

ABSTRACT
Pancreatic cancer remains largely unresponsive to immune modulatory therapy attributable in part to an immunosuppressive, desmoplastic tumor microenvironment. Here, we analyze mechanisms of cancer cell-autonomous resistance to T cells. We used a 3D co-culture model of cancer cell spheroids from the KPC (LSL-KrasG12D/+/LSL-Trp53R172H/+/p48-Cre) pancreatic ductal adenocarcinoma (PDAC) model, to examine interactions with tumor-educated T cells isolated from draining lymph nodes of PDAC-bearing mice. Subpopulations of cancer cells resistant to these tumor-educated T cells were isolated from the in vitro co-culture and their properties compared with sensitive cancer cells. In co-culture with resistant cancer cell subpopulations, tumor-educated T cells showed reduced effector T cell functionality, reduced infiltration into tumor cell spheroids and decreased induction of apoptosis. A combination of comparative transcriptomic analyses, cytometric and immunohistochemistry techniques allowed us to dissect the role of differential gene expression and signaling pathways between sensitive and resistant cells. A decreased expression of the chemokine CXCL12 (SDF-1) was revealed as a common feature in the resistant cell subpopulations. Adding back CXCL12 reversed the resistant phenotype and was inhibited by the CXCR4 inhibitor AMD3100 (plerixafor). We conclude that reduced CXCL12 signaling contributes to PDAC subpopulation resistance to T cell-mediated attack.
KEYWORDS: pancreatic cancer, T cell resistance, cytotoxicity, co-culture, chemokines
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with a 5-y overall survival rate of 10%.1 PDAC has been described as an immune-privileged tumor due to its immune suppressed tumor microenvironment (TME) and prominent desmoplasia resulting in resistance to immunotherapy.2 Still, the PDAC microenvironment is replete with immune cells,3,4 T cells being the most prevalent immune cell population in the majority of human PDACs.5,6 However, these T cells were not effective in killing autologous PDAC xenografts after ex vivo expansion5 or were located in the stroma at the invasive tumor margin and not infiltrating into the juxtatumoral compartment.3 As one arm of the adaptive immune system, T cells play a crucial role in executing the cytotoxic response and it is puzzling how PDAC resistance to T cell-mediated killing is maintained. One unresolved question is whether PDAC cancer subpopulations drive T cell resistance, particularly in regard to impaired T cell functionality in spite of their abundance (see Table S2). Previous studies have reported the role of tumor cell-intrinsic factors in shaping the immune TME and immunotherapy response in PDAC and other types of cancer.7–10 Tumor cell-derived CXCL1 and ubiquitin-specific protease 22 (USP22) were shown to decrease T cell infiltration and response to immunotherapy in PDAC.7,8 Also, driver mutations and altered gene expression patterns of cancer cell subpopulations may contribute to antitumor immunity, treatment resistance and poor survival of patients.11,12 However, most studies use cancer cell lines and animal models of unknown heterogeneity making it difficult to pinpoint immune resistance mechanisms in a physiological environment in the context of complex interplay with the TME.7,8,13,14 To date, no studies have explored the cytotoxic effects of isolated T cells on PDAC cancer cell subpopulations. Such an approach could allow for a better understanding and unbiased selection of resistant cancer cell populations to T cell attack. We have recently established a 3D cancer spheroid/T cell co-culture model to probe cancer cell – T cell interactions and used this model to dissect mechanisms that mediate sensitivities of cancer cell subpopulations to T cell killing.15 To generate appropriately educated T cells, we implanted PDAC cells from KPC (LSL-KrasG12D/+/LSL-Trp53R172H/+/p48-Cre) mice16 into appropriate syngeneic hosts and harvested T cells from tumor-draining lymph nodes (TdLNs) of allograft-bearing mice. Clonally selected PDAC cell lines exposed to these tumor-educated T (edT) cells in 3D spheroid culture identified T cell-sensitive cancer cell clones and resulted in selection of resistant cancer cell populations. The comparative analysis of differentially expressed genes revealed candidate pathways that have the potential to modulate responses of cancer cells to T cell attack, and we show that reduced CXCL12 signaling contributes to the resistance phenotype.
Materials and methods
Cell lines
The primary KPC pancreatic tumor cell line KPC 5991 was derived from a 65-d-old male KPC mouse.16 KPC 5991 cancer cells at cell culture passage 24–26 were utilized for s.c. injections into mice. The clonal cell lines C5, C8, D10, G8, G9 and F5 were established via single-cell cloning from a different primary KPC pancreatic tumor.17 The paired parental/resistant cell lines C8/C8R and D10/D10R1/D10R2 at cell culture passage no. 24–26 were utilized for all assays. All cancer cell lines were maintained in DMEM supplemented with 10% fetal bovine serum. T cells for co-culture assays were freshly isolated from mice on the day of co-culture. Co-cultures including T cells were maintained in RPMI supplemented with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin.
Animal models
All mice procedures were conducted in accordance with recommendations by the Georgetown University Institutional Animal Care and Use Committee. The animal study protocols for the procedures conducted in this study were approved by the Georgetown University InstitutionalAnimal Care and Use Committee. The transgenic KPC mouse model was originally described by Hingorani et al.16 For the allograft mouse model, 2- to 4-month-old male wild-type mice (littermates of the KPC mice) were utilized to establish allograft tumors subcutaneously in both flanks. Subcutaneously implanted allografts form dedifferentiated, desmoplastic lesions that are typical for pancreatic adenocarcinoma and are histologically indistinguishable from orthotopically implanted allografts.18
KPC-derived pancreatic cancer cell line culture
Fresh mouse pancreatic tumor tissue was minced for 5 minutes and shaken at 150 rpm for 1 h at 37°C in a Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12, Gibco Life) with 5% fetal bovine serum (FBS), 2 mg/mL collagenase (Sigma-Aldrich), 4 mg/mL trypsin (Sigma-Aldrich), 50 µg/mL gentamicin (Gibco Life), and 1 IU/mL penicillin/streptomycin (Gibco Life). The cell pellet was washed and centrifuged at 600 g and 4°C in DMEM/F-12, four times. The cell pellet was suspended in primary cell culture media (F-12, 10% FBS, 16 µg/mL insulin (Gibco Life), 10 ng/mL epidermal growth factor, 1 µg/mL hydrocortisone (Sigma-Aldrich), 4 ng/mL cholera toxin (Sigma-Aldrich), 50 µg/mL gentamicin, and 0.5 IU/mL penicillin/streptomycin. The cells were placed in a 37°C, 5% CO2, humidified incubator on a collagen-1 coated 10 cm culture dish (Corning BioCoat) in primary cell culture media for 60 minutes to let fibroblasts attach. Subsequently, the unattached cancer cells were transferred to a regular 10 cm dish. Primary cell culture media was changed every 48 h. After 1 week, the primary cancer cells were trypsinized and expanded to establish a cell line. After another week, the KPC 5991 cell line was grown in DMEM supplemented with 10% fetal bovine serum.
Tumor immunization of immunocompetent mice
KPC tumor-bearing mice show a spontaneous adaptive immune response to shared antigens across different PDAC tumors.19 We thus immunized mice with the above-described heterogeneous primary KPC PDAC cancer cell line (KPC 5991). Two- to 4-month-old male wild-type C57BL/6 × SV129 mice (littermates of the KPC mice) were utilized for immunization with cancer cells. Mice were anesthetized via isoflurane inhalant anesthesia until lack of response to toe pinch. All hair at both flanks including the back was shaved, and surgical scrub was performed. Four million KPC 5991 cells were suspended in 100 µL of serum-containing cell culture medium and injected subcutaneously into the right and left flank of each mouse. For each experiment, 2–6 mice were tumor immunized, dependent on the total amount of T cells needed for the respective experiment. On average, six to eight million T cells can be isolated from tumor-draining lymph nodes (TdLNs) of a tumor-immunized mouse. For in vivo studies, one million of KPC 5991 cells were suspended in 100 µl of serum-containing culture medium and injected subcutaneously into the right and left flank of each mouse. After 11 d, one million parental or resistant clonal PDAC cells were injected subcutaneously about 1 cm away from the KPC 5991 tumors on either the left or right side of the flank. At the same time, parental and resistant cells were injected into naïve mice without KPC 5991 tumors as controls. Three to eight mice were used per group. Tumor growth was monitored every third to fourth day by using a caliper. Tumors were harvested 8 or 12 d after tumor cell injections.
Lymph node/spleen excision and T cell isolation
Fourteen days after cancer cell injection, mice were euthanized using Carbon Dioxide-Standard Procedure followed by cervical dislocation. Longitudinal midline incision was performed to flip open the skin on both sides. Once the subcutaneous tumors with the adjacent inguinal and axillary lymph nodes were exposed (Suppl. Fig. S8A-C), tumors as well as draining lymph nodes were carefully removed. After opening the abdominal cavity via left transverse incision, the spleen was mobilized and carefully removed. In naïve mice, cervical lymph nodes were removed as well.
