

Summary
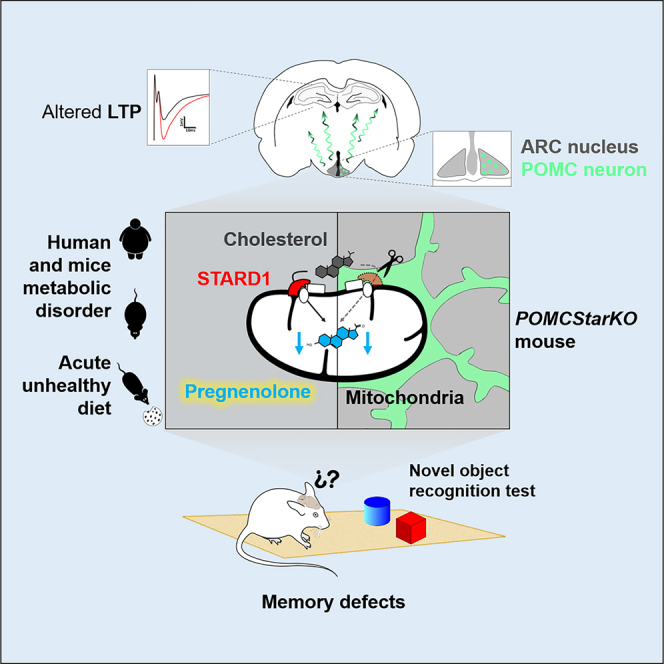
Obesity and type 2 diabetes are associated with cognitive dysfunction. Because the hypothalamus is implicated in energy balance control and memory disorders, we hypothesized that specific neurons in this brain region are at the interface of metabolism and cognition. Acute obesogenic diet administration in mice impaired recognition memory due to defective production of the neurosteroid precursor pregnenolone in the hypothalamus. Genetic interference with pregnenolone synthesis by Star deletion in hypothalamic POMC, but not AgRP neurons, deteriorated recognition memory independently of metabolic disturbances. Our data suggest that pregnenolone’s effects on cognitive function were mediated via an autocrine mechanism on POMC neurons, influencing hippocampal long-term potentiation. The relevance of central pregnenolone on cognition was also confirmed in metabolically unhealthy patients with obesity. Our data reveal an unsuspected role for POMC neuron-derived neurosteroids in cognition. These results provide the basis for a framework to investigate new facets of POMC neuron biology with implications for cognitive disorders.
Keywords: hypothalamus, obesity, diabetes, cognitive function, recognition memory, neurosteroids, POMC neurons, pregnenolone, Stard1, mitochondria
Graphical abstract
Highlights
-
•
Acute western diet impairs memory performance, which is reversed by pregnenolone
-
•
Pregnenolone synthesis interference in POMC neurons causes cognitive dysfunction
-
•
POMC pregnenolone mediates memory function via hippocampal mechanisms
-
•
Pregnenolone in the CSF of individuals with unhealthy obesity correlates with cognitive score
Ramírez et al. show that reduced hypothalamic pregnenolone levels, in the context of metabolic diseases, are associated with cognitive deterioration. POMC neuron-derived pregnenolone is a key mediator of cognition, but not metabolism, acting via hippocampal mechanisms. These results identify an unsuspected role for POMC neuron-derived pregnenolone in memory performance.
Introduction
Obesity and type 2 diabetes mellitus (T2D) have reached epidemic dimensions, thus becoming major global health issues (Blüher, 2019). Epidemiological and clinical studies denote that, among the several complications related to these metabolic disorders, cognitive dysfunction is a prominent comorbidity. Indeed, evidence shows that obesity and T2D are associated with diverse forms of cognitive decline, including subtle cognitive decrements, mild cognitive impairment, and even severe dementia (Biessels and Despa, 2018; Tanaka et al., 2020).
An emerging concept posits brain insulin resistance as a relevant condition linking metabolic disorders with cognitive deterioration (Biessels and Despa, 2018; Sripetchwandee et al., 2018; Tanaka et al., 2020). In this context, multiple early risk factors (poor glycemic control, hypertension, and dyslipidemia) have been proposed to participate in this pathological process (Biessels and Despa, 2018; Feinkohl et al., 2015). However, these studies were unable to dissociate confounding metabolic alterations or to dissect out their specific contribution to cognitive decline. Therefore, our current understanding of the initial events and mechanisms triggering cognitive dysfunction within the framework of metabolic abnormalities remains largely enigmatic.
In recent years, a complex bidirectional interplay between the disruption of brain cholesterol metabolism and insulin resistance has been revealed. Indeed, murine models of diabetes and aging display a reduction in brain cholesterol biosynthesis associated with cognitive phenotypes (Martín-Segura et al., 2019; Suzuki et al., 2010, 2013). In this regard, recent findings have disclosed a role for the disruption of intracellular cholesterol trafficking in neurodegeneration, with the identification of steroidogenic acute regulatory protein (Stard1) as a potential preclinical marker associated with early stages of Alzheimer disease (Arenas et al., 2020). Cholesterol is the precursor for the biogenesis of neurosteroids, which are steroid hormone derivatives locally synthesized within the brain. In this process, Stard1 mediates the trafficking of cholesterol to the mitochondria, which is subsequently metabolized by CYP11A1 (P450Scc) into the neurosteroid precursor pregnenolone (Arenas et al., 2017; Manna et al., 2016).
Since its breakthrough description in the early 1990s, alterations in neurosteroid production or action have been implicated in the etiology of a wide range of neuropsychiatric and neurological disorders: anxiety, depression, epilepsy, addiction, neurodegeneration, learning, and memory (Ratner et al., 2019). Numerous brain regions are able to synthetize and respond to neurosteroids, including the hypothalamus and classical memory-related structures (hippocampus, amygdala, or cortex), thus influencing the performance of different types of memory (Mellon et al., 2001; Ratner et al., 2019).
Despite receiving much less attention in the field of cognitive science, the hypothalamus is crucially positioned to connect metabolic status with cognition for diverse reasons. First, because distinct hypothalamic nuclei and neuronal types (namely, POMC and AgRP neurons) play fundamental roles in the regulation of systemic energy balance and in the development of obesity and T2D (Jais and Brüning, 2021). Second, because the hypothalamus has been historically related to memory disorders (Vercruysse et al., 2018), and third, because evidence from recent studies shows that it may act as an interface for diverse types of cognition (Burdakov and Peleg-Raibstein, 2020). Despite this evidence, the role of the hypothalamus as a nexus connecting metabolism with cognitive disturbances and the underlying molecular and cellular underpinnings remains unknown.
In the present study, we hypothesized that hypothalamic neuronal populations that are fundamental for energy homeostasis control are at the core of a functional axis that links metabolism and cognitive performance.
Results
Cognitive impairment caused by short-term obesogenic diet, in the absence of overt metabolic disturbances, is reversed by central pregnenolone administration
Obesity and T2D have emerged as risk factors for the development of cognitive deficits, but the precise basis of this relationship is masked by robust disease-related phenotypical traits (i.e., adiposity and hyperglycemia) (Biessels and Despa, 2018; Tanaka et al., 2020). To dissociate the impact of overt metabolic alterations on cognitive dysfunction, we fed C57Bl/6J mice with high-fat high-sucrose western diet (WD) for just 4 days (4d-WD; Figure S1A). This dietary regime did not cause noticeable metabolic perturbations (Figures S1B–S1E), thus excluding obesity and altered systemic glucose homeostasis per se as confounding factors. We next assessed spatial and recognition memory via Barnes maze tests (BMTs; Figure 1A) and novel object recognition test (NORT; Figure 1B), respectively. Short-term WD neither interfered with task learning (Figure S1F) nor caused cognitive impairments in spatial memory (Figures 1C–1F). However, it significantly deteriorated recognition memory when compared with chow-fed counterparts (Figures 1G–1J) in the absence of exploration or locomotor alterations (Figures S1G–S1J).
Figure 1.

Cognitive impairment caused by short-term obesogenic diet is reversed by central pregnenolone administration.
(A) Schematic illustration of Barnes maze test (BMT) and experimental timeline. The white circle and the rectangle represent the scape hole and scape chamber location, respectively. The position of the scape chamber remained constant on each trial. On the test day, the scape hole was closed and the chamber removed.
(B) Schematic illustration of the novel object recognition test (NORT) and experimental chronology.
(C–F) Recorded parameters to assess BMT performance in mice fed with either chow or western diet for 4 days (4d-WD) during the test phase: (C) latency to target hole, (D) time spent in target quadrant, (E) total distance traveled, and (F) representative traces of mouse movement trajectories (n = 11/diet).
(G–J) Recorded parameters to assess NORT performance in mice fed with either chow or 4d-WD during the test phase: (G) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (H) exploration time (time exploring novel object + time exploring familiar object), (I) total distance traveled, and (J) representative traces of mouse movement trajectories (n = 9–10/diet).
(K–N) Hippocampal long-term potentiation (LTP) study in mice fed with chow or 4d-WD. (K) Schematic illustration of the experimental strategy. The Schaffer collateral pathway (SC, red) was stimulated (Stim), and field potentials were recorded (Rec) in the CA1 region of the hippocampus. LTP was induced by theta-burst stimulation (TBS). DG, dentate gyrus; CA, cornu ammonis.
(L) Representative traces of individual recordings showing baseline fEPSPs before (black traces) and after (red traces) LTP induction.
(M) Time course of fEPSP recordings demonstrating robust changes in fEPSP slope in chow-fed animals after TBS (arrow) (n = 5 recordings from 4 animals). In 4d-WD animals, LTP induction was markedly impaired (n = 7 recordings from 4 animals).
(N) Quantification of fEPSP slope change over the last 45 min of recording period shown in (M).
(O–Q) Quantification of pregnenolone concentration in the (O) perirhinal cortex, (P) hippocampus, and (Q) arcuate-enriched mediobasal hypothalamus in mice fed with either chow or 4d-WD (n = 7–8/diet).
(R) Schematic illustration of central acute delivery of pregnenolone and subsequent NORT. Red arrow shows injection phase.
(S) Discrimination index during the test phase of NORT in mice fed with either chow or 4d-WD treated with vehicle (Veh) or pregnenolone (Preg) (n = 6–8/group).
All studies were performed on male mice at 8–9 weeks of age. Dots in panels represent individual samples. Data are presented as mean ± SEM. ∗p < 0.05.
The hippocampus plays a pivotal role in memory formation and flexibility. Long-term potentiation (LTP) is a form of synaptic plasticity that is closely linked with memory function in mammals. It has been reported that energy-dense diets reduce LTP magnitude in mice (Davis et al., 2021; Heyward et al., 2016). We conducted field potential recordings in the dendritic area of the CA1 region after stimulation of the Schaffer collaterals of CA3 neurons in hippocampal slices (Figure 1K) from C57Bl/6J mice fed with either chow or WD for 4 days. Input-output relationship, at diverse stimuli intensities (Figure S1K), and paired-pulse facilitation (Figure S1L) were comparable between both dietary conditions, suggesting analogous basal synaptic properties in terms of excitability and presynaptic calcium-dependent neurotransmitter release. However, acute WD feeding significantly reduced the field excitatory postsynaptic potential (fEPSP) slope after theta-burst stimulation (TBS), indicating impaired hippocampal LTP (Figures 1L–1N).
Neurosteroids have been implicated in a broad spectrum of neurological diseases (Ratner et al., 2019). The cholesterol metabolite pregnenolone is the first intermediate of the mitochondrial steroid synthesis pathway and precursor of virtually all steroid hormones (Figure S1M). We sought to determine whether acute exposure to WD was able to make an impact on neurosteroid concentration in brain regions classically involved in memory (perirhinal cortex and hippocampus) or metabolic control (arcuate nucleus of the hypothalamus; ARC). Notably, we observed a significant reduction in pregnenolone content in ARC-enriched samples but not in the perirhinal cortex or hippocampus of 4d-WD mice (Figures 1O–1Q). These results demonstrate that overt manifestations of obesity/diabetes per se are not the primary cause of hypothalamic pregnenolone synthesis shortage and cognition failure.
We next explored the potential causality of hypothalamic deficiency of pregnenolone biosynthesis upon cognitive impairment. To this end, we acutely administered pregnenolone intracerebroventricularly (i.c.v.) (adjacent to the ARC) to 4d-WD mice after the NORT training phase. Notably, this strategy normalized memory (Figures 1R, 1S, S1N, and S1O). This evidence causally associates pregnenolone action in the hypothalamus with cognitive function.
Interference with pregnenolone biosynthesis in hypothalamic POMC neurons, but not AgRP neurons, results in cognitive dysfunction
Intrigued by these findings, we next investigated the function of pregnenolone in the ARC with regard to cognition. To this aim, we generated mice lacking Star in AgRP or POMC neurons (Figures 2A and 2B) (hereafter referred to as AgRPStarKO and POMCStarKO mice, respectively). This gene encodes for Stard1, a protein that mediates mitochondrial transfer of cholesterol, which is the rate-limiting step in neurosteroid production (Arenas et al., 2017; Manna et al., 2016) (Figure S1M).
Figure 2.

Genetic deletion of Star exclusively in POMC neurons impairs recognition memory
(A and B) Schematic illustration of the generation of transgenic mice: (A) AgRPStarKO and (B) POMCStarKO mice.
(C–E) Pregnenolone concentration in the (C) perirhinal cortex, (D) hippocampus, and (E) arcuate-enriched mediobasal hypothalamus from control and AgRPStarKO mice (n = 5–9/genotype).
