

Abstract
Alzheimer’s disease (AD) is the most prevalent cause of dementia. While there is no effective treatment for AD, passive immunotherapy with monoclonal antibodies against amyloid beta (Aβ) is a promising therapeutic strategy. Meningeal lymphatic drainage plays an important role in Aβ accumulation in the brain, yet it is unknown if modulating meningeal lymphatic function can influence the outcome of immunotherapy in AD. Ablation of meningeal lymphatics in 5xFAD mice exacerbated Aβ deposition, microgliosis and neurovascular function, affecting the outcome of anti-Aβ passive immunotherapy and behavior. In contrast, therapeutic delivery of vascular endothelial growth factor C improved Aβ clearance by monoclonal antibodies. Notably, a significant overlap was found between the gene signature of microglia from 5xFAD mice with dysfunctional meningeal lymphatics and the transcriptional profile of activated microglia from the human AD brain. Overall, our data demonstrate that impaired meningeal lymphatic drainage impacts the microglial inflammatory response in AD and that enhancement of meningeal lymphatics combined with immunotherapies could lead to better clinical outcomes.
Alzheimer’s disease (AD) is characterized by accumulation of toxic Aβ aggregates in the brain1, in part, due to progressive impairment of cleansing mechanisms2–6. Passive immunotherapy, using monoclonal antibodies against Aβ, is a promising therapeutic strategy aimed at enhancing the clearance of toxic Aβ7–10. Two clinical trials with anti-Aβ monoclonal antibody Aducanumab10, EMERGE and ENGAGE, have recently yielded inconsistent results, since cognitive decline was significantly slowed down in patients receiving the highest dose (10 mg/kg) in the EMERGE cohort but not in the ENGAGE cohort11. This highlights the need for a better understanding of factors that influence the outcome of immunotherapy in AD.
Here, using transcriptional profiling of mouse meningeal lymphatic endothelial cells (LECs), microglia and brain blood endothelial cells (BECs), we show that meningeal lymphatic function is coupled to microglia activation and brain vascular response in AD. We also demonstrate a synergistic effect of combining meningeal lymphatic drainage enhancement and anti-Aβ monoclonal antibodies in plaque clearance from the brain in AD mouse models. Interestingly, we identified microglia with similar activation signatures in the brains of AD patients and of AD transgenic mice with ablated meningeal lymphatics, supporting the potential role of the meningeal lymphatic system in modulating the neuroinflammatory response in AD.
Meningeal lymphatics deteriorate with age in 5xFAD mice
Young-adult (~3 months old) 5xFAD mice do not exhibit meningeal lymphatic dysfunction5. Here, we investigated whether changes in meningeal lymphatic vasculature and immune response would emerge with aging in AD mice. No changes were observed at 5–6 months (Extended Data Fig. 1a–e), but a significant decrease in lymphatic vessel coverage along the superior sagittal sinus (SSS), transverse sinus (TS) and the confluence of sinuses (COS) was evident at 13–14 months in the meninges of 5xFAD mice (Extended Data Fig. 1c, d). No changes in meningeal lymphatic coverage around the petrosquamosal (PSS) and sigmoid (SS) sinuses were observed between the two groups (Extended Data Fig. 1e). The deterioration of the lymphatic vasculature at the dorsal meninges observed in 13–14-month-old 5xFAD mice was accompanied by a significant increase in Aβ deposition throughout the meninges (Extended Data Fig. 1c–e), with extensive Aβ deposition along the blood and lymphatic vasculature (Extended Data Fig. 1a, insets) at anatomical locations previously shown as ‘hot spots’ along meningeal lymphatics12,13.
We also found that LECs isolated from the meninges of 6-month-old 5xFAD mice showed significant changes in the expression of genes involved in processes such as exocytosis, phospholipase D signaling, and response to low-density lipoprotein (Extended Data Fig. 1f–k), suggesting that meningeal Aβ deposition might affect meningeal LECs early on through deleterious processes previously implicated in AD14,15. Mass cytometric analysis of different meningeal leukocyte populations in 5xFAD mice at 5–6 and 11–12 months revealed a significant increase in the numbers of B cells, CD4+ T cells and CD8+ T cells in the older mice (Extended Data Fig. 2a–e) that can also be indicative of an accelerated impairment of meningeal lymphatic vasculature12.
Effects of meningeal lymphatic impairment on anti-Aβ immunotherapy
We hypothesized that modification of meningeal lymphatic drainage would affect the efficacy of monoclonal antibodies to clear Aβ aggregates. To test this hypothesis, we used mAducanumab16 and mAb1587, the murine chimeric analogs of Aducanumab10 and BAN24019,17, respectively. Meningeal lymphatic dysfunction was induced by a previously described method of photoablation5,12 that results in the loss of lymphatic vasculature lining the transverse sinus (in the dorsal meninges; Extended Data Fig. 3a, b). This method does not affect the lymphatic vessels around the SS and PSS (in the basal meninges; Extended Data Fig. 3a, c). Lymphatic photoablation results in significant reduction in drainage of CSF tracers into the deep cervical lymph nodes (dCLNs) (Extended Data Fig. 3d, e and Supplementary Videos 1, 2). These results emphasize the importance of initial lymphatics at the dorsal brain meninges in CSF drainage5,12,18.
Adult 5xFAD mice (2 months old) with intact or ablated meningeal lymphatics were subjected to a regimen of weekly intraperitoneal (i.p.) injections of mAducanumab or control mIgG (each at 40 mg/kg), for a total of eight weeks (Fig. 1a). In parallel, older 5xFAD mice (3–3.5 months old) were injected (i.p.) with mAb158 or mIgG (each at 40 mg/kg) following the same regimen (Extended Data Fig. 3f). Treatments with mAducanumab or mAb158 proved to be efficient in reducing the density of Aβ plaques (Fig. 1b, c and Extended Data Fig. 3g, h). Neither mAducanumab nor mAb158 affected the average size of Aβ plaques (Extended Data Fig. 3i, j). However, 5xFAD mice with dysfunctional meningeal lymphatics (Vis./photo. groups) that received mAducanumab or mAb158 showed significantly higher Aβ plaque coverage, when compared to their controls with intact meningeal lymphatics (Vis. groups; Fig. 1d and Extended Data Fig. 3k). LAMP1+ dystrophic neurites were significantly increased in 5xFAD mice with dysfunctional meningeal lymphatics (Fig. 1b, e and Extended Data Fig. 3g, l). Accordingly, measurements of IBA1+ cell coverage, number of peri-Aβ plaque IBA1+ cells, levels of CD68 on IBA1+ cells and fibrinogen coverage revealed an aberrant activation of myeloid cells and increased fibrinogen levels in the brain of mice with impaired meningeal lymphatic drainage (Fig. 1f–j and Extended Data Fig. 3m–q). Notably, mice with impaired meningeal lymphatics spent less time in the center of the open field arena (Extended Data Fig. 4a–d and h–k) and took more time to find the submerged platform in the acquisition of the Morris water maze test (Extended Data Fig. 4e–g and l–n), when compared to their control counterparts. Prolonged treatments with mAducanumab or mAb158 did not improve the performance of 5xFAD mice in the open field or the Morris water maze (Extended Data Fig. 4a–n). Altogether, this data is indicative of a deleterious effect of reduced meningeal lymphatic drainage on brain fibrinogen levels and neuroinflammation in 5xFAD mice, which impact neuronal function, as previously shown19, and translate into heightened anxiousness and accelerated cognitive decline.
Figure 1 |. Compromised meningeal lymphatic function in 5xFAD mice limits brain Aβ clearance by chimeric murine Aducanumab and modulates the microglial and neurovascular responses.
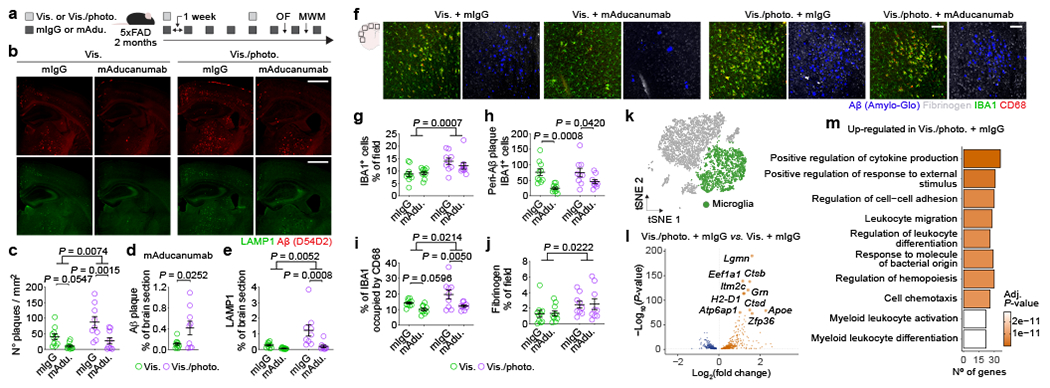
a, Experimental scheme involving 2-month-old male 5xFAD mice (behavior testing in Extended Data Fig. 4); intra-cisterna magna (i.c.m.), intraperitoneal (i.p.), Visudyne plus photoconversion (Vis./photo.) or without photoconversion (Vis.). b, Representative images of brain stained for Aβ (red, with D54D2 antibody) and LAMP1 (green; scale bars, 1 mm). c-e, Graphs showing c) number of Aβ plaques per mm2, d) Aβ coverage and e) LAMP1 coverage. f, Representative images of brain stained for Aβ (blue, with Amilo-Glo), fibrinogen (grey), IBA1 (green) and CD68 (red; scale bars, 100 μm). g-j, Graphs showing g) IBA1 coverage, h) peri-Aβ IBA1+ cells, i) percentage of IBA1 occupied by CD68 and j) fibrinogen coverage. Results in c-e and g-j are presented as mean ± s.e.m.; n = 10 in Vis. groups and n = 9 in Vis./photo. groups; in c, e and g-j, two-way ANOVA with Holm-Sidak’s multiple comparisons test; in d, two-tailed unpaired Student’s T test; data in a-j is representative of one experiment; see Extended Data Fig. 3f–q for similar data using chimeric mAb158. k, Transcriptomes of enriched brain cells analyzed by single-cell RNA-seq. t-Distributed stochastic neighbor embedding (tSNE) plot highlighting microglia (in green). l, Volcano plot with significantly down-regulated and up-regulated genes in microglia from Vis./photo. plus mIgG versus Vis. plus mIgG. m, Top ten up-regulated GO terms. Data in k-m resulted from a single experiment (see also Extended Data Fig. 6); differentially expressed genes plotted in l were determined using a F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P-values; Gene Ontology analyses used over-representation test and scale bars in m represent Benjamini-Hochberg adjusted P-values for each pathway.
Based on recent experimental evidence for an impaired perivascular CSF influx (via the glymphatic pathway) in mouse models of meningeal lymphatic dysfunction5,20, we postulated that meningeal lymphatic ablation in 4–4.5-month-old 5xFAD mice would affect the clearance of Aβ plaques by monoclonal antibodies delivered directly into the CSF (Extended Data Fig. 5a). Prolonged ablation of meningeal lymphatics led to significantly higher Aβ burden in the meninges and brain parenchyma, interfered with Aβ plaque clearance by CSF-injected mAb158 (Extended Data Fig. 5b–g) and resulted in increased ferric iron deposits (Prussian blue staining) (Extended Data Fig. 5h, i). In an attempt to explain the reduced efficacy of mAb158 observed in the mice with impaired meningeal lymphatic drainage, we administered the antibody into the CSF and, one hour later, measured its colocalization with vascular Aβ (overlap with CD31+ vessels) or with Aβ plaques (marked with Amilo-Glo RTD). Interestingly, although there were no differences in the amount of mAb158 localized to brain vascular Aβ (Extended Data Fig. 5j, k), significantly less mAb158 was found to be colocalized with brain parenchymal Aβ aggregates in 5xFAD mice with ablated meningeal lymphatics (Extended Data Fig. 5j, l). These findings suggest that impairment of meningeal lymphatic drainage in 5xFAD mice leads to decreased influx of mAb158 from the CSF into the brain, resulting in less targeting of Aβ plaques for clearance.
Meningeal lymphatic dysfunction impacts the microglial and brain vascular responses
Our data, suggesting that meningeal lymphatic dysfunction accelerates cognitive decline and affects the removal of Aβ from the brain by monoclonal antibodies, led us to explore the specific cell populations closely involved in AD pathogenesis, namely microglia21–23 and the BECs that form the blood-brain barrier4,24,25. Brain cell suspensions, enriched for live myeloid (CD45+CD11b+Ly6G−), mural (CD45−CD11b−CD13+CD31−) and BECs (CD45−CD11b−CD31) (Extended Data Fig. 6a), were analyzed by single-cell RNA sequencing (Fig. 1k–m and Extended Data Fig. 6b–p). In silico removal of undesired cell contaminants, shared nearest neighbor clustering and t-distributed stochastic neighbor embedding, resulted in nine clusters of brain cells, including microglia, capillary, arterial and venous BECs (Extended Data Fig. 6b–d). We observed significant changes in gene expression between microglia isolated from the brains of 5xFAD mice with ablated meningeal lymphatics (Vis./photo plus mIgG) and control 5xFAD mice with intact meningeal lymphatic function (Vis. plus mIgG; Fig. 1k–m and Supplementary Table 1). Up-regulated genes in microglia from mice with impaired meningeal lymphatics were involved in Gene Ontology (GO) pathways such as positive regulation of cytokine production (GO:0001819), leukocyte migration (GO:0050900), cell chemotaxis (GO:0060326) and myeloid leukocyte activation (GO:0002274; Fig. 1m). On the other hand, regulation of antigen processing and presentation of peptide antigen via major histocompatibility complex class II (GO:0002586) and T cell activation (GO:0042110) were among the down-regulated pathways in microglia from 5xFAD mice with reduced meningeal lymphatic drainage (Extended Data Fig. 6g).
Analysis of homeostatic and disease-associated microglia (DAM) gene signatures in microglia from 5xFAD mice with impaired meningeal lymphatics revealed higher expression of Apoe, Lyz2, Fth1, Timp2, H2-d1, Axl, Cst7, Spp1 and Lpl (Extended Data Fig. 6e, f and Supplementary Table 2), which are involved in the transition from homeostatic to activated microglia21,26. Interestingly, treatments with mAducanumab reduced the expression of the aforementioned disease-related genes in microglia, and significantly increased the expression of the homeostatic P2ry12, Tmem119 and Selplg (Extended Data Fig. 6f and Supplementary Table 2). These observations suggest that meningeal lymphatic ablation leads to a loss of homeostatic microglia, and acquisition of the DAM signature (and/or the neurodegenerative microglial signature)21,22,26, all of which is counteracted by passive immunotherapy.
Transcriptional changes were also induced in brain BECs by meningeal lymphatic ablation in 5xFAD mice (Extended Data Fig. 6h–p and Supplementary Table 1). Regardless of their arterial, capillary or venous origin, up-regulated genes in BECs from the Vis./photo. plus mIgG group were involved in similar pathways linked to vascular activation, endothelial cell migration and proliferation (Extended Data Fig. 6i, l, n). Interestingly, down-regulated gene-sets in brain capillary BECs from 5xFAD mice with impaired meningeal lymphatics included response to interferon-gamma (GO:0034341), chemokine- and cytokine-mediated signaling pathways (GO:0070098 and GO:0019221) and monocyte chemotaxis (GO:0002548; Extended Data Fig. 6j). Altogether, transcriptional profiling of cells from the brains of 5xFAD mice with ablated meningeal lymphatics pointed to a deleterious microglia activation and aberrant induction of vascular repair and leukocyte transmigration mechanisms that follow the surge in brain amyloidosis. We next asked whether increased Aβ accumulation due to reduced meningeal lymphatic drainage could be attributed to changes in vascular transport mechanisms that promote Aβ efflux from the brain4,27. To address this, we compared the expression levels of Abcg2, Lrp1, Picalm, Rab5a, Rab7 and Rab11a between the different groups of capillary and venous BECs (Extended Data Fig. 6o, p). We observed similar gene expression levels, except for Abcg2 and Rab7, which were up-regulated in groups treated with mAducanumab, and Rab11a, which was downregulated in venous BECs in one of the mAducanumab groups (Extended Data Fig. 6o, p). Interestingly, the gene expression values were also comparable between brain BECs from the Vis. plus mIgG and the Vis./photo plus mIgG groups, which suggests that reducing meningeal lymphatic drainage does not significantly affect the expression of genes involved in Aβ efflux/transport by brain BECs.
Enhancing meningeal lymphatics modulates anti-Aβ immunotherapy and neuroinflammation
To explore the therapeutic potential of meningeal lymphatics, we combined passive mAb158 immunotherapy with viral-mediated expression of vascular endothelial growth factor-C (mVEGF-C). Injections of mVEGF-C-expressing virus and mAb158 into the CSF of 4–5-month-old 5xFAD mice (Extended Data Fig. 7a) had a synergistic effect on Aβ plaque clearance (Extended Data Fig. 7b, c). Mice treated with mVEGF-C and mAb158 also presented significantly less peri-Aβ microgliosis and decreased levels of CD68 in IBA1+ cells (Extended Data Fig. 7d–g). The reduction in Aβ load and microgliosis in 5xFAD mice treated with mVEGF-C and mAb158 was accompanied by an expansion of lymphatic vasculature around the transverse sinus (Extended Data Fig. 7h–m).
