction using this material in primates [41]. Using this strategy, encapsulation of hPSC-derived β cells, followed by intraperitoneal transplantation, resulted in long-term glycemic control of chemically induced diabetes in immune competent mice [42■]. It will be paramount to perform similar experiments in model systems of autoimmune diabetes to further this approach. Taken together, both macro and microencapsulation have recently demonstrated significant advances in material composition and encapsulation strategies, but the efficient survival and functionality of encapsulated cells seems to remain challenging. Detailed studies following encapsulated grafts immediately after transplant are required to determine if transplantation of functional β cells compared with more resilient progenitor cells is feasible. Alternatively, transplantation of naked cells could be envisioned, circumventing β cell survival and function challenges currently posed by encapsulation devices. However, in such scenario effective protection from the host immune system, including autoimmunity, need to be present.
IMMUNE-PRIVILEGED DESIGNER β CELLS
In parallel with studies to differentiate hPSCs into functional β cells followed by encapsulation, the field of T1D immunology has been investigating the immune-b cell interface within human islets from T1D organ donors [43]. Several groups have reported that the polymorphic HLA-A,B,C molecules are upregulated within β cells in T1D organ donors [44,45]. These specific HLA class I molecules occur in many different subtypes and differ among individuals to allow the presentation of a broad repertoire of peptides to CD8+ T cells or cytotoxic T lymphocytes, which have the requisite intracellular machinery to kill β cells (Fig. 1A). Cytotoxic CD8+ T cells specific for islet proteins have been identified and visualized within islets from organ donors with recent onset and established T1D [46]. Interestingly, residual β cells within normal appearing islets of T1D individuals lack this HLA-A,B,C overexpression [47,48]. Thus, β cells used for replacement therapy should not be able to express HLA-A, B, and C helping to abrogate both allo- and autoimmune responses. (Fig. 1B).
FIGURE 1.
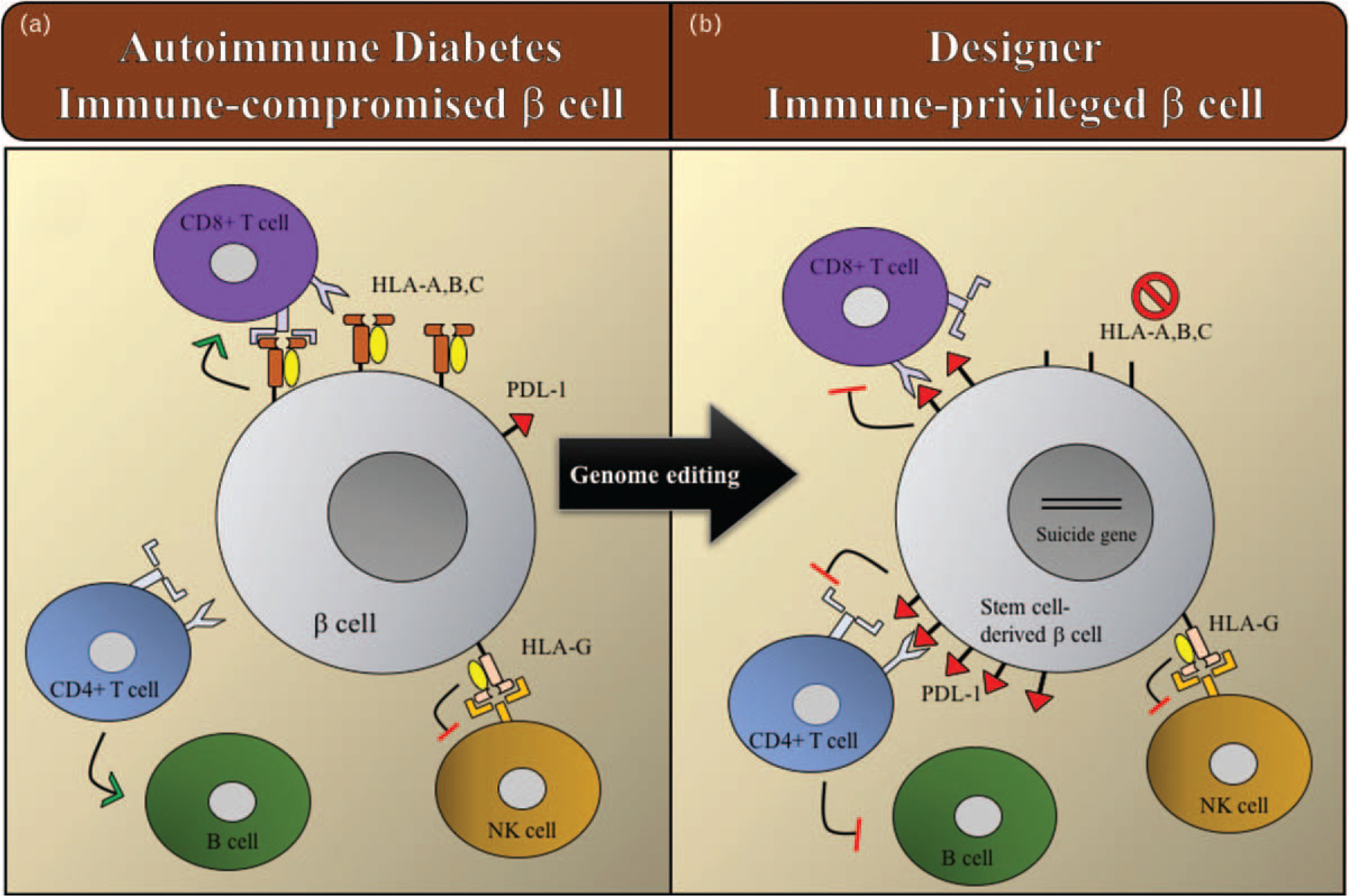
Schematic of proposed immune-privileged designer β cells for type 1 diabetes. (A) Autoimmune attack on β cells in which there is hyperexpression of polymorphic HLA-A,B,C molecules, which present antigens to cytotoxic CD8+ T cells. (B) Mechanisms to block the autoimmune attack with engineered immune-privileged β cells that lack HLA-A,B,C and express PD-L1 to deactivate specific immune cells. Expression of ‘self’ molecules such as HLA-G remain intact to protect cells from immune destruction by Natural Killer (NK) cells. Green arrow represents activation and the red line deactivation of immune cells.