T cell isolation from lymph nodes and spleens was performed as negative selection using magnetic activated cell sorting (MACS) using Dynabeads™ Untouched™ Mouse T Cells Kit (# 11413D, Invitrogen), according to the manufacturer’s protocol. In brief, superparamagnetic beads (4.5 µm diameter) are coated with a secondary polyclonal antibody that binds rat IgGs. The antibody mix contains a cocktail of rat IgGs that bind mouse B cells, NK cells, monocytes/macrophages, dendritic cells, erythrocytes and granulocytes. After adding the antibody mix to the sample to bind unwanted non-T cells, the beads are then added. After a short incubation, the bead-bound cells are separated using a magnet. The purified T cells in the supernatant were transferred for subsequent co-culture assays. Overall, a purification of 85–99% CD3+ cells was reached (Suppl. Fig. S1C, first panel from left).
PDAC spheroid generation and 3D co-culture with T cells
A detailed description and protocol of the technical details of the 3D co-culture system is described in Lin et al.15 In short, 1000 cancer cells/spheroid were seeded in 35- or 81-microwell agarose casts (# A6013, Sigma-Aldrich) generated from 3D Petri Dishes®, a spheroid formation device from Microtissues® (Microtissues Inc., RI, USA). 15 min after cell seeding, 1 mL (for 35-microwell cast) or 2 mL (for 81-microwell cast) of cell culture medium were added and cancer spheroid were formed for 2 d at 37°C and 5% CO2 in a scaffold-free environment. At d 2, freshly isolated T cells from mice were added to the cancer spheroids in a 10:1 T cell to cancer cell ratio and co-culture was maintained in RPMI supplemented with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin (Suppl. Fig. S8D). A 10:1 T cell to cancer cell ratio was chosen because a lower cancer cell killing rate was observed with a ratio of 5:1. A ratio of 20:1 did not increase the killing rate (data not shown). This matches with other studies which reported tumor cell killing at a similar ratio of T cells to cancer cells, i.e. 10:1 to 25:1.20–22 After incubation for another 2 d, interleukin 2 (30 U/mL) and interleukin 7 (0.5 ng/mL) were added to the co-culture and controls. Anti-mouse-PD-1 was added in the co-culture or control +treatment groups at 100 μg/mL (clone BE0146, BioXCell).
3D spheroid invasion assay
The technical details of the 3D spheroid invasion assay into type 1 collagen, and subsequent analyses are described in Lin et al.15 In brief, after 2 d of cancer spheroid – T cell co-culture, the invasion into type 1 collagen (# 08–115, Millipore) was initiated (Suppl. Fig. S8D). First, collagen was neutralized to a pH of 7.0–8.0 and the co-culture was embedded in neutralized collagen within the 35-microwell agarose casts. After 4 min of incubation, the agarose casts including the collagen-embedded co-culture were inverted incubated for 1 h. Thereafter, the casts were flipped back and RPMI medium containing 5% FBS, 1% Pen/Strep, including 30 U/mL interleukin 2 (# 402-ML-020, R&D) and 0.5 ng/mL interleukin 7 (# 407-ML-005, R&D) were added. The invasion assay was performed for 2 d until images were taken and survived cells recovered from the collagen matrix. Co-culture of each cancer cell line in each condition was represented in at least two different agarose casts.
2D cancer cell proliferation assay
To monitor cancer cell proliferation in 2D, an electric cell-substrate impedance sensing (ECIS) system was used, in which cells were seeded in a 96-well format plate of an E-plate 33 instrument from xCELLigence.23,24 The cells were grown for in average 48 h (10.000 cells per well) until a complete monolayer was formed with a steady-state impedance reading. Changes in the electric impedance of the monolayer were measured at 15-min intervals as real-time readout, until total closure of the wound. Data analysis was performed using simple linear regression analysis in Prism 9.0 to determine the slope of cell index/hours within the proliferation phase.23
Cancer cell wound-healing assay
The wound-healing assay as an ECIS (electric cell-substrate impedance sensing)-based assay has been described in Sharif et al.23 In brief, PDAC cells (150.000 cells) were plated in the 8W10E array ECIS system for 4 h until cells were settled and formed a cell monolayer at the bottom of the wells. Cell culture media was changed in media ± mitomycin C (5 μg/mL, Sigma-Aldrich) to inhibit cell proliferation. After the formation of an intact monolayer with a steady-state impedance reading, pulses of high voltage were sent through the electrode array for 5 min to kill the cells growing directly on the electrodes. The impedance dropped by >80% until the cells neighboring the electrodes repaired the monolayer defect, covering the denuded area on the electrodes and restoring the original level of impedance. The kinetics of the wound closure was monitored in real-time, and the effect of mitomycin C on the migratory behavior was examined. Data analysis as performed on normalized resistance and using simple linear regression analysis in Prism 9.0 to determine the slope of resistance (ohm)/hours after wounding.23
Image analysis
Image analysis of the cancer spheroid invasion into type 1 collagen has been described in Lin et al.15 Cell culture images were acquired using the brightfield mode of an Olympus IX −71 inverted microscope with 10x magnification (Olympus, Tokyo, Japan). Invasion was defined as the invasion area relative to the size of the spheroid by using the Freehand Draw Tool from ImageJ (NIH, Bethesda, MD, USA). The invasion area was calculated as the ratio of total area (invasion + spheroid area) to spheroid area. At least ten images were analyzed for each cell line and each condition.
Cell recovery from collagen matrix
Surviving cancer cells were recovered from the collagen matrix after co-culture, according to Lin et al.15 In summary, collagen matrices including survived PDAC cells were separated from the agarose casts and digested with 1 mg/mL Collagenase 4 (# LS004188, Worthington) in cell culture medium. Cell clusters were broken up by pipetting up and down with a bevel-cut and 2.5% BSA pre-coated P1000 tip before and after a 15–20 min incubation step at 37°C. After complete dissolving of the collagen matrices, the cells were resuspended in serum-containing cell culture media, followed by three subsequent centrifugation steps (washing steps). Thereafter, TrypLETM (# 12604013, Thermo Fisher) was added to perform single-cell dissociation by incubating the cells for 20–30 min at 37°C. The dissociation reaction was stopped by adding serum-containing cell culture media followed by another centrifugation step. Afterward, cells were either maintained in culture or further processed for subsequent analysis.
Apoptosis assay
Cancer spheroids and T cells were co-cultured for 4 d until all cells were collected from the agarose casts.15 After single-cell dissociation with TrypLETM (# 12604013, ThermoFisher), Annexin V staining was performed. T cells were separated from the cancer cells by gating on CD3+ (# 100209, BioLegend) population. Annexin V FITC (# 640945, BioLegend) or PE/Cyanine 7 (# 640951, BioLegend) was used according to the manufacturer’s recommendation. All tests were performed in duplicates or triplicates.
Drug compounds and conditioned media
CXCR4 inhibitor (AMD3100) was purchased from Abcam (# ab120718) and recombinant murine CXCL12 (SDF-1α) from PeproTech (# 250–20A). AMD3100 was reconstituted in 1X PBS, diluted in cell culture medium to 10 μg/mL and replenished after 48 h co-culture. CXCL12 was reconstituted in ddH20, diluted in cell culture medium to 50 ng/mL and replenished after 48 h co-culture. For conditioned media, parental cells were seeded to be 30–40% confluent the next day. After 3 h, the media was substituted by RPMI + 10% FBS (“T cell media”) and after further 19 h incubation, the media was collected and filtered for subsequent assays. For AMD3100 rescue studies, CXCL12 and AMD3100 were simultaneously added to the co-culture media and replenished after 48 h.
Flow cytometry analysis
Protein cell surface expression was confirmed by flow cytometry. For flow cytometry analysis, T cells were labeled with fluorescence-conjugated CD3 (# 100209, BioLegend – dilution 1/200), CD4 (# 100549, Biolegend – dilution 1/200), CD8a (# 301035, BioLegend – dilution 1/200), CD25 (# 101915, BioLegend – dilution 1/200), PD-1 (# 135205, BioLegend – dilution 1/200), FOXP3 (# 15–5773-80, Invitrogen – dilution 3/200), CXCR4 (# 146505, BioLegend – dilution 1/200) antibodies, and cancer cells were labeled with fluorescence-conjugated PD-L1 (# 124315 – dilution 1.5/200), H-2Kb/H-2 Db (# 114607 – dilution 1/200) and CXCR4 (# 146505 – dilution 3/200) antibodies (BioLegend) and analyzed by FACS (BD LSRFortessa Cell Analyzer, # 647177).
Histopathology and immunohistochemistry
The details of the procedure for 3D spheroid co-culture have been described in Lin et al.15 In brief, cells are co-cultured for 4 d until they were embedded in HistoGelTM (# HG-4000-012, ThermoFisher) within agarose casts. The casts were dehydrated through an ethanol series and embedded in paraffin for subsequent sectioning at 5 μm per section starting from the bottom of the agarose cast and deposited on glass slides. Paraffin-embedded sections were de-paraffinized in xylene. For histological analysis, sections were stained with Harris hematoxylin and eosin (H&E). For immunohistochemistry, sections were rehydrated in graded alcohol and antigen retrieval was done by incubating the sections in low pH citrate solution and blocking endogenous peroxidases with H2O2. The sections were blocked with normal goat serum and stained with anti-CD4 (# 25229 – dilution 1/60) and anti-CD8 (# 98941 – dilution 1/25) antibodies (Cell Signaling) overnight. Anti-rabbit-HRP conjugated secondary antibodies were applied and the staining was developed using 3,3’-diaminobenzidine (DAB) chromagen. Nuclei were counterstained with Harris hematoxylin. For allograft tumor samples, the same procedure was used. For CD8 staining (Cell Signaling, # 98941 – dilution 1/25) in allograft tumors tissue sections were incubated for 1 h at room temperature. Images of stained tissues were captured using an Olympus BX40 microscope. Quantification of tumor areas and positive stainings was carried out using ImageJ (NIH, Bethesda, MD, USA).