Aged mice treated with mVEGF-C have been shown to exhibit enhanced glymphatics and improved cognition5. Here we show that transcriptional signature of meningeal LECs in 20–24-month-old mice is altered following viral-mediated mVEGF-C treatment, which is linked to the activation of gene pathways such as mitochondrial gene expression (GO:0140053), fatty acid homeostasis (GO:0055089) and smoothened signaling pathway (GO:0007224; Extended Data Fig. 7n–q). These results led us to investigate the effects of enhancing meningeal lymphatic drainage on Aβ clearance by monoclonal antibodies in aged AD transgenic mice. We were unable to detect changes in Aβ plaques upon peripheral administration of mAducanumab to 22–26-month-old APPswe mice (Extended Data Fig. 7r–t), but CSF delivery of antibodies along with mVEGF-C-expressing virus resulted in a significant decrease in Aβ plaque coverage in APPswe (26–30 months; Extended Data Fig. 8a–e) and J20 (14–16 months; Extended Data Fig. 8f–j) mice.
The gene expression profile in the hippocampi of aged APPswe mice revealed treatment-specific effects (Fig. 2a, b). Treatment with mAducanumab induced an overexpression of genes involved in synapse organization (GO:0050808), regulation of membrane potential (GO:0042391) and neurotransmitter transport (GO:0006836; Extended Data Fig. 8k) pathways, which might be attributed to the enhanced clearance of soluble Aβ seeds28 from the brain of aged APPswe mice. The top three GO pathways induced in the hippocampus by mVEGF-C treatment alone (in the mVEGF-C plus mIgG group; Fig. 2c) were dendrite development (GO:0016358), protein folding (GO:0006457) and learning and memory (GO:0007611). Treatment-specific effects were corroborated by the gene expression patterns depicted in four of the most altered GO pathways. In the dendrite development and protein folding pathways (Fig. 2d, e), hippocampal gene up-regulation was only obvious in the groups that received mVEGF-C. However, in the learning or memory pathway (Extended Data Fig. 8l), gene upregulation was amplified when mVEGF-C was administered alone and, in some instances, counteracted when combined with mAducanumab. The opposite was observed in the synapse organization pathway (Extended Data Fig. 8m), where mAducanumab treatment alone drove gene up-regulation.
Figure 2 |. Combining mVEGF-C with immunotherapy reprograms the hippocampal transcriptional profile and shapes the microglial and neurovascular responses in aged APPswe mice.

a, Aged male APPswe mice (22–26 months old) were treated with AAV1 expressing enhanced green fluorescent protein (eGFP) or murine VEGF-C (mVEGF-C), combined with mIgG or mAducanumab (regimen in Extended Data Fig. 7r). The right hippocampus was dissected (3 mice per group) and total RNA was isolated, purified and sequenced. b, Principal component analysis (PCA) plot. c, Top ten up-regulated GO terms for highlighted group comparison. d, e, Heatmaps depicting the d) dendrite development (GO:0016358) and e) protein folding (GO:0006457) gene pathways. Data in a-e result from a single experiment; PCA in b computed with singular value decomposition; Gene Ontology analyses used over-representation test and scale bar in c represents Benjamini-Hochberg adjusted P-values for each pathway; heatmaps in d and e depict counts-per-million normalized expression minus per-gene mean expression. f, Transcriptomes of enriched brain cells analyzed by single-cell RNA-seq. g, tSNE representation highlighting identified brain cell clusters. h, i, Volcano plots with significantly down-regulated and up-regulated genes in h) microglia or i) capillary BECs from Vis./photo. plus mIgG versus Vis. plus mIgG. j, k, Ten up-regulated Gene Ontology terms (selected from top 20 terms) in j) microglia or k) capillary BECs. Data in f-k resulted from a single experiment (see also Extended Data Fig. 8n–t); differentially expressed genes plotted in h and i were determined using a F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P-values; Gene Ontology analyses used over-representation test and scale bars in j and k represent Benjamini-Hochberg adjusted P-values for each pathway.
The response of specific brain cell types to the treatment with mVEGF-C was further investigated by single-cell RNA-seq (Fig. 2f, g and Extended Data Fig. 8n). After cell type annotation and cluster identification (Extended Data Fig. 8o), we focused on the transcriptional profile of microglia and brain BECs (Fig. 2h–k and Extended Data Fig. 8p–s). Interestingly, the majority of the top GO terms induced in microglia (Fig. 2j) and BECs (Fig. 2k and Extended Data Fig. 8q, s) from the brains of APPswe mice treated with mVEGF-C, pointed to an induction of interferon responses and of antigen processing and presentation via major histocompatibility complex class I. Although we cannot rule out a direct effect of mVEGF-C on brain BECs, which show some expression of Flt4, microglia are unlikely to respond to mVEGF-C due to the low frequency of Flt4-expressing cells (Extended Data Fig. 8t).
Meningeal lymphatic dysfunction is linked to mouse and human microglial activation
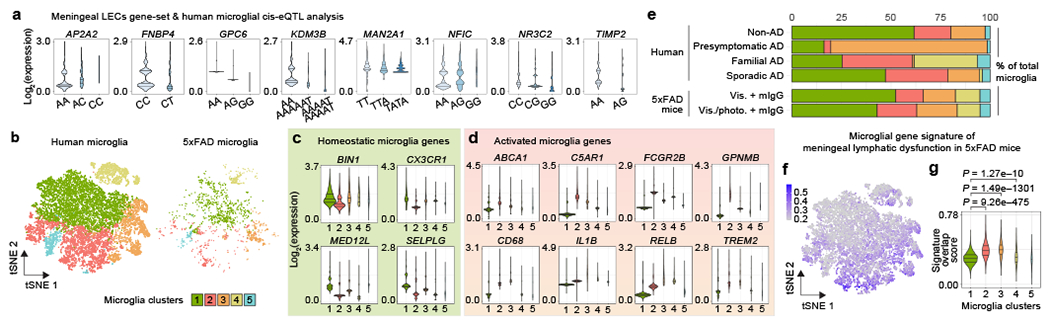
Genome-wide association studies have revealed numerous genes containing single nucleotide polymorphisms (SNPs) linked to AD and other disorders29–32. Interestingly, a considerable fraction of genes that have been associated with Parkinson’s, multiple sclerosis and Alzheimer’s are highly expressed in mouse LECs (Extended Data Fig. 9a–i). Mouse meningeal LECs and brain BECs presented a higher proportion of highly expressed AD-associated genes (Extended Data Fig. 9g–k), when compared to microglia from the 5xFAD cortex (Extended Data Fig. 9l–n). In fact, 39 AD-associated genes were found within the 10th percentile of highly expressed genes in meningeal LECs, including Apoe, which is intrinsically linked to altered risk for late onset AD15,31 and was one of the few highly expressed genes found at the intersection between all three cell types (Extended Data Fig. 9o).
We next determined the strength of the associations between the top 10% highly expressed genes in mouse meningeal LECs, brain BECs and microglia, and the latest summary statistics for AD amyloid imaging32, CSF soluble TREM233, Aβ42, Tau and phospho-Tau34, age at onset35, risk36 and progression37 (Supplementary Table 3). A strong association was observed between the brain BECs’ gene-set, amyloid imaging (P = 0.0089) and age at onset (P = 1.04E-10). Significant connections were observed between the microglia gene-set and age at onset (P = 4.82E-12), and CSF soluble TREM2 (P = 3.59E-07), Aβ42 (P = 0.021), phospho-Tau (P = 0.029) and Tau (P = 0.005) levels. A significant association was also observed between the set of meningeal LECs’ highly expressed genes and the levels of Aβ42 in the CSF (P = 0.021; Supplementary Table 3).
Using single-nucleus RNA-seq data from the brains of AD and neuropathology-free donors, we show that gene SNPs in the 1 Mb region of AD-associated genes highly expressed in meningeal LECs are associated with altered microglial gene expression in the human parietal cortex (Fig. 3a). We performed an integrated transcriptional analysis of microglia from human donor brains (from non-AD, presymptomatic sporadic AD, familial AD and sporadic AD patients; Supplementary Table 4) and from the brains of 5xFAD mice with intact or defective meningeal lymphatics (Fig. 3b and Extended Data Fig. 10a). Based on the human microglia nuclei gene expression profiles, we identified a resting-state phenotype in cluster 1 with higher expression of the homeostatic genes BIN1, CX3CR1, MED12L, SELPLG, P2RY12, P2RY13 and TMEM119 (Fig. 3c and Extended Data Fig. 10b). Cluster 2 microglia presented low expression of the homeostatic genes and higher levels of ABCA1, C5AR1, FCGR2B, GPNMB, CD68, CD83, JUN, LGMN, LPL and TNFAIP3 (Fig. 3d and Extended Data Fig. 10c), indicative of an activated phenotype. Cluster 3 microglia denoted an alternative activation phenotype characterized by high expression of both homeostatic and activation genes, and clusters 4 and 5 were composed by activated microglia that expressed lower levels of the homeostatic SELPLG, P2RY13 and TMEM119 genes and intermediate expression levels of GPNMB, IL1B, JUN, LPL and TNFAIP3 (Fig. 3c, d and Extended Data Fig. 10b, c). More than half of the microglial nuclei from the brains of non-AD patients presented a resting-state phenotype, whereas the samples from presymptomatic AD patients (patients showing characteristic AD brain pathology but a clinical dementia rating of 0.5 or less) were enriched in alternatively activated cluster 3 microglia (Fig. 3e). Cluster 2 microglia composed the biggest fraction of activated microglia isolated from the brains of familial and sporadic AD patients (Fig. 3e). Concerning mouse microglial phenotypes, there was an obvious enrichment for homeostatic in cluster 1, activated in cluster 2 (showing a DAM signature) and alternatively activated in cluster 3 (Extended Data Fig. 10d). Noticeably, the brains of 5xFAD mice with ablated meningeal lymphatics showed a significantly lower fraction of cluster 1 resting-state microglia (P = 0.00026) and higher fractions of clusters 2 (P = 0.00191) and 3 (P = 0.03535) activated microglia, when compared to mice with intact meningeal lymphatics (Fig. 3e). Moreover, there was a significantly stronger overlap between the signature of microglia from 5xFAD mice with dysfunctional meningeal lymphatics (composed of 54 genes; Supplementary Table 5) and the transcriptional profile of human microglia from clusters 2, 3 and 4 compared to cluster 1 (Fig. 3f, g). Furthermore, gene-set based analyses for genome-wide associations of AD-related phenotypes and risk revealed significant correlations between the set of up-regulated genes in microglia isolated from 5xFAD mice with dysfunctional meningeal lymphatics and amyloid imaging (P = 0.017), CSF Tau levels (P = 0.036), age at onset (P = 0.027) and AD risk (P = 0.011; Supplementary Table 6). On the other hand, up-regulated genes in microglia isolated from aged APPswe mice treated with mVEGF-C were associated with AD age at onset (P = 0.014) and risk (P = 0.045; Supplementary Table 6).
Figure 3 |. Gene-set analysis uncovers a link between impaired meningeal lymphatic vasculature and microglial activation in Alzheimer’s disease.
a, Single nucleotide polymorphisms in the 1 Mb region of genes highly expressed in meningeal LECs are in cis-expression quantitative expression loci (cis-eQTL) in microglia from the parietal cortex of human brains (non-AD and sporadic AD cases only). Violin plots with the distribution of gene expression and the first, median and third quantiles depicted for each genotype. b, tSNE plots showing the segregation of microglia from human donors or 5xFAD mice into 5 distinct clusters upon cross-species RNA-seq data integration and analysis (see also Extended Data Fig. 10). c, d, Violin plots with expression levels of c) homeostatic genes and d) activation genes in the different human microglia clusters. e, Graph showing the cluster proportions in the human non-AD, presymptomatic AD, familial AD, sporadic AD microglia groups, and in the 5xFAD mouse Vis. plus mIgG and Vis./photo. plus mIgG microglia groups. Statistically significant differences were observed between the Vis. plus mIgG and Vis./photo. plus mIgG groups regarding the proportions of microglia clusters 1 (P = 0.00026), 2 (P = 0.00191) and 3 (P = 0.03535). f, tSNE representation of the scaled average expression (range in scale bar) of the module of 54 human gene orthologs of the 5xFAD microglial gene signature of meningeal lymphatic dysfunction, subtracted by an aggregated expression of randomly chosen control feature gene-sets. g, Violin plot showing each microglia cluster’s signature score obtained by partial residuals from linear mixed models. Data in b-g were obtained from the integrated analysis of a total of 5,462 non-AD, 618 presymptomatic AD, 4,548 familial AD and 6,461 sporadic AD microglia (single-nucleus RNA-seq data) from human brain parietal lobes, and from a total of 781 and 770 microglia (single-cell RNA-seq data) from the brains of 5xFAD mice of the Vis. plus mIgG and the Vis./photo. plus mIgG groups, respectively; results involving human microglia were corrected for age of death, sex and disease status; statistically significant differences were calculated by two-proportion Z-test in e and by linear mixed models in g (individual comparisons between the score in cluster 1 and the other scores).
Discussion
Our data provided here suggests that it might be possible to devise strategies to therapeutically target both microglia and the brain blood vasculature, two important players in AD pathophysiology4,21,23,38–40, by modulating the function of the lymphatic vasculature at the brain borders. These findings also underscore the importance of early diagnosis and therapeutic intervention in AD patients, preferably at a stage when the meningeal lymphatic system is still operational. The advanced stage of disease (or simply the advanced age) at which antibody-based therapies are administered, might explain part of their marginal beneficial effects (and potential deleterious side effects), which could be attributed to a compromised meningeal lymphatic function.
Future studies should investigate whether specific combinations of gene SNPs can be used to infer information about the integrity of the brain-draining lymphatic system, predict meningeal lymphatic dysfunction, and target meningeal lymphatics therapeutically as early as possible. Ultimately, development of a gene signature of early meningeal lymphatic dysfunction, which is potentially aligned with the degree of microglial activation and angiopathy, could allow patient stratification for immunotherapies and to facilitate personalized treatments to improve brain lymphatic drainage and ameliorate AD progression.
METHODS
Mouse strains and housing.
Adult (2–3 months old) male C57BL/6J wild type (WT) mice were purchased from the Jackson Laboratory (JAX stock #000664, Bar Harbor, Maine, USA). Aged male (20–24 months old) WT mice were provided by the National Institutes of Health/National Institute on Aging (Bethesda, MD, USA). All mice were maintained in the animal facility for habituation for at least one week prior to the start of the experiment. Male hemizygous B6.Cg-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax (5xFAD, JAX stock #008730), B6.Cg-Tg(PDGFB-APPSwInd)20Lms/2Mmjax (J20, JAX stock #006293) and B6.Cg-Tg(APP695)3Dbo (APPswe, JAX stock #005866) were purchased from the Jackson Laboratory and bred in-house on a C57BL/6J background. In-house bred male transgene carriers and non-carrier (WT) littermates were used at different ages. The genotype and age of mice from different strains are indicated in figure schemes or legends throughout the manuscript. Male mice were used in all the different experiments. Mice of all strains were housed in an environment with controlled temperature and humidity and on a 12-hour light/dark cycle (lights on at 7:00). All mice were fed with regular rodent’s chow and sterilized tap water ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Virginia.
Treatments with anti-Aβ monoclonal antibodies.
The protein sequences for murine chimeric Aducanumab (mAducanumab) variable heavy chain and light chain (WO 2016/087944) were codon-optimized, generated as double-stranded DNA fragments (Integrated DNA Technologies), and cloned into expression vectors with mouse IgG2ab and mouse Kappa constant regions, respectively. Recombinant antibody was produced by transient co-transfection of heavy and light chain vectors in Expi293 cells (Thermo Fisher) followed by purification by Protein G chromatography16. The generation of the control murine IgG2ab (mIgG), recognizing human PLD3 sequences not found in mice, was performed as previously described16. The mAducanumab and respective control mIgG antibodies were produced by the Holtzman laboratory and can be obtained by academic investigators with a Material Transfer Agreement from the Washington University in St. Louis. Murine mAb158 and respective control IgG2a (mIgG) antibodies were manufactured by Absolute Antibody Ltd., Oxford Centre for Innovation, United Kingdom. Anti-Aβ monoclonal antibodies and respective mIgG controls were administered via intraperitoneal (i.p.) injection, at a dose of 40 mg/kg. Antibody dosages are also specified in the main text, illustrated in schemes and in each figure legend. Alternatively, mAb158 and mIgG antibodies were injected directly into the CSF, following the methodology described in the next section.
Intra-cisterna magna injections of drugs, viral vectors and antibodies.