(F–H) Pregnenolone concentration in (F) perirhinal cortex, (G) hippocampus, and (H) arcuate-enriched mediobasal hypothalamus from control and POMCStarKO mice (n = 8–6/genotype).
(I–L) Recorded parameters to assess Barnes maze performance in AgRPStarKO mice during the test phase: (I) latency to target hole, (J) time spent in target quadrant, (K) total distance traveled, and (L) representative traces of mouse movement trajectories (n = 8/genotype).
(M–P) Recorded parameters to assess NORT performance in AgRPStarKO mice during the test phase: (M) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (N) exploration time (time exploring novel object + time exploring familiar object), (O) total distance traveled, and (P) representative traces of mouse movement trajectories (n = 8/genotype).
(Q–T) Recorded parameters to assess Barnes maze performance in POMCStarKO mice during the test phase: (Q) latency to target hole, (R) time spent in target quadrant, (S) total distance traveled, and (T) representative traces of mouse movement trajectories (n = 9–10/genotype).
(U–X) Recorded parameters to assess NORT performance in POMCStarKO mice during the test phase: (U) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (V) exploration time (time exploring novel object + time exploring familiar object), (W) total distance traveled, and (X) representative traces of mouse movement trajectories (n = 7–8/genotype).
(Y–AA) Hippocampal long-term potentiation (LTP) study comparing control and POMCStarKO mice. (Y) Representative traces of individual recordings showing baseline fEPSPs before (black traces) and after (red traces) LTP induction. Slope and peak amplitude of fEPSPs increased after theta-burst stimulation (TBS) in control mice but was attenuated in POMCStarKO animals.
(Z) Time course of fEPSP recordings demonstrating that LTP induction was markedly impaired in POMCStarKO animals (n = 6 recordings from 5 animals) when compared with controls (n = 9 recordings from 6 animals).
(AA) Quantification of the fEPSP slope change over the last 45 min of recording period shown in (Z).
All studies were performed in male mice between 10 and 16 weeks of age. Dots in panels represent individual samples.
Data are presented as mean ± SEM. ∗p < 0.05; ∗∗∗p < 0.001.
Tissue- and cell-specific deletion of Star was demonstrated by recombination of the floxed alleles (Figures S2A and S2B), fluorescent in situ hybridization (Figures S2C and S2D), and qPCR (Figure S2E). The nucleus of the solitary tract (NTS) harbors a small population of POMC neurons, and consistently, Star expression was also significantly reduced in this brain region (Figure S2E). Neuronal population size was unaltered in mutant mice (Figures S2F and S2G), indicating that Stard1 deficiency did not affect neuron ontogeny or viability.
Although the POMC promoter also recombines in the pituitary, the expression of genes involved in mitochondrial cholesterol transport (Star, Cyp11a1, and Tspo) or in pituitary gland development and function (Crhr1, Gh, Pit1, Pomc, Tbx19, and Tshb) was analogous between controls and mutant mice (Figure S2H). Accordingly, pregnenolone concentration in the pituitary, as well as basal and stress-induced corticosterone circulating levels, was comparable between that of control and POMCStarKO mice (Figures S2I and S2J). Together, these results indicate that genetic recombination in the pituitary of POMCStarKO mice did not hinder pituitary-adrenal axis function.
Hypothalamic AgRP and POMC neurons are fundamental nexus for the physiological control of energy balance and glucose metabolism (Jais and Brüning, 2021). Thus, we conducted detailed metabolic phenotyping of AgRPStarKO and POMCStarKO mice. Interestingly, Stard1 deficiency in AgRP or POMC neurons did not make an impact on energy balance and glucose metabolism (Figures S3A–S3J).
Next, we tested whether genetic deletion of Star in AgRP or POMC neurons translated into reduced hypothalamic content of pregnenolone. The concentration of pregnenolone was not altered in any of the brain regions analyzed from AgRPStarKO mice (Figures 2C–2E). In POMCStarKO animals, pregnenolone content was unaltered in the perirhinal cortex, hippocampus, or NTS (Figures 2F, 2G, and S3K), but it was significantly decreased in ARC-enriched samples (Figure 2H). This divergence was likely the consequence of the higher neurosteroidogenic capacity of ARC POMC neurons, as suggested by the analysis of publicly available single-cell RNA-seq data (Campbell et al., 2017) (Figure S3L). Accordingly, while AgRPStarKO mice did not exhibit cognitive alterations (Figures 2I–2P and S3M), POMCStarKO animals exhibited unaltered spatial memory (Figures 2Q–2T and S3N) but significant impairments in object recognition memory (Figures 2U–2X). This memory defect was corroborated in a second paradigm based on olfactory cues (Figures S3O–S3Q). Evaluation of the neurosensory status (Figures S3R and S3S), as well as anxiety- and depression-like states (Figures S4A–S4Q), demonstrated the full capability of POMCStarKO mice to perform the behavioral tests.
It has been reported that hypothalamic POMC neurons project to the hippocampal subiculum (Wang et al., 2015). Using a tracing virogenetic approach (Figure S5A), we observed that ARC POMC neurons (Figures S5B–S5E) established synapses in this brain region (Figures S5F–S5I), thus confirming previous findings. Next, we evaluated the impact of Stard1 deficiency on the synaptic plasticity of the hippocampus. Hippocampal slices from control and POMCStarKO mice showed comparable input-output relationship (Figure S5J) and paired-pulse facilitation (Figures S5K–S5M). In contrast, LTP was significantly impaired in mutant mice as reflected by the reduced fEPSP slope (Figures 2Y–2AA), confirming perturbed synaptic plasticity and congruent with the cognitive impairments observed. Hippocampal CREB-activated pathways are crucial for learning, memory, and synaptic plasticity (Belgacem and Borodinsky, 2017). Consistently, we observed a significant reduction in the number of pCREB-positive cells in the dentate gyrus of cognitive-deficient POMCStarKO mice (Figures S5N and S5O).
Together, these results confirm a causal link between POMC neuron neurosteroid deficiency and memory dysfunction via hippocampal mechanisms.
Insulin signaling in POMC neurons does not mediate cognitive performance
It has been reported that cognitive impairment is associated with central insulin resistance (Biessels and Despa, 2018; Tanaka et al., 2020). Thus, we next assessed the insulin signaling status of our POMCStarKO mice. While AKT phosphorylation was equivalent in the perirhinal cortex, hippocampus, and peripheral tissues, it was blunted in ARC-enriched samples from POMCStarKO mice (Figures 3A–3E).
Figure 3.

Insulin signaling in POMC neurons does not mediate memory performance
(A–E) Assessment of insulin signaling via AKT phosphorylation (pS473-AKT) in the (A) perirhinal cortex, (B) hippocampus, (C) arcuate-enriched mediobasal hypothalamus, (D) skeletal muscle, and (E) liver from control and POMCStarKO mice. Representative blots and quantification (normalized to total AKT) are shown (n = 3–7/genotype).
(F) Schematic illustration of the generation of POMCInsrKO mice.
(G–I) Pregnenolone content in the (G) perirhinal cortex, (H) hippocampus, and (I) arcuate nucleus-enriched mediobasal hypothalamus from control and POMCInsrKO mice (n = 9/genotype).
(J–M) Locomotor and exploratory activity of control and POMCInsrKO mice in an open-field paradigm: (J) time spent in the center, (K) entries in the center, (L) total distance traveled, and (M) global activity (n = 9/genotype).
(N) Task learning curve for the latency to reach the scape hole during the training phase of the Barnes maze test (BMT). Results show the average of 2 trials per day during 5 consecutive days (n = 8–9/genotype).
(O–R) Recorded parameters to assess Barnes maze performance in control and POMCInsrKO mice during the test phase: (O) latency to target hole, (P) time spent in target quadrant, (Q) total distance traveled, and (R) representative traces of mouse movement trajectories (n = 8–9/genotype).
(S–V) Recorded parameters to assess NORT performance in control and POMCInsrKO mice during the test phase: (S) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (T) exploration time (time exploring novel object + time exploring familiar object), (U) total distance traveled, and (V) representative traces of mouse movement trajectories (n = 8/genotype).
All studies were performed in male mice between 10 and 16 weeks of age. Dots in panels represent individual samples. Data are presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns, not significant.
To further investigate the relevance of insulin signaling in POMC neurons upon cognition, we generated conditional mice lacking insulin receptors (POMCInsrKO; Figure 3F) (Könner et al., 2007). No changes in pregnenolone content in the relevant brain areas were observed (Figures 3G–3I). Overall, locomotor activity, exploratory drive, and memory fitness of POMCInsrKO mice were indistinguishable from those of control counterparts (Figures 3J–3V). Together, these results indicate that insulin signaling in POMC neurons is irrelevant for memory performance maintenance and suggest that defective insulin signaling in POMCStarKO mice is likely a secondary effect.
POMC neuron-derived pregnenolone mediates cognitive performance likely via autocrine mechanisms
To ascertain if reduced POMC neuron-derived neurosteroid precursor synthesis underlies the cognitive impairment observed in our mutant mouse line, we i.c.v. injected pregnenolone into POMCStarKO mice (Figure 4A). Remarkably, this strategy was able to restore memory of POMCStarKO mice (Figures 4B–4D) and to enhance hippocampal synaptic plasticity (Figures 4E–4G). Equivalent results were obtained when pregnenolone was directly infused into the ARC of mutant mice (Figures S6A–S6D), thus confirming a causal link between ARC neurosteroid deficiency and cognitive dysfunction. These results also validated the i.c.v. route as an adequate experimental strategy in the context of our research.
Figure 4.

Central pregnenolone administration recovers cognitive function in POMCStarKO mice
(A) Schematic illustration of central acute delivery of pregnenolone and subsequent NORT in POMCStarKO mice. Red arrow shows the injection phase.
(B–D) Recorded parameters to assess NORT performance in POMCStarKO mice, after central administration of vehicle (Veh) or pregnenolone (Preg), during the test phase: (B) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (C) exploration time (time exploring novel object + time exploring familiar object), and (D) total distance traveled (n = 3–9/genotype).
(E–G) Hippocampal long-term potentiation (LTP) study after central administration of either vehicle (Veh) or pregnenolone (Preg) in POMCStarKO mice. (E) Representative traces of individual recordings showing baseline fEPSPs before (black traces) and after (red traces) LTP induction. Slope and peak amplitude of fEPSPs improved after pregnenolone treatment.
(F) Time course of the fEPSP slope change in vehicle-treated (n = 4 recordings from 4 animals) and pregnenolone-treated POMCStarKO mice (n = 5 recordings from 3 animals) after theta-burst stimulation (TBS; arrow).
(G) Quantification of the fEPSP slope change over the last 45 min of recording period shown in (F).
(H) Schematic illustration of central acute delivery of pregnenolone and subsequent analysis of FOS expression in control C57Bl/6J mice. Red arrow shows the injection phase.
(I) Representative confocal images of FOS and POMC colocalization after central administration of vehicle or pregnenolone in control C57Bl/6J mice. White arrows show colocalization staining. Scale bar, 20 μm.
(J) Quantification of FOS-positive POMC neurons in arcuate nucleus sections from control C57Bl/6J mice after central administration of vehicle or pregnenolone (n = 3/genotype).
(K) Quantification of FOS positivity in non-POMC cells in arcuate nucleus sections from control C57Bl/6J mice after central administration of vehicle or pregnenolone (n = 3/genotype).
(L) Schematic illustration of the activatory and inhibitory chemogenetic strategy, coupled with central pregnenolone administration, in POMCcre/+ and POMCStarKO mice. Orange and red arrows show CNO and pregnenolone (Preg) injection, respectively.
(M and N) Recorded parameters to assess NORT performance in mice after chemogenetic activation of POMC neurons, combined with central administration of vehicle (V) or pregnenolone (Preg), during the test phase: (M) discrimination index of control POMCcre/+ mice (n = 4–5/genotype) and (N) POMCStarKO mice (n = 4–6/genotype).
(O and P) Recorded parameters to assess NORT performance in mice after chemogenetic repression of POMC neuron activity, combined with central administration of vehicle (V) or pregnenolone (Preg), during the test phase: (O) discrimination index of control POMCcre/+ mice (n = 4–6/genotype) and (P) POMCStarKO mice (n = 5–6/genotype).
All studies were performed in male mice between 10 and 16 weeks of age. Dots in panels represent individual samples. Data are presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant.
To identify the cellular targets of neurosteroids in the ARC, we i.c.v. injected C57Bl/6J mice with pregnenolone and analyzed FOS positivity as a marker of neuronal activity (Figure 4H). Colocalization of POMC and FOS was increased upon pregnenolone administration, suggesting neurosteroid-mediated enhanced activation of this neuronal population (Figures 4I and 4J). The proportion of activated non-POMC cells was similar in vehicle and pregnenolone-injected mice (Figure 4K), denoting that in the ARC, pregnenolone mainly targets POMC neurons.
Given that pregnenolone was able to activate POMC neurons, we investigated whether stimulation of POMC neuron activity was sufficient to mediate recognition memory. With this aim, we bilaterally delivered activatory (AAV-hM3Dq) designer receptors exclusively activated by designer drugs (DREADDs) into the ARC of control POMCCre/+ and POMCStarKO mice (Figures 4L and S6E). This permits Cre-mediated specific activation of POMC neurons by clozapine N-oxide (CNO) injection (Figure S6F). Notably, no changes in the cognitive capacity, exploratory drive, or distance traveled were promoted by chemogenetic activation of POMC neurons in control or mutant mice (Figures 4M, 4N, and S6G–S6J).