T cell infiltration analysis
T cell infiltration into cancer spheroids was analyzed after cancer spheroid – T cell co-culture and IHC staining for CD4 and CD8 T cells in agarose casts, as described above. An average of 20 co-culture images of comparably sized cancer spheroids within one agarose cast per cell line and condition were taken with an inverted Olympus BX20 light microscope at 40× magnification. CD4+ and CD8+ T cells, respectively, were manually counted and divided into “adherent” and “infiltrated” T cells dependent on their localization in relation to the cancer spheroid. For analysis of T cell infiltration into allograft tumors, intratumoral CD8+ T cells were manually counted using Olympus BX20 light microscope at 10× magnification. Infiltrated CD8+ T cells are shown relative to the tumor area (pixels from ImageJ; NIH, Bethesda, MD, USA).
RNA extraction and RNA sequencing
RNA extraction was performed from PDAC cells grown in cell culture flasks (2D) and as spheroids embedded in collagen I. The RNA extraction of cancer spheroids in 3D is described in Lin et al.15 In brief, 12 collagen matrices including spheroids from each cell line were collected from 81-microwell casts (3000 cells/cast). Denaturation and separation of the phases were achieved by TRIzol (# 15596026, ThermoFisher) – chloroform procedure, followed by precipitation of the RNA in the aqueous phase with 70% ethanol. The total RNA was extracted using RNeasy Mini kit (Qiagen) with on-column DNAse digestion following a homogenization step using the MagNA Lyser. The RNA extracts were analyzed by a Nanodrop ND-1000 spectrophotometer and quantified by Qubit RNA BR Assay (ThermoFisher Scientific, Waltham, MA). Integrity of isolated RNA samples was validated using an Agilent RNA 6000 Nano Assay on the 2100 Bioanalyzer TapeStation (Agilent Technologies, Santa Cruz, CA, USA) and all samples sent for RNA-sequencing (Novogene, Inc., Sacramento, CA) if the RNA Integrity Value (RIN) was >9.0. A reference index was generated using GTF annotation from GENCODEv28. Raw FASTQ files were aligned to GRCm38 with HISAT2 and normalized and background-corrected using FeatureCounts and EdgeR. Resulting differential expression tables quantifying gene expression in counts per million and P value or comparisons were used to create a rank ordered list, which was then analyzed by GSEA. Heatmaps were generated by the pheatmap package (Novogene, Inc., Sacramento, CA) in the RStudio environment.
Quantitative RT-PCR (qRT-PCR)
RNA was isolated from co-cultured cancer spheroids with educated T cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany) with on-column DNAse digestion following a homogenization step using the MagNA Lyser, and quantified by Qubit RNA BR Assay (Thermo Fisher, Waltham, MA). Reverse transcription was done using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) according to the manufacturer’s protocol. Real-time quantitative RT-PCR was performed with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). Primers used for RT–qPCR were purchased from Integrated DNA Technologies, and their sequences are provided in Supplementary Table S1. Fold change was calculated by 2−ΔΔCT normalized to mouse actin as a housekeeping gene. All qRT-PCR assays were done in triplicates.
Statistical analysis
All statistical tests and graphing were carried out using GraphPad Prism 9.2.0, Microsoft Excel, GSEA, Ingenuity® Pathway Analysis and EdgeR package in R. ANOVA was used for multiple comparisons and t tests was used for paired comparisons, with P < .05 as the threshold for statistical significance in all tests.
Software
Spheroid invasion area in collagen I and allograft tumor area were measured with ImageJ (NIH, Bethesda, MD, USA). Flow cytometry data was processed using the FCS Express 7 software (De Novo Software).
Compliance with ethical standards
Studies involving the use of animals and cell lines were approved by the Georgetown University Institutional Animal Care and Use Committee (IACUC).
Results
T cell-mediated cancer cell killing distinguishes PDAC clonal populations
To probe the effect of T cells to curtail the invasion of PDAC tumor cells, we used a recently established 3D cancer spheroid co-culture model that monitors in vitro invasion of cancer cells in co-culture (Figure 1(a)).15 Wildtype littermates of the KPC mice were injected subcutaneously with KPC cells that were generated from a separate KPC donor and kept as a heterogeneous cell population (named KPC 5991). Tumor-educated (edT) cells were then isolated from tumor-draining lymph nodes (TdLNs) or spleen, and co-cultured with PDAC clonal cancer cells (C8 or D10) or with KPC 5991 for 4 d.
Figure 1.
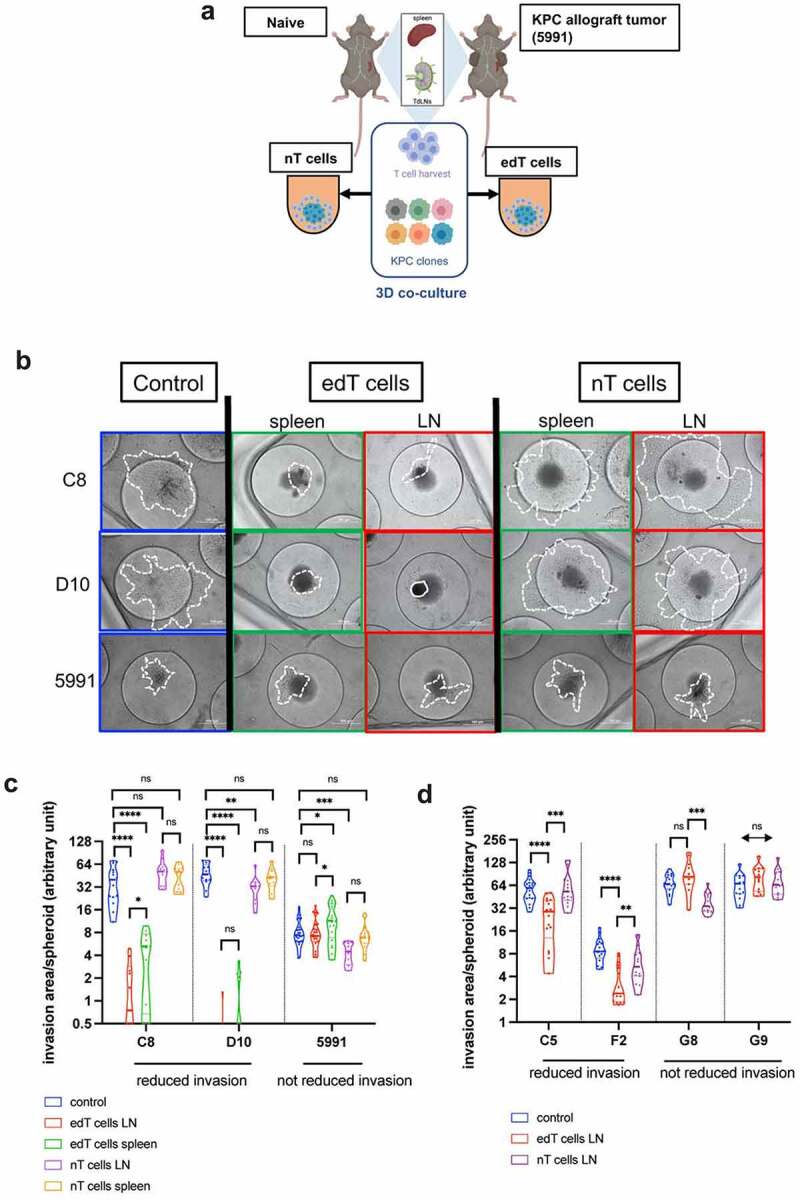
T cell-mediated PDAC cancer spheroid killing is specific for PDAC subpopulations. (a) Schematic of the approach. T cells from lymph nodes or spleens of naïve and KPC PDAC (KPC 5991 cells) allograft carrying mice were harvested. Six clonal cell lines from a primary KPC PDAC were used for 3D co-culture with naïve and edT cells. TdLNs = tumor-draining lymph nodes. (b) Representative images of cell lines from (A) in 3D co-culture with PDAC tumor-educated (edT) or naïve (nT) cells from spleen or tumor-draining lymph nodes (LN). Cancer spheroids without T cells served as controls. Area of cell invasion is outlined by a white dashed line; cell debris by a continuous line. Scale bars, 100 μm. (c–d) Violin plots of Log2 invasion area per cancer spheroid of the PDAC cell lines after co-culture with T cells. Cell lines with decreased invasion area after edT cell co-culture are grouped on the left. (Student t test, *P < .05, **P < .01, ***P < .001, ****P < .0001, n.s., not significant). Schematic in A created in BioRender.com.