In order to surgically expose the cisterna magna for injections, mice were anaesthetized by i.p. injection of a mixed solution of ketamine (100 mg/kg) and xylazine (10 mg/kg) in saline. The skin of the neck was shaved and cleaned with iodine and 70% ethanol, ophthalmic solution placed on the eyes to prevent drying and the mouse’s head was secured in a stereotaxic frame. After making a small (4-5 mm) skin incision, the muscle layers were retracted and the atlantooccipital membrane of the cisterna magna was exposed. Using a Hamilton syringe (coupled to a 33-gauge needle), the volume of the desired solution was injected into the CSF-filled cisterna magna compartment at a rate of ~2.5 μL per minute. After injecting, the syringe was left in place for at least 2 min to prevent back-flow of CSF. The neck skin was then sutured, the mice were allowed to recover in supine position on a heating pad until fully awake and subcutaneously injected with ketoprofen (2 mg/kg). This method of intra-cisterna magna (i.c.m.) injection was used to administer 5 μL of either 0.5 μm microspheres (FluoSpheres™ carboxylate-modified microspheres, Thermo Fisher Scientific), Visudyne® (verteporfin for injection, Valeant Ophtalmics), adeno associated viral vectors (AAV1-CMV-mVEGF-C-WPRE or AAV1-CMV-eGFP, at 1012 or 1013 genome copies per μL, purchased from Vector BioLabs, Philadelphia) or murine mAb158 or mIgG (each at 1 μg/μL). Antibody and viral vector dosages/titers are also specified in the main text, illustrated in schemes and in each figure legend.
Meningeal lymphatic vessel ablation.
Selective ablation of the meningeal lymphatic vessels was achieved by injection of Visudyne (Vis.) and consecutive transcranial photoconversion (photo.) steps following previously described methodology and regimens5,12. Visudyne was reconstituted following manufacturer’s instructions, aliquoted and kept at −20°C until further used. Immediately upon being thawed, Visudyne was injected into the CSF (i.c.m.) and 15 min later, an incision was performed in the skin to expose the skull bone and photoconvert the drug by pointing a 689-nm-wavelength nonthermal red light (Coherent Opal Photoactivator, Lumenis) on 5 different spots above the intact skull (close to the injection site, above the superior sagittal sinus close to the rostral rhinal vein, above the confluence of sinuses and above each transverse sinus). Each spot was irradiated with a light dose of 50 J/cm2 at an intensity of 600 mW/cm2 for a total of 83 s. Controls were injected with the same volume of Visudyne only, without the photoconversion step. The scalp skin was then sutured, the mice were allowed to recover on a heating pad until fully awake and subcutaneously injected with ketoprofen (2 mg/kg).
In vivo measurement of CSF outflow into dCLNs.
Upon i.c.m. injection of 5 μL of a suspension of 0.5 μm yellow-green fluorescent (505/515 nm) microspheres (Thermo Fisher Scientific) diluted in artificial CSF (1:1 v/v, Harvard Apparatus, #597316) following the procedure described in the previous section entitled “intra-cisterna magna injections of drugs, viral vectors and antibodies.”, the syringe was left in place for 10 min to prevent backflow and then the mouse was prepared for live imaging of microsphere drainage from the CSF into the dCLNs using a stereo microscope (M205 FA, Leica Microsystems). The mouse was positioned supine with the head held in position with a length of suture behind the upper incisors and the upper limbs held in place with medical tape. Incisions were made from the center of the clavicle, anterior to the top of the salivary gland and lateral approximately 1 cm. The further preparation was performed on the right side, however in some instances moved to the left side when anatomical variation prevented imaging. The salivary gland was carefully separated at its lateral extent and gently retracted medially. The omohyoid and sternomastoid muscles were retracted laterally, exposing the dCLN. Imaging began approximately 15 min after i.c.m. injection. Images were acquired at 25-30 frames per second for a total of 60 seconds. After imaging, mice were euthanized by injection of Euthasol (10% v/v in saline). Fluorescent microsphere drainage was analyzed in FIJI software by drawing a line demarcating the draining lymphatic vessel afferent to the dCLN and manually counting the microspheres passing the line by a blinded experimenter. Mice were discarded from the analysis due to prior complications during the surgical procedure (e.g. hemorrhages) or due to improper injection and failure in detecting microspheres draining into the dCLNs during image acquisition. In representative images, microspheres were tracked using TrackMate41 to show the cumulative tracks over a 20 sec interval. Examples of sequentially acquired images using mice with intact (Vis.) or ablated (Vis./photo.) meningeal lymphatic vessels can be found in Supplementary Videos 1 and 2, respectively.
Open field test.
Mice were habituated to the behavior room, including the background white noise, for at least 30 min prior to starting the test. Individual mice were then placed into the open field arena (made of opaque white plastic material, 35 cm × 35 cm) by a blinded experimenter and allowed to explore it for 15 min. Total distance (in cm), velocity (in mm per second) and % time spent in the center (22 cm × 22 cm area) were quantified using video tracking software (TopScan, CleverSys, Inc.) and analyzed in Microsoft Excel and Prism 8.3.4 (GraphPad Software, Inc.).
Morris water maze test.
Mice were habituated to the behavior room, including the background white noise, for at least 30 min before starting the test. The MWM test consisted of 4 days of acquisition, 1 day of probe trial and 2 days of reversal. In the acquisition, mice performed four trials per day, for 4 consecutive days, to find a hidden 10-cm diameter platform located 1 cm below the water surface in a pool 1 m in diameter. Tap water was made opaque with nontoxic white paint (Prang ready-to-use washable tempera paint) and the water temperature was kept at 25 ± 1°C by addition of warm water. A dim light source was placed within the testing room and only distal visual cues were available above each quadrant of the swimming pool to aid in the spatial navigation and location of the submerged platform. The latency to platform, i.e., the time required by the mouse to find and climb onto the platform, was recorded for up to 60 seconds. Each mouse was allowed to remain on the platform for ~15 seconds and was then moved from the maze to its home cage. If the mouse did not find the platform within 60 seconds, it was manually placed on the platform and returned to its home cage after ~15 seconds. The inter-trial interval for each mouse was of at least 5 min. On day 5, the platform was removed from the pool, and each mouse was tested in a probe trial for 60 seconds. On days 1 and 2 of the reversal, without changing the position of the visual cues, the platform was placed in the quadrant opposite to the original acquisition quadrant and the mouse was retrained for four trials per day. All MWM testing was performed between 10 a.m. and 6 p.m., during the lights-on phase, by a blinded experimenter. During the acquisition, probe and reversal, data were recorded using the EthoVision automated tracking system (Noldus Information Technology). The mean latency (in seconds) of the four trials for each day of test and the % of time in the platform quadrant during the probe trial were calculated in Excel and statistically analyzed in Prism 8.3.4.
Tissue collection and processing.
Mice were given a lethal dose of anesthetics by i.p. injection of Euthasol (10% v/v in saline) and transcardially perfused with ice cold phosphate buffer saline (PBS, pH 7.4) with heparin (10 U/mL). After stripping the skin and muscle from the bone, the entire head was collected and drop fixed in 4% paraformaldehyde (PFA) for 24 hours at 4°C. After removal of the mandibles and nasal bone, the top of the skull (skull cap) was removed with fine surgical curved scissors (Fine Science Tools) by cutting clockwise, beginning and ending inferior to the right post-tympanic hook and kept in PBS 0.02% azide at 4°C until further use. Fixed meninges (dura mater and arachnoid) were carefully dissected from the skullcaps with Dumont #5 and #7 fine forceps (Fine Science Tools) and kept in PBS 0.02 % azide at 4°C until further use. Alternatively, the skull was cut along the sagittal median plane and, after removing the brain, the skull pieces with the attached meningeal layers were kept in PBS 0.02 % azide at 4°C until further use. The brains were kept in 4% PFA for additional 24 hours (48 hours in total). Fixed brains were washed with PBS, cryoprotected with 30% sucrose and frozen in Tissue-Plus® O.C.T. compound (Thermo Fisher Scientific). Fixed and frozen brains were sliced (50 μm-thick sections) with a cryostat (Leica) and kept in PBS 0.02% azide at 4°C.
Immunohistochemistry, imaging and quantifications.
The following steps were generally applied for free floating fixed brain sections and meningeal whole mounts. To stain ferric iron deposits by Prussian blue, brain slices were mounted to gelatin type A-coated Superfrost™ Plus slides (Thermo Fisher Scientific) and allowed to dry. Slides were then washed twice for 3 min with ddH2O to remove buffer salts and incubated in a 1:1 mixture of 5% 18N HCl in ddH2O (v/v) and 5% potassium hexacyanoferrate (II) trihydrate (MilliporeSigma, 60279) in ddH2O (w/v) for 30 min at room temperature (RT). Slides were rinsed twice in ddH2O, dehydrated in gradients of 50%, 70%, 90% and 100% ethanol (Thermo Fisher Scientific, 04-355-223) for 3 min each and cleared twice in xylenes (MilliporeSigma, XX0055) for 1 minute. Slides were mounted with Histomount Mounting Solution (Thermo Fisher Scientific, 008030) and glass coverslips. Images were acquired on a Keyence BZ-X800 microscope and the amount of Prussian blue foci per brain slice were analyzed and quantified using the FIJI software. Alternatively, when appropriate, brain sections were stained for amyloid deposits with the Amylo-Glo® RTD™ reagent (Biosensis, Fine Bioscience Tools, South Australia), following manufacturer’s instructions. For immunofluorescence staining, tissue was rinsed in PBS and incubated with PBS 0.5% Triton X-100 (Thermo Fisher Scientific, PBS-T) for 30 min, followed by PBS-T containing 0.5% of normal serum (either goat or chicken) or 0.5% bovine serum albumin (BSA) for 30 min at RT. This blocking step was followed by incubation with appropriate dilutions of primary antibodies: rat anti-LYVE-1-eFluor660 or anti-LYVE-1-Alexa Fluor®488 (eBioscience, clone ALY7, 1:200), Armenian hamster anti-CD31 (MilliporeSigma, MAB1398Z, clone 2H8, 1:200), goat anti-IBA1 (Abcam, ab5076, polyclonal, 1:200), rat anti-CD68 (BioLegend, 137002, clone FA-11, 1:100), rat anti-LAMP-1 (Abcam, ab25245, clone 1D4B, 1:300), rabbit anti-Fibrinogen (Dako, A0080, polyclonal, 1:200), rabbit anti-Aβ1-37/42 (Cell Signaling, 8243S, clone D54D2, 1:400) in PBS-T containing 0.5% BSA overnight at 4°C. Meningeal whole mounts or brain sections were then washed 3 times for 10 min at RT in PBS-T followed by incubation with the appropriate rat, chicken, goat or donkey eFluor570 or Alexa Fluor® 488, 594, or 647 conjugated anti-rat, -goat, -rabbit, -mouse or -Armenian hamster IgG antibodies (Thermo Fisher Scientific, 1:500) for 1 hour at RT in PBS-T. After an incubation for 10 min with DAPI (0.5 μg/mL, Thermo Fisher Scientific) in PBS, the tissue was washed 3 times for 5 min with PBS, left to dry at RT (~20 min) and mounted with Shandon™ Aqua-Mount (Thermo Fisher Scientific) and glass coverslips. To stain lymphatic vasculature in the intact skull cap meninges, the same skull hemisphere was incubated in PBS-T 0.5% BSA for 2 hours and then with anti-LYVE-1 eFluor 660 (1:100) in PBS-T 0.5% BSA for 48 hours. Skull caps were then washed 3 times for 1 hour with PBS-T and left washing in PBS-T overnight at 4°C. Skull caps were washed once with PBS kept in PBS at 4°C. Preparations were stored at 4°C for no more than one week until images were acquired. A stereo microscope (M205 FA, Leica Microsystems) was used to image the meningeal lymphatic vessels within the skull caps. A widefield microscope (DMI6000 B with Adaptive Focus Control, Leica Microsystems; MetaMorph v7.10.1.161 software, Molecular Devices, LLC) was used to acquire images of Aβ deposits in brain sections and a confocal microscope (FV1200 Laser Scanning Confocal Microscope, Olympus; Olympus Fluoview v4.2a software) to acquire all the other images. Upon acquisition, images were opened in the FIJI software for quantification. The ROI (region of interest) manager, simple neurite tracer and cell counter plugins were used to measure total lymphatic vessel length and branching points in a particular region of the meningeal whole mount. The threshold (45-255 for Aβ in widefield images of brain sections; 55-255 for LAMP-1 in widefield images of brain sections; 900-4095 for Aβ in meningeal whole mounts; 500-4095 for LYVE-1 in meningeal whole mounts; 1200-4095 for fibrinogen; 1400-4095 for IBA1) and measure plugins were used to measure the coverage (as % of ROI) by Aβ or LAMP-1 in the brain (in delineated hippocampus, cortex/striatum/amygdala, or whole brain section; plotted values resulted from the average of 3 representative sections per sample), Aβ or LYVE-1 in meningeal whole mounts, as well as fibrinogen and IBA1 in images of the brain cortex (plotted values resulted from the average of 3-5 representative images taken from 2-3 brain sections per sample). The analyze particles plugin (size, 4-infinity μm2; circularity, 0.05-1) was used to measure the Aβ plaque density (number per mm2) and average size of the plaques (μm2) in each brain region/section. The cell counter plugin was also used to quantify the number of peri-Aβ plaque IBA1+ cells (cell body within 20 μm of plaque). The threshold (800-4095 for mAb158 and Aβ; 1000-4095 for CD31; 1400-4095 for IBA1 and CD68) and image calculator plugins were used to determine the % of colocalization between the signals of mAb158 and CD31, mAb158 and Aβ (Amylo-Glo RTD), or IBA1 and CD68 in brain images acquired using the confocal microscope. All measurements were performed by a blinded experimenter, Microsoft Excel was used to calculate average values in each experiment and statistical analysis performed using Prism 8.3.4.
Mass cytometry and high-dimensional data analysis.
Prior to the start of the experiment, metal isotope-labeled antibodies were purchased from Fluidigm or conjugated in-house with Maxpar (MP) antibody conjugation kits (Fluidigm) following the manufacturer’s protocol. Mice were injected with Euthasol (i.p.) and were transcardially perfused with ice cold PBS with heparin. Individual meninges were immediately dissected from the mouse’s skull cap in ice-cold RPMI 1640 (Gibco). The meningeal tissues were digested for 20 min at 37°C with 1 mg/mL of Collagenase VIII (MilliporeSigma), 1 mg/mL of Collagenase D (MilliporeSigma) and 50 U/mL of DNAse I (all from MilliporeSigma) in RPMI 1640. The same volume of RPMI with 10% FBS (Atlas Biologicals) and 10 mM EDTA (Thermo Fisher Scientific) was added to the digested tissue, which was then filtered through a 70 μm cell strainer (Fisher Scientific). The cell pellets from each sample were washed and resuspended in ice-cold fluorescence-activated cell sorting (FACS) buffer (pH 7.4; 0.1 M PBS; 1 mM EDTA and 1% BSA), transferred into a 96-well plate and washed with MP PBS. Aliquots (20 μL) from each individual unstained single cell suspension were set aside and incubated with ViaStain™ AOPI Staining Solution (CS2-0106, Nexcelom Bioscience) to provide accurate counts using Cellometer Auto 2000 (Nexcelom Bioscience). Unless stated otherwise, all washes and incubations were performed with MP Cell Staining Buffer (MP CSB). Individual samples were incubated with 50 μL of 2.5 μM cisplatin (Fluidigm) in MP PBS for 5 min at RT, followed by two washes, and preincubated with Fc-receptor blocking solution (anti-CD16/32 in MP PBS supplemented with 0.5% BSA) for 15 min at 4°C. Cells were then stained for fixation-sensitive surface markers for 30 min at 4°C with: anti-Ly6C-Nd-142 (clone HK1.4, Maxpar Ready, Biolegend, conjugated in-house), anti-CD169-Sm-147 (clone H1.2F3, Maxpar Ready, conjugated in-house), anti-XCR1-Eu-153 (clone ZET, BioLegend, conjugated in-house), anti-Siglec-H-Gd-160 (clone 551, Biolegend, conjugated in-house), anti-FcER1-Dy-161 (clone MAR-1, Maxpar Ready, conjugated in-house), anti-PD-1-Er-166 (clone CHBC0116081, R&D systems, conjugated in-house), anti-H-2Kb/Db-Yb-173 (clone 28-8-6, BioLegend, conjugated in-house), anti-CCR2-FITC (clone 475301, R&D Systems) and anti-Thy1.2-PE (clone 30-H12, eBioscience). After washing twice, cells were fixed in 1.6% PFA in MP PBS for 10 min at RT. Individual samples were barcoded using six palladium metal isotopes according to the manufacturer’s instructions (Cell-ID 20-plex Pd barcoding kit, Fluidigm) to reduce tube-to-tube variability. All individual samples were combined in the same tube and subsequently stained as multiplexed samples by incubating for 30 min at RT with the following antibodies: anti-CD45-Yb-89 (clone 30-F11, Fluidigm 3089005B), anti-CD11b-Nd-143 (clone M1/70, Fluidigm 3143015B), anti-FITC-Nd-144 (clone FIT22, Fluidigm 3144006B), anti-CD4-Nd-145 (clone RM4-5, Fluidigm 3145002B), anti-F4/80-Nd-146 (clone BM8, Fluidigm 3146008B), anti-Ly6G-Nd-148 (clone 1A8, Maxpar Ready, conjugated in-house), anti-CD19-Sm-149 (clone 6D5, Fluidigm 3149002B), anti-CD24-Nd-150 (clone M1/69, Fluidigm 3150009B), anti-CD64-Eu-151 (clone X54-5/7.1, Fluidigm 3151012B), anti-CD3ε-Sm-152 (clone 145-2C11, Fluidigm 3152004B), anti-Ter119-Sm-154 (clone Ter119, Fluidigm 3154005B), anti-PE-Gd-156 (clone PE001, Fluidigm 3156005B), anti-CD103-Dy-162 (clone FIB504, Fluidigm 3162026B), anti-CD14-Dy-163 (clone M14-23, Biolegend, Ultra-LEAF, conjugated in-house), anti-CD62L-Dy-164 (clone MEL-14, Fluidigm 3164003B), anti-CD8α-Er-168 (clone SK1, Fluidigm 3168002B), anti-TCRβ-Tm-169 (clone H57597, Fluidigm 3169002B), anti-NK1.1-Er-170 (clone PK136, Fluidigm, 3170002B), anti-CD44-Yb-171 (clone IM7, Fluidigm 3171003B), anti-CD86-Yb-172 (clone GL1, Fluidigm 3172016B), anti-I-A/I-E-Yb-174 (clone M5/114.15.2, Fluidigm 3174003B), anti-CD127-Lu-175 (clone A7R34, Fluidigm 3175006B), anti-B220-Yb-176 (clone RA3-682, Fluidigm 3176002B) and anti-CD11c-Bi-209 (clone N418, Fluidigm 3209005B). The fixed cells were washed and stained for intracellular antigens in MP Nuclear Antigen Staining Buffer Set (Fluidigm) for additional 30 min at RT with: anti-TNF-Pr-141 (clone MP6-XT22, Fluidigm 3141013B), anti-FOXP3-Gd-158 (clone FJK-16s, Fluidigm 3158003A) anti-RORγt-Tb-159 (clone B2D, Fluidigm 3159019B), anti-T-bet-Ho-165 (clone 4B10, Maxpar Ready, conjugated in-house) and anti-GATA3-Er-167 (clone TWAJ, Fluidigm 3167007A). Samples were washed twice in Nuclear Antigen Staining Buffer and incubated in 125 nM Ir-191/193 DNA intercalator solution (Cell-ID Intercalator-Ir in Maxpar Fix/Perm buffer, Fluidigm) overnight at 4°C. Before acquisition on a Helios Mass Cytometer (available at the University of Virginia Flow Cytometry Core Facility), samples were washed twice with MP Fix/Perm buffer, MP water and MP Cell acquisition solution. Raw data were normalized for detector sensitivity by adding five element beads to the sample and processed as described previously42. Samples were debarcoded using the Zunder Lab single-cell debarcoder (https://github.com/zunderlab/single-cell-debarcoder) in MATLAB (9.7 version) and files uploaded in Cytobank (7.2 version). Raw data were subjected to arcsinh transformation (cofactor = 5), manually gated to exclude debris, doublets, dead cells, normalization beads, and include 191/193Ir_DNA+, 195Pt_Cisplatin−, 140Ce_EQbeads−, 89Yb_CD45+ events that were corrected for Mahalanobis distance (Extended data Fig. 2a). Gating was followed by dimensionality reduction and t-distributed stochastic neighbor embedding-based visualization (viSNE) on the combined data set. Individual fcs files containing viSNE plots of single live CD45+ events were exported from Cytobank and read into R as a flowset using the flowCore package (R package version 1.48.1). Clustering was performed using the default settings for Rphenograph (Cytofkit package for version 3.5 of Bioconductor)43 and all markers in the panel with the exception of CD45. For the generation of heatmaps showing median marker expression, the median quantile scaled expression value among cells from each cluster was visualized. Initial Rphenograph nodes depicting the median marker expression values within each cluster were then examined and clusters were merged to reflect biologically meaningful populations. A subset of cells was selected for viSNE visualization by randomly sampling an equal number of cells from each replicate, totaling 12,000 cells from each condition. Frequencies were calculated as the number of live CD45+ cells from each sample belonging to each cluster divided by the total number of live CD45+ cells in that sample. Frequencies were then used to calculate the number of cells in each cluster for each group. Data processing was done with Excel and statistical analysis performed using Prism 8.3.4.