In view of these results, together with the observation that central administration of neurosteroids did not improve the otherwise normal cognitive function of control mice (Figures 1S, 4B, and 4M), we reasoned that inhibition of POMC neurons should impair cognitive performance. For this purpose, we chemogenetically inhibited POMC neurons via AAV-hM4Di DREADDs (Figure 4L). As anticipated, acute repression of POMC neurons caused a significant reduction in recognition memory of control POMCCre/+ mice (Figures 4O, S6K, and S6L).
It has been speculated that neurosteroids largely exert their effects via autocrine mechanisms (Ratner et al., 2019), and our study assessing POMC neuron activity after central pregnenolone treatment (Figures 4H–4K) is consistent with this idea. To further explore the potential autocrine effects of neurosteroids in POMC neurons upon cognition, we injected pregnenolone into the third ventricle of POMCCre/+ or POMCStarKO mice with prior chemogenetic repression of POMC neurons. Earlier inhibition of POMC neuronal activity negated the beneficial effects of pregnenolone with regard to cognitive performance in both models (Figures 4O, 4P, and S6K–S6N), suggesting that these effects are mediated via an autocrine axis on POMC neurons.
The collective interpretation of these results indicates that POMC neuron activity is necessary for adequate memory function and for the beneficial effects of pregnenolone. Furthermore, POMC neuron stimulation fails to improve memory in the context of genetic deficiency of pregnenolone, but it is rescued by its central delivery. Hence, it is likely that adequate memory performance requires both responsive POMC neurons and the release of pregnenolone.
Obesity-related metabolic complications associate with impaired cognitive function and reduced hypothalamic neurosteroid concentration in mice
Our observations in POMCStarKO mice indicated that primary alterations in pregnenolone signaling affected cognitive performance without causing metabolic alterations. Thus, we next questioned the reverse, that is, whether established metabolic conditions associated with cognitive impairments progressed with inadequate central pregnenolone levels. To examine this, we conducted studies on obese mice and human patients. We fed C57Bl/6J mice with either regular chow or WD for 16 weeks (16w-WD; Figure 5A). Predictably, this long-term diet regime resulted in dramatic metabolic perturbations, including obesity (Figure 5B) and defective glucose metabolism (Figures 5C–5E). Cognitive scrutiny revealed that chronic WD feeding did not alter the latency to localize the target hole in the BMT (Figures 5F–5J). Nevertheless, 16w-WD mice spent more time in the target quadrant, but the interpretation of this result is intriguing (Figure 5H). The NORT evidenced a significant reduction in the discrimination index of 16w-WD mice, denoting an impairment in recognition memory (Figures 5K–5N). The increased exploration time during the test phase of NORT (Figure 5L) was attributed to enhanced exploration of the familiar object rather than the novel one, as it can be inferred from the discrimination index data (Figure 5K). Locomotor or exploration drive in an open field was not affected by the diet, ruling out physical inability to perform the behavioral paradigms (Figures 5O–5R).
Figure 5.

Obesity-related metabolic complications associate with impaired cognitive function and reduced hypothalamic neurosteroid concentration in mice
(A) Schematic illustration of the experimental strategy. Eight-week-old C57Bl/6J mice were fed with either chow or western diet for 16 consecutive weeks (16w-WD).
(B) Body weight profile (n = 10/genotype).
(C) Basal blood glucose concentration and (D) glucose tolerance test (n = 7/genotype).
(E) Insulin sensitivity test (n=10/genotype).
(F) Task learning curve for the latency to reach the scape hole during the training phase of the Barnes maze test (BMT) in mice fed with either chow or 16w-WD. Results show the average of 2 trials per day during 5 consecutive days (n = 7–8/diet).
(G–J) Parameters recorded to assess Barnes maze performance in mice, fed with either chow or 16w-WD, during the test phase: (G) latency to target hole, (H) time spent in target quadrant, (I) total distance traveled, and (J) representative traces of mouse movement trajectories (n = 7–8/genotype).
(K–N) Parameters recorded to assess NORT performance in mice, fed with either chow or 16w-WD, during the test phase: (K) discrimination index (time exploring novel object − time exploring familiar object)/(time exploring novel object + time exploring familiar object), (L) exploration time (time exploring novel object + time exploring familiar object), (M) total distance traveled, and (N) representative traces of mouse movement trajectories (n = 8–10/genotype).
(O–R) Recorded parameters to assess open-field performance in mice fed with either chow or 16w-WD: (O) time spent in the center, (P) number of entries in the center, (Q) total distance traveled, and (R) global activity (n = 10/genotype).
(S–U) Quantification of pregnenolone concentration in the (S) perirhinal cortex, (T) hippocampus, and (U) arcuate-enriched mediobasal hypothalamus in mice fed with either chow or 16w-WD (n = 9/genotype).
All studies were performed on male mice at 24–26 weeks of age. Dots in panels represent individual samples. Data are presented as mean ± SEM. ∗p < 0.01; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant.
Next, we aimed to evaluate whether diet-induced cognitive deterioration was related to deficient pregnenolone concentration in the relevant brain regions. Interestingly, similar to our previous findings, pregnenolone content was reduced in ARC-enriched samples but not in perirhinal cortex or hippocampus of 16w-WD mice (Figures 5S–5U). These results indicate that diet-induced metabolic perturbations are associated with cognitive impairments and reduced pregnenolone content in the hypothalamus.
Pregnenolone concentration in the cerebrospinal fluid of metabolically unhealthy patients with obesity positively correlates with cognitive score
To examine the relevance of central neurosteroids upon cognition in the context of human biology, we determined the concentration of pregnenolone in the cerebrospinal fluid (CSF) from a clinically characterized cohort of metabolically healthy (MHO) and unhealthy (MUO) patients with obesity for which cognitive status was assessed by the Mini-Mental Status Examination (MMSE) (Figure 6A; Table S1). The MMSE is a commonly used screening test for the evaluation of cognitive impairments in adults that assesses several domains: orientation, registration, attention and calculation, recall, and language. In MHO subjects, pregnenolone levels did not correlate with any of the diverse anthropometric, clinical, or cognitive parameters (Figure 6B). However, in MUO patients, CSF pregnenolone was negatively associated with body mass index (BMI) and positively correlated with MMSE (Figures 6C and 6D). These results evidenced a mechanism linking central pregnenolone with cognitive function in metabolically unhealthy obese individuals.
Figure 6.

Pregnenolone concentration in the cerebrospinal fluid of metabolically unhealthy patients with obesity positively correlates with cognitive score
(A) Schematic illustration of sample processing from the cohort of patients.
(B) Analysis of Spearman’s rank correlation between cerebrospinal fluid (CSF) pregnenolone concentrations and diverse anthropometric, clinical, and cognitive parameters in a cohort of metabolically healthy (MHO) and unhealthy obese (MUO) patients. Preg, pregnenolone; MMSE, mini-mental state examination; BMI, body mass index; HOMA, homeostatic model assessment-insulin resistance; FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; TG, triglyceride; SBP, systolic blood pressure; DBP, diastolic blood pressure; CHO, cholesterol; LDL, low-density lipoprotein; HDL, high-density lipoprotein.
(C and D) Correlation between CSF pregnenolone concentrations and BMI (C) or MMSE (D) in MUO patients. Dots in panels represent individual samples.
Discussion
Previous studies have placed brain cholesterol metabolism as a bridge between metabolic and cognitive disorders (Martín-Segura et al., 2019; Suzuki et al., 2010, 2013). In light of this evidence, we hypothesized that neurosteroids, which are synthesized from cholesterol and have been widely related to cognition and memory processes, could be implicated in the cognitive impairment observed in detrimental metabolic contexts. In this study, we reveal that key appetite-regulating POMC neurons also mediate recognition memory via a pregnenolone-based mechanism.
Extensive evidence has demonstrated that obesity and T2D are associated with cognitive dysfunction (Biessels and Despa, 2018; Tanaka et al., 2020). Our patient cohort study indicated that pregnenolone concentration in the CSF positively correlated with the MMSE and negatively correlated with BMI but solely in MUO patients. This observation suggests a neuroprotective role for neurosteroids and supports the notion that obesity itself is insufficient to perturb the pregnenolone-cognition axis. In this regard, it is likely that the combination of additional metabolic complications of obesity prompts such cognitive phenotypes. Our data provide conceptual evidence that central pregnenolone may be relevant in cognitive impairments associated with metabolic disorders in humans.
Our studies in preclinical models confirmed that recognition memory was impaired after a chronic diet-induced obesity intervention. Similarly, a brief period (4 days) of western-style diet administration, which advanced without causing noticeable metabolic disturbances, was sufficient to specifically deteriorate recognition memory. In contrast to other reports, our dietary studies did not perturb spatial memory (Beilharz et al., 2016; Bocarsly et al., 2015). This discrepancy may be the consequence of variations in the experimental procedures and nutrient composition of diets. Importantly, under both chronic and acute energy-dense dietary regimes, defective recognition memory was paralleled by a reduced content of pregnenolone in the hypothalamus but not in memory-related regions such as the perirhinal cortex and hippocampus. Nevertheless, we cannot exclude that western-style diet feeding also alters neurosteroid content in other brain structures. This cognitive phenotype was rescued via central delivery of pregnenolone, demonstrating the causality of the hypothalamic deficiency of this neurosteroid in the development of cognitive dysfunction. Another important conclusion of these findings, consistent with human data, is that overt manifestations of obesity/diabetes per se (i.e., overweight and hyperglycemia) are not the primary causes of hypothalamic pregnenolone synthesis shortage and cognition decline. Instead, these observations pointed toward rapid yet undetermined effects of nutrient excess and/or their metabolites that attenuate hypothalamic pregnenolone production. It is reasonable to speculate that, similar to related biological responses (Sergi and Williams, 2019), an excessive influx of saturated fatty acids in the hypothalamus might participate in this context.
To further understand the triangular connection between neurosteroids, cognitive function, and metabolism, we generated mice lacking Stard1 in hypothalamic AgRP or POMC neurons. This strategy impinged upon cholesterol transport into the mitochondria, thus disrupting the first and limiting step of neurosteroid production in the aforementioned populations of neurons. Analysis of publicly available single-cell sequencing data suggested a higher neurosteroidogenic potential for POMC neurons in comparison with AgRP neurons. This notion was supported by the unaltered hypothalamic pregnenolone content and normal cognitive performance of AgRPStarKO mice. In contrast, loss of Stard1 in POMC neurons impaired recognition memory performance in the face of normal metabolic and energy balance status. These findings indicate that inadequate neurosteroid production by POMC neurons is fundamentally implicated in cognition, but it is dispensable for energy homeostasis control. This suggests the existence of divergent, yet coordinated, pathways influencing memory and metabolism within POMC neurons.
Brain insulin resistance has long been considered as one of the mechanisms underlying cognitive dysfunction. For example, it has been reported that insulin signaling is altered in preclinical models and patients with neurodegenerative diseases (Kellar and Craft, 2020). Research has also evidenced that insulin and insulin sensitizers have neuroprotective effects under these pathological conditions (Kellar and Craft, 2020). However, it is debatable whether brain insulin desensitization is the cause or the consequence of cognitive dysfunction. The cognitive deterioration observed in POMCStarKO mice progressed with attenuated insulin signaling exclusively in the hypothalamus. However, our results show that genetic blockage of insulin signaling in POMC neurons is insufficient to cause detectable cognitive deficits in mice. While it is very likely that brain insulin dysfunction can contribute to further deterioration of cognitive capacity once cognitive decline emerges or is established, our data do not support the concept of disturbed brain insulin signaling as a primary trigger of the pathology in our mutant mice.
DREADD-mediated inhibition of POMC neurons negatively influenced recognition memory in control mice, indicating that excitable POMC neurons are required for adequate memory function. Synaptic plasticity is an energy-demanding process that is required for learning and memory formation. Consistent with this, recognition memory was repressed when a negative energy balance state was simulated through chemogenetic inhibition of POMC neurons. From an energetic perspective, this could be part of a cost-effective program to limit an energy-consuming process (such as memory formation) or enhance permissiveness to certain food resources under conditions of scarcity. In contrast, the activation of POMC neurons during feeding conditions would support energy-consuming processes related to the formation and storage of memories. The fact that chemogenetic activation of POMC neurons did not make an impact on cognition capacity in control mice may be a reflection of the inability to further improve the otherwise normal cognition. Given the absence of pregnenolone synthesis in POMCStarKO mice, the chemogenetic activation of POMC neurons in this model cannot stimulate its release. These results reinforce the importance of POMC neuron-derived pregnenolone as a key mediator of memory performance.
The beneficial effects of POMC neuron-derived pregnenolone upon cognitive function are likely mediated by an autocrine axis, as demonstrated by preferential activation of POMC neurons after central pregnenolone delivery and a sequential combination of chemogenetics and pharmacological rescue. Given the lipophilic nature of neurosteroids, paracrine-autocrine signaling has been postulated as a common mechanism underlying their diverse neurobiological effects (Ratner et al., 2019). Neurosteroids can act through multiple receptors, including N-methyl-D-aspartate glutamate receptors (NMDARs) (Tuem and Atey, 2017). The genes encoding the diverse NMDAR subunits (GRIN family) are expressed in POMC neurons (Lam et al., 2017), and thus, it is reasonable to speculate that neurosteroids might exert autocrine effects on POMC neurons via NMDARs. Nevertheless, we cannot rule out that pregnenolone also exerts part of its effects on POMC neurons via indirect pathways.