PDAC clonal cell lines C8 and D10 showed a drastic decrease of spheroid invasive spreading in the presence of edT cells compared to spheroids without T cells (control) and naïve controls (nT cells) (Figure 1(b,c)). As a result of the edT cell killing, an accumulation of cell debris from the C8 and D10 cell co-cultures was observed (Suppl. Fig. S1A). This effect is specific for edT cells from the syngeneic host since no cell debris was observed in edT cell co-culture with control KPC 5991 cells. Also, KPC 5991 cell spheroids did not show a significantly decreased invasion in co-culture with edT cells compared to control and nT cells (Figure 1(b,c)). To test if there is a secondary lymphatic organ-specific function during tumor development, T cells from both spleen and lymph nodes (LNs) were used to examine their effects on the invaded area of the cancer cell spheroids. Similar to edT cells from LNs, splenic edT cells also significantly decreased in the area invaded by C8 and D10 cancer cell spheroids (Figure 1(b,c)). However, edT cells from LN showed a stronger effect on decreasing spheroid invasion and caused more cell debris than spleen edT cells (Figure 1(b,c), Suppl. Fig. S1A), suggesting that TdLNs of PDAC allograft mice contain more cancer cell-specific edT cells.
To examine whether the observed T cell-mediated cancer cell killing was distinct for different clonal cell lines, four other KPC PDAC clonal cell lines generated from the same tumor (C5, F2, G8, G9) were utilized for T cell co-culture experiments. Indeed, the extent of invasion into collagen by these clonal cell lines was impacted differently. While C5 and F2 clones showed significantly decreased cancer spheroid invasion in edT cell co-cultures, G8 and G9 invasion was not impacted (Figure 1(d)). These data indicate that KPC PDAC clonal cells show distinct responses to co-culture with PDAC-educated T cells.
T cell populations in the spheroids were analyzed via flow cytometry before (Suppl. Fig. S1B) and after (Suppl. Fig. S1C) co-culture. The ratio of CD8+/CD4+ T cells was the highest in edT cells from LNs (Suppl. Fig. S1B, second panel from left), and in LN edT cells co-cultured with either C8 or D10 cancer cells (Suppl. Fig. S1C, second panel from left). We thus only used LN edT cells for subsequent co-culture experiments. Regulatory T cells (Tregs) and programmed cell death protein-1 (PD-1) expression on T cells can be indicators for immunosuppression and inhibited cytotoxic T cell function.25 We observed a significantly higher percentage of Tregs (CD4+ CD25+ FoxP3+) and PD-1 positive T cells in the spleen of PDAC-exposed mice compared to TdLNs and to T cells from naive mice (Suppl. Fig. S1B-C, first and second panel from right). However, the differences in percentage of Tregs and PD-1 positive T cells from LNs after co-culture were small between the clonal cells and KPC 5991 cells. This suggests that immune suppressive cells and checkpoint proteins were not responsible for causing the differential effects between the clonal cells and KPC 5991 cells in co-culture.
Selection of edT cell-resistant cancer cell subpopulations
We observed that a small fraction of cancer cells survived the exposure to edT cells. We recovered these edT cell-resistant (“R”) C8 and D10 cells from the collagen matrix to examine their resistance mechanisms to T cell cytotoxicity (Figure 2(a)). Fewer resistant cancer cells were recovered from clone D10 after edT co-culture (D10R) than from clone C8 (C8R) (Figure 2(b)). Cell morphology (Suppl. Fig. S2A), proliferation (Suppl. Fig. S2B, E), 2D migration (Suppl. Fig. S2C, D) and 3D invasion (Suppl. Fig. S2F) were examined.
Figure 2.
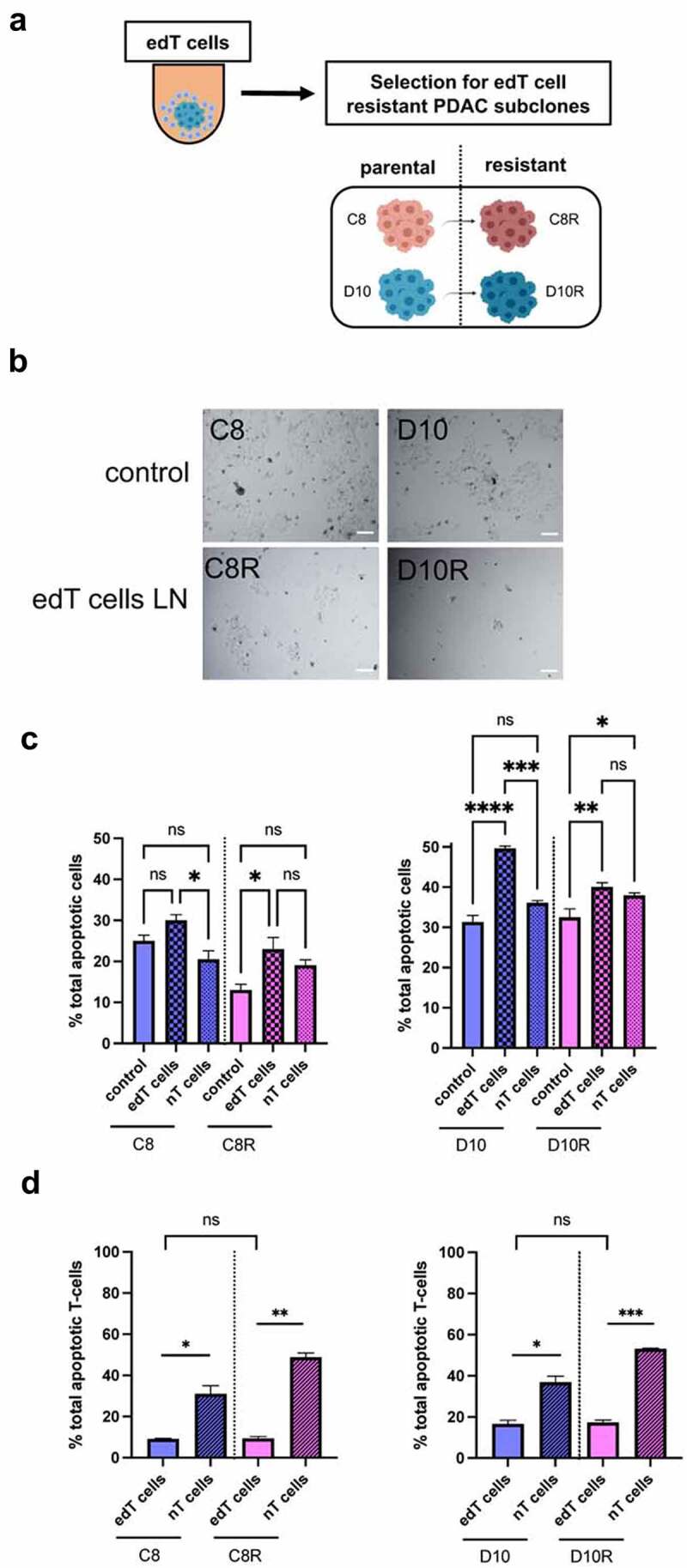
Selection of PDAC cells resistant to edT cell-mediated cytotoxicity. (a) Schematic of the isolation of resistant cells. (b) Images of a representative area within a cell culture dish after isolation of resistant PDAC cells from edT cell co-cultures or control (cancer spheroids without T cells). Scale, 100 μm. (c and d) Monitoring apoptosis via FACS with propidium iodide (PI) and Annexin V staining: (c) Percentage of total apoptotic cancer cells after co-culture with edT and nT cells. Control is cancer spheroids only. (d) Percentage of total apoptotic T cells after co-culture with cancer spheroids. Data represent the mean ± SD from at least three biological replicates. (Student t test, *P < .05, **P < .01, ***P < .001, ****P < .0001, n.s., not significant). Schematic in A created in BioRender.com.
The data show a distinct growth phenotype between the cell lines in 2D and 3D culture settings. Most importantly, there were no differences in invasive behavior between parental and edT cell-resistant cells in the absence of T cells, indicating that the invasive phenotype was not responsible for the resistance to edT cell co-culture.
Next, we evaluated cancer cell killing by edT cell co-culture by monitoring apoptosis via FACS with propidium iodide (PI) and Annexin V staining (Figure 2(c,d)). Both parental PDAC cell lines showed a significantly higher apoptosis rate after edT cell co-culture than the resistant cell lines (Figure 2(c)). Also, edT cells in co-culture showed less apoptosis than nT cells as expected from the stimulation edT cells receive from the engagement of the targeted cancer cells (Figure 2(d)). To investigate if different cell surface expression of MHC-1 and PD-L1 is responsible for edT cell resistance, flow cytometry analysis was performed (Suppl. Fig. S2G). Most notably, D10R showed a distinctly higher cell surface expression of PD-L1 in 3D culture compared to C8R and both parental PDAC cell lines. However, anti-PD-1 treatment did not reverse resistance of D10R cells to T cell-mediated killing (Suppl. Fig. S3A), suggesting that the elevated PD-L1 is not the driver of resistance.