There are also other HLA class I molecules such as HLA-E, F, and G, which are not polymorphic and function to regulate the activation of immune cells [49,50]. HLA-G in particular, regulates natural killer cell activation and has been shown to be present in human islets with expression levels parallel to their secretory activity [51]. Advances in genome engineering, such as with clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, allows efficient modification of hPSC cells. Indeed, hESCs with a genetic knock out of the β-2microglobulin chain, shared among all HLA class I molecules, results in an effective abrogation of all HLA surface molecules [52]. This approach has been shown to decrease CD8+ T lymphocyte-mediated lysis but increase of natural killer-mediated death of different hES-derived cell types. We believe that specifically targeting HLA-A,B,C for inactivation and not the invariant light chain, β2-microglobulin that abrogates all HLA class I molecules, will be an effective way to protect hPSC-derived β cells from immune-mediated destruction.
To construct the ideal immune-privileged β cell, other subsets of immune cells also need to be regulated, namely, activated CD4+ T cells and B cells. We hypothesize that this could be accomplished through an immune checkpoint pathway, the program cell death protein 1, and ligand (PD1/PD-L1) axis (Fig. 1B). Indeed, a recent study showed that overexpression of PD-L1 in hematopoietic stem and progenitor cells inhibits an autoimmune attack in vitro, providing a proof of principle for this approach [53■]. In the normal adaptive immune response, the program cell death protein (PD1) is expressed on T cells and B cells, whereas its ligand PD-L1 is expressed on a variety of cells including antigen presenting cells. The interaction between PD1 and PD-L1 results in dampening an immune response. Interestingly, cancer immunotherapies have targeted this axis and there are a number of clinically approved monoclonal antibodies that block PD1 or PD-L1, thereby resulting in activated lymphocytes that target tumors [54]. A number of malignancies (melanoma, lung cancer, renal cell cancer, and others) have evolved to line themselves with PD-L1 that downregulate an immune response [55–64]. Despite great success in tumor regression and enhanced survival, inhibition of immune checkpoints also causes immune-related side-effects, especially autoimmunity directed toward self-tissues [65,66]. The development of T1D has been reported from a few weeks to several months after the start of treatment [67,68■,69]. Evidence exists that these cases are indeed the immune-mediated form of diabetes as there are case reports with the presence of T1D-associated autoantibodies and diabetes-specific T-cells in the peripheral circulation [70]. Notably, T1D has only been reported with PD1/PD-L1 blockade and not with other immune checkpoint inhibitors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade, highlighting the importance of this regulatory mechanism in T1D development.