Sorting of mouse meningeal LECs and RNA isolation.
This procedure was performed as described in previous publications5,12. Adult WT or 5xFAD mice (6 months of age) or old mice (20-24 months of age, one month upon injection with different AAV1 vectors), were euthanized by Euthasol injection (i.p.) and transcardially perfused with ice cold PBS with heparin. To obtain a suspension of meningeal LECs, skull caps were quickly collected, and meninges (dura mater and arachnoid) were dissected using Dumont #5 and #7 fine forceps in complete media composed of DMEM (Gibco) with 2% FBS, 1% L-glutamine (Gibco), 1% penicillin/streptomycin (Gibco), 1% sodium pyruvate (Gibco), 1% non-essential amino-acids (Gibco) and 1.5% Hepes buffer (Gibco). Individual brain meninges were then incubated with 1 mL of DMEM with 1 mg/mL of Collagenase VIII and 35 U/mL of DNAse I for 15 min at 37°C. Cell suspensions from 10 individual meninges were then pooled into a single tube after filtration through a 70 μm nylon mesh cell strainer. Suspensions of meningeal LECs were pelleted, resuspended in ice-cold FACS buffer containing DAPI (0.1 μg/mL), anti-CD45-BB515 (1:200, clone 30-F11, BD Biosciences), anti-CD31-Alexa Fluor® 647 (1:200, clone 390, BD Biosciences) and anti-PDPN-PE (1:200, clone 8.1.1, eBioscience) and incubated for 15 min at 4°C. Cells were then washed and resuspended in ice-cold FACS buffer. Briefly, singlets were gated using the pulse width of the side scatter and forward scatter. The live LECs were then gated as DAPI−CD45−CD31+PDPN + (using the exact same gating strategy as described in a previous publication5) and sorted into a 96-well plate containing 100 μL of RNA extraction lysis buffer using the Influx™ Cell Sorter (BD Biosciences) that is available at the University of Virginia Flow Cytometry Core Facility. Total RNA isolation was immediately performed following the manufacturer’s instructions (Arcturus PicoPure RNA Isolation Kit, Thermo Fisher Scientific). RNA samples were stored at −80°C until further use.
Hippocampus dissection and RNA isolation.
This procedure was performed as previously described5, with minor modifications. The whole hippocampus was macrodissected from the right brain hemisphere in ice-cold advanced DMEM/F12 (Gibco, 12634010) using Dumont #5 and #7 fine forceps and immediately snap-frozen in dry ice and stored at −80 °C until further use. After defrosting on ice, samples were mechanically dissociated in the appropriate volume of extraction buffer from the RNA isolation kit (RNeasy mini kit, 74106, QIAGEN). Total RNA from each sample was isolated and purified using the kit components according to the manufacturer’s instructions.
Bulk RNA sequencing.
The Illumina TruSeq Stranded Total RNA Library Prep Kit was used for cDNA library preparation from total RNA samples isolated from the hippocampus. Sample quality control was performed on an Agilent 4200 TapeStation Instrument, using the Agilent D1000 kit, and on the Qubit Fluorometer (Thermo Fisher Scientific). For RNA sequencing (RNA-seq), libraries were loaded on to a NextSeq 500 (Illumina) using Illumina NextSeq High Output (150 cycle, #FC-404-2002) and Mid Output (150 cycle, #FC-404-2001) cartridges. Processing of total RNA extracted from mouse meningeal LECs (including linear RNA amplification and cDNA library generation) and RNA-seq was performed by HudsonAlpha Genomic Services Laboratory (Huntsville, AL). For meningeal LECs’ bulk RNA-seq experiments, the fastq files were downloaded using HudsonAlpha’s provided software and fastq files were merged by lane. The merged fastq files were then chastity filtered to remove low quality bases, trimmed using the trimmomatic software, and mapped to the UCSC mm10 genome using Salmon44,45. The resulting transcript abundances were read into R and summarized using tximport46. Ensembl identifiers were mapped to gene symbols where possible and kept as Ensembl ID’s where no mapping was identified. Additionally, counts for Ensembl ID’s mapping to the same gene symbol were summed. Genes were filtered to remove those with fewer than five counts in at least two samples as well as those with high intra-sample standard deviations. The remaining counts were analyzed using DESeq2 for normalization, differential expression, and principal component analysis (PCA)47. The Benjamini-Hochberg correction was used to adjust the associated P-values for differentially expressed genes in all datasets and those with adjusted P-values below 0.05 were considered to be significant. Significantly differentially expressed gene-sets were used as input for functional enrichment of Gene Ontology (GO) terms as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using the ClusterProfiler Bioconductor package48,49. Highly variable genes were defined as those with the largest variance across samples using the log transformed expression values. Heatmaps of differentially expressed genes, highly variable genes, and gene-sets enriched for specific pathways were visualized using the R package pheatmap and expression values for each sample were represented as standard deviations from the mean across each gene. For bulk RNA sequencing from whole hippocampus, the fastq files were downloaded using the provided software and merged by lane, trimmed using TrimGalore (https://github.com/FelixKrueger/TrimGalore), and mapped to the UCSC mm10 genome using STAR50. Gene counts were then read into R and analyzed with the edgeR package51. Filtering out genes with low expression was done with the filterByExpr function which calculates the optimal number of genes to retain based on dataset characteristics. Normalization factors were calculated with calcNormFactors and then counts were normalized as log counts per million before undergoing PCA via Singular Value Decomposition. Raw counts were then used for differential expression analysis. Dispersion was estimated with estimateGLMRobustDisp to increase robustness against outliers and a quasi-likelihood model was fit with glmQLFit. Expression differential was tested with empirical Bayes quasi-likelihood F-test and results were then filtered using as threshold a Benjamini-Hochberg adjusted P-value < 0.05 (statistically significant). Volcano plots were made with the EnhancedVolcano package (available at https://github.com/kevinblighe/EnhancedVolcano). Genes matching this significance threshold were divided on the basis of up- or down-regulation52 and used as input for GO analysis using the ClusterProfiler Bioconductor package48,49.
Whole brain hemisphere blood vascular and myeloid cell sorting.
Adult 5xFAD or aged APPswe mice were euthanized by i.p. injection of Euthasol and transcardially perfused with ice cold PBS with heparin. The skulls were collected, and the whole right brain hemisphere was carefully dissected into ice-cold advanced DMEM/F12 (Gibco, 12634010). Brain tissue was mechanically dissociated with sterile sharp scissors and digested for 45 min at 37°C in advanced DMEM/F12 with 1 mg/mL of Collagenase VIII, 50 U/mL of DNAse I and 1% FBS. Every 15 min, the tissue suspensions were sequentially passed through a 10 mL serological pipette (10 times), followed by two passages through a 5 mL serological pipette (10 times each). Cellular suspensions were filtered through a 70 μm cell strainer, thoroughly mixed with an equal volume of 22% BSA in PBS and centrifuged at 1,000 × g for 10 min at RT (without break), to remove the myelin. After discarding the upper layer and supernatant containing the myelin and cell debris, the cell pellets were washed with advanced DMEM/F12 supplemented with 10 % FBS, resuspended in ice-cold FACS buffer, preincubated for 10 min at 4°C with Fc-receptor blocking solution (anti-CD16/32, 101302, BioLegend, 1:200 in ice-cold FACS buffer) and stained for extracellular markers with the following antibodies (at 1:200 in ice-cold FACS buffer, unless stated otherwise): anti-CD13-FITC (at 1:50, 558744, BD Biosciences), anti-Ly6G-PE (127608, BioLegend), anti-CD11b PerCP-Cy5.5 (550993, BD Biosciences), anti-CD31-APC (17-0311-80, eBioscience) and anti-CD45-APC-Cy7 (103116, BioLegend). DAPI (0.1 μg/mL) was also added to access cell viability. After an incubation period of 25 min at 4°C, cells were washed and resuspended in ice-cold FACS buffer. Cells were gated on DAPI− singlets (based on height, area and the pulse width of the forward and side scatters), CD45+CD11b+Ly6G− (myeloid cells), CD45−CD11b−CD13+CD31− (mural cells) and CD45−CD11b−CD31+ (blood endothelial cells, Extended Data Fig. 6a) and sorted using the Influx™ Cell Sorter at the University of Virginia Flow Cytometry Core Facility. The aforementioned enriched single cell populations pertaining to the same group were sorted into the same 1.5 mL tube previously coated (overnight) with 0.1% ultrapure non-acetylated BSA (Thermo Fisher Scientific, AM2616) in PBS and containing 500 μL of ice-cold advanced DMEM/F12. Single cells were centrifuged at 350 × g for 5 min at 4°C and resuspended in 0.1% ultrapure non-acetylated BSA in PBS at 1,000 cells per μL estimated from counting on a hemocytometer and Trypan blue (MilliporeSigma) staining. Single-cell suspensions were kept on ice until further use.
Brain cortical myeloid cell sorting.
5xFAD mice (5.5 months old) with intact or ablated meningeal lymphatics were euthanized by i.p. injection of Euthasol and transcardially perfused with ice cold PBS with heparin. The skulls were collected, and the brain was carefully extracted into ice-cold HBSS medium (14025, Thermo Fisher Scientific). Under an articulated stereo microscope, pia and choroid plexus tissue contaminants were carefully discarded and the brain cortices were collected by macrodissection. Brain tissue was passed through a 5 mL pipette tip (10 times) and digested for 30 min at 37°C in HBSS with 1 mg/mL of Collagenase VIII, 1 mg/mL of Collagenase D, 50 U/mL of DNAse I and 25 μg/mL Actinomycin D to inhibit transcription mediated by all RNA polymerases (all from MilliporeSigma). The same volume of DMEM/F12 (Gibco) with 5% FBS and 10 mM EDTA was added to the digested tissue, which was then passed through a 1 mL pipette tip (10 times) and filtered through a 70 μm cell strainer. The cell pellets were washed, resuspended in ice-cold FACS buffer, preincubated for 10 min at 4°C with Fc-receptor blocking solution (anti-CD16/32, 1:200 in FACS buffer) and stained for extracellular markers with the following antibodies (all at 1:200 in FACS buffer): anti-CD11b-PerCP-Cy5.5 (550993, BD Biosciences), anti-CD45-A700 (560510, BD Biosciences) and anti-Ly6G-BV421 (562737, BD Biosciences). After an incubation period of 25 min at 4°C, cells were washed and resuspended in FACS buffer with SYTOX™ Green Nucleic Acid Stain following the manufacturer’s instructions (Thermo Fisher Scientific) to determine the cell viability. Cells were gated on singlets (based on height, area and the pulse width of the forward and side scatters) and SYTOX−Ly6G−CD45+CD11b+ (myeloid cells) and sorted using the Influx™ Cell Sorter at the University of Virginia Flow Cytometry Core Facility. Single myeloid cells from samples pertaining to the same group were sorted into the same 1.5 mL tubes previously coated with and containing 500 μL of ice-cold 0.1% ultrapure non-acetylated BSA in PBS. Single cells were centrifuged at 350 × g for 5 min at 4°C and resuspended in the same buffer to 1,000 cells per μL estimated from counting on a hemocytometer and Trypan blue (MilliporeSigma) staining. Single-cell suspensions were kept on ice until further use.
Murine single-cell RNA sequencing.
For the experiments involving sorted blood vascular and myeloid cells from whole brain hemispheres (n = 3 mice per group), ~10,000 cells per sample were loaded onto a Chromium™ Single Cell A Chip (PN-120236, 10x Genomics) placed in a 10x™ Chip Holder and run on a 10x™ platform Chromium™ Controller to generate cDNAs carrying cell- and transcript-specific barcodes. For the experiment involving sorted myeloid cells only from the brain cortex (n = 6 mice), ~2,000 cells per sample were loaded onto the Chromium™ Single Cell A Chip and run on a 10x™ platform Chromium™ Controller to generate the cDNAs. Sequencing libraries were constructed using the Chromium™ Single Cell 3’ Library & Gel Bead Kit v2 (PN-120237, 10x Genomics). After a quality control (QC) step performed on an Illumina MiSeqNano system, libraries were sequenced on the Illumina NextSeq platform using paired-end sequencing, with 100,000 reads targeted per cell. All murine single-cell RNA-seq data were generated in the Genome Analysis and Technology Core of the University of Virginia (RRID:SCR_018883). Binary base call (bcl) files were converted to fastq format using the Illumina bcl2fastq2 software. Fastq files were then mapped to the mm10 transcriptome using the Cellranger 3.0.2 pipeline, specifically the count function. The resulting gene by count matrices were then read into R and filtered to remove low quality cells based on unique molecular counts, unique genes, and percent mitochondrial gene expression. Additionally, genes were filtered to retain only those which were expressed more than five cells. The remaining cells were then normalized, and log transformed using the scran normalization package53,54. The resulting normalized counts were then transformed from log2 scale to the natural log scale for compatibility with Seurat v355 and the effects of sequencing depth per cell, number of unique features, and percentage of mitochondrial genes were regressed out. Highly variable genes were determined using the variance stabilizing transformation and the top 2,000 most variable genes were used as input for PCA. Principal components were selected based on an elbow plot. Alternatively, for the cortical myeloid single-cell dataset, the significance of the first twenty principal components was evaluated using the Jackstraw test. Based on these results, on the percentage of variance explained by each component and the number of cells in the dataset, the first five principal components were used for clustering and t-Stochastic Neighbor Embedding (tSNE) analysis. Cluster membership was determined using shared nearest neighbor graph embedding and optimization of the Louvain algorithm as implemented in the Seurat FindNeighbors and FindClusters functions with a resolution of 0.6. All differentially expressed genes were calculated using the Wilcoxon Rank Sum test as implemented in Seurat. For cluster markers, the FindAllMarkers function was used to test genes showing a minimum log fold change of 0.25 (for the whole brain vascular and microglia single-cell experiments) or 0.1 (for the cortical microglia single-cell experiment) in each cluster versus all other clusters and expressed in a minimum of 30% of cells in the cluster of interest. In all datasets, cluster annotation was performed manually based on the expression of canonical markers and clusters collapsed based on common cell types. A final number of 7286 cells, including 2625 microglia, 1958 capillary blood endothelial cells (BECs), 1412 venous BECs and 545 arterial BECs was obtained in the whole brain vascular and myeloid single-cell dataset obtained using adult 5xFAD mice. A final number of 7739 cells, including 2345 microglia, 2934 capillary BECs, 602 venous BECs and 766 arterial BECs, was obtained in the whole brain vascular and myeloid single-cell dataset obtained using aged APPswe mice. In the cortical myeloid single-cell dataset obtained using 5xFAD mice, small populations of potentially contaminating oligodendrocyte precursor cells, oligodendrocytes, neurons and astrocytes were identified and removed based on the expression levels of Cspg4, Mbp, Cam2ka, and Gfap genes, respectively. Additionally, after an initial clustering, one population of cells was removed based on cluster markers, leading to a final number of 649 microglia. For analysis of differentially expressed genes between groups in the whole brain BECs and microglia datasets, each cluster was filtered to include genes that had at least 5 transcripts in at least 5 cells, then the top 2,000 highly variable genes were determined and included for further analysis using the SingleCellExperiment modelGeneVar and getTopHVGs functions. After filtering, observational weights for each gene were calculated using the ZINB-WaVE zinbFit and zinbwave functions56. These were then included in the edgeR model, which was created with the glmFit function, by using the glmWeightedF function51. Results were then filtered using as threshold a Benjamini-Hochberg adjusted P-value < 0.05 (statistically significant). Volcano plots were made with the EnhancedVolcano package (https://github.com/kevinblighe/EnhancedVolcano). Genes matching this significance threshold were divided on the basis of up- or down-regulation52 and used as input for GO Analysis using the ClusterProfiler package48,49. For analysis of differentially expressed genes between groups in the cortical microglia dataset, the FindMarkers function was used to test all genes expressed in at least 10% of cells within each group. P-values were adjusted using the Bonferroni correction method and genes were considered to be significantly differentially expressed if the adjusted P-values < 0.0557.