How this putative autocrine loop into POMC neurons influences hippocampal LTP remains enigmatic. Despite POMC neurons projecting to specific regions of the hippocampus (Wang et al., 2015; Figure S5A), we did not observe changes in hippocampal pregnenolone content in any of the models investigated. Hence, it seems unlikely that ARC POMC neurons release pregnenolone in this brain region. Instead, the involvement of POMC neuron-derived bioactive molecules is plausible. For example, the neuropeptide alpha-melanocyte-stimulating hormone (α-MSH) influences synaptic plasticity in the hippocampus. In this regard, it has been reported that the hippocampus expresses melanocortin 4 receptor (MC4R; the receptor for α-MSH) (Gantz et al., 1993; Liu et al., 2003). Furthermore, α-MSH has been shown to exert neuroprotective and neurogenesis effects on the hippocampus to enhance LTP and improve cognitive function in models of neurodegenerative diseases (Giuliani et al., 2015; Gonzalez et al., 2009; Ma and McLaurin, 2014; Shen et al., 2013).
Eating behavior is intimately connected with multiple and complex neuro-cognitive processes. Diverse memory systems contribute to food-related decision-making to support nutritional needs. For example, it is crucial to locate, identify, and associate food items with particular outcomes (taste, nutritional value, toxicity, etc.) to sustain life (Higgs and Spetter, 2018). Given the vital relationship between ingestive behavior and memory, it is evolutionarily plausible that fundamental biological systems or neurons influencing appetite and energy balance may also participate (directly or indirectly) in related cognitive processes (Kosse and Burdakov, 2019; Suarez et al., 2019; Zimmer et al., 2019). Our results show that POMC neuron-derived neurosteroid signaling principally mediates recognition memory, conceivably facilitating the identification and acquisition of food rather than contributing to spatial navigation to locate food resources. Nevertheless, we cannot rule out that POMC neurons also modulate spatial memory via neurosteroid-independent routes. It is noteworthy that POMC neurons also influence emotional states such as stress, anxiety, or fear (Greenman et al., 2013, Lee et al., 2020, Liu et al., 2007, Mandela et al., 2014, Qu et al., 2020). These intermingled neurobiological mechanisms may represent complex adaptive responses to coordinate appetitive and consummatory behaviors, thus maximizing survival in a constantly changing environment. This novel POMC neuron functional axis at the interface of metabolism and cognition lays the grounds for a conceptual framework to understand the fundamental co-regulation of appetite and memory processes.
In this study, we show that while metabolic disease is associated with reduced hypothalamic pregnenolone and cognitive deterioration, genetic interference with pregnenolone synthesis in POMC neurons impacts on cognition but not on metabolism. This emphasizes the primary role of hypothalamic pregnenolone in maintaining cognitive function. Our findings suggest that ARC POMC neurons integrate cognitive and metabolic health via distinct pathways and advocate a wider role for this population of neurons beyond appetite and metabolic control. This novel facet of POMC neuron biology may have relevant implications for cognitive disorders.
Limitations of study
Our study defines a novel role for hypothalamic pregnenolone in memory performance. However, we cannot rule out that a part of these effects is mediated by downstream derivates as pregnenolone is the precursor for the biosynthesis of many neurosteroids. The use of non-metabolizable pregnenolone (or other neurosteroids) would provide further insights into this aspect of the research. The microinjection of pregnenolone into the ARC will inevitably spread, up to a certain extent, to neighboring areas. Although we believe that the volume/injection rate used is largely restricted to the ARC, we cannot fully assure the exclusive implication of the ARC in the observed pregnenolone effects.
The combination of FOS, as a correlate of neuronal activity, and chemogenetic studies suggests that the beneficial effects of pregnenolone on memory are mediated, at least in part, by autocrine or paracrine effects on POMC neurons. This would be consistent with the chemical nature of neurosteroids and other reports suggesting this type of signaling (Ratner et al., 2019). Nevertheless, it should be noted that our chemogenetic studies do not exclude potential alternative pathways that still converge on POMC neurons but via indirect routes.
The analysis of pregnenolone in human patients has been performed in the CSF, which likely reflects overall biochemical changes in the brain. For translational significance, it would be pertinent to measure pregnenolone content in human post-mortem hypothalamic samples under diverse pathophysiological conditions.
STAR★Methods
Key resources table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Phospho-Akt (Ser473) | Cell Signaling | Cat# 9271S; RRID: AB_329825 |
| Rabbit Akt (pan) mAB | Cell Signaling | Cat# C67E7; RRID: AB_915783 |
| Rabbit polyclonal anti-c-FOS | Santa Cruz | Cat# Sc-52; RRID: AB_2106793 |
| Rabbit polyclonal anti-POMC | Phoenix Pharmaceuticals | Cat# H-029-30; RRID: AB_2307442 |
| Rabbit Phospho-CREB (Ser133) (87G3) mAB | Cell Signaling | Cat# 9198S; RRID: AB_2561044 |
| Chicken Anti-GFP | Aves Labs | Cat#GFP-1010, RRID: AB_2307313 |
| Rabbit Anti-RFP | Rockland | Cat#600-401-379, RRID: AB_2209751 |
| Goat Anti-Rabbit IgG-Peroxidase | GE Healthcare | Cat# A0545; RRID: AB_257896 |
| Alexa 488, Chicken anti-rabbit | Life Technologies | Cat# A21441; RRID: AB_2535859 |
| Alexa 555, Donkey anti-rabbit A21207 | Life Technologies | Cat# A21207; RRID: AB_141637 |
| Alexa 488, Goat anti-Chicken | Life Technologies | Cat# A-11039, RRID: AB_2534096 |
| Alexa 594, Donkey anti-rabbit | Life Technologies | Cat# A-21207, RRIF: AB_141637 |
| Bacterial and virus strains | ||
| AAV8-hSYN-DIO-hM3D(Gq)-mCherry | Addgene | Cat# 44361 |
| AAV8-hSYN-DIO-hM4D(Gi)-mCherry | Addgene | Cat# 44362 |
| AAV1-phSyn1(S)-FLEX-tdTomato-T2A-SypEGFP | Addgene | Cat# 51509 |
| Chemicals, peptides, and recombinant proteins | ||
| Glucose 40% | Fresenius kabi | Cat# 620724 |
| Human insulin (Humulin) | Lilly | Cat# C.N 710008.9 |
| 2-hydroxypropyl beta-cyclodextrin | Sigma-Aldrich | Cat# H5784 |
| Pregnenolone | Sigma-Aldrich | Cat# P162 |
| Artificial cerebrspinal fluid (aCSF) | Tocris Bioscience | Cat# 3525 |
| Clozapine-N-oxide (CNO) | Tocris Bioscience | Cat# 4936 |
| Trizol reagent | Invitrogen | Cat# 15596026 |
| Gelatin | Sigma-Aldrich | Cat# G-2500 |
| Triton X-100 | Sigma-Aldrich | Cat# X-100 |
| Sodium Chloride (NaCl) 0.9% | Merck | Cat# 1064040500 |
| N-hexane | Sigma-Aldrich | Cat# 296090 |
| Paraformaldehyde aqueous solution 16% | Electron Microscopy Sciences | Cat# 15710 16 |
| Ethylene glycol | Sigma-Aldrich | Cat# 1096211000 |
| Bovine serum albumin | Sigma-Aldrich | Cat# A9647 |
| Phosphate buffered saline 1x | Sigma-Aldrich | Cat# D8537 |
| Ethanol | VWR | Cat# 1.009.831.000 |
| Sucrose | Sigma-Aldrich | Cat# S0389 |
| Acetonitrile | Sigma-Aldrich | Cat# 360457 |
| Critical commercial assays | ||
| Corticosterona EIA | Immunodiagnostic systems | Cat# AC-14F1 |
| Pregnenolone ELISA | LDN | Cat# FR E-2700 |
| Enhanced chemiluminescence reagent | Pierce | Cat# 32105 |
| 4–12% Pre-cast protein Gel | Bio-Rad | Cat# 345-0124 |
| Protease and Phosphatase Inhibitor Cocktail | Sigma-Aldrich | Cat# PPC1010 |
| Stripping buffer | Thermo Fisher | Cat# 21059 |
| High Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368813 |
| Premix Ex Taq mastermix | Takara | Cat# RR39LR |
| RNAscope HybEz hybridization system | Advanced Cell Diagnostics | Cat# 321720 |
| Bradford Reagent | Sigma-Aldrich | Cat# B6916 |
| Experimental models: Organisms/strains | ||
| POMC-Cre mouse | Xu et al., 2005 | N/A |
| AgRP-Cre mouse | Tong et al., 2008 | N/A |
| Starflox/flox mouse | Torres et al., 2019 | N/A |
| Insrflox/flox mouse | Könner et al., 2007 | N/A |
| C57BL/6J mouse | Janvier Labs | N/A |
| Oligonucleotides | ||
| RNAscope probe Pomc | Advanced Cell Diagnostics | Cat# 314081-C1 |
| RNAscope probe Agrp | Advanced Cell Diagnostics | Cat# 400711-C2 |
| RNAscope probe Star | Advanced Cell Diagnostics | Cat# 591531-C3 |
| Taqman probe Star | Thermo Fisher | Mm00441558_m1 |
| Taqman probe Cyp11a1 | Thermo Fisher | Mm00490735_m1 |
| Taqman probe Tpso | Thermo Fisher | Mm00437828_m1 |
| Taqman probe Crhr1 | Thermo Fisher | Mm00432670_m1 |
| Taqman probe Gh | Thermo Fisher | Mm01258409_g1 |
| Taqman probe Pit1 | Thermo Fisher | Mm00476852_m1 |
| Taqman probe Pomc | Thermo Fisher | Mm00435874_m1 |
| Taqman probe Tbx19 | Thermo Fisher | Mm00453377_m1 |
| Taqman probe Tshβ | Thermo Fisher | Mm00437190_m1 |
| Software and algorithms | ||
| Image J software | NIH, Open source | http://imagej-nih-gov.sire.ub.edu/ij/ |
| Prism | GraphPad Softwate | http://www.graphpad.com/scientific-software/prism/ |
| Axon pClamp 10.6 | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite#gref |
| SMART v3.0, video tracking software | Panlab | https://www.panlab.com/en/products/smart-video-tracking-software-panlab |
| Other | ||
| Regular chow diet | Tecklad, Envigo | Cat# 2014C |
| RD Western diet 43% Kcal from CH and 40% Kcal from fat | Research Diets | Cat# D12079B |
| Microvette CB-300 | Sarstedt | Cat# NC9141704 |
| Glucometer | Arkray | Cat# GT-1910 |
| SuperFrost UltraPlus Adhesion slides | Thermo Fisher | Cat# 322000 |
| ImmEdge hydrophobic barrier pen | Thermo Fisher | Cat# 10417002 |
| ProLong Gold Antifade Mountant | Thermo Fisher | Cat# P36930 |
| Vetbond | 3M | Cat# 1469SB |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Marc Claret (mclaret@clinic.cat).
Materials availability
All newly generated materials are available from the corresponding author upon reasonable request.
Experimental model and subject details
Animal care and diets
All animal studies were performed with approval of the University of Barcelona Ethics Committee, complying with current Catalan, Spanish and European legislation. Male mice were maintained on a temperature controlled (20-24°C), 12-hour light/dark cycle with free access to water and standard chow (Teklad maintenance diet 14% protein; Envigo). In particular studies, Western diet (40% Kcal from fat and 43% Kcal from carbohydrates; Research Diets) was provided ad libitum for 4 days or 16 weeks (starting at 8 weeks of age). Health status of the mice were checked regularly. The age and number of mice analyzed for each experiment is detailed in the figure legends.
Mouse lines
C57BL/6J mice were purchased from Janvier Labs. Mice with a floxed allele for Star (Starflox/flox) (Torres et al., 2019) were crossed with POMC-Cre (Xu et al., 2005) or AgRP-Cre (Tong et al., 2008) mice to generate animals selectively lacking Stard1 in either POMC neurons (POMCStarKO) or AgRP neurons (AgRPStarKO). Mice with a floxed allele for Insuslin receptor (Insrflox/flox) (Könner et al., 2007) were crossed with POMC-Cre mice to generate animals selectively lacking Insr in POMC neurons (POMCInsrKO). As control mice we used Cre negative, floxed littermates. Mice were on a C57BL/6 background.