We propose to develop a cell replacement therapy approach that generates well tolerated, immune-privileged β cells from hPSCs using genome engineering prior to transplantation (Fig. 1B). Potential concern with this approach of designing immune-privileged β cells is that such cells might be prone to viral infection and cancerogenesis. To directly address these issues and provide an emergency ‘exit switch’ in case transplanted cells become erratic, inclusion of an inducible ‘suicide gene’ will be necessary [71].
CONCLUSION
After reviewing the current literature and research landscape for β cell replacement therapies, we concluded with several key observations: stem cell-derived β cells can now be generated on a large scale effectively removing the shortage of cadaveric islets previously preventing wide spread implementation of cell therapy for T1D patients. However, three main challenges remain, namely, (1) how and where to deliver these cells without great loss, (2) how to protect grafts from allogenic rejection without systemic immune suppression and from re-occurring autoimmunity, and (3) how to provide sufficient safety measurements to allow clinical trials. Addressing these remaining challenges will allow for well tolerated and specific β cell replacement for T1D patients in the ensuing years.
KEY POINTS.
hPSC can provide an abundant source of insulin-producing cells for βcell replacement therapy in diabetes.
β cell survival after transplantation can be improved by reducing ischemic-induced death.
Transplantation of encapsulated stem cell-derived pancreatic cells is under current clinical investigation in T1D patients.
Using knowledge of the immune-β cell interface in human T1D combined with state of the art genome editing technologies provides an avenue to generate ‘immune-privileged β cells’ which holds promise for improved outcomes for β cell replacement therapy.
Acknowledgements
A.W.M. is an inventor on a patent titled ‘Compounds that modulate autoimmunity and methods of using the same,’ licensed to ImmunoMolecular Therapeutics (US patent number 9,629,848). AWM is a scientific cofounder of ImmunoMolecular Therapeutics and own shares in the company. H.A.R. serves on the Scientific Advisory Board of Prellis Biologics Inc. and as a Consultant for Sigilon Therapeutics.
Financial support and sponsorship
This work was supported by NIH Grants (DK108868, DK110845, DK032083, DK104223, AI1019905), the Children’s Diabetes Foundation and the Barbara Davis Center Translational Research Unit.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014; 383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer-Davis EJ, Lawrence JM, Dabelea D, et al. Incidence trends of type 1 and type 2 diabetes among youths, 2002–2012. N Engl J Med 2017; 376: 1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro AJ, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000; 343:230–238. [DOI] [PubMed] [Google Scholar]
- 4.Brennan DC, Kopetskie H, Sayre P, et al. Long-term follow-up of the Edmonton protocol of islet transplantation in the United States. Am J Transplant 2016; 16:509–517. [DOI] [PubMed] [Google Scholar]
- 5.Rangel EB. Tacrolimus in pancreas transplant: a focus on toxicity, diabetogenic effect and drug–drug interactions. Expert Opin Drug Metab Toxicol 2014; 10:1585–1605. [DOI] [PubMed] [Google Scholar]
- 6.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 2007; 117:2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niclauss N, Bosco D, Morel P, et al. Rapamycin impairs proliferation of transplanted islet β cells. Transplantation 2011; 91:714–722. [DOI] [PubMed] [Google Scholar]
- 8.Barton FB, Rickels MR, Alejandro R, et al. Improvement in outcomes of clinical islet transplantation: 1999–2010. Diabetes Care 2012; 35:1436–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes-Walker DJ, Gunton JE, Hawthorne W, et al. Islet transplantation provides superior glycemic control with less hypoglycemia compared with continuous subcutaneous insulin infusion or multiple daily insulin injections. Transplantation 2017; 101:1268–1275. [DOI] [PubMed] [Google Scholar]
- 10.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. cell 2007; 131:861–872. [DOI] [PubMed] [Google Scholar]
- 12.D’Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006; 24:1392–1401. [DOI] [PubMed] [Google Scholar]
- 13.Kroon E, Martinson LA, Kadoya K, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008; 26:443–452. [DOI] [PubMed] [Google Scholar]