CD8+ T cell infiltration into edT cell-resistant PDAC spheroids is decreased
Previous studies have identified a correlation between CD8+ T cell infiltration and response to immunotherapy in melanoma and PDAC.8,26 To examine whether resistance to edT cell-mediated PDAC cell killing is related to T cell access to cancer cells, we analyzed PDAC 3D co-cultures for the distribution of cancer and T cells by staining for CD4+ and CD8+ T cells. This analysis allowed us to quantify T cells that only adhere to the cancer spheroid periphery and those which infiltrated into the spheroid (Figure 3). Only spheroids with comparable sizes were subjected to the comparative analysis. The number of CD8+ edT cells that adhered to the spheroids showed no significant differences between parental and resistant cancer cells although D10 cancer cells attracted a higher number of edT cells per spheroid than C8 cancer cells. For both C8 and D10 cancer cell lines there was significantly more CD8+ edT cell infiltration into spheroids from parental cancer cells than spheroids generated from resistant cancer cells (Figure 3(b)). Because the D10 cell line showed more infiltrated CD8+ edT cells than the C8 cell line, we also stained for CD4+ edT cells (Suppl. Fig. S3B). The number of infiltrated or adherent CD4+ edT cells was not significantly different between parental and resistant cells and there were much less infiltrated CD4+ than CD8+ edT cells (Suppl. Fig. S3C).
Figure 3.
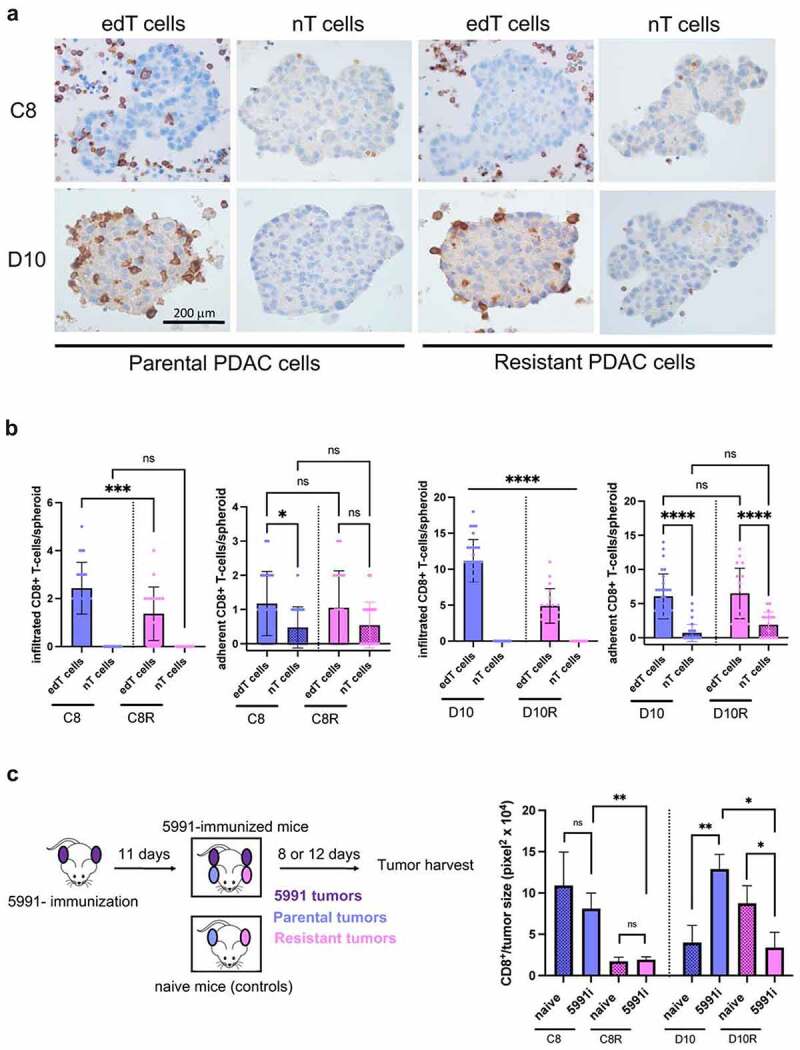
CD8+ edT cell infiltration into resistant PDAC cancer spheroids and allograft tumors is decreased. (a) Representative 3D co-culture sections of paired parental/resistant cells from C8 and D10 stained by immunohistochemistry (IHC) for CD8+ T cells. Scale, 200 μm. (b) Quantification of infiltrated and adherent CD8+ T cells co-cultured with cancer cell spheroids. (c) Schematic of the experimental setup for the in vivo studies (left). Quantification of tumor-infiltrated CD8+ T cells (right). 5991i = immunized with KPC 5991 cells. Data represent the mean ± SD from at least two biological replicates and n ≥ 3 mice per group for the in vivo experiments. (One-way ANOVA for multiple comparison and Student t test otherwise, *P < .05, **P < .01, ***P < .001, ****P < .0001, n.s., not significant).
To validate the T cell-sensitive and -resistant phenotype that was selected in vitro also in vivo, mice were pre-immunized with KPC 5991 cancer cells prior to injection of the parental (sensitive) and resistant cells (Figure 3(c), left). The analysis of parental and resistant tumors from KPC 5991 cells-immunized mice revealed significantly higher CD8+ edT cell infiltration into both parental cell lines C8 and D10 compared to resistant cell lines C8R and D10R, respectively (Figure 3(c), right). While C8 parental cells showed higher CD8+ T cell infiltration than C8R resistant cells independent of immunization, CD8+ T cell infiltration was significantly increased in the D10 parental cell line after immunization. In contrast, D10R tumors showed decreased CD8+ edT cell infiltration. Accordingly, D10 tumor size was significantly decreased in the immunized group compared to naïve mice without immunization (Suppl. Fig. S3D). From the above data, we conclude that CD8+ edT cell infiltration is reduced in spheroids grown from edT cell-resistant cancer cells and that this decreased T cell infiltration is also observed in vivo, thus supporting validating the in vitro selection approach.
Differential gene expression of CXCL12 distinguishes parental from resistant PDAC cells
RNA sequencing was used to examine altered gene expression patterns of parental and edT cell-resistant PDAC cell lines. Overall, the RNA-seq analysis showed a distinct gene expression pattern of cancer cells grown in 2D and 3D (Suppl. Fig. S4A). Gene set enrichment analysis (GSEA) revealed that hallmark pathways for Inflammatory Response and Protein Secretion were inhibited in edT cell-resistant PDAC cells (Suppl. Fig. S4B-D). From the 50 most downregulated genes in edT cell-resistant PDAC cells, overlapping genes that were shared between paired cell lines are shown in Figure 4(a). Interestingly, the chemokine CXCL12 was the only overlapping gene significantly downregulated in all resistant cell lines compared to parental cell lines in 2D and 3D (Figure 4(a-c)). Since CXCL12 is known to mediate T cell recruitment and infiltration into tumors,27,28 we examined the expression levels of other genes encoding chemokines with T cell-recruiting functions. The expression levels of CCL2 and CCL7, both T cell chemoattractant chemokines,29 were downregulated in edT cell-resistant PDAC cells as well (Suppl. Fig. S5A). However, Th1-type chemokines, such as CCL5, CXCL10 and CXCL11, did not show any differential expression in edT cell-resistant PDAC cells compared to the parental cell lines (Suppl. Fig. S5B). Taken together, differential gene expression of the chemokines CXCL12, CCL2 and CCL7 distinguishes parental from edT cell-resistant PDAC cells.
Figure 4.