Human subjects and cerebrospinal fluid samples
We studied 40 patients with obesity (BMI>35) aged ≥ 40 years, with a sex distribution of 45% female and 55% male, belonging to an ongoing multicentric longitudinal cohort study (Hospital de la Santa Creu i Sant Pau and Hospital Clinic de Barcelona-IDIBAPS) aimed at assessing the effects of bariatric surgery on biochemical and neuroimaging Alzheimer’s disease biomarkers. Anthropometric, clinical and cognitive parameters were recorded from all individuals. Cerebrospinal fluid (CSF) samples were obtained by lumbar puncture and stored at -80°C until analysis. Pregnenolone concentration in CSF was determined using a commercially available ELISA kit (LDN) following manufacturer’s instructions. The reliability of the kit has been previously validated by liquid chromatography tandem mass spectrometry (Mahata et al., 2014). Patients were divided into metabolically healthy obese (MHO) or metabolically unhealthy obese (MUO) based on the criteria established by Aguilar-Salinas and collaborators (Aguilar-Salinas et al., 2008). According to these criteria, MHO patients exhibited a BMI >30 kg/m2 together with plasma HDL cholesterol of at least 40 mg/dl and the absence of type-2 diabetes and arterial hypertension. Conversely, MUO patients were defined by the presence of one or more of the following metabolic abnormalities: plasma HDL cholesterol ≥40 mg/dl, type 2 diabetes, and arterial hypertension. Type 2 diabetes was considered present in subjects with a random glucose concentration of ≥200 mg/dl, a fasting glucose of ≥126 mg/dl, or those receiving glucose-lowering medication. Arterial hypertension was defined as the use of antihypertensive treatment and/or blood pressure >140 mm Hg systolic and/or >90 mm Hg diastolic. Glucose levels 2 hours after an oral glucose tolerance test were not included in the criteria. The local Ethical Committee approved the studies, and all study participants provided informed consent to donate blood and CSF samples.
Method details
Physiological tests
Body weights were monitored using a precision scale. For feeding studies, mice were singly-housed and acclimatized for 1 week prior to study. Daily food intake was manually measured for 5 consecutive days using a precision scale. Blood samples were collected via tail vein using a capillary collection system with EDTA (Sarstedt). Blood glucose concentration was measured using a glucometer (Glucocard X-meter, Arkray). Glucose tolerance tests (2 g/Kg) were performed on overnight (16 hour) fasted mice. Insulin sensitivity tests (0.2 IU/Kg; Humulin, Lilly) were conducted on 6-hour food-deprived mice.
Intracerebroventricular (i.c.v.) surgery and cannulation
I.c.v. surgery was performed as described (Schneeberger et al., 2013). Briefly, adult (8-10 weeks old) mice were anaesthetized with a Ketamine/xylazine cocktail (i.p., 100 mg/Kg and 10 mg/Kg) and positioned in a stereotaxic frame (Kopf Instruments). The skull was exposed and a 26-gauge stainless steel guide cannula (PlasticsOne) was implanted into the third ventricle (midline 0 mm, 0.82 mm posterior from bregma, depth 4.8 mm from skull surface). The cannula was secured to the skull with screws and dental cement and temporarily occluded with a dummy cannula (PlasticsOne). After surgery, the mice were singly-housed and given at least 1 week to recover. I.c.v. injections were performed using a 30-gauge needle that extended 0.5 mm below the guide cannula (PlasticsOne), connected by cannula connector to a 5 μl Hamilton syringe and infused using a microinfusion pump (Harvard Apparatus).
ARC surgery and cannulation
Adult (8-10 weeks old) mice were anaesthetized with a Ketamine/xylazine cocktail (i.p., 100 mg/Kg and 10 mg/Kg) and positioned in a stereotaxic frame (Kopf Instruments). The skull was exposed and 26-gauge stainless-steel bilateral cannula (center-to-center distance 0.8 mm; PlasticsOne) was implanted into the ARC parenchyma using stereotaxic procedures (coordinates from bregma −1.6 mm, lateral ±0.4 mm, ventral −5.0 mm from skull surface). The cannula was secured to the skull with stainless steel screws and dental cement, and temporarily occluded with a 33-gauge stainless-steel obturator. Animals were allowed to recover for at least 7 days before being studied. Treatments were given over 5 minutes via a 33-gauge needle extending 0.8 mm beyond the tip of the cannula (5.8 mm from dura) using a microinfusion pump (Harvard Apparatus), and followed by a 2-minute delay before needle withdrawal.
Corticosterone measurements
A standard restraint protocol was used to measure corticosterone-mediated stress response in control and POMCStarKO mice. The study was performed between 10:00 A.M. and 11:00 A.M. to minimize circadian corticosterone fluctuations. Immediately after collecting the first blood sample (baseline levels, time=0), mice were placed into a restrainer for 30 minutes. Afterwards, a second blood sample was taken (stress levels). Blood samples were collected via tail vein using a capillary collection system with EDTA (Sarstedt). Plasma corticosterone was measured using a commercially available EIA kit (Immunodiagnostic systems) following manufacturer’s instructions.
Pregnenolone extraction and quantitative analysis
Animals were killed by decapitation and the perirhinial cortex, hippocampus, arcuate-enriched mediobasal hypothalamus, nucleus of the solitary tract and pituitary were dissected and stored at −80°C until analyzed. Tissues were homogenized in 150 μl of acetonitrile using an electric hand-held homogenizer. Tissue lysates were centrifuged for 10 minutes at 10200 g at 4°C and the pellet was discarded. Next, we performed a liquid-liquid extraction with 300 μl of n-hexane in order to separate the neurosteroid fraction from the other components of the brain tissue. During this step, homogenates were vigorously shaken during 30 minutes at room temperature. Then we allowed the two phases to separate and recovered the neurosteroid fraction. Samples were dried in a preheated vacuum centrifuge (SpeedVac Savant SPD1010, Thermo Scientific) at 40°C. The dried steroid residues were kept at -20°C until they were resuspended (in PBS + 0.1% gelatin + 5% ethanol) for quantification with a validated pregnenolone ELISA test (LDN) (Mahata et al., 2014) following vendor’s instructions. The readings from the optical densities at 450 nm were compared to a standard curve that was also diluted in resuspension buffer in order to reduce the interference of the matrix.
In vivo insulin signaling analysis by immunoblot
Over-night (16 hour) fasted mice were anesthetized with ketamine/xylacine (i.p., 100 mg/Kg and 10 mg/Kg) and subsequently injected with 5U of insulin (humulin, Lilly) via the inferior cava vein. Relevant tissues were collected at specific time-points: 30 seconds (liver), 1 minute (skeletal muscle), 10 minutes (mediobasal hypothalamus), 11 minutes (perirhinal cortex) and 12 minutes (hippocampus). Tissues were immediately frozen in liquid nitrogen and stored at -80°C. Tissue samples were homogenized in a buffer containing 25 mM Tris-HCl (pH 7.4), 10 mM Na3VO4, 100 mM NaF, 50 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, and 1% nonidet-P40 supplemented with a protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Tissue lysates were centrifuged for 15 minutes at 12000 rpm. Supernatants were collected and quantified for protein concentration using Bradford reagent (Bio-Rad) and 10 μg of protein were subjected to gradient 4-12% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad). Proteins were electrotransferred onto PVDF membranes (Millipore), blocked in 5% BSA buffer for 1 hour at room temperature and probed over-night (4°C) with pAKT (Ser473; 1:1000) or after 30 minutes of stripping (Thermo Scientific) with total AKT (1:1000). Both antibodies were from Cell Signaling Technology. After washing, membranes were incubated 1hour at room temperature with a secondary HPRT antibody (anti-rabbit IgG HPRT linked; 1:5000; GE Healthcare). Detection was performed by enhanced chemiluminescence (Pierce) and images were obtained using a LAS4000 imaging system (GE Healthcare). Protein expression was quantified by densitometry using ImageJ software (NIH).
Quantitative polymerase chain reaction (qPCR)
Tissues were harvested, immediately frozen in liquid nitrogen and stored at -80°C until use. Tissues were homogenized and total mRNA was isolated using Trizol (Invitrogen) using standard protocols. Retrotranscription was performed using reagents from Applied Biosystems. Quantitative PCR was conducted using Premix Ex Taq mastermix (Takara) in an ABI Prism 7900 HT system (Applied Biosystems). Proprietary Taqman Gene Expression assay FAM/TAMRA probes (Applied Biosystems) used for qPCR analysis are listed in the key resources table. Gene expression was expressed relative to Hprt levels. Data was analyzed using the standard curve method.
Fluorescent in situ hybridization and quantification
Animals were transcardially perfused with saline followed by ice-cold 4% phosphate-buffered paraformaldehyde (PFA; pH 7.4). The brains were dissected and post-fixed in 4% PFA at 4°C for 24 hours, and cryoprotected by a sequence of 10-30% sucrose in 1x phosphate buffered saline (PBS, pH 7.4) at 4°C until the tissue sunk to the bottom of the tube. Brains were cut at 25 μm-thick on a freezing microtome and collected in 24-well plate containing cryoprotectant solution (30% ethylene glycol; 30% sucrose in PBS) and subsequently stored at -80°C to preserve RNA until further processing.
Fluorescent in situ hybridization for the simultaneous detection of Pomc and Star or Agrp and Star mRNA was performed using RNAScope. All reagents were purchased from Advanced Cell Diagnostics. The Pomc probe contained 10 oligo pairs and targeted region 19-995 (Acc. No. NM_008895.3) of the Pomc transcript. The Agrp probe constituted 16 oligo pairs and targeted region 11-764 of the Agrp transcript (Acc. No. NM_001271806.1). The Star probe constituted 12 oligo pairs and targeted region 199-726 of the Star transcript (Acc. No. NM_011485.5). All incubation steps were performed at either at 60°C or 40°C using the ACD HybEz hybridization system.
On the day before the assay, one every 4th sections throughout the ARC was mounted on SuperFrost UltraPlus Adhesion slides (ThermoFisher), air-dried at room temperature and stored at -20°C overnight. From each animal, one section from the same region of the brain was also mounted for assessment of negative control probe to allow subsequent calculation of background.
On the day of the assay, one series section throughout the ARC was selected and equilibrated to room temperature. From each animal, one section from the same region of the brain was also mounted to assess the negative control probe to enable subsequent calculation of background. Slides were washed in PBS, baked at 60°C for 30 minutes and post-fixed with 4%PFA for 15 minutes. Sections were then dehydrated, baked for 30 minutes at 60°C and subsequently submerged into boiling (98.5 - 100°C) Target Retrieval reagent for 5 minutes, followed by two brief rinses in MilliQ-purified water. The slides were quickly dehydrated in 100% ethanol and allowed to air dry for 5 minutes. A hydrophobic barrier was then created around the sections using an ImmEdge hydrophobic barrier pen (Vector laboratories). Slides were then placed into RNAScope holder and sections were treated with Protease III for 30 minutes at 40°C.
The subsequent steps, i.e., hybridization of the probes and the amplification and detection steps, were performed according to the manufacturer’s protocol for RNAscope Multiplex Fluorescent Assay. The color module chosen for each experiment finally labeled the Pomc probe with Atto 550, the Agrp probe with Atto 647, and the Star probe with Atto 488. Sections were counterstained with DAPI and coverslipped with ProLong Gold Antifade Mountant (ThermoFisher) and stored in the dark at 4°C until imaging.
Images were captured using a confocal Leica DM 2500 microscope, equipped with a 40x/1.15 oil objective and using a zoom of 2x. Z-stacks of 1 μm of the ARC were captured bilaterally from 4 rostral to caudal sections per animal. Laser intensities for the probe channels were kept constant throughout the entire image acquisition. Images were imported into Fijii (NIH) where maximum intensity projections were made. In order to acquire the minimum intensity value for analyzing the expression of Star, the threshold for probe recognition was calculated as the mean cell intensity present in the negative control sections +3xSD. All labeling above this value was considered to be true signal as published by previous reports (Timper et al., 2018). Brightness and threshold were adjusted uniformly across the sections. For quantification, each POMC positive neuron per section was manually selected and the amount of Star particles were analyzed.
Double POMC and FOS immunofluorescence after pregnenolone delivery
C57Bl/6J mice were i.c.v injected with 2 μl of either vehicle (20% of 2-hydroxypropyl beta- cyclodextrin in NaCl 0.9%, Sigma-Aldrich) or pregnenolone (5 μg/μl, Sigma-Aldrich). Ninety minutes later, mice were transcardially perfused with saline followed by ice-cold 4% phosphate-buffered PFA. Brains were cryoprotected, frozen and subsequently cut into 30 μm-thick slices. Brain sections were blocked with 2% donkey serum in KPBS + 0.1% Triton X-100 + 3% BSA and incubated with rabbit anti FOS antibody (1:100; Santa Cruz) in blocking solution for 16 hours at 4°C. As secondary antibody, a donkey anti-rabbit Alexa Fluor 555 (1:200; Life Technologies) in KPBS + 0.1% Triton X-100 + 3% BSA was used (90 minutes at room temperature). After extensive washing, slices were blocked with 2% chicken serum in KPBS + 0.4% Triton X-100 and incubated with rabbit anti-POMC precursor antibody (1:1000; Phoenix Pharmaceuticals) for 16 hours at 4°C. Subsequently, slices were incubated with chicken anti-rabbit Alexa Fluor 488 (1:200; Life Technologies) for 90 minutes at room temperature. Imaging was performed using a Leica DMI 6000B microscope with a 20x objective. A minimum of 388 POMC neurons from 3 mice per genotype were included in the analysis.