Neurological disease-associated gene expression.
Summary statistics were downloaded from the NHGRI-EBI GWAS Catalog for the following experimental factor ontologies: EFO_0000249, EFO_0003756, EFO_0003885, EFO_0002508, and EFO_0000692 on 10/24/201930. For each disease the set of reported genes were used for comparison with our LECs’ bulk RNA-seq datasets and HGNC symbols were mapped to their MGI counterparts using the bioMart database58,59. Normalized counts from each of our LECs’ datasets were used to calculate average expression of each gene across samples. Reported disease-associate genes were determined to be in the top percentiles based on their average expression. RNA-seq data of LECs from diaphragm or from ear skin were included in the heatmap visualizations for reference but were not used for calculation of the average expression. Heatmaps were visualized using the log2 transformed expression values and the pheatmap package. Genes falling within the top 25th percentile of highly expressed genes across our LECs’ datasets were used as gene-sets for functional enrichment of GO terms and KEGG pathways using Fischer’s exact test as implemented in the ClusterProfiler package48,49. Additionally, the normalized count matrix from GEO study GSE9881625 was downloaded and cells labelled as BECs were extracted for further analysis in Seurat. The normalized counts were scaled, principal components analysis was applied using the top 2,000 highly variable genes, and the top nine significant principal components were used for shared nearest neighbor clustering and tSNE. Five out of the seven identified clusters matched a similar transcriptional profile to the arterial, capillary, and venous BECs identified by Vanlandewijck et al25 and were subset and re-clustered. This analysis resulted in 4 clusters of brain BECs: capillary 1, capillary 2, arterial and venous (in Extended Data Fig. 9j). The average normalized expression of each gene was calculated within each cluster and the percentage of disease-associated genes falling within each quantile was determined based on average expression of the gene within the cluster. The union of the sets of disease-associated genes falling within the top 2nd percentile of highly expressed genes for each cluster was visualized in the heatmap using log2 average normalized expression values. Finally, we looked for the AD-associated genes in our cortical microglia single-cell RNA-seq dataset. The average normalized expression of each gene was calculated within each microglial cluster (clusters 1-4, in Extended Data Fig. 9m) and the percentage of AD-associated genes falling within each quantile was determined based on average expression of the gene within the cluster. The union of the sets of AD-associated genes falling within the top 2nd percentile of highly expressed genes for each cluster was visualized in a heatmap using log2 average normalized expression values. For the venn diagram (Extended Data Fig. 9o) comparing the AD-associated genes expressed in the top 10th percentile of meningeal LECs, brain BECs (including all 4 clusters) and microglia (including all 4 clusters), the gene-set was determined by ranking genes for that cell type based on average expression across all cells in the dataset rather than by cluster.
Gene-set associations with AD-related phenotypes and risk.
We determined if the genes and gene-sets identified in murine bulk and single-cell RNA-seq analyses were enriched for common genetic variants (1 Mb upstream and downstream of the gene) associated with AD-related phenotypes (amyloid imaging32, CSF soluble TREM233, Aβ42, Tau and phospho-Tau34) or AD age at onset35, risk36 and progression37. Single variant association was determined by using the latest summary statistics for each AD-related phenotype or risk. We also ran gene-based and gene-sets analyses using MAGMA60, which mapped every single-nucleotide polymorphism (SNP) to the nearest gene, taking into account linkage disequilibrium structures, and used multiple regression analyses to provide a P-value for the association between each gene and the tested phenotypes. This gene-set analysis used a regression structure to allow generalization analysis of continuous properties of genes and simultaneous analysis of multiple gene-sets and other gene properties and provided a P-value for each gene in the genome, taking into account all the independent SNPs in the gene region. The top 10th percentile of AD-associated genes highly expressed in murine meningeal LECs (resulting in 74 human gene orthologs), brain BECs (resulting in 58 human gene orthologs) or microglia (resulting in 29 human gene orthologs) were used in the gene-set analysis depicted in Supplementary Table 3. The up-regulated genes in microglia from 5xFAD mice with ablated meningeal lymphatic vasculature (in the Vis./photo. plus mIgG group versus the Vis. plus mIgG group; resulting in 507 human gene orthologs) and in aged APPswe mice treated with mVEGF-C-expressing AAV1 (in the mVEGF-C plus mIgG group versus the eGFP plus mIgG group; resulting in 46 human gene orthologs) were used in the gene-set analysis depicted in Supplementary Table 6.
Human microglia single-nucleus RNA-seq.
The Neuropathology Cores of the Charles F. and Joanne Knight Alzheimer’s Disease Research Center (Knight-ADRC) and the Dominantly Inherited Alzheimer Network (DIAN) provided the parietal lobe tissue of postmortem brains for each sample. These samples were obtained with informed consent for research use and were approved by the review board of Washington University in St. Louis. AD neuropathological changes were assessed according to the criteria of the National Institute on Aging-Alzheimer’s Association (NIA-AA). Their demographic, clinical severity, and neuropathological information are presented in Supplementary Table 4. From the 70 frozen human parietal lobe samples, approximately 500 mg of tissue were cut and weighed on dry ice using sterile disposable scalpels. The parietal tissue was homogenized in ice-cold homogenization buffer (0.25 M sucrose, 150 mM KCl, 5 mM MgCl2, 20 mM tricine-KOH pH 7.8, 0.15 mM spermine, 0.5 mM spermidine, EDTA-free protease inhibitor, and recombinant RNase inhibitors) with a dounce homogenizer. Homogenates were centrifuged for 5 min at 500 × g, at 4°C, to pellet the nuclear fraction. The nuclear fraction was mixed with an equal volume of 50% iodixanol and added on top of a 35% iodixanol solution for 30 min at 10,000 × g, at 4°C. After myelin removal, the nuclei were collected at the 30%-35% iodixanol interface. Nuclei were resuspended in nuclei wash and resuspension buffer (1% BSA and recombinant RNase inhibitors in PBS) and pelleted for 5 min at 500 × g and 4°C. Nuclei were passed through a 40 μM cell strainer to remove cell debris and large clumps. Nuclei concentration was manually determined using DAPI counterstaining and hemocytometer. Nuclei concentration was adjusted to 1,200 nuclei per μL and processed immediately following the 10x Genomics® Single Cell Protocol instructions. We generated single-nucleus RNA-seq libraries using the 10x Chromium single cell reagent Kit v3, for 10,000 cells per sample and sequenced 50,000 reads per cell from 70 frozen human parietal lobe samples. The CellRanger (v3.0.2 10x Genomics) software was employed to align the sequences and quantify gene expression. We used the GRCH38 (3.0.0) reference to prepare a pre-mRNA reference according to the steps detailed by 10x Genomics. The software was packaged into a Docker container (https://hub.docker.com/r/ngicenter/cellranger3.0.2), allowing us to launch it within the McDonnell Genome Institute (MGI) infrastructure, reducing the computing time for generating the BAM files. Filtering and QC was done using Seurat package (3.0.1). Each raw gene expression matrix for each sample was plotted, using BarcodeInflectionsPlot to calculate the inflection points derived from the barcode-rank distribution. Once the thresholds were determined, a subset of the data was isolated. We removed nuclei with high mitochondria gene expression following a previously proposed dynamic model61. Briefly, the nuclei were grouped by their percentage of mitochondria values using k = 2 clustering and the group with the higher percentage values was removed. Genes that were not expressed in at least 10 nuclei were removed from the final matrix. To detect and discard doublets, we used Doublet Finder62, which removes nuclei with expression profiles that resemble synthetically mixed nuclei from the dataset. The gene expression matrices from all samples were combined in R for further processing using the Seurat protocol. The expression matrix was normalized using the SCtransform protocol by Seurat. This function calculates a model of technical noise in single-cell RNA-seq data using ‘regularized negative binomial regression’ as described previously63. We regressed, during the normalization, against number of Genes, number of UMI and the percentage of mitochondria. The principal components (PC) were calculated using the first 3,000 variable genes and the Uniform Manifold Approximation and Projection (UMAP) analysis was performed with the top 14 PCs. The clustering was done using a resolution of 0.2. We employed a list of marker genes that we had previously curated64 to annotate brain single-nucleus RNA-seq data. We used the DotPlot function (Seurat package) to visualize the average expression of genes related to specific cell types. This approach enabled the labelling of cell-types based on the overall expression profile of the nuclei, regardless of dropout events. In addition, we employ a supervised method termed Garnett65 that leverages machine learning to classify each of the nuclei and estimate cluster homogeneity. This method also provides a metric of gene ambiguity, which enables further optimization of the marker genes to be included in the classification process. For this method, we employed SYT1, SNAP25, GRIN1 to generally classify neurons, and used NRGN, SLC17A7 and CAMK2A for excitatory and GAD1 and GAD2 for inhibitory neurons; AQP4 and GFAP for astrocytes; CSF1R, CD74 and C3 for microglia; MOBP, PLP1 and MBP for oligodendrocytes; PDGFRA, CSPG4 and VCAN for oligodendrocyte progenitor cells; CLDN5, TM4SF1 and CDH5 for endothelial cells, and ANPEP for pericytes. We employed the function check_markers (Garnett package) to evaluate the ambiguity score and the relative number of cells for each cell type. A classifier was then trained using the marker file, with num_unknown set to 50. This classifier was used to annotate cells with cell type assignments extended to nearby cells using the clustering-extended type labeling option. The microglia nuclei were then isolated from the full dataset for further analysis.
Microglial cis-eQTL analysis.
We tested whether the top 10% meningeal LECs’ genes harbored SNPs in the local proximity that regulate their expression in microglia isolated from brain samples of neuropathologically-confirmed AD donors without familial mutations (24 sporadic AD cases) and neuropathology-free donors (7 non-AD cases). We selected the 23,337 SNPs in a 500 kb window up/downstream of the transcription start site for the genes of interest and kept those SNPs with a minor allele frequency (MAF) > 0.05. To perform the expression quantitative trait analyses we applied a linear mix model correcting for sex, age of death (AOD), AD status, and subject of origin with a zero-inflated negative binomial distribution using the R package ‘glmmTMB’: UMI ~ genotype + SEX + AOD + ADstatus + (1|subjs), ziformula=~1, family=nbinom2. We created gene-SNP pairs by extracting the most significant SNP for each gene. We followed an approach previously employed in bulk RNA-seq expression quantitative trait loci (eQTL) analyses66–69 to correct for multiple testing while accounting for linkage disequilibrium. We performed permutation analyses70 on the top 40 most significant pairs, shuffling expression levels and phenotypic variables. We learned an empirical null distribution by running 1,000 iterations for each gene, sampling without replacement and ensuring no duplicate iterations. We employed violin plots to provide a visual representation of genes expression that have the first, median and third quantiles indicated by black lines (ggplot2 R package). We used partial residuals from linear mixed models to provide a depiction of the relationship of interest corrected for covariates. For example, in the eQTL analysis, genotype is the term of interest in the following model: count = b1genotype + b2sex + b3AOD + b4ADstatus + b5(1|subjs)+ e. Using the values from fitting the model above, the partial residual or corrected expression (countc) is: countc = b1genotype + e. The partial residual can frequently be negative, so to allow for log2 transformation, we then added 1 and subtracted the minimum corrected expression value before plotting: countlog2(c) = log2(countc + 1 − min(countc)).
Cross-species microglia RNA-seq analysis.
Mouse microglia (see also results in Fig. 1k–m and Extended Data Fig. 7e–g) identified using the methodology described above (in the Murine single-cell RNA sequencing section) were isolated and genes were mapped to their human orthologs with the biomaRt package in R. Non-protein coding genes as well as genes without a one-to-one conversion between species were removed from the dataset resulting in the retention of 12,689 genes. Seurat was used for all subsequent integration and analysis. Each condition was normalized with SCTransform separately, to match human processing. The harmonization of transcriptomic data was performed following Seurat platform best practices. The top 3,000 most variable features from human and mouse cells were identified (SelectIntegrationFeatures function) and then the datasets were directly integrated with stepwise PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData functions. PCA was run and principal components were selected based on an elbow plot. The first eight principal components were used for clustering and tSNE analysis. Cluster membership was determined using shared nearest neighbor graph embedding and optimization of the Louvain algorithm as implemented in the Seurat FindNeighbors and FindClusters functions with a resolution of 0.2. The percent of each sample belonging to each cluster was calculated and a two-proportions Z-test was run to determine any statistically significant differences between the mouse conditions. A microglial gene signature of meningeal lymphatic dysfunction (Supplementary Table 5) was created by selecting the 54 genes that showed a log2 (fold change) > 1 and were significantly overexpressed (false discovery rate q-value < 0.05) in the Vis./photo. plus mIgG group, when compared to the Vis. plus mIgG group (Fig. 1l), and mapped to a human gene ortholog. A score of the gene signature was calculated in the human microglia using Seurat’s AddModuleScore function. The pairwise significance difference between clusters were calculated using linear mixed model moduleScore ~ cluster + SEX + AOD + (1|subjs), ziformula=~0, family=gaussian and the glmmTMB function previously described. Visualization of the module scores was performed using the same partial residual technique employed for the Microglia cis-eQTL plots. Briefly, the cluster effect on the module scores, or the partial residual, was isolated representing the module score corrected for AOD, sex, and subject. The above-mentioned partial residual technique, correcting for AOD, sex and subject was also used to represent the expression levels of specific human genes by microglial cluster in the form of violin plots.
Statistical analysis and reproducibility.
Sample sizes were chosen on the basis of standard power calculations (with α = 0.05 and power of 0.8) performed for similar experiments that were previously published5,12. In general, statistical methods were not used to re-calculate or predetermine sample sizes. Animals from different cages, but within the same experimental group, were selected to assure randomization. Experimenters were blinded to the identity of experimental groups from the time of euthanasia until the end of data collection and analysis for all the independent experiments. The Kolmogorov-Smirnov test was used to assess the distribution of the data. Variance was similar within independent groups of the same experiment and between groups from independent experiments. The ROUT test was used to identify and discard potential outliers. No exclusion criteria were pre-established. An outlier was identified and excluded in Extended Data Fig. 3e, in the Visudyne group (the outlier is indicated in the correspondent source data file). Data in graphs was always presented as mean ± standard error mean (s.e.m.). Two-group comparisons were made using two-tailed unpaired Student’s T test. For comparisons of multiple factors (for example, lymphatic vessel ablation vs antibody treatment), two-way ANOVA with Holm-Sidak’s multiple comparisons test was used. Repeated measures two-way ANOVA with Tukey’s multiple comparisons test was used to analyze data acquired during the acquisition and reversal of the Morris water maze test. Statistical analysis in experiments involving mouse models, tissues and cells was performed in Prism version 8.3.4 (GraphPad Software, Inc.) or in R software (version 3.5.0). Statistical tests used for group or cluster comparisons in bulk, single-cell or single-nucleus RNA-seq experiment analysis are specified in the respective methods’ sections.
Reporting summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Code and data availability.