- 14.Russ HA, Parent AV, Ringler JJ, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J 2015; 34:1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rezania A, Bruin JE, Arora P, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014; 32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 16.Pagliuca FW, Millman JR, Gürtler M, et al. Generation of functional human pancreatic β cells in vitro. Cell 2014; 159:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Z, Li QV, Lee K, et al. Genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of pancreatic development and diabetes. Cell Stem Cell 2016; 18:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng H, Guo M, Zhou T, et al. An isogenic human ESC platform for functional evaluation of genome-wide-association-study-identified diabetes genes and drug discovery. Cell stem cell 2016; 19: 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Z-D, Lee K, Yang D, et al. Genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic development. Cell Stem Cell 2017; 20: 675.e67–688.e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo M, Zhang T, Dong X, et al. Using hESCs to probe the interaction of the diabetes-associated genes CDKAL1 and MT1E. Cell Rep 2017; 19: 1512–1521. [DOI] [PubMed] [Google Scholar]
- 21.Ameri J, Borup R, Prawiro C, et al. Efficient generation of glucose-responsive beta cells from isolated GP2(+) human pancreatic progenitors. Cell Rep 2017; 19:36–49. [DOI] [PubMed] [Google Scholar]
- 22.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 23.Carlsson P-O, Liss P, Andersson A, Jansson L. Measurements of oxygen tension in native and transplanted rat pancreatic islets. Diabetes 1998; 47:1027–1032. [DOI] [PubMed] [Google Scholar]
- 24.Delaune V, Berney T, Lacotte S, Toso C. Intraportal islet transplantation: the impact of the liver micro-environment. Transpl Int 2017; 30:227–238. [DOI] [PubMed] [Google Scholar]
- 25.Carlsson P-O, Palm F, Andersson A, Liss P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes 2001; 50:489–495. [DOI] [PubMed] [Google Scholar]
- 26. Faleo G, Russ HA, Wisel S, et al. Mitigating ischemic injury of stem cell derived insulin-producing cells after transplant. Stem Cell Reports 2017; 9:807–819.28803916 ■ This article shows that stem cell derived beta cells suffer ischemic injury upon transplant. A simple strategy is provided to reduce these effects resulting in improved transplantation outcomes.
- 27.Mora C, Serzanti M, Consiglio A, et al. Clinical potentials of human pluripotent stem cells. Cell Biol Toxicol 2017; 33:351–360. [DOI] [PubMed] [Google Scholar]
- 28.Desai T, Shea LD. Advances in islet encapsulation technologies. Nat Rev Drug Discov 2017; 16:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang R, Faleo G, Russ HA, et al. Nanoporous immunoprotective device for stem-cell-derived β-cell replacement therapy. ACS Nano 2017; 11:7747–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yasunami Y, Nakafusa Y, Nitta N, et al. A novel subcutaneous site of islet transplantation superior to the liver. Transplantation 2018; doi: 10.1097/TP.0000000000002162. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Schmidt C Pancreatic islets find a new transplant home in the omentum. Nature Biotechnol 2017; 35:8. [DOI] [PubMed] [Google Scholar]
- 32.Cantarelli E, Citro A, Pellegrini S, et al. Transplant site influences the immune response after islet transplantation: bone marrow versus liver. Transplantation 2017; 101:1046–1055. [DOI] [PubMed] [Google Scholar]
- 33.Brady A-C, Martino MM, Pedraza E, et al. Proangiogenic hydrogels within macroporous scaffolds enhance islet engraftment in an extrahepatic site. Tissue Eng Part A 2013; 19:2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujikawa T, Oh S-H, Pi L, et al. Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am J Pathol 2005; 166:1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuch BE, Keogh GW, Williams LJ, et al. Safety and viability of microencapsulated human islets transplanted into diabetic humans. Diabetes Care 2009; 32:1887–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider S, Feilen PJ, Brunnenmeier F, et al. Long-term graft function of adult rat and human islets encapsulated in novel alginate-based microcapsules after transplantation in immunocompetent diabetic mice. Diabetes 2005; 54:687–693. [DOI] [PubMed] [Google Scholar]
- 37.Foster JL, Williams G, Williams LJ, Tuch BE. Differentiation of transplanted microencapsulated fetal pancreatic cells. Transplantation 2007; 83: 1440–1448. [DOI] [PubMed] [Google Scholar]
- 38.Elliott R, Escobar L, Tan P. Intraperitoneal alginate-encapsulated neonatal porcine islets in a placebo-controlled study with 16 diabetic cynomolgus primates. Transplant Proc 2005; 37:3505–3508. [DOI] [PubMed] [Google Scholar]
- 39.Elliott R, Escobar L, Calafiore R. Transplantation of micro-and macroencapsulated piglet islets into mice and monkeys. Transplant Proc 2005; 37:466–469. [DOI] [PubMed] [Google Scholar]
- 40.Duvivier-Kali VF, Omer A, Parent RJ, et al. Complete protection of islets against allorejection and autoimmunity by a simple barium-alginate membrane. Diabetes 2001; 50:1698–1705. [DOI] [PubMed] [Google Scholar]
- 41.Vegas AJ, Veiseh O, Doloff JC, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotechnol 2016; 34:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vegas AJ, Veiseh O, Gürtler M, et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat Med 2016; 22:306–311.26808346 ■ First demonstration of rescue from diabetes in an immune competent mouse model by microencapsulated hPSC derived beta cells.