POMC and AgRP neuron count
The number of neurons were determined as described (Ramírez et al., 2017). In brief, brains from 10 week-old control, POMCStarKO and AgRPStarKO mice were collected. Mouse perfusion and brain slicing was performed as described above. POMC and AgRP neurons were identified by immunofluorescence or fluorescent in situ hybridization (color module Atto 488) respectively, following the protocols detailed above. Sections throughout the ARC (–1.1 mm to –2.7 mm from bregma) were collected in 3 series. The number of POMC and AgRP neurons were determined in 1 series and multiplied the neuron count by 3 to estimate the total cell number. Imaging was performed using confocal Leica DM 2500 microscope, equipped with a 20x objective. The number of cells from 3 animals per genotype was counted manually using ImageJ software. We analyzed at least 2,409 POMC and 912 AgRP neurons per mouse.
pCREB analysis by immunofluorescence
Control and POMCStarKO mice under random-fed conditions were anesthetized and transcardially perfused with saline followed by ice-cold 4% PFA (pH 7.4). The brains were dissected and post-fixed in 4% PFA at 4°C for 24 hours, and cryoprotected by a sequence of 10-30% sucrose in 1x PBS (pH 7.4) at 4°C until the tissue sunk to the bottom of the tube. Brains were cut at 30 μm-thickness on a freezing microtome and collected. Serial hippocampal sections were blocked with 2% chicken serum in KPBS + 0.4% Triton X-100 and 3% BSA and incubated with rabbit anti pCREB antibody (1:200; Cell signaling) in blocking solution overnight (16 hours) at 4°C. As secondary antibody, a chicken anti-rabbit Alexa Fluor 488 (1:250; Life Technologies) in KPBS + 0.4% Triton X-100 was used (2 hours at room temperature). Imaging was performed using a Leica DMI 6000B microscope with a 20x objective. Three mice per genotype were included for the analysis of the number of pCREB-positive cells in the dentate gyrus of the hippocampus. For each mouse, the mean number was calculated by averaging the number of pCREB-positive cells counted from four images.
General behavioral procedures
Before each test, mice were acclimatized to the behavioral room and arena for 1 hour (or otherwise stated). The arena was cleaned with 70% ethanol before and after every trial. Light intensity was adapted to each task. The investigator remained outside of the behavioral room during testing. A video camera, positioned directly above the arena, was used to track the movement of each animal. Videos were recorded on a computer and analyzed with a video-tracking software (SMART v3.0, Panlab).
Neurosensory screen
Prior to behavioral studies, control and mutant mice were submitted to a basic sensorimotor assessment to ensure equivalent capacity to perform subsequent tasks. This consisted in a battery of 10 score-based standardized measures to evaluate several parameters, including scope response, vision, motor coordination, balance, orientation, reflexes or nociception. This protocol was based upon the behavioral screen previously described by Rogers and collaborators (Rogers et al., 1997). The test was performed as follows. A) Balance. Assessed by placing the mice on a 1-meter-long wooden beam, with 15 mm of flat surface, resting 50 cm from the surface. The latency to fall was recorded. Score: 0=falls off; 1=holds immobile; 2=walks slowly; 3=walks across the beam. B) Tail elevation. Scoring: 0=flaccid; 1=horizontal; 2=elevated. C) Piloerection. Scoring: 0=absence; 1=presence. D) Paw withdrawal reflex. Pressure was applied to the interdigital area of the right hind limb using a modified forceps (to standardize the amount of pressure). The movement of the leg was assessed. Scoring: 0=no reaction; 1=soft retraction; 2=moderate retraction; 3=quick retraction; 4=quick and repeated retractions. E) Ear withdrawal reflex. The outer ear of the mouse was gently touched using a swab stick. The movement of the ear was assessed. Scoring: 0=no reaction; 1=moderate retraction; 2=multiple retractions. F) Corneal reflex. A swab stick was progressively approached to the eye of the mouse. Scoring: 0=no reaction; 1=active eye blinking; 2=multiple eye blinking. G) Negative geotaxis. Mice were placed on a wire mesh grid that was lifted vertically, with the subject facing down. The latency to fall and overall performance was scored: 0=turns and climbs up; 1=turns but does not climb; 2=moves but does not turn; 3=does not move for 30 seconds; 4=falls off. H) Twisting. Mice were held by the tail, 20 cm above the surface, for a few seconds. Scoring: 0=no twisting; 1=twisting. I) Visual acuity. Mice were held by the tail, 20 cm above the surface, while a pencil was progressively approached to the mouse’s face. Mice should expand their forepaws as soon as they see the pencil. Scoring: 0=forepaws are not extended; 1=forepaws are extended after contact with pencil; 2=forepaws are extended before contact with whiskers. J) Scope response. Mice were held by the tail, 20 cm above the surface, and progressively lowered. Mice should expand their forepaws as they approach the surface. Scoring: 0=paws not extended; 1=paws extended after contact with the surface; 2=paws extended after contact with the whiskers; 3=paws extended before contact with the whiskers; 4=paws extended after starting movement. In addition, we also examined the olfactory capacity of mice as described in previous studies (Yang and Crawley, 2009). It consisted in the sequential presentation of diverse odors: water, non-social odor #1 (high-fat diet), non-social odor #2 (green tea) and social odor #3 (bedding from other cages). Mice were acclimated for 30 minutes to the arena (35 x 35 x 35 cm). Each odor was presented in a non-accessible clean container (separated by a grid) for 2 minutes in three consecutive video recorded trials. Inter-trial interval was 1 minute (time needed to change the odor stimulus). Sniffing time was scored when the mouse was directly oriented towards the odor. Typically, mice exhibit a decrease in sniffing time over repeated presentations of the odor (habituation) followed by increased sniffing when a new odor is offered (dishabituation).
Barnes maze test
The Barnes maze is a widely used paradigm for the measurement of hippocampal-dependent spatial memory. Our protocol was based on original studies (Bach et al., 1995; Barnes, 1979). In brief, the maze consisted of a white elevated circular platform (85 cm stand height and 92 cm in diameter) with 20 equidistant holes located around the circumference. A black Plexiglas escape chamber (17.5 x 7.5 x 8.0 cm), that serves as refuge, was placed underneath one of the holes (target hole). The reference cues (a thick green cable, a white sliding door and a black cross) were presented in the walls surrounding the maze. Animals were tested under high-intensity light (>30 lux) to create an aversive environment in the surface of the apparatus. The protocol consisted of five days of training (acquisition phase), two days resting period and the test day. On the first training day, a mouse was placed in the center of the maze and given 3 minutes to find the target hole and enter into the attached escape cage. If they did not enter on their own during that time, the researcher would gently place the mouse to the side of the hole and help them enter. By doing so, the mouse realizes that the escape cage exists and gives them practice in stepping down to the platform in the cage. The mice were allowed to stay in the escape cage for 1 minute before being returned to the home cage. All mice performed two trials per day, with a 1-hour inter-trial interval, for five consecutive days. The position of the escape hole remained constant for each mouse during the acquisition phase. After these trials, rodents typically remember which hole contains the escape chamber and quickly proceed in a direct path towards the hole. Improved performance over sessions or trials reflects adequate learning. Mice were allowed to rest on days 6 and 7. The test trial was performed on day 8, in which the escape chamber was removed and animals were allowed to explore the maze for 2 minutes. During the test phase we scored the following parameters: a) latency to target hole (time spent from the center of the circular platform to the precise hole where the escape chamber was located); b) time in quadrant (time spent in the quadrant of the platform where the escape hole was located); c) total distance (total distance travelled by the mouse during the 2-minute test). All trials were video recorded. Analysis of the latency to find the target hole, time spent in the target quadrant and total distance were automatically measured by the video-tracking software.
Novel object recognition test (NORT)
The novel object recognition test (NORT) is a highly validated task for recognition memory. Our protocol was adapted from Leger and collaborators (Leger et al., 2013). The test was conducted in a methacrylate arena (35 x 35 x 35 cm) under a low-intensity light (20 lux) environment. NORT consisted in three phases: habituation, training and test phase. During habituation (days 1-3), mice were allowed to explore the arena for 10 minutes. We considered object exploration when the mice were directing the nose toward the object, while actively sniffing and/or pawing, at a distance <3 cm. Sitting on top of the object while sniffing the surrounding air or chewing the object were not considered exploration, unless that action was accompanied with a nose-directing behavior toward the object. In the training phase (day 4), mice were allowed to explore two identical objects (equidistantly spaced in the arena) for 10 minutes and returned to their home cages immediately after training. The test phase was conducted 2 hours after the training phase. At this time interval, between training and testing periods, the involvement of the hippocampus in memory retention is expected (Antunes and Biala, 2012). In the test phase, one of the objects was replaced by a new one (novel object) and the mouse was allowed to explore them for 10 minutes. We kept the position of the 2 objects constant across sessions. Discrimination indices were calculated as: (Time exploring novel object – Time exploring familiar object) / (Time exploring novel object + Time exploring familiar object). In addition, we also scored total exploration time (Time exploring novel object + Time exploring familiar object) and total distance travelled during the test session. Animals that showed freezing behavior or exhibited <5 seconds of exploration behavior were excluded from the analyses. To reduce experimenter interference, the trial was video-recorded and off-line analyzed in a blind manner using SMART v3.0 software (Panlab). Total distance was automatically scored by the software.
Novel odor recognition test (NODR)
The protocol was adapted from previous studies (Feinberg et al., 2012; Flores-Montoya et al., 2015). Similar to NORT, the test consisted in three phases: habituation, training and test phase. During habituation (days 1-3), mice were allowed to explore the arena for 10 minutes. The arena contained two identical leaky containers equidistantly positioned. In the training phase (day 4), mice were allowed to sniff two identical non-social odors (high-fat diet) placed in the leaky containers for 10 minutes, and returned to their home cage immediately after training. The test phase was conducted 2 hours after the training session. In this phase, one of the odors was replaced by a new one (Western diet) and the mouse was allowed to sniff for 10 minutes. We kept the position of the 2 leaky containers constant across sessions. Discrimination indices were calculated as: (Time exploring novel odor – Time exploring familiar odor) / (Time exploring novel odor + Time exploring familiar odor). In addition, we also scored total exploration time (Time exploring novel odor + Time exploring familiar odor) and total distance travelled. The non-social odors selected were two types of novel diets for the mice (40% high fat diet and western diet) due to their low level of anxiety generation. To exclude a preference effect of the diets, we performed an odor preference test with these two diets prior to the actual experiment in a different cohort of mice. Animals that showed freezing behavior or exhibited <5 seconds of exploratory behavior were excluded from the analyses. To reduce experimenter interference, the trial was video-recorded and off-line analyzed in a blind manner using SMART v3.0 software (Panlab). Total distance was automatically scored by the software.
Open field test
Open field test is commonly used for measuring the exploratory behavior and general activity of animals. Our protocol was based on previous studies (Fan et al., 2019). Mice were placed in the center of a dark methacrylate arena (35 x 35 x 35 cm) and allowed to freely explore it for 10 minutes. Trials were video recorded. Animals were tested under a low-intensity light (< 30 lux) to create a safe environment and avoid stress. Total distance and the time spent in the center of the arena, compared with time spent in the edges, was scored using SMART v3.0 software (Panlab).
Light–Dark box test
Dark-light box test assesses anxiety-like behavior. It is based on the innate aversion of rodents to brightly illuminated areas. Our protocol was based on previous studies (Fan et al., 2019). The test apparatus consisted in a methacrylate arena (35 x 35 x 35 cm) divided into a small dark (safe) compartment and a large strongly-illuminated (200 lux; aversive) compartment. A door connected the two compartments. Mice were placed in the dark compartment and allowed to freely move between the two chambers for 5 minutes. Trials were video recorded. Video tracking data was analyzed using SMART v3.0 software (Panlab) to calculate the time spent in each compartment and the latency to enter the illuminated compartment. Typically, anxiety models spent less time in the bright chamber.
Elevated plus maze test
Elevated plus maze test is also widely used for measuring anxiety-like behavior. This task takes advantage of the innate aversion of rodents to open spaces. Our protocol was based on previous studies (Fan et al., 2019). The apparatus used was a cross-shaped 4-arm maze, with 2 open arms and 2 closed (wall sheltered) arms (25 x 5 cm). This structure was elevated 60 cm above the floor. Mice were placed in the center of the apparatus, facing towards a closed arm, and their behavior was video recorded for 5 minutes. Time spent in the closed arms and open arms was analyzed via video-tracking software (SMART v3.0 software; Panlab). The preference for being in closed arms is an indicator of anxiety-like behavior.
Tail suspension test
Tail suspension test is commonly used for measuring depression-like behavior. It is based on the fact that rodents subjected to short-term inescapable stress eventually give up and take an immobile position. Our protocol was based on previous studies (Fan et al., 2019). Mice were suspended during 6 minutes by their tails using adhesive tape and behavior was video recorded. The periods of agitation and immobility during the test were scored off-line manually using SMART v3.0 software (Panlab). Longer immobility times is an indicator of depression-like behavior.
Marble burying test
The marble burying test is used to evaluate anxiety and/or obsessive-compulsive behaviors (Deacon, 2006). It is based on the innate proclivity of mice to bury objects in their bedding. The test was done under low-intensity lighting (<30 lux). The arena (35 x 35 x 35 cm) was filled with bedding to a depth of 5.5 cm. Each mouse was habituated to the arena for 15 minutes. After the habituation phase, mice were returned to their home cages and 25 pied glass marbles (Ø 1 cm) were placed equidistantly in a 5 x 5 arrangement. Mice were allowed to explore the arena and the marbles during 15 minutes. The number of marbles buried (at least 2/3 of its surface area covered by bedding) were scored manually just after the test. The number of marbles buried reflects anxiety-like and/or obsessive-compulsive behavior.