Source data files depicting the quantification values mentioned in the text or plotted in graphs shown in Figs. 1 and 3 and Extended Data Figs. 1, 2, 3, 4, 5, 7, 8 and 9, and the gene lists used for gene-set andalysis in Supplementary Table 3 and Supplementary Table 6 are available in the online version of this paper at http://www.nature.com/nature. New RNA-seq data sets have been deposited online in the Gene Expression Omnibus (GEO database) under the accession number GSE141917. Previously published RNA-seq data sets can be found under the accession numbers GSE99743 and GSE104181. Custom code used to analyze the RNA-seq data and datasets generated and/or analyzed in the current study are available from the corresponding authors upon request.
Extended Data
Extended Data Figure 1 |. Morphological assessment and functional enrichment analysis of differentially expressed genes show accelerated meningeal lymphatic dysfunction in 5xFAD mice.
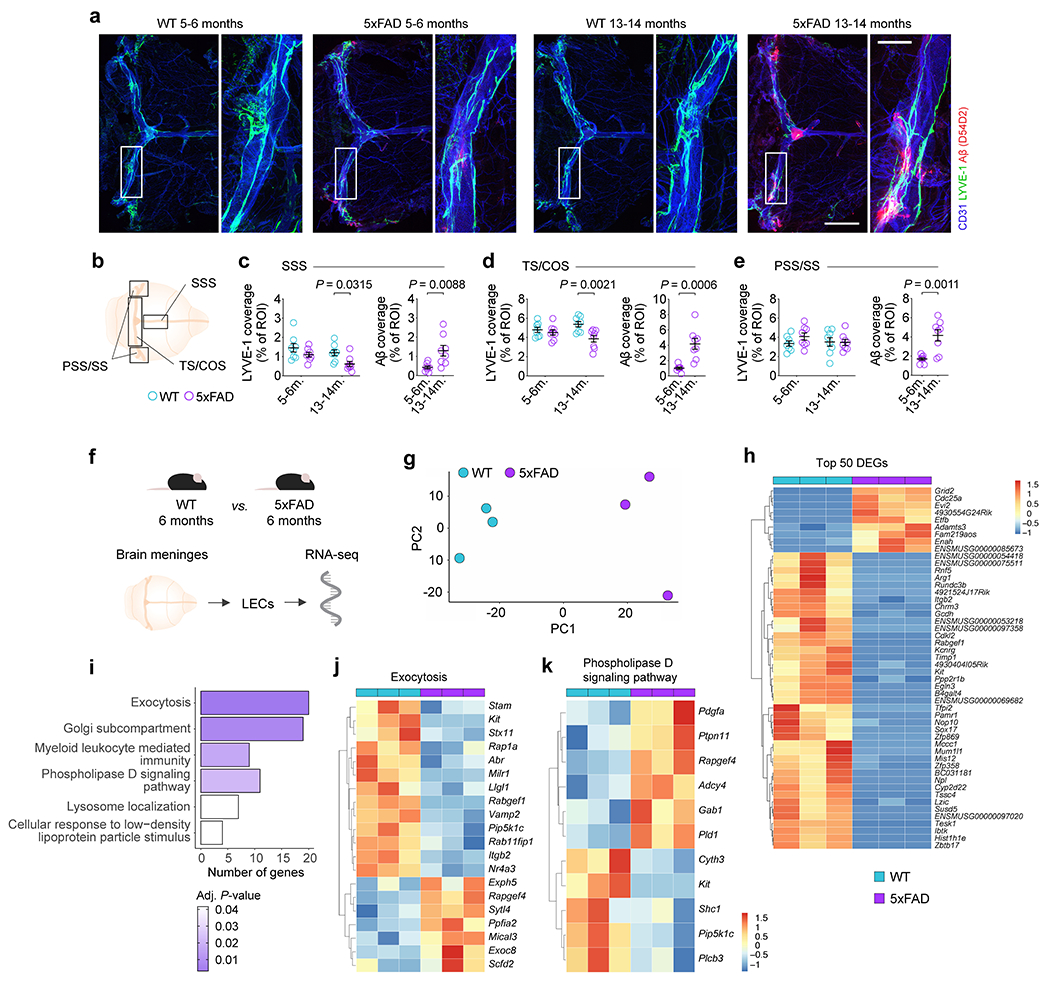
a, Representative images of meningeal whole mounts from 5xFAD mice at 5–6 and 13–14 months of age, stained for CD31 (blue), LYVE-1 (green) and Aβ (red, stained with the D54D2 antibody; scale bar, 2 mm; inset scale bar, 500 μm). b, Scheme depicting the compartmentalization of the meningeal whole mount for quantification of LYVE-1 and Aβ coverage. c-e, Graphs showing the coverage by LYVE-1+ vessels and Aβ as a percentage of the region of interest (% of ROI) at the c) superior sagittal sinus (SSS), d) transverse sinus and confluence of sinuses (TS/COS), and e) petrosquamosal and sigmoid sinuses (PSS/SS). Results in a-e are presented as mean ± s.e.m.; n = 8 per group; two-way ANOVA with Holm-Sidak’s multiple comparisons test (for LYVE-1+ vessels) and two-tailed unpaired Student’s T test (for Aβ coverage); data result from 2 independent experiments. f, Lymphatic endothelial cells (LECs) were isolated from the brain meninges of male 5xFAD mice and WT littermate controls at 6 months of age, total RNA was extracted and sequenced. g, Principal component analysis (PCA) plot showing segregation between WT (blue) and 5xFAD (purple) meningeal LEC transcriptomes. h, Heatmap of top 50 differentially expressed genes in 5xFAD meningeal LECs at 6 months of age. i, Gene-sets obtained by functional enrichment of differentially expressed genes in meningeal LECs from 5xFAD mice. j, k, Exocytosis (GO:0006887) and phospholipase D signaling pathway (KEGG:mmu04072) gene-sets obtained by functional enrichment analysis, with corresponding differentially expressed genes. Data in f-k consists of n = 3 per group; individual RNA samples result from LECs pooled from 10 meninges over 3 independent experiments; the Benjamini-Hochberg correction was used to adjust the associated P-values in i (adj. P-value < 0.05); functional enrichment of differential expressed genes in i was determined with Fisher’s exact test; color scale bar in h and k represents expression values for each sample as standard deviations from the mean across each gene.
Extended Data Figure 2 |. Meningeal immune cell profiling by mass cytometry in 5xFAD mice at 5–6 and 11–12 months.
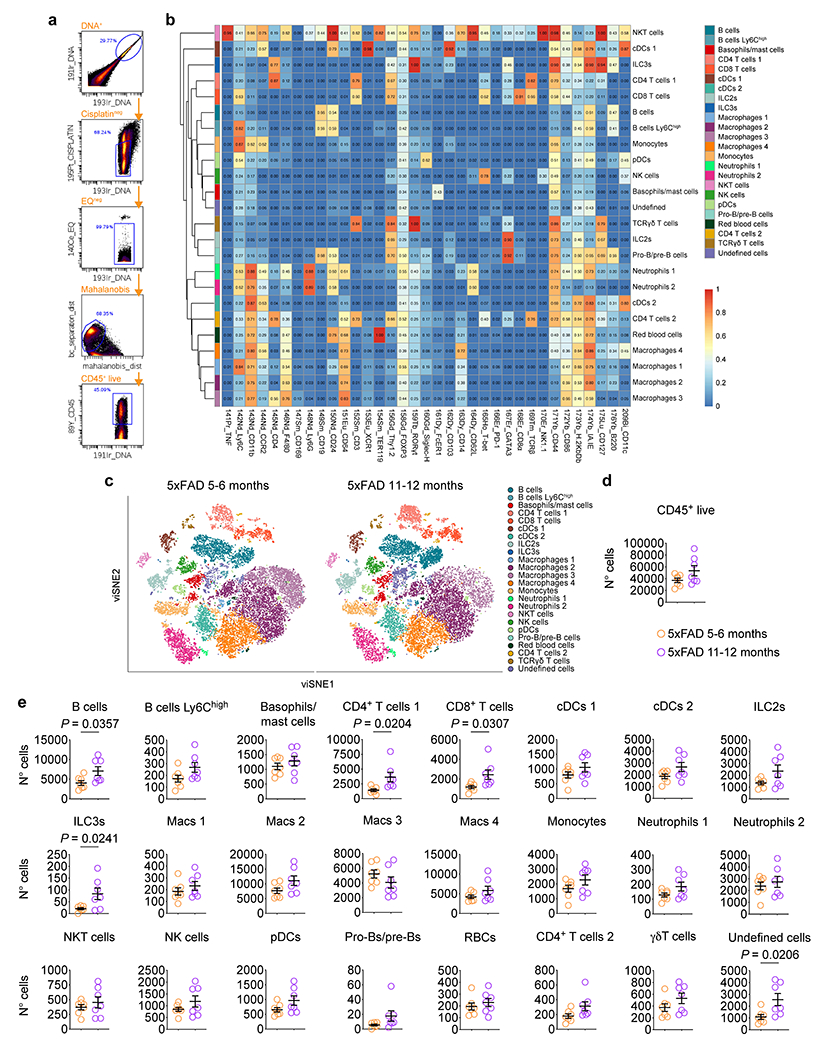
a, Meningeal single-cell suspensions were obtained from male 5xFAD mice at 5–6 months and 11–12 months and processed for mass cytometry. Representative mass cytometry dot plots depicting gating strategy used to select CD45+ live cells used in further high-dimensional analysis. b, Heatmaps of the median marker expression values for each immune cell cluster identified using Rphenograph. Median marker expression values are indicated by color intensity depicted in the scale bar. c, t-distributed stochastic neighbor embedding-based visualization (viSNE) plots showing unsupervised clustering of CD45+ live immune cells. d, Number of CD45+ live meningeal leukocytes at 5–6 (orange) and 11–12 (purple) months in 5xFAD mice. e, Numbers of different meningeal immune cells showing a statistically significant increase in B cells, CD4+ T cells 1, CD8+ T cells, type 3 innate lymphoid cells (ILC3s) and undefined cells in the meninges of 5xFAD mice at 11-12 months of age. Data in a-e are representative of a single experiment; results in d and e are presented as mean ± s.e.m.; n = 7 per group; two-tailed unpaired Student’s T test.
Extended Data Figure 3 |. Compromising meningeal lymphatic function in 5xFAD mice limits brain Aβ clearance by mAb158 and modulates neuritic dystrophy, microglial activation and fibrinogen levels.
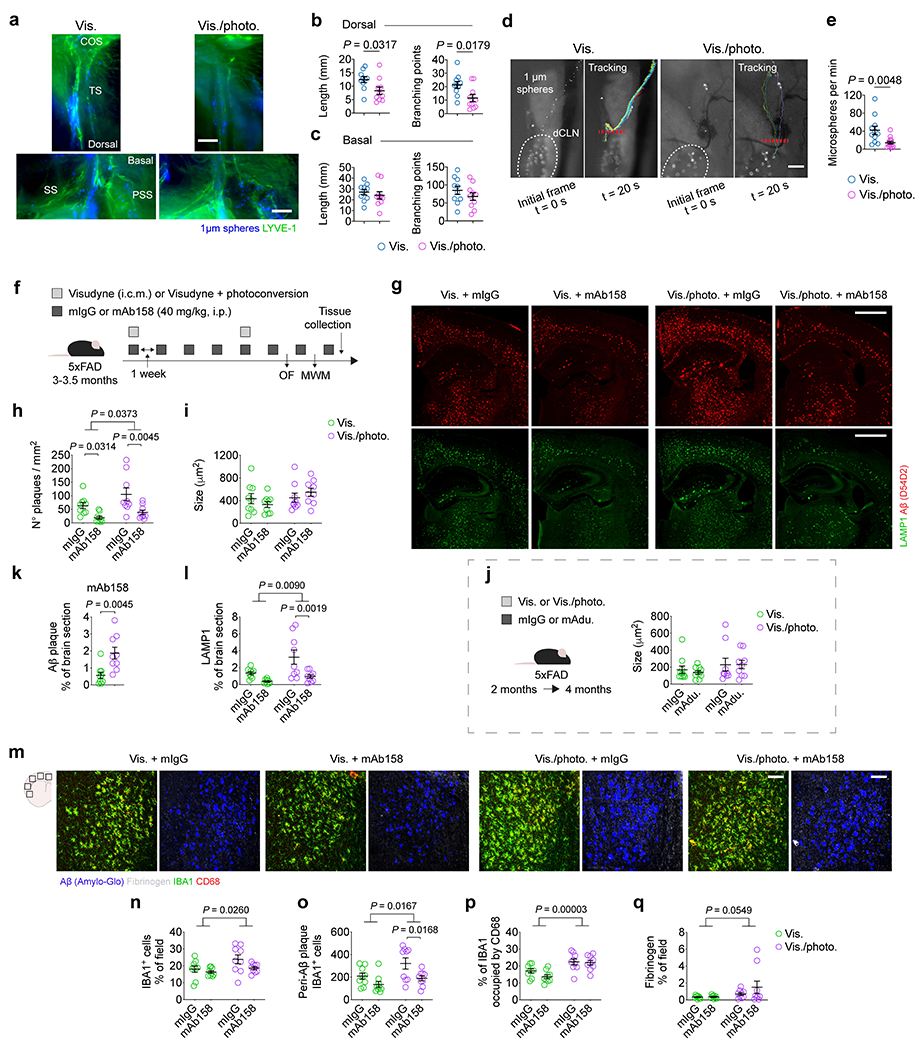
a-e, Adult 2-month-old male WT mice were injected with 5 μL of Visudyne (intra-cisterna magna, i.c.m.) followed by a transcranial photoconversion step (Vis./photo.) to ablate meningeal lymphatic vessels at the dorsal meninges. Control mice were injected with Visudyne without photoconversion (Vis.). One week later, each mouse was injected with 5 μL of a suspension of fluorescent microspheres (1 μm in diameter) into the CSF and 15 min later the lymphatic vessel afferent to the deep cervical lymph node (dCLN) was imaged by live in vivo fluorescence stereomicroscopy. a, Representative images of skull caps with attached meningeal layers showing microspheres (blue) and lymphatic vessels stained for LYVE-1 (in green) around the confluence of sinuses (COS) and transverse sinus (TS) at the dorsal meninges or around the sigmoid (SS) and petrosquamosal (PSS) sinuses at the basal meninges (scale bars, 500 μm). b, c, Graphs showing LYVE-1+ vessels’ total length (in mm) and number of branching points in lymphatics at the b) dorsal and c) basal meninges. Results in b and c are presented as mean ± s.e.m.; n = 10 per group; two-tailed unpaired Student’s T test; data are representative of 2 independent experiments. d, Representative frames showing microspheres flowing through the lymphatic vessel afferent to the dCLN, or cumulative sphere tracking (for 20 sec), in mice with intact or ablated meningeal lymphatic vessels. e, Graph with quantifications of microsphere flow (number of microspheres per minute) in mice from each group. Results in e are presented as mean ± s.e.m.; n = 11 in Vis. group and n = 14 in Vis./photo. group; two-tailed unpaired Student’s T test; data result from 2 independent experiments. f, Adult 3–3.5-month-old male 5xFAD mice were injected (i.c.m.) with Visudyne (5 μL) plus photoconversion (Vis./photo.) or Visudyne without photoconversion (Vis.). Upon recovery, mice received intraperitoneal (i.p.) injections of mAb158 (a murine antibody against Aβ protofibrils) or the control murine IgG (mIgG) antibodies, each at a dose of 40 mg/kg. Antibodies were injected weekly for a total of four weeks. Additional steps of meningeal lymphatic vessel ablation or control interventions were followed by four weekly injections with antibodies. Mice were tested in the open field and Morris water maze (see Extended Data Fig. 4). g, Representative images of brain sections from 5xFAD mice stained for Aβ (red, stained with the D54D2 antibody) and for LAMP1 (green; scale bars, 1 mm). h-l, Graphs showing h) number of Aβ plaques per mm2 of brain section, i) average size of Aβ plaques (μm2) in mAb158 cohort, j) average size of Aβ plaques (μm2) in mAducanumab cohort (related to Fig. 1a–e), k) coverage by Aβ plaques and l) coverage by LAMP1+ dystrophic neurites (as % of brain section) in each group. m, Representative images of the brain cortex stained for Aβ (blue, stained with Amilo-Glo), fibrinogen (grey), IBA1 (green) and CD68 (red; scale bar, 100 μm). n-q, Graphs showing the n) coverage by IBA1+ cells (% of field), o) number of peri-Aβ plaque IBA1+ cells, p) percentage of IBA1 occupied by CD68 and q) fibrinogen coverage (% of field) in each group. Data in f-q are representative of a single experiment; results in h-l and n-q are presented as mean ± s.e.m.; n = 9 in each group; in h, i, j, l and n-q, two-way ANOVA with Holm-Sidak’s multiple comparisons test; in k, two-tailed unpaired Student’s T test.
Extended Data Figure 4 |. Meningeal lymphatic dysfunction leads to anxious-like behavior and worsened spatial learning and memory in 5xFAD mice.