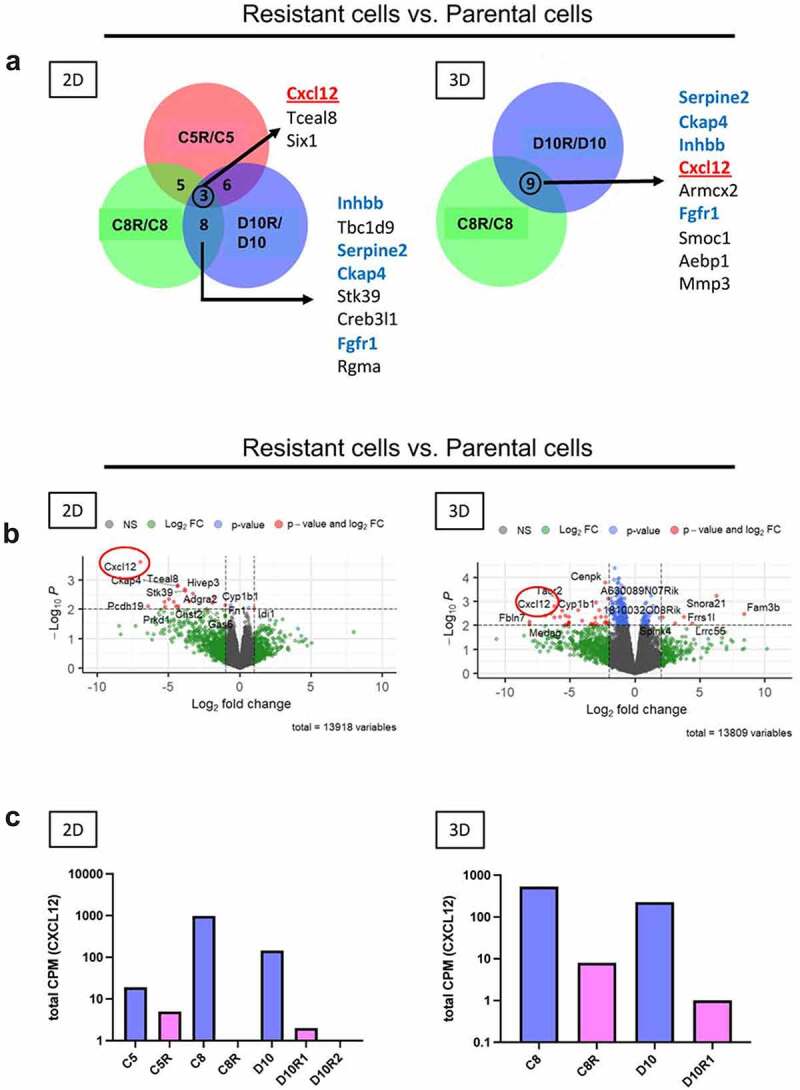
CXCL12 expression is decreased in edT cell-resistant PDAC cells. (a) Venn diagrams showing the 50 most downregulated genes in resistant vs. parental cell lines. Gene expression rankings were derived from comparisons of fold differences between RNA-sequencing data from resistant and parental cell lines. CXCL12 is present in the overlap of all three paired cell lines in 2D and in both paired cell lines in 3D. Distinctly regulated genes shared between C8R/C8 and D10R/D10 in 2D and in 3D growth conditions are highlighted in blue. The edT cell-resistant cell line C5R has been isolated from the PDAC clonal cancer cell line C5 in a separate co-culture experiment. C5/C5R were included in the transcriptomic analysis. (b) Volcano plots of differentially expressed genes based on RNA-sequencing analysis of resistant vs. parental cell lines in 2D and 3D. Highlighted is CXCL12, being downregulated in resistant cell lines in 2D and 3D. Dashed lines represent p = .01 and z-score = −2 and +2. (c) Gene expression level of CXCL12 in paired parental/resistant cell lines based on RNA-sequencing analysis. D10R1 and D10R2 are two different resistant cell lines derived from D10 from two independent co-culture experiments. D10R1 is also referred to as D10R in 3D co-culture experiments. CPM = transcript counts per million.
CXCL12 increases T cell killing of resistant cells through CXCR4 signaling
The transcriptomic analysis suggested that soluble factors contribute to edT cell sensitivity in PDAC cells, with CXCL12 being a major contributor. To test this hypothesis, resistant PDAC spheroids in edT cell co-culture were treated with conditioned media from the respective parental cell line (CMP) and CXCL12 (Suppl. Fig. S6A, left). Both resistant PDAC cell lines C8R and D10R showed a significant increase in apoptosis compared to untreated resistant cells (by 39.6% in C8R and by 16.1% in D10R). However, adding conditioned media from resistant cells (CMR) and blocking the CXCL12 receptor CXCR4 with AMD3100 (plerixafor), did not render parental PDAC cells more resistant (Suppl. Fig. S6A, right). To isolate the effect of CM from the effect of CXCL12, resistant cells in edT cell co-culture were treated either with CM or CXCL12 alone (Figure 5(a)) and the data indicate that CXCL12 is sufficient to increase edT cell-mediated PDAC cell apoptosis in C8R cells. To determine whether CXCL12 treatment improves T cell activation and effector functions, mRNA expression levels of Tumor Necrosis Factor alpha (TNFa), Granzyme b (Gzmb), Perforin-1 (Prf1), Interleukin-2 (IL-2) and Interferon-gamma (IFN-γ) were analyzed. IFN-γ is an important effector of cytotoxic CD8+ T cells. Indeed, IFN-γ expression of CXCL12-treated resistant cell co-cultures was higher compared to untreated groups (Figure 5(b)). The CXCL12 effect was statistically significant for C8R, whilst CXCL12 treatment of D10R induced the IFN-γ expression to trend higher but did not reach the level of parental D10 cell co-culture. There was also a trend toward increased expression of other T cell activation markers upon CXCL12 treatment, such as TNFa, Gzmb, Prf-1 and IL-2 (Suppl. Fig. S6B). Accordingly, CXCL12 treatment increased CD8+ T cell infiltration into cancer spheroids of resistant PDAC cell lines (Figure 5(c)).
Figure 5.
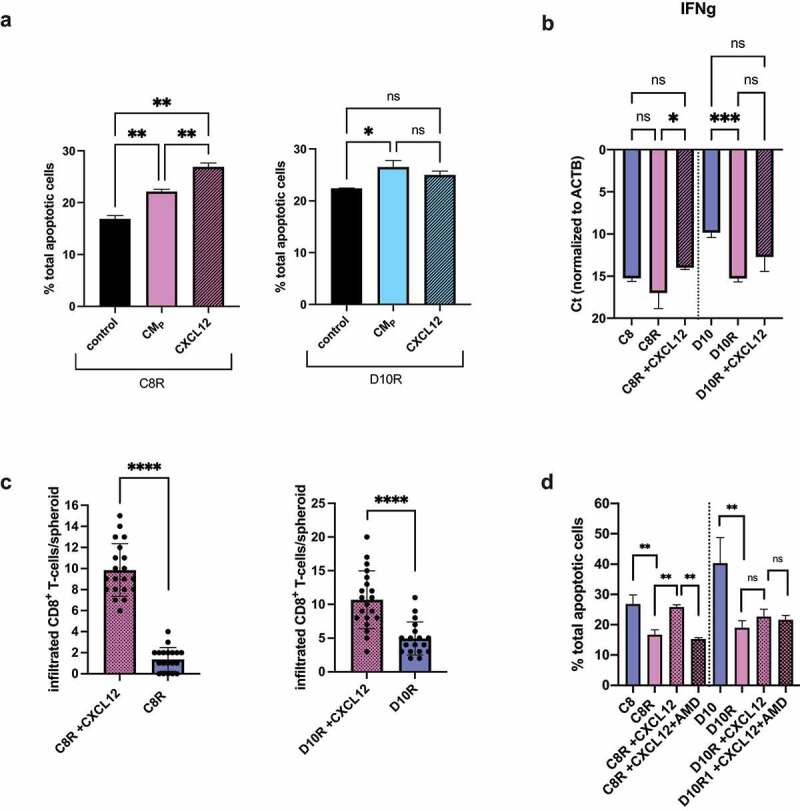
CXCL12 increased edT cell-mediated killing of resistant PDAC cells and effector edT cell functionality. (a) Percentage of total apoptotic-resistant cancer cells with parental cell conditioned media (+CMP) or +CXCL12 after co-culture with edT cells. Controls are cancer spheroid co-culture with edT cells without treatment. (b) Expression of IFN-γ by qRT-PCR from cancer spheroid/T cell co-culture. Ct-values normalized to beta-actin are shown. (c) Quantification of infiltrated CD8+ T cells into CXCL12-treated resistant cancer spheroids compared to untreated resistant spheroids. (d) Apoptotic cancer cells ±CXCL12 ± AMD3100 after co-culture with edT cells. All data represent the mean ± SD from at least three biological replicates and two technical replicates. (Student t test, *P < .05, **P < .01, ***P < .001, ****P < .0001, n.s., not significant). CMP = conditioned media of parental cells.
CXCL12 is a ligand for CXCR4 and CXCR7.30–32 To examine whether the CXCL12 effect is CXCR4-mediated, resistant PDAC cell spheroids in edT cell co-culture were treated with both CXCL12 and the antagonist AMD3100 (plerixafor). In the presence of AMD3100 we observed a significant decrease of cancer apoptotic rate to a similar level as the untreated group in C8R cells (Figure 5(d)). Consistent with D10R cells not showing a significant CXCL12 effect, AMD3100 treatment also did not significantly decrease cancer cell apoptosis in edT cell co-culture. The lack of an effect was not related to an altered CD8+/CD4+ ratio, or altered T cell apoptosis rate (Suppl. Fig. S6C). Considering that CXCR4 can be expressed on both cancer and T cells,32 CXCR4 protein expression was analyzed via flow cytometry and we found that CXCR4 is expressed on both resistant PDAC cell lines and edT cells (Suppl. Fig. S7A-B). However, CXCR4 surface expression was >3-fold and >10-fold higher on edT CD4+ and CD8+ cells, respectively, relative to PDAC cells (Suppl. Fig. S7B vs A). It is noteworthy that CXCL12 did not impact resistant PDAC spheroid apoptosis in the absence of edT cells (Suppl. Fig. S7C). We conclude from these data that CXCL12 treatment increased killing of resistant PDAC cells through improved effector T cell functionality and increased CD8+ T cell infiltration into spheroids via CXCL12/CXCR4 signaling in CD8+ edT cells.