NORT performance after i.c.v. or intra-ARC administration of pregnenolone
NORT was essentially performed as described above. Cannulated mice into the third ventricle were infused with 2μl of either vehicle (20% of 2-hydroxypropyl beta-cyclodextrin in NaCl 0.9%, Sigma-Aldrich) or pregnenolone (5 μg/μl, Sigma-Aldrich) right after the training phase. Intra-ARC cannulated mice were bilaterally microinfused with 250 nl of either vehicle or pregnenolone (at a rate of 50 nl/min) followed by a 2-min delay before needle withdrawal. The test phase was conducted 2 hours later.
NORT performance after chemogenetic modulation of POMC neuron activity and i.c.v pregnenolone administration
Ten-week-old POMCCre/+ and POMCStarKO mice were anesthetized with a Ketamine/Xylazine cocktail (i.p., 100 mg/Kg and 10 mg/Kg) and placed in a stereotaxic frame (Kopf Instruments) for subsequent injection of AAV vectors encoding for activatory (AAV8-hSYN-DIO-hM3D(Gq)-mCherry; 7.53x1012 gc/ml, Addgene) or inhibitory (AAV8-hSYN-DIO-hM4D(Gi)-mCherry; 1.10x1013 gc/ml, Addgene) Designer Receptors Exclusively Activated by Designer Drugs (DREADDs). Viruses were bilaterally injected into the ARC (300 nl/injection site) using a 33-gauge needle connected to a 5 μl Neuro-Syringe (Hamilton). AAVs were delivered over 8 minutes according to the following coordinates: 1.5 mm posterior to the bregma, ± 0.3 mm lateral to midline, and 5.8 mm below the surface of the skull. Immediately after the procedure, the same mice were cannulated into the third ventricle as described above. The incision was sutured by VetBond tissue adhesive (3M) and mice were placed in a heated cage until they recovered from anesthesia. Three weeks later, mice were submitted to the NORT habituation phase (days 1-3). Right after the training phase on the fourth day, mice were i.p. injected with Clozapine-N-oxide (CNO; 1.5 mg/Kg for the activation and 1 mg/Kg for the inhibition; Tocris Bioscience) and returned to their home cage. Ninety minutes after CNO injection, 2 μl of vehicle (20% of 2-hydroxypropyl betacyclodextrin in NaCl 0.9%, Sigma-Aldrich) or pregnenolone (5 μg/μl, Sigma-Aldrich) was administered via the implanted cannula. The test phase was conducted 30 minutes later.
Synaptophysin studies
Ten-week-old POMCCre/+ mice were anesthetized with a Ketamine/Xylazine cocktail (i.p., 100 mg/Kg and 10 mg/Kg) and placed in a stereotaxic frame for subsequent injection of Cre-mediated AAV vectors that express cytoplasmic tdTomato and presynaptic (synaptophysin-fused) EGFP (AAV1-phSyn1(S)-FLEX-tdTomato-T2A-SypEGFP; 4x1012 gc/ml, Addgene). Viruses were bilaterally injected into the ARC (300 nl/injection site) using a 33-gauge needle connected to a 5 μl Neuro-Syringe (Hamilton). AAVs were delivered over 8 minutes according to the following coordinates: 1.5 mm posterior to the bregma, ± 0.3 mm lateral to midline, and 5.8 mm below the surface of the skull. After the procedure, the incision was sutured by 30 VetBond tissue adhesive (3M) and mice were placed in a heated cage until they recovered from anesthesia. Six weeks after the injections, mice were transcardially perfused with saline followed by ice-cold 4% phosphate-buffered PFA. Brains were cryoprotected, frozen and subsequently cut into 30 μm-thick sections. Brain sections were blocked in 2% donkey serum, 2% goat serum, 0.05% tween in 1xPBS. The EGFP signal was amplified by staining with a chicken anti-GFP antibody (1:500; Aveslab). To amplify the tdTomato signal slices we incubated with a rabbit anti-RFP antibody (1:1000; Rockland). Some sections were not incubated with primary antibodies in order to assess the specificity of the staining (negative control). After extensive washes, sections were then incubated with secondary antibodies: goat anti-chicken 488 (1:300; ThermoFisher) and donkey anti-rabbit 594 (1.300; ThermoFisher) during 2 hours at room temperature. Images were acquired in a Zeiss LSM700 confocal microscope. POMC neurons located in the ARC were identified with a 532nm laser. POMC projections into the hippocampal subiculum were recognized with a 488nm laser. For each section and area, image stacks with 1 μm distance interval were taken. Tile scans were acquired for whole section visualization. The representative images shown were taken with a 63x objective and maximum intensity stacks were made in FIJI (NIH) equally adjusted for brightness and contrast.
Electrophysiological studies
Electrophysiological studies were conducted in mice belonging to three experimental settings: a) chow-fed vs. 4d-WD; b) control vs. POMCStarKO; c) POMCStarKO + i.c.v. vehicle (2 μl of 20% 2-hydroxypropyl betacyclodextrin in NaCl 0.9%, Sigma-Aldrich) vs. POMCStarKO + pregnenolone (2 μl at 5 μg/μl, Sigma-Aldrich). Mice were deeply anesthetized with isofluorane and decapitated. In the case of vehicle vs. pregnenolone studies, mice were sacrificed 2 hours after the i.c.v injection. Brains were removed in ice-cold, high-sucrose extracellular artificial cerebrospinal fluid (aCSF1, in mM; 206 sucrose, 1.3 KCl, 1 CaCl2, 10 MgSO4, 26 NaHCO3, 11 glucose, 1.25 NH2PO4, purged with 95% CO2/5% O2, pH 7.4) and subdivided into the hemispheres. Thick (380 μm) coronal slices of hippocampus were obtained with a vibratome (VT1000S; Leica) and transferred into an incubation beaker with extracellular aCSF appropriate for neurophysiological recordings (aCSF2, in mM; 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.25 NaH2PO4, 1.5 MgSO4, 25 NaHCO3, 11 glucose, purged with 95 % CO2/5 % O2, pH 7.4). Slices were kept at 32°C for 1 hour and subsequently at room temperature for at least 1 additional hour. For field potential measurements, single slices were then transferred into a measurement chamber perfused with aCSF2 at 2 ml/minute at 28-30°C. A bipolar stimulation electrode (Platinum-Iridium stereotrode, PI2ST30.1A5, Science Products) was placed in the Schaffer collateral pathway. Recording electrodes were made with a puller (P-1000, Shutter Instrument Company) from thick-walled borosilicate glass with a diameter of 1.5 mm (Sutter Instruments). The recording electrode filled with aCSF2 was placed in the dendritic branching of the CA1 region for local field potential measurement (field excitatory postsynaptic potential, fEPSP). A stimulus isolation unit A385 (World Precision Instruments) was used to elicit stimulation currents between 25-700 μA. Before baseline recordings for long-term potentiation (LTP), input-output (IO) curves were recorded for each slice at 0.03 Hz. The stimulation current was then adjusted in each recording to evoke fEPSP at which the slope was at 50-60 % of maximally evoked fEPSP slope value. After baseline recording for 30 minutes with 0.03 Hz, LTP was induced by theta-burst stimulation (TBS; 10 theta bursts of 4 pulses of 100 Hz with an interstimulus interval of 200 ms repeated 7 times with 0.03 Hz). After LTP induction, fEPSPs were recorded for 1 additional hour with 0.03 Hz. Paired-pulse fEPSPs in the test pathway were measured before baseline recordings with an interstimulus interval of 50 ms. All recordings were amplified and stored using amplifier AxoClamp 2B (Molecular Devices). Traces were analyzed using Axon pClamp software (Molecular Devices, version 10.6).
Quantification and statistical analysis
Data are expressed as mean ± SEM. Statistical analysis were performed using GraphPad Prism software. Two group-one factor comparisons were performed using a two-tailed unpaired Student’s t test. Datasets with two factors-one dependent variable were analyzed using two-way ANOVA followed by Sidak’s post-hoc test, except for the IO curve data that was analyzed with a two-way ANOVA followed by Tukey’s post hoc test. One-sample t tests were performed to determine whether the NORT discrimination indexes observed in control/vehicle groups were significantly different from chance/0. In all cases p < 0.05. Correlations were assessed by Spearman coefficient. p < 0.05 was considered statistically significant. Symbols used are: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Statistical parameters can be found in the Figures and Figure legends. No statistical methods were used to determine whether the data met assumptions of the statistical approach.
Acknowledgments
We thank Josep Dalmau for financial support; Olga Jauregui, Albert Ferrés-Coy, and Analía Bortolozzi for scientific advice; and Servier Medical Art for illustrations. This work was supported by the Swiss National Science Foundation (no.176206; NCCR Synapsy grant no.185897) to C.S.; the European Research Council (ERC) advanced grant SYNEME to J.C.B.; Instituto de Salud Carlos III (ISCIII)—Fondo Europeo de Desarrollo Regional (FEDER) (PI17/00296), RETICs Oftared (RD16/0008/0014), and Generalitat de Catalunya (2017SGR737) to X.G.; Ministerio de Ciencia e Innovación (BFU2017-83317-P) to D.S.; Ministerio de Economia, Industria y Competitividad, Maria de Maeztu (MDM-2017-0729) to Institut de Neurociencies, Universitat de Barcelona; ISCIII-FEDER (PI14/01126, PI17/01019), the National Institutes of Health (NIA grants 1R01AG056850-01A1, R21AG056974, and R01AG061566), Fundació La Marató de TV3 (20141210), and Generalitat de Catalunya (SLT006/17/00119) to J.F.; ISCIII-FEDER (PI17/00279 and PI20/0042), Fundació La Marató de TV3 (201614.31), and Generalitat de Catalunya (SLT008/18/00127) to A.J.; Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and FEDER (PID2019-111669RB-I00 and PID2020-115055RB-I00), CIBEREHD, the center grant P50AA011999 Southern California Research Center for ALPD and Cirrhosis funded by NIAAA/NIH, Generalitat de Catalunya (SGR-2017-1112), the European Cooperation in Science & Technology (COST) ACTION CA17112, FUNDACIÓN BBVA (“ER stress-mitochondrial cholesterol axis in obesity-associated insulin resistance and comorbidities”), and Red Nacional 2018-102799-T de Enfermedades Metabólicas y Cáncer and Fundació La Marató de TV3 (201916/31) to J.C.F.-C.; and ERC consolidator grant MITOSENSING (725004), ISCIII-FEDER (PI16/00963), “la Caixa” Foundation (ID100010434) under agreement LCF/PR/HR19/52160016, and CERCA Programme/Generalitat de Catalunya to M.C. D.A. is supported by ISCIII (INT19/00016) and Generalitat de Catalunya PERIS program (SLT006/17/125), A.P. is supported by Hospital Clínic de Barcelona (“Ajut Josep Font”), A.O. is supported by a Miguel Servet contract (CP19/00083) from ISCIII-FEDER, and R.H.-T. is supported by a Marie Skłodowska-Curie Action fellowship (H2020-MSCA-IF) and NEUROPREG (891247). S.R. is a recipient of Juan de la Cierva Formación (FJCI-2016-28911) and Incorporación (IJC2018-037341-I) programs from the Spanish Ministry of Science and Innovation. This work was carried out in part at Esther Koplowitz Centre.
Author contributions
S.R. conceived the study, designed and conducted experiments, analyzed results, and contributed to the manuscript preparation. R.H.-T., M.T., M.P., A.O., A.G.G.-V., I.C., T.V.E., I.Z., C.B., M.I., G.G., R.N., and C.S. contributed to the performance of experimental and behavioral procedures. M.M.-G. contributed to the welfare of the mice and performed all the genotyping. J.A. and E.E. conducted biostatistical and bioinformatics analysis. M.R., D.S., and X.G. performed and analyzed electrophysiological studies. C.B. and J.C.B. provided POMCIRKO animals. V.R., C.G.-R., and J.C.F.-C. generated Starflox/flox animals. A.P., D.A., J.F., and A.J. participated in the recruitment and curation of the human cohort. M.C. conceived the study; designed, coordinated, and supervised experiments; wrote the manuscript; and secured funding. All authors discussed the results, commented on the manuscript before submission, and agreed with the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.12.023.
Contributor Information
Sara Ramírez, Email: ramirez@clinic.cat.
Marc Claret, Email: mclaret@clinic.cat.
Supplemental information
Data and code availability
-
•
The data generated in this study will be shared by the lead contact upon reasonable request.