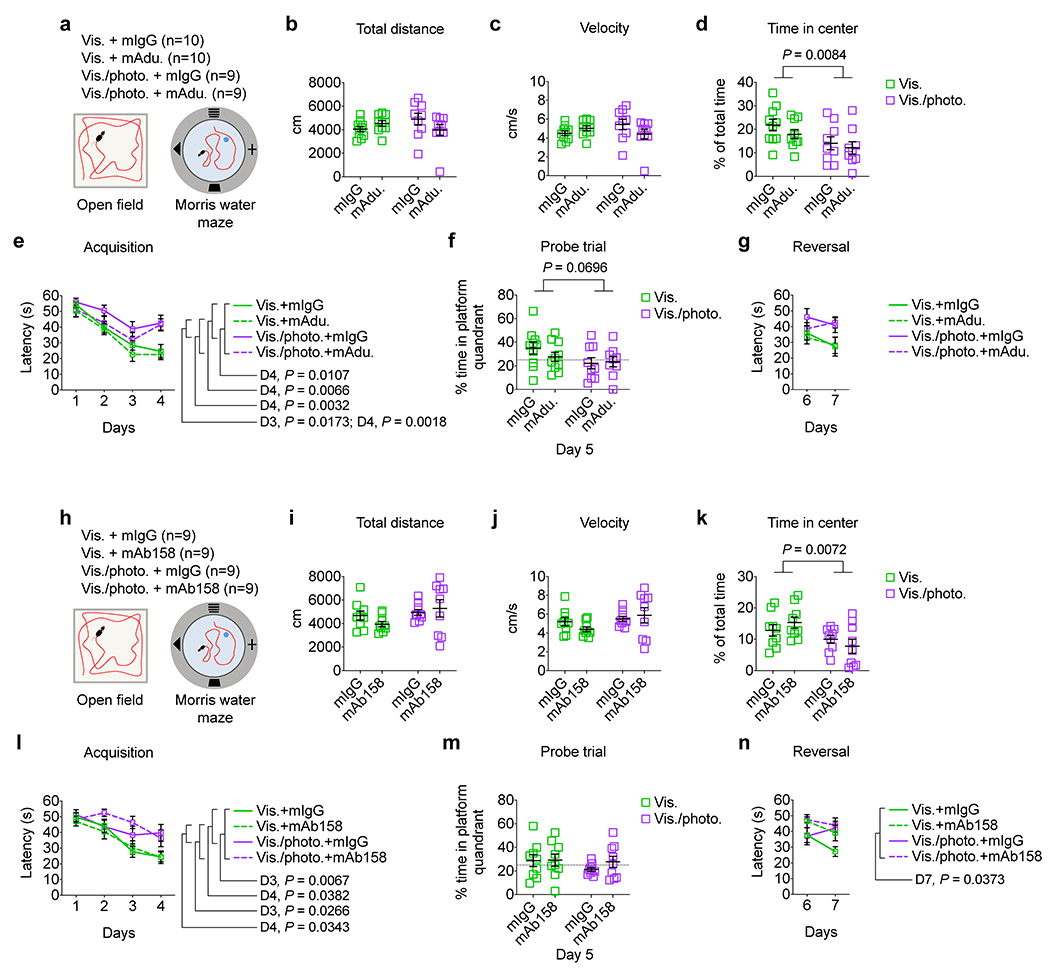
a, Adult 5xFAD mice with intact or ablated meningeal lymphatics and treated with mAducanumab (mAdu.) or control mIgG antibodies (see Fig. 1 for experimental scheme and more results) were tested in the open field arena and in the Morris water maze. b-d, Graphs showing b) total distance (in centimeters), c) velocity (in centimeters per second) and d) percentage of time in the center of the open field arena (% of total time). e-g, Graphs showing e) latency to platform in acquisition (in seconds), f) percentage of time in the platform quadrant in the probe trial and g) latency to platform in reversal (in seconds). Data in a-g result from a single experiment; results in b-g are presented as mean ± s.e.m.; n = 10 in Vis. groups and n = 9 in Vis./photo. groups; two-way ANOVA with Holm-Sidak’s multiple comparisons test in b-d and f; repeated measures two-way ANOVA with Tukey’s multiple comparisons test in e and g; statistically significant differences between groups in days 3 and 4 of the Morris water maze test are indicated as D3 and D4, respectively. h, Adult 5xFAD mice with intact or ablated meningeal lymphatics and treated with mAb158 or control mIgG antibodies (see Extended Data Fig. 3f–q for experimental scheme and more results) were tested in the open field arena and in the Morris water maze. i-k, Graphs showing i) total distance (in centimeters), j) velocity (in centimeters per second) and k) percentage of time in the center of the open field arena (% of total time). l-n, Graphs showing l) latency to platform in acquisition (in seconds), m) percentage of time in the platform quadrant in the probe trial and n) latency to platform in reversal (in seconds). Data in h-n result from a single experiment; results in i-n are presented as mean ± s.e.m.; n = 9 in each group; two-way ANOVA with Holm-Sidak’s multiple comparisons test in i-k and m; repeated measures two-way ANOVA with Tukey’s multiple comparisons test in l and n; statistically significant differences between groups in days 3, 4 and 7 of the Morris water maze test are indicated as D3, D4 and D7, respectively.
Extended Data Figure 5 |. Meningeal lymphatic vessel ablation precludes brain Aβ plaque clearance by mAb158 administered into the CSF.
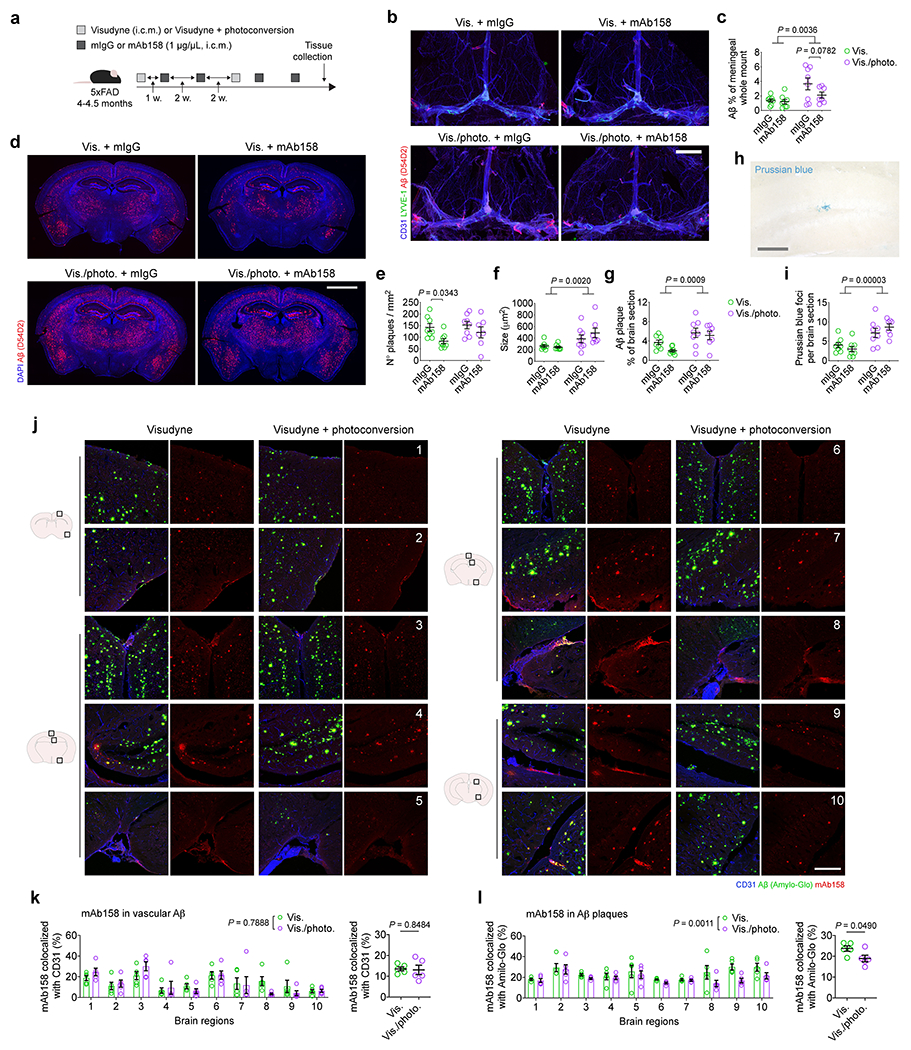
a, Adult 4–4.5-month-old male 5xFAD mice were injected (i.c.m.) with Visudyne (5 μL) plus photoconversion (Vis./photo.) or Visudyne without photoconversion (Vis.). One week later, 5 μL of mAb158 antibodies or the same volume of the control murine IgG (mIgG) antibodies were directly injected into the CSF (i.c.m.), both at 1 μg/μL. Injections with antibodies were repeated two weeks later. Additional steps of meningeal lymphatic vessel ablation or control interventions were followed by two more i.c.m. injections with antibodies according to the scheme. b, Representative images of meningeal whole mounts stained for CD31 (blue), LYVE-1 (green) and Aβ (red, stained with the D54D2 antibody; scale bar, 2 mm). c, Graph showing the coverage by Aβ as a percentage of the meningeal whole mount. d, Representative images of brain sections from 5xFAD mice stained for Aβ (red, stained with the D54D2 antibody) and with DAPI (blue; scale bar, 2 mm). e-g, Graphs showing e) number of Aβ plaques per mm2 of brain section, f) average size of Aβ plaques (μm2) and g) total coverage of Aβ plaques (% of brain section) in each group. h, Representative inset showing an example of a Prussian blue focus in a brain tissue section of a 5xFAD mouse (scale bar, 100 μm). i, Graph showing the quantifications of Prussian blue foci per brain section in each group. Data in a-i are representative of 2 independent experiments; results in c, e-g and i are presented as mean ± s.e.m.; n = 8 in Vis. plus mIgG, Vis. plus mAb158 and Vis./photo. plus mIgG, n = 7 in Vis./photo. plus mAb158; two-way ANOVA with Holm-Sidak’s multiple comparisons test. j, 5xFAD mice (5 months old) with intact or ablated meningeal lymphatic vasculature were injected (i.c.m.) with 5 μL of mAb158 (at 1 μg/μL). One hour later, mice were transcardially perfused and the brains were collected for assessment of mAb158 linked to Aβ in blood vasculature or Aβ plaques. Images of ten different regions of the brain of 5xFAD mice from each group showing blood vessels stained for CD31 (blue), Aβ plaques (green, stained with Amylo-Glo) and mAb158 (red; scale bar, 200 μm). k, Graphs with colocalization between mAb158 and CD31 (% of total CD31 coverage) in each brain region (1 to 10) or presented as the average of all regions. l, Graphs with colocalization between mAb158 and Amilo-Glo (% of total Amilo-Glo-stained Aβ plaques) in each brain region (1 to 10) or presented as the average of all regions. Data in j-l result from a single experiment; results in k and l are presented as mean ± s.e.m.; n = 5 per group; two-way ANOVA with Holm-Sidak’s multiple comparisons test (for comparisons between groups in each brain region) and two-tailed unpaired Student’s T test (for comparisons between the two groups).
Extended Data Figure 6 |. Effects of meningeal lymphatic vessel ablation and mAducanumab immunotherapy on the microglial and blood endothelial cell transcriptomes in 5xFAD mice.
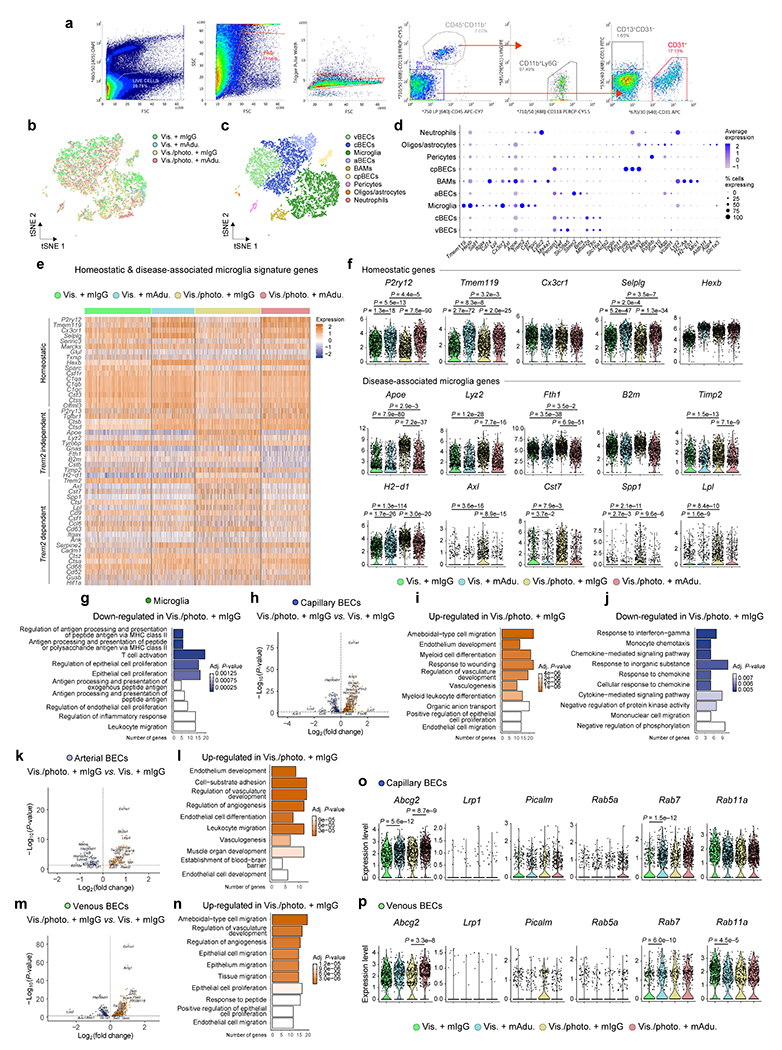
a, Representative flow cytometry dot plots showing gating strategy used to sort (and enrich for) live (DAPI−) singlets that were CD45+CD11b+Ly6G− (macrophages/microglia), CD45−CD11b−CD31+ (blood endothelial cells) and CD45−CD11b−CD13+CD31− (mural cells). b, c, Representations of the t-distributed stochastic neighbor embedding (tSNE) plots highlighting the brain cells identified by single-cell RNA-seq discriminated by b) group or c) type. d, Dot plot depicting the average scaled expression levels of specific genes (in the x axis) used to identify the brain cell populations, as well as the percentage of cells expressing those genes within each population; choroid plexus blood endothelial cells (cpBECs), border-associated macrophages (BAMs), arterial BECs (aBECs), capillary BECs (cBECs), venous BECs (vBECs). e, Heatmap showing expression levels of genes involved in the transition from homeostatic to Trem2-independent and Trem2-dependent disease-associated microglia phenotypes in the different groups. f, Violin plots showing the expression levels of the homeostatic P2ry12, Tmem119, Cx3cr1, Selplg and Hexb genes, and disease-associated microglia Apoe, Lyz2, Fth1, B2m, Timp2, H2-d1, Axl, Cst7, Spp1 and Lpl genes in each group. g, Top ten Gene Ontology terms obtained after analyzing significantly down-regulated genes in microglia from the Vis./photo. plus mIgG group, when compared to the Vis. plus mIgG group. h, Volcano plot with significantly down-regulated (in blue) and up-regulated (in orange) genes after comparing the transcriptomes of cBECs from the Vis./photo. plus mIgG and the Vis. plus mIgG groups. i, j, Top ten Gene Ontology terms obtained after analyzing significantly i) up-regulated or j) down-regulated genes in cBECs from the Vis./photo. plus mIgG group, when compared to the Vis. plus mIgG group. k-n, Volcano plots, with significantly down-regulated (in blue) and up-regulated (in orange) genes, and top ten Gene Ontology terms (using up-regulated genes) obtained after comparing the transcriptomes of aBECs (k and l) and vBECs (m and n) from the Vis./photo. plus mIgG and the Vis. plus mIgG groups. o, p, Violin plots showing the expression levels of Abcg2, Lrp1, Picalm, Rab5a, Rab7 and Rab11a in o) cBECs and p) vBECs from each group. Data in a-p resulted from a single experiment where the transcriptomes of 7,286 cells (isolated from brain hemispheres of 3 mice per group) were analyzed, including 2,625 microglia, 1,958 cBECs, 545 aBECs and 1,412 vBECs; scale bar in e represents scaled expression at the single-cell level; differentially expressed genes plotted in h, k and m were determined using a F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P-values; Gene Ontology analyses used over-representation test and scale bars in g, i, j, l and n represent Benjamini-Hochberg adjusted P-values for each pathway; gene expression comparison in f, o and p was done using Wilcoxon Rank-Sum test with Bonferroni’s adjusted P-values reported.
Extended Data Figure 7 |. Improved brain Aβ plaque clearance by delivery of mVEGF-C and mAb158 into the CSF is correlated with lymphatic vessel expansion at the dorsal meninges and transcriptional reprogramming of meningeal LECs.