- 43.Pugliese A, Yang M, Kusmarteva I, et al. The juvenile diabetes research foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) program: goals, operational model and emerging findings. Pediatr Diabetes 2014; 15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson SJ, Rodriguez-Calvo T, Gerling IC, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016; 59:2448–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bottazzo GF, Dean BM, McNally JM, et al. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med 1985; 313:353–360. [DOI] [PubMed] [Google Scholar]
- 46.Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 2012; 209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Calvo T, Suwandi JS, Amirian N, et al. Heterogeneity and lobularity of pancreatic pathology in type 1 diabetes during the prediabetic phase. J Histochem Cytochem 2015; 63:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nejentsev S, Howson JM, Walker NM, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007; 450:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pazmany L, Mandelboim O, Valés-Gómez M, et al. Protection from natural killer cell-mediated lysis by HLA-G expression on target cells. Science 1996; 274:792–795. [DOI] [PubMed] [Google Scholar]
- 50.Gornalusse GG, Hirata RK, Funk SE, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 2017; 35:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cirulli V, Zalatan J, McMaster M, et al. The class I HLA repertoire of pancreatic islets comprises the nonclassical class Ib antigen HLA-G. Diabetes 2006; 55:1214–1222. [DOI] [PubMed] [Google Scholar]
- 52.Wang D, Quan Y, Yan Q, et al. Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl Med 2015; 4:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nasr MB, Tezza S, D’addio F, et al. PD-L1 genetic overexpression or pharmacological restoration in hematopoietic stem and progenitor cells reverses autoimmune diabetes. Science Transl Med 2017; 9:; eaam7543. ■ Overexpression of PD-L1 in hematopoietic stem and progenitor cells abrogates immune responses in vitro and reverted diabetes in new onset NOD mice in vivo.
- 54.Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/ PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget 2017; 8:24031–24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 2017; 377:1824–1835. [DOI] [PubMed] [Google Scholar]
- 56.Shoushtari AN, Friedman CF, Navid-Azarbaijani P, et al. Measuring toxic effects and time to treatment failure for nivolumab plus ipilimumab in melanoma. JAMA Oncol 2018; 4:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017; 18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol 2017; 10:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noman MZ, Janji B, Abdou A, et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017; 6:e1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med 2016; 374: 2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol 2017; 18:1202–1210. [DOI] [PubMed] [Google Scholar]
- 62.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katz H, Wassie E, Alsharedi M. Checkpoint inhibitors: the new treatment paradigm for urothelial bladder cancer. Medical Oncol 2017; 34:170. [DOI] [PubMed] [Google Scholar]
- 64.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med 2015; 373:1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med 2018; 378: 158–168. [DOI] [PubMed] [Google Scholar]
- 66.Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy: immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol 2017; 13:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gauci A-L, Laly P, Vidal-Trecan T, et al. Autoimmune diabetes induced by PD-1 inhibitor: retrospective analysis and pathogenesis: a case report and literature review. Cancer Immunol Immunother 2017; 66:1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Akturk HK, Michels AW. Adverse events associated with immune checkpoint blockade. N Engl J Med 2018; 378:1163–1164. ■ This letter compares and contrasts prototypical childhood onset type 1 diabetes with autoimmune diabetes induced by immune checkpoint blockade.
- 69.Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One 2016; 11:e0160221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hughes J, Vudattu N, Sznol M, et al. Precipitation of autoimmune diabetes with anti-PD-1 immunotherapy. Diabetes Care 2015; 38:e55–e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sulkowski M, Konieczny P, Chlebanowska P, Majka M. Introduction of exogenous HSV-TK suicide gene increases safety of keratinocyte-derived induced pluripotent stem cells by providing genetic ‘emergency exit’ switch. Int J Mol Sci 2018; 19:197. [DOI] [PMC free article] [PubMed] [Google Scholar]