Discussion
Understanding immune resistance mechanisms of PDAC have important therapeutic implications. Using a recently developed 3D cancer spheroid/T cell co-culture model,15 we were able to isolate edT cell-resistant subpopulations from PDAC clonal cancer cells, and uncover T cell resistance mechanisms in cancer cell subpopulations (Figure 6). The rationale for this experimental approach is to examine edT cell-mediated effects on PDAC cancer cell subpopulations under well-defined conditions. This approach provides a platform for the discovery of PDAC cell-intrinsic resistance mechanisms to edT cell-mediated attack as shown here and can be expanded to more complex model systems including human PDAC. The model system is designed to resemble tumor pathophysiology: First, we used the KPC mouse model as the source for our primary cancer cell lines since it mimics many features of the human disease.16 Second, we performed co-culture in a 3D setting instead of 2D culture conditions. Our transcriptomic data showed significant difference in gene expression pattern between cells grown in 2D and 3D culture environment (Suppl. Fig. S4A), highlighting the importance of utilizing 3D culture systems to create a more physiological environment. Third, we utilized T cells from draining lymph nodes of tumor-bearing mice instead of nonspecifically activated T cells or genetically engineered T cells designed to recognize exogenous antigens on cancer cells.22,33,34 Notably, the resistance phenotype was confirmed in in vivo studies, corroborating the significance of the in vitro approach.
Figure 6.
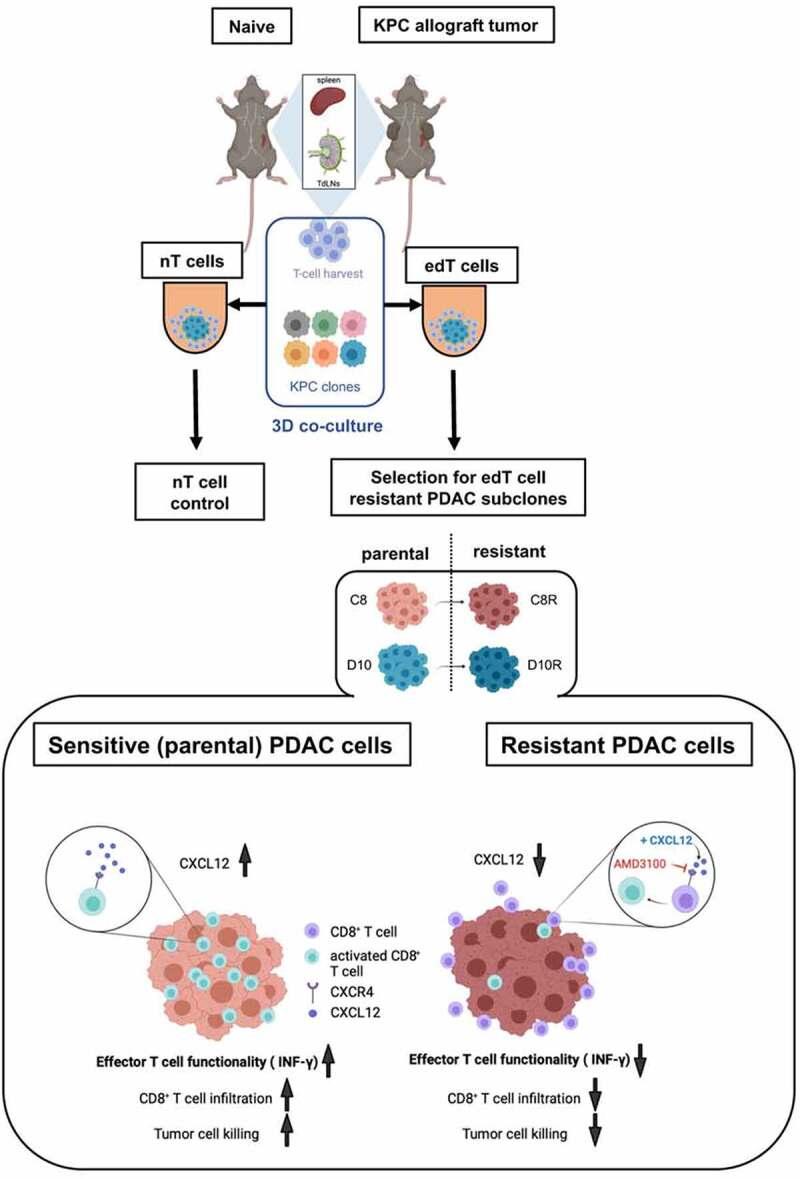
Graphical abstract. CXCL12 sensitizes resistant KPC PDAC cells to T cell-mediated cytotoxicity through CXCR4 signaling. Overview of the experimental design and main findings. Mice were subcutaneously tumor-immunized with KPC PDAC cells. After 14 d, T cells from tumor-draining lymph nodes (TdLNs) and spleens of naïve and KPC PDAC tumor-immunized mice were harvested. Using different KPC PDAC clonal cell lines, 3D co-culture with T cells was performed. Naïve (nT) cells from non-tumor bearing mice served as controls. From two clonal cell lines, C8 and D10, tumor-educated (edT) cell resistant subpopulations C8R and D10R were isolated after co-culture. edT cell-resistant PDAC cells showed less CXCL12 expression than parental cells, resulting in decreased tumor cell killing due to impaired effector T cell functionality (IFN-γ expression) and decreased CD8+ edT cell infiltration. CXCL12 add-back improved effector T cell functionality and killing of resistant tumor cells. AMD3100 is a selective CXCR4 antagonist. Figure created in BioRender.com.
Here we evaluated cancer cell vulnerabilities to T cell resistance at a subclonal cell level and by using edT cells from tumor-draining lymph nodes (TdLNs) of PDAC-immunized mice. Since edT cells have been primed in vivo after tumor engraftment, TdLNs accumulate tumor antigen-specific T cells.35 Compared to tumor-infiltrating lymphocytes (TILs), T cells from TdLNs are less exhausted and immune tolerant,25,36,37 and provide a more robust edT cell source for co-culture experiments. There is a growing understanding in how tumor burden perturbs the peripheral immune landscape.36 Secondary lymphoid organs, such as spleen and TdLNs, form an immunological network in continuous communication during tumor growth. While TdLNs are closely connected with the tumor site, the spleen represents immune alterations in the circulation and we thus compared edT cell from TdLNs and the spleens in PDAC cell co-culture. Overall, there was no qualitative difference in the effect of edT cells from the two lymphoid sources although cells from TdLN showed a stronger effect on spheroid invasion and increased cancer cell cytotoxicity. Thus, using edT cells from TdLNs enabled a more specific selection of cancer cell-reactive T cells for the co-culture experiments. Importantly, co-culture with splenic edT cells from spontaneous PDAC-bearing KPC mice resulted in similar effects on the cancer cells compared to edT cells from mice with subcutaneous tumors (Suppl. Fig. S9, Figure 1(c)). Hence, our allograft model is a valid model for T cell education in pancreatic cancer.
Our focus on CXCL12 (SDF-1) as a regulator of effector T cell activity was based on an unbiased analysis of transcriptomic data that showed significantly lower CXCL12 expression in edT cell-resistant PDAC cells compared to the respective parental control cells. CXCL12 add-back resulted in improved effector T cell functionality with increased CD8+ T cell infiltration of both resistant subclones and significantly sensitized the resistant subclone C8R to T cell cytotoxicity. This suggests that additional factors contribute to D10R cell survival and the studies with conditioned media (CM) indicate that additional soluble factors contribute to the sensitivity to D10R cells (Figure 5(a)). Since the RNA-sequencing data showed downregulated expression of CCL2 and CCL7 in D10R compared to D10 cells (Suppl. Fig. S5), these chemokines might have contributed to sensitize D10R cells in the CM experiments.
Interestingly, inhibition of the CXCL12 receptor CXCR4 with AMD3100 (plerixafor) did not decrease baseline T cell sensitivity in both parental cell lines, indicating that it might be sufficient for the rescue but is not the only rate-limiting factor. Altogether, our data suggest that in resistant subclones CXCL12 effects CD8+ T cell cytotoxic activity through its receptor CXCR4, resulting in increased effector T cell activation and T cell infiltration in cancer spheroids, and thus increased PDAC cell cytotoxicity.
Thus far, clinical data have been inconclusive concerning the role of CXCR4 inhibition in cancer patients: combination of nivolumab (anti-PD-1) and ulocuplumab (anti-CXCR4 monoclonal antibody) failed to demonstrate efficacy against PDAC (clinical trial #NCT02472977).38 A phase I study of the CXCR4 inhibitor LY2510924 and durvalumab (anti-PD-L1) showed modest clinical activity.39 These data indicate that CXCL12/CXCR4 signaling in cancer has yet to be completely understood and this lack of understanding limits the successful application of drug combinations that modulate nodes of this pathway. Previous studies have implicated CXCL12 as promoting immune evasion in PDAC,3,19,40 as well as stimulating PDAC progression and metastasis through direct effects on the tumor cells.41 However, the effects were indirectly shown by inhibiting its receptor CXCR4 with AMD3100. Furthermore, in these studies animal models and bulk tumors have been exclusively used. Nonetheless, the exact mechanism of CXCL12-mediated immune modulation in PDAC has not yet been elucidated. It is hypothesized that high mobility group box 1 (HMGB1), being overexpressed and secreted by metabolically stressed cancer cells,42 captures CXCL12 by forming a high-affinity heterocomplex.43 This phenomenon ought to explain the paradoxical localization of CXCL12 on cancer cells despite their lack of expression of CXCL12.19 In contrast to these studies, our data reveal that PDAC cancer subpopulations do show distinct CXCL12 expression. We used a different model system and examined interaction of cancer cell subpopulations with tumor-educated T cells. Thus, our studies cannot be directly compared with those previous studies. Furthermore, CXCL12 is known as a bidirectional chemokine, and immunologic response to antigenic challenge is regulated by its concentration and cell-intrinsic dependent properties.44 Functional studies revealed cancer cells moving away from stromal CXCL12 sources in physiologic microenvironments.44 In this regard, previous studies showing increased immune response after CXCR4 inhibition might be due to fine-tuning of CXCL12 concentration toward movement of cytotoxic immune cells to cancer cells.40
In conclusion, the co-culture of in vivo cancer-educated T cells and PDAC cell subpopulations revealed that differential expression of CXCL12 and CXCL12/CXCR4 signaling can modulate the sensitivity of PDAC cells to T cell cytotoxicity. Thus, our data contribute to a more differentiated understanding of this pathway to T cell responses. Also, we provide a platform to study cancer cell/T cell crosstalk. Future work will be required to expand this to other cell types present in the TME and to human PDAC to delineate the contribution of subclonal crosstalk to resistance to immune targeting.