-
•
This study did not generate new code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aguilar-Salinas C.A., García E.G., Robles L., Riaño D., Ruiz-Gomez D.G., García-Ulloa A.C., Melgarejo M.A., Zamora M., Guillen-Pineda L.E., Mehta R., et al. High adiponectin concentrations are associated with the metabolically healthy obese phenotype. J. Clin. Endocrinol. Metab. 2008;93:4075–4079. doi: 10.1210/jc.2007-2724. [DOI] [PubMed] [Google Scholar]
- Arenas F., Garcia-Ruiz C., Fernandez-Checa J.C. Intracellular cholesterol trafficking and impact in neurodegeneration. Front. Mol. Neurosci. 2017;10:382. doi: 10.3389/fnmol.2017.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M., Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn. Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas F., Castro F., Nuñez S., Gay G., Garcia-Ruiz C., Fernandez-Checa J.C. STARD1 and NPC1 expression as pathological markers associated with astrogliosis in post-mortem brains from patients with Alzheimer’s disease and Down syndrome. Aging (Albany, NY) 2020;12:571–592. doi: 10.18632/aging.102641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach M.E., Hawkins R.D., Osman M., Kandel E.R., Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–915. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Barnes C.A. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Beilharz J.E., Maniam J., Morris M.J. Short-term exposure to a diet high in fat and sugar, or liquid sugar, selectively impairs hippocampal-dependent memory, with differential impacts on inflammation. Behav. Brain Res. 2016;306:1–7. doi: 10.1016/j.bbr.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Belgacem Y.H., Borodinsky L.N. CREB at the crossroads of activity-dependent regulation of nervous system development and function. Adv. Exp. Med. Biol. 2017;1015:19–39. doi: 10.1007/978-3-319-62817-2_2. [DOI] [PubMed] [Google Scholar]
- Biessels G.J., Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018;14:591–604. doi: 10.1038/s41574-018-0048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blüher M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019;15:288–298. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- Bocarsly M.E., Fasolino M., Kane G.A., LaMarca E.A., Kirschen G.W., Karatsoreos I.N., McEwen B.S., Gould E. Obesity diminishes synaptic markers, alters microglial morphology, and impairs cognitive function. Proc. Natl. Acad. Sci. USA. 2015;112:15731–15736. doi: 10.1073/pnas.1511593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D., Peleg-Raibstein D. The hypothalamus as a primary coordinator of memory updating. Physiol. Behav. 2020;223:112988. doi: 10.1016/j.physbeh.2020.112988. [DOI] [PubMed] [Google Scholar]
- Campbell J.N., Macosko E.Z., Fenselau H., Pers T.H., Lyubetskaya A., Tenen D., Goldman M., Verstegen A.M.J., Resch J.M., McCarroll S.A., et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 2017;20:484–496. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.A., Paul J.R., Yates S.D., Cutts E.J., McMahon L.L., Pollock J.S., Pollock D.M., Bailey S.M., Gamble K.L. Time-restricted feeding rescues high-fat-diet-induced hippocampal impairment. iScience. 2021;24:102532. doi: 10.1016/j.isci.2021.102532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon R.M.J. Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat. Protoc. 2006;1:122–124. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- Fan K.-Q., Li Y.-Y., Wang H.-L., Mao X.-T., Guo J.-X., Wang F., Huang L.-J., Li Y.-N., Ma X.-Y., Gao Z.-J., et al. Stress-induced metabolic disorder in peripheral CD4+ T cells leads to anxiety-like behavior. Cell. 2019;179:864–879.e19. doi: 10.1016/j.cell.2019.10.001. [DOI] [PubMed] [Google Scholar]
- Feinberg L.M., Allen T.A., Ly D., Fortin N.J. Recognition memory for social and non-social odors: differential effects of neurotoxic lesions to the hippocampus and perirhinal cortex. Neurobiol. Learn. Mem. 2012;97:7–16. doi: 10.1016/j.nlm.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Feinkohl I., Price J.F., Strachan M.W.J., Frier B.M. The impact of diabetes on cognitive decline: potential vascular, metabolic, and psychosocial risk factors. Alzheimers. Res. Ther. 2015;7:46. doi: 10.1186/s13195-015-0130-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Montoya M.G., Alvarez J.M., Sobin C. Olfactory recognition memory is disrupted in young mice with chronic low-level lead exposure. Toxicol. Lett. 2015;236:69–74. doi: 10.1016/j.toxlet.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz I., Miwa H., Konda Y., Shimoto Y., Tashiro T., Watson S.J., DelValle J., Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- Giuliani D., Neri L., Canalini F., Calevro A., Ottani A., Vandini E., Sena P., Zaffe D., Guarini S. NDP-α-MSH induces intense neurogenesis and cognitive recovery in Alzheimer transgenic mice through activation of melanocortin MC4 receptors. Mol. Cell. Neurosci. 2015;67:13–21. doi: 10.1016/j.mcn.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Gonzalez P.V., Schiöth H.B., Lasaga M., Scimonelli T.N. Memory impairment induced by IL-1β is reversed by α-MSH through central melanocortin-4 receptors. Brain Behav. Immun. 2009;23:817–822. doi: 10.1016/j.bbi.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Greenman Y., Kuperman Y., Drori Y., Asa S.L., Navon I., Forkosh O., Gil S., Stern N., Chen A. Postnatal ablation of POMC neurons induces an obese phenotype characterized by decreased food intake and enhanced anxiety-like behavior. Mol. Endocrinol. 2013;27:1091–1102. doi: 10.1210/me.2012-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyward F.D., Gilliam D., Coleman M.A., Gavin C.F., Wang J., Kaas G., Trieu R., Lewis J., Moulden J., Sweatt J.D. Obesity weighs down memory through a mechanism involving the neuroepigenetic dysregulation of Sirt1. J. Neurosci. 2016;36:1324–1335. doi: 10.1523/JNEUROSCI.1934-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs S., Spetter M.S. Cognitive control of eating: the role of memory in appetite and weight gain. Curr. Obes. Rep. 2018;7:50–59. doi: 10.1007/s13679-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jais A., Brüning J.C. Arcuate nucleus-dependent regulation of metabolism - pathways to obesity and diabetes mellitus. Endocr. Rev. Published online September. 2021;7:2021. doi: 10.1210/endrev/bnab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellar D., Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19:758–766. doi: 10.1016/S1474-4422(20)30231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner A.C., Janoschek R., Plum L., Jordan S.D., Rother E., Ma X., Xu C., Enriori P., Hampel B., Barsh G.S., et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kosse C., Burdakov D. Natural hypothalamic circuit dynamics underlying object memorization. Nat. Commun. 2019;10:2505. doi: 10.1038/s41467-019-10484-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam B.Y.H., Cimino I., Polex-Wolf J., Nicole Kohnke S., Rimmington D., Iyemere V., Heeley N., Cossetti C., Schulte R., Saraiva L.R., et al. Heterogeneity of hypothalamic pro-opiomelanocortin-expressing neurons revealed by single-cell RNA sequencing. Mol. Metab. 2017;6:383–392. doi: 10.1016/j.molmet.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.J., Hanchate N.K., Kondoh K., Tong A.P.S., Kuang D., Spray A., Ye X., Buck L.B. A psychological stressor conveyed by appetite-linked neurons. Sci. Adv. 2020;6:eaay5366. doi: 10.1126/sciadv.aay5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger M., Quiedeville A., Bouet V., Haelewyn B., Boulouard M., Schumann-Bard P., Freret T. Object recognition test in mice. Nat. Protoc. 2013;8:2531–2537. doi: 10.1038/nprot.2013.155. [DOI] [PubMed] [Google Scholar]
- Liu J., Garza J.C., Truong H.V., Henschel J., Zhang W., Lu X.-Y. The melanocortinergic pathway is rapidly recruited by emotional stress and contributes to stress-induced anorexia and anxiety-like behavior. Endocrinology. 2007;148:5531–5540. doi: 10.1210/en.2007-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Kishi T., Roseberry A.G., Cai X., Lee C.E., Montez J.M., Friedman J.M., Elmquist J.K. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J. Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K., McLaurin J. α-Melanocyte stimulating hormone prevents GABAergic neuronal loss and improves cognitive function in Alzheimer’s disease. J. Neurosci. 2014;34:6736–6745. doi: 10.1523/JNEUROSCI.5075-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahata B., Zhang X., Kolodziejczyk A.A., Proserpio V., Haim-Vilmovsky L., Taylor A.E., Hebenstreit D., Dingler F.A., Moignard V., Göttgens B., et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep. 2014;7:1130–1142. doi: 10.1016/j.celrep.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandela P., Yan Y., LaRese T., Eipper B.A., Mains R.E. Elimination of Kalrn expression in POMC cells reduces anxiety-like behavior and contextual fear learning. Horm. Behav. 2014;66:430–438. doi: 10.1016/j.yhbeh.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna P.R., Stetson C.L., Slominski A.T., Pruitt K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine. 2016;51:7–21. doi: 10.1007/s12020-015-0715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Segura A., Ahmed T., Casadomé-Perales Á., Palomares-Perez I., Palomer E., Kerstens A., Munck S., Balschun D., Dotti C.G. Age-associated cholesterol reduction triggers brain insulin resistance by facilitating ligand-independent receptor activation and pathway desensitization. Aging Cell. 2019;18 doi: 10.1111/acel.12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon S.H., Griffin L.D., Compagnone N.A. Biosynthesis and action of neurosteroids. Brain Res. Brain Res. Rev. 2001;37:3–12. doi: 10.1016/s0165-0173(01)00109-6. [DOI] [PubMed] [Google Scholar]
- Qu N., He Y., Wang C., Xu P., Yang Y., Cai X., Liu H., Yu K., Pei Z., Hyseni I., et al. A POMC-originated circuit regulates stress-induced hypophagia, depression, and anhedonia. Mol. Psychiatry. 2020;25:1006–1021. doi: 10.1038/s41380-019-0506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez S., Gómez-Valadés A.G., Schneeberger M., Varela L., Haddad-Tóvolli R., Altirriba J., Noguera E., Drougard A., Flores-Martínez Á., Imbernón M., et al. Mitochondrial dynamics mediated by mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metab. 2017;25:1390–1399.e6. doi: 10.1016/j.cmet.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Ratner M.H., Kumaresan V., Farb D.H. Neurosteroid actions in memory and neurologic/neuropsychiatric disorders. Front. Endocrinol. 2019;10:169. doi: 10.3389/fendo.2019.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.C., Fisher E.M.C., Brown S.D.M., Peters J., Hunter A.J., Martin J.E. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm. Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Schneeberger M., Dietrich D.O., Sebastián S., Imbernón M., Castaño C., Garcia A., Esteban Y., Gonzalez-Franquesa A., Castrillón Rodríguez I., Bortolozzi A., et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;26:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergi D., Williams L.M. Potential relationship between dietary long-chain saturated fatty acids and hypothalamic dysfunction in obesity. Nutr. Rev. 2019;78:261–277. doi: 10.1093/nutrit/nuz056. [DOI] [PubMed] [Google Scholar]
- Shen Y., Fu W.-Y., Cheng E.Y.L., Fu A.K.Y., Ip N.Y. Melanocortin-4 receptor regulates hippocampal synaptic plasticity through a protein kinase A-dependent mechanism. J. Neurosci. 2013;33:464–472. doi: 10.1523/JNEUROSCI.3282-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripetchwandee J., Chattipakorn N., Chattipakorn S.C. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front. Endocrinol. 2018;9:496. doi: 10.3389/fendo.2018.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez A.N., Noble E.E., Kanoski S.E. Regulation of memory function by feeding-relevant biological systems: following the breadcrumbs to the hippocampus. Front. Mol. Neurosci. 2019;12:101. doi: 10.3389/fnmol.2019.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., Lee K., Jing E., Biddinger S.B., McDonald J.G., Montine T.J., Craft S., Kahn C.R. Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab. 2010;12:567–579. doi: 10.1016/j.cmet.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., Ferris H.A., Chee M.J., Maratos-Flier E., Kahn C.R. Reduction of the cholesterol sensor SCAP in the brains of mice causes impaired synaptic transmission and altered cognitive function. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Gourley D.D., Dekhtyar M., Haley A.P. Cognition, brain structure, and brain function in individuals with obesity and related disorders. Curr. Obes. Rep. 2020;9:544–549. doi: 10.1007/s13679-020-00412-y. [DOI] [PubMed] [Google Scholar]
- Timper K., Paeger L., Sánchez-Lasheras C., Varela L., Jais A., Nolte H., Vogt M.C., Hausen A.C., Heilinger C., Evers N., et al. Mild impairment of mitochondrial OXPHOS promotes fatty acid utilization in POMC neurons and improves glucose homeostasis in obesity. Cell Rep. 2018;25:383–397.e10. doi: 10.1016/j.celrep.2018.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q., Ye C.-P., Jones J.E., Elmquist J.K., Lowell B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres S., Baulies A., Insausti-Urkia N., Alarcón-Vila C., Fucho R., Solsona-Vilarrasa E., Núñez S., Robles D., Ribas V., Wakefield L., et al. Endoplasmic reticulum stress-induced upregulation of STARD1 promotes acetaminophen-induced acute liver failure. Gastroenterology. 2019;157:552–568. doi: 10.1053/j.gastro.2019.04.023. [DOI] [PubMed] [Google Scholar]
- Tuem K.B., Atey T.M. Neuroactive steroids: receptor interactions and responses. Front. Neurol. 2017;8:442. doi: 10.3389/fneur.2017.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercruysse P., Vieau D., Blum D., Petersén Å., Dupuis L. Hypothalamic alterations in neurodegenerative diseases and their relation to abnormal energy metabolism. Front. Mol. Neurosci. 2018;11:2. doi: 10.3389/fnmol.2018.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., He X., Zhao Z., Feng Q., Lin R., Sun Y., Ding T., Xu F., Luo M., Zhan C. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front. Neuroanat. 2015;9:40. doi: 10.3389/fnana.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A.W., Kaelin C.B., Takeda K., Akira S., Schwartz M.W., Barsh G.S. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J. Clin. Invest. 2005;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Crawley J.N. Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci. 2009;Chapter 8 doi: 10.1002/0471142301.ns0824s48. Unit 8.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M.R., Schmitz A.E., Dietrich M.O. Activation of Agrp neurons modulates memory-related cognitive processes in mice. Pharmacol. Res. 2019;141:303–309. doi: 10.1016/j.phrs.2018.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data generated in this study will be shared by the lead contact upon reasonable request.
-
•
This study did not generate new code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.