Supplementary Material
Acknowledgments
We thank Drs Marta Catalfamo, Maha Moussa and Sarah M. Roth for their thoughtful input and Dr Robin D. Tucker for instruction on lymph node dissection. Drs Michael B. Atkins, Samir N. Khleif and Louis M. Weiner (all Georgetown University) kindly reviewed early versions of the manuscript and provided helpful suggestions.
Funding Statement
This research was supported by grants from the DFG (Deutsche Forschungsgemeinschaft) to YL (LI 2547/4-1) and the National Institutes of Health to AW (P30 CA51008R01 CA231291), and to ATR (R01 CA205632). Shared Resources used include TCRBSR, FCCSSR, MIAMSR and HTSR are supported by P30 CA051008.
Author contributions
YL and AW conceived the project and wrote the manuscript. EEV generated and characterized the clonal cell lines. YL and MOS performed the research. All authors discussed and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A.. Cancer statistics, 2021. Ca Cancer J Clin. 2021;71(1):7–15. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4(6):418–428. doi: 10.1016/j.trecan.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A, Vignali M, Park JO, Daniel SK, Pollack SM, et al. Mobilization of CD8+T Cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res. 2019;25(13):3934–3945. doi: 10.1158/1078-0432.CCR-19-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Brit J Cancer. 2013;108(4):914–923. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, Hinz U, Hank T, Ehrenberg R, Volkmar M, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(12):e1240859. doi: 10.1080/2162402X.2016.1240859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, Whiting SH, Yeh MM, Yee C, Riddell SR, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One. 2014;9(5):e96565. doi: 10.1371/journal.pone.0096565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, Stanger BZ. Tumor Cell–Intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol Res. 2020;8(3):282–291. doi: 10.1158/2326-6066.CIR-19-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. 2018;49(1):178–193.e7. doi: 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18(3):139–147. doi: 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest. 2019;129(9):3594–3609. doi: 10.1172/JCI127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxena M, van der BSH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21(6):360–378. doi: 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 12.Beatty GL, Werba G, Lyssiotis CA, Simeone DM. The biological underpinnings of therapeutic resistance in pancreatic cancer. Gene Dev. 2021;35(13–14):940–962. doi: 10.1101/gad.348523.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuptrine CW, Ajina R, Fertig EJ, Jablonski SA, Lyerly HK, Hartman ZC, Weiner LM. An unbiased in vivo functional genomics screening approach in mice identifies novel tumor cell-based regulators of immune rejection. Cancer Immunol Immunother. 2017;66(12):1529–1544. doi: 10.1007/s00262-017-2047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou P, Shaffer DR, Arias DAA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506(7486):52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin YN, Nasir A, Camacho S, Berry DL, Schmidt MO, Pearson GW, Riegel AT, Wellstein A. Monitoring Cancer Cell Invasion and T-Cell Cytotoxicity in 3D Culture. J Vis Exp. 2020;160: doi: 10.3791/61392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, et al. Downstream of Mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal. 2014;7(324):ra42–ra42. doi: 10.1126/scisignal.2005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vietsch EE. Intratumoral heterogeneity and tumor-host crosstalk alter drug sensitivity of clonal subpopulation in a pancreatic cancer model [PhD Thesis]. Erasmus University Rotterdam; 2018. [Google Scholar]
- 19.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc National Acad Sci. 2013;110(50):20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courau T, Bonnereau J, Chicoteau J, Bottois H, Remark R, Assante Miranda L, Toubert A, Blery M, Aparicio T, Allez M, et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J Immunother Cancer. 2019;7(1):74. doi: 10.1186/s40425-019-0553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. Generation of Tumor-Reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174(6):1586–1598.e12. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khandelwal N, Breinig M, Speck T, Michels T, Kreutzer C, Sorrentino A, Sharma AK, Umansky L, Conrad H, Poschke I, et al. A high-throughput RNAi screen for detection of immune-checkpoint molecules that mediate tumor resistance to cytotoxic T lymphocytes. EMBO Mol Med. 2015;7(4):450–463. doi: 10.15252/emmm.201404414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharif GM, Schmidt MO, Yi C, Hu Z, Haddad BR, Glasgow E, Riegel AT, Wellstein A. Cell growth density modulates cancer cell vascular invasion via Hippo pathway activity and CXCR2 signaling. Oncogene. 2015;34(48):5879–5889. doi: 10.1038/onc.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abassi YA, Xi B, Zhang W, Ye P, Kirstein SL, Gaylord MR, Feinstein SC, Wang X, Xu X. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem Biol. 2009;16(7):712–723. doi: 10.1016/j.chembiol.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T cell dysfunction in cancer immunity and Immunotherapy. Front Immunol. 2019;10:1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Wang Z, Wu H, Xue M, Lin P, Wang S, Lin N, Huang X, Pan W, Liu M, et al. Epigenetic regulation of CXCL12 plays a critical role in mediating tumor progression and the immune response in osteosarcoma. Cancer Res 2018;78(14):3801. doi: 10.1158/0008-5472.CAN-17-3801. 2017. [DOI] [PubMed] [Google Scholar]
- 28.Dunussi-Joannopoulos K, Zuberek K, Runyon K, Hawley RG, Wong A, Erickson J, Herrmann S, Leonard JP. Efficacious immunomodulatory activity of the chemokine stromal cell-derived factor 1 (SDF-1): local secretion of SDF-1 at the tumor site serves as T-cell chemoattractant and mediates T-cell-dependent antitumor responses. Blood. 2002;100(5):1551–1558. doi: 10.1182/blood.V100.5.1551.h81702001551_1551_1558. [DOI] [PubMed] [Google Scholar]
- 29.Berencsi K, Rani P, Zhang T, Gross L, Mastrangelo M, Meropol NJ, Herlyn D, Somasundaram R. In vitro migration of cytotoxic T lymphocyte derived from a colon carcinoma patient is dependent on CCL2 and CCR2. J Transl Med. 2011;9(1):33. doi: 10.1186/1479-5876-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horuk R. Chemokine receptors. Cytokine Growth Factor Rev. 2001;12(4):313–335. doi: 10.1016/S1359-6101(01)00014-4. [DOI] [PubMed] [Google Scholar]
- 31.Sánchez-Martín L, Sánchez-Mateos P, Cabañas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med. 2013;19(1):12–22. doi: 10.1016/j.molmed.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Balabanian K, Lagane B, Infantino S, Chow KYC, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F. The Chemokine SDF-1/CXCL12 Binds to and Signals through the Orphan Receptor RDC1 in T Lymphocytes*. J Biol Chem. 2005;280(42):35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 33.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T Cell–Mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams JB, Li S, Higgs EF, Cabanov A, Wang X, Huang H, Gajewski TF. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat Commun. 2020;11(1):602. doi: 10.1038/s41467-020-14290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeanbart L, Ballester M, de Titta A, Corthésy P, Romero P, Hubbell JA, Swartz MA. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol Res. 2014;2(5):436–447. doi: 10.1158/2326-6066.CIR-14-0019-T. [DOI] [PubMed] [Google Scholar]
- 36.Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. 2021;21(6):345–359. doi: 10.1038/s41568-021-00347-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer — clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17(9):527–540. doi: 10.1038/s41571-020-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Hara MH, Messersmith W, Kindler H, Zhang W, Pitou C, Szpurka AM, Wang D, Peng S-B, Vangerow B, Khan AA, et al. Safety and pharmacokinetics of CXCR4 peptide antagonist, LY2510924, in combination with durvalumab in advanced refractory solid tumors. J Pancreat Cancer. 2020;6(1):21–31. doi: 10.1089/pancan.2019.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JED, Basu B, Magiera L, Johnson TI, Bax L, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc National Acad Sci. 2020;117(46):28960–28970. doi: 10.1073/pnas.2013644117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de VEGE, Walenkamp AME. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49(1):219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29(38):5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209(3):551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poznansky MC, Olszak IT, Foxall R, Evans RH, Luster AD, Scadden DT. Active movement of T cells away from a chemokine. Nat Med. 2000;6(5):543–548. doi: 10.1038/75022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.