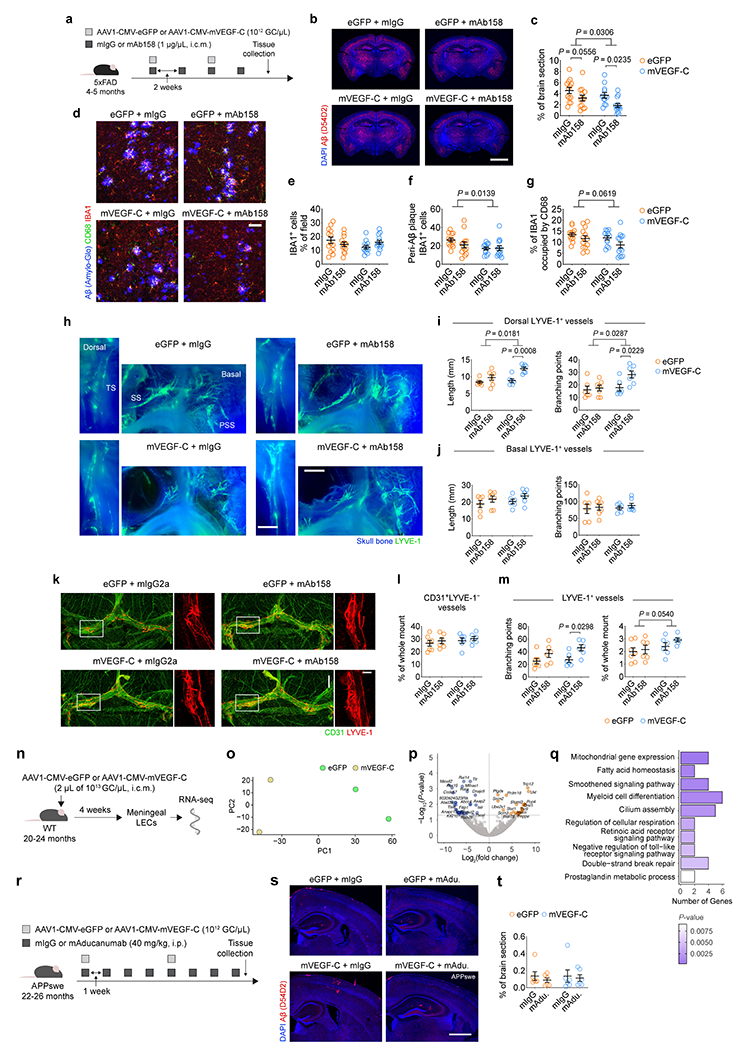
a, Adult 4–5-month-old male 5xFAD mice were injected with 5 μL (i.c.m.) of AAV1 expressing enhanced green fluorescent protein (eGFP) or murine VEGF-C (mVEGF-C), under the cytomegalovirus (CMV) promoter (each at 1012 GC/μL), in combination with either mAb158 antibodies or the respective mIgG control (each at 1 μg/μL) antibodies. Antibody injections (5 μL at 1 μg/μL, i.c.m.) were repeated two weeks later. The same regimen of the aforementioned i.c.m. injections was repeated as indicated in the scheme and tissue was collected two weeks after the last injection. b, Representative images of brain sections stained for Aβ (red, stained with the D54D2 antibody) and with DAPI (blue; scale bar, 2 mm). c, Graph showing coverage of Aβ as percentage of brain section in each group. d, Representative images from the brain cortex stained for Aβ (blue, stained with the Amilo-Glo), CD68 (green) and IBA1 (red; scale bar, 50 μm). e-g, Graphs showing the e) coverage by IBA1+ cells (% of field), f) number of peri-Aβ plaque IBA1+ cells and g) percentage of IBA1 occupied by CD68 in each group. Results in c and e-g are presented as mean ± s.e.m.; n = 12 in mVEGF-C plus mIgG and n = 13 in eGFP plus mIgG, eGFP plus mAb158 and mVEGF-C plus mAb158; two-way ANOVA with Holm-Sidak’s multiple comparisons test; data in a-g result from 2 independent experiments. h, Representative fluorescence stereomicroscopy images of skull caps (skull bone signal in blue) and attached meningeal lymphatic vessels stained for LYVE-1 (in green) around the transverse sinus (TS) at the dorsal meninges or around the sigmoid (SS) and petrosquamosal (PSS) sinuses at the basal meninges (scale bars, 500 μm). i, j, Graphs showing LYVE-1+ vessels’ total length (in mm) and number of branching points in lymphatics at the i) dorsal and j) basal meninges. Results in h-j are presented as mean ± s.e.m.; n = 6 in mIgG groups and n = 7 in mAb158 groups; two-way ANOVA with Holm-Sidak’s multiple comparisons test; data in h-j are representative of 2 independent experiments. k, Representative images of meningeal whole mounts stained for CD31 (green) and LYVE-1 (red; scale bar, 1 mm; inset scale bar, 300 μm). l, m, Graphs showing the l) coverage by CD31+LYVE-1− vessels (% of meningeal whole mount) and the m) number of branching points and coverage by LYVE-1+ vessels (% of meningeal whole mount). Results in l and m are presented as mean ± s.e.m.; n = 7 in eGFP plus mIgG and n = 6 in eGFP plus mAb158, mVEGF-C plus mIgG and mVEGF-C plus mAb158; two-way ANOVA with Holm-Sidak’s multiple comparisons test; data in k-m are representative of 2 independent experiments. n, Aged WT mice (20–24 months of age) were injected with 2 μL (i.c.m.) of AAV1 expressing eGFP or mVEGF-C, under the CMV promoter (each at 1013 GC/μL). One month later, mice were transcardially perfused, skull caps were collected, meninges harvested and LECs were sorted by FACS for bulk RNA-seq. o, PCA plot showing segregation between eGFP (orange) and mVEGF-C (blue) meningeal LEC transcriptomes. p, Volcano plot showing the significantly down-regulated (in blue) and up-regulated (in orange) genes between meningeal LECs from the mVEGF-C and eGFP groups. q, Ten Gene Ontology terms (selected from the 30 most altered) obtained after analysis of the differentially expressed genes between meningeal LECs from the mVEGF-C and eGFP groups. Data in n-q consists of n = 2 per group; individual RNA samples result from LECs pooled from 10 meninges over 2 independent experiments; differentially expressed genes (P < 0.05) plotted in c were determined using a F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model; Gene Ontology analysis in q used over-representation test and the scale bar represents the P-value for each pathway. r, Aged APPswe (22–26 months of age) were treated with viral vectors expressing eGFP or mVEGF-C (via i.c.m. injections) and with mIgG or mAdu. antibodies (via i.p. injections, according to the regimen in the scheme; related to Fig. 2). s, Representative images of brain sections stained for Aβ (red, stained with the D54D2 antibody) and with DAPI (blue; scale bar, 1 mm). t, Graph showing coverage of Aβ (as percentage of the brain sections) in each group. Results in t are presented as mean ± s.e.m.; n = 6 per group; two-way ANOVA with Holm-Sidak’s multiple comparisons test; data in r-t resulted from a single experiment.
Extended Data Figure 8 |. Therapeutic effects of mVEGF-C on the clearance of Aβ by antibodies in old APPswe mice and the gene expression profile of brain cells.
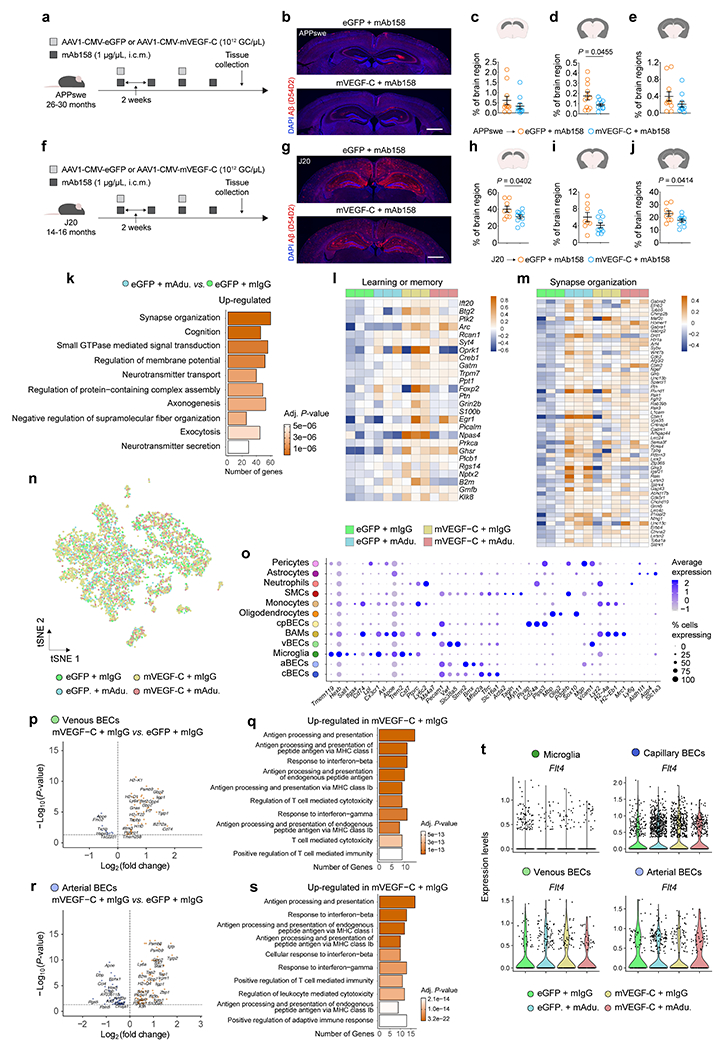
a, Aged APPswe mice (26–30 months old) were injected with 5 μL (i.c.m.) of AAV1 expressing eGFP or mVEGF-C (each at 1012 GC/μL) in combination with mAb158 (at 1 μg/μL) as indicated in the scheme. b, Representative images of brain sections from APPswe mice stained for Aβ (red, stained with the D54D2 antibody) and with DAPI (blue; scale bar, 1 mm). c-e, Graphs showing coverage of Aβ (% of brain region) in the c) hippocampus, d) cortex/striatum/amygdala and e) combined regions. Results in c-e are presented as mean ± s.e.m.; n = 11 per group; two-tailed unpaired Student’s T test; data in a-e result from a single experiment. f, Aged J20 mice (14–16 months old) were injected with 5 μL (i.c.m.) of AAV1 expressing eGFP or mVEGF-C (each at 1012 GC/μL) in combination with mAb158 (at 1 μg/μL) as indicated in the scheme. g, Representative images of brain sections from J20 mice stained for Aβ (red, stained with the D54D2 antibody) and with DAPI (blue; scale bar, 1 mm). h-j, Graphs showing coverage of Aβ (% of brain region) in the h) hippocampus, i) cortex/striatum/amygdala and j) combined regions. Results in h-j are presented as mean ± s.e.m.; n = 8 in eGFP plus mAb158 and n = 10 in mVEGF-C plus mAb158; two-tailed unpaired Student’s T test; data in f-j result from a single experiment. k, Top ten Gene Ontology terms obtained after analyzing significantly up-regulated genes in hippocampi (n = 3 per group) from the eGFP plus mAdu. group when compared to the eGFP plus mIgG group (related to Fig. 2a–c). l, m, Heatmaps depicting the expression profile of genes comprised in the l) learning and memory (GO:0007611) and m) synapse organization (GO:0050808) Gene Ontology pathways (related to Fig. 2d, e). Data in k-m is from a single experiment; Gene Ontology analyses used over-representation test and scale bar in k represents Benjamini-Hochberg adjusted P-values; heatmaps in l and m depict counts-per-million normalized expression minus per-gene mean expression. n, Representation of the tSNE plot highlighting sequenced brain cells by group. o, Dot plot depicting the average scaled expression levels of specific genes (in the x axis) used to identify the brain cell populations, as well as the percentage of gene-expressing cells within each population; smooth muscle cells (SMCs) choroid plexus blood endothelial cells (cpBECs), border-associated macrophages (BAMs), venous BECs (vBECs), arterial BECs (aBECs) and capillary BECs (cBECs). p-s, Volcano plots, with significantly down-regulated (in blue) and up-regulated (in orange) genes, and Gene Ontology terms (obtained using up-regulated genes; selected from top 20 terms) obtained after comparing the transcriptomes of vBECs (p and q) and aBECs (r and s) from the mVEGF-C plus mIgG group and the eGFP plus mIgG group. t, Violin plots showing the expression levels of the Flt4 gene in microglia, capillary, venous and arterial BECs in each group. Data in n-t are related to Fig. 2f–k and resulted from a single experiment where the transcriptomes of 7,739 cells (isolated from brain hemispheres of 3 mice per group) were analyzed, including 2,345 microglia, 2,934 cBECs, 602 vBECs and 766 aBECs; differentially expressed genes plotted in p and r were determined using an F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P-values; Gene Ontology analyses used over-representation test and scale bars in q and s represent Benjamini-Hochberg adjusted P-values for each pathway; gene expression comparison in t was done using Wilcoxon Rank-Sum test with Bonferroni’s P-value adjustment.
Extended Data Figure 9 |. Expression profile of genes associated with Parkinson’s disease, multiple sclerosis and AD in meningeal LECs, brain blood endothelial cells and microglia from different mouse models.
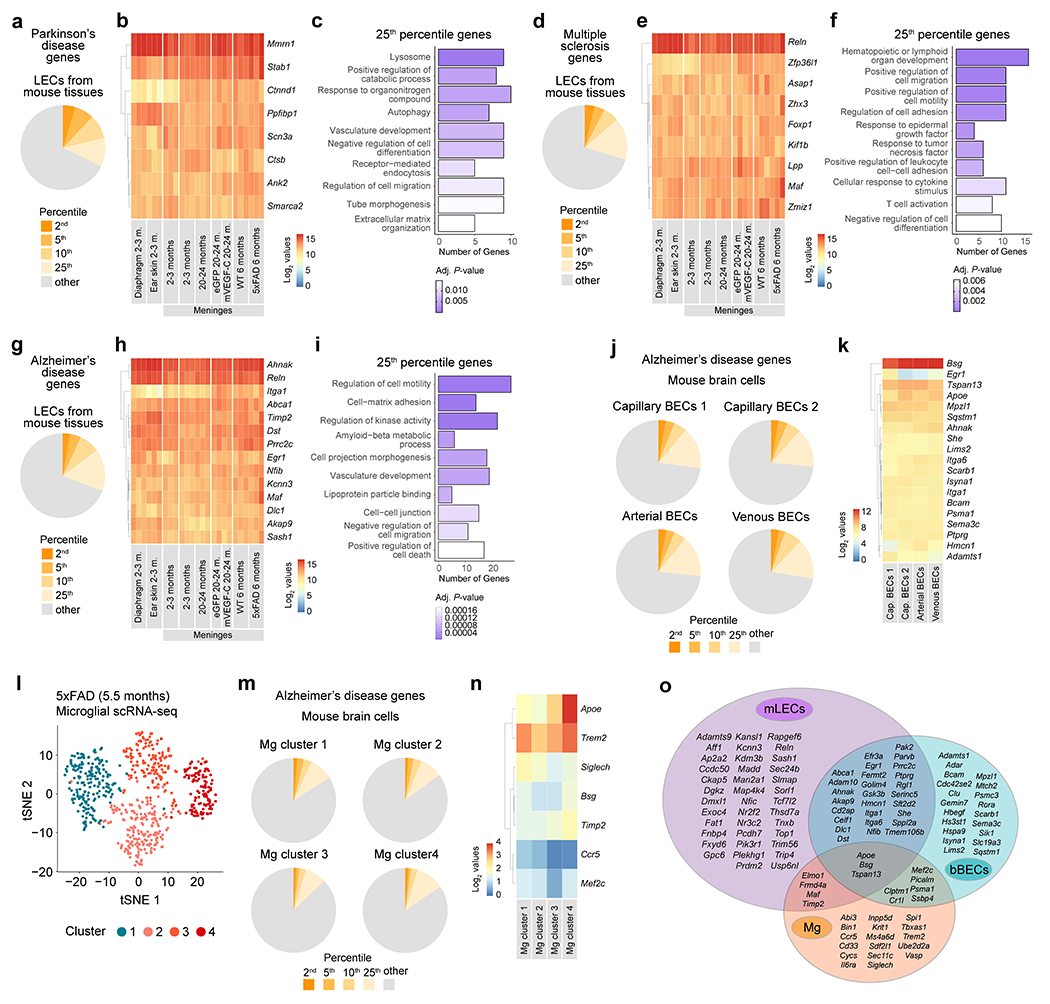
a, Pie chart showing the proportion of Parkison’s disease-associated genes, for which the average expression across all RNA-seq datasets of lymphatic endothelial cells (LECs) was in the top 2nd, 5th, 10th, or 25th percentile out of all genes. b, Heatmap showing the log2-normalized expression values (depicted in the color scale bar) for Parkinson’s disease-associated genes whose average expression values fall within the top 2nd percentile of all genes expressed across all LECs’ RNA-seq datasets. c, Gene-sets obtained by functional enrichment of 25th percentile Parkinson’s disease-associated genes expressed across all LECs’ RNA-seq datasets. d, Pie chart showing the proportion of multiple sclerosis-associated genes, for which the average expression across all LECs’ RNA-seq datasets was in the top 2nd, 5th, 10th, or 25th percentile out of all genes. e, Heatmap showing the log2-normalized expression values (depicted in the color scale bar) for multiple sclerosis-associated genes whose average expression values fall within the top 2nd percentile of all genes expressed across all LECs’ RNA-seq datasets. f, Gene-sets obtained by functional enrichment of 25th percentile multiple sclerosis-associated genes expressed across all LECs’ RNA-seq datasets. g, Pie chart showing the proportion of AD-associated genes, for which the average expression across all LECs’ RNA-seq datasets was in the top 2nd, 5th, 10th, or 25th percentile out of all genes. h, Heatmap showing the log2-normalized expression values (depicted in the color scale bar) for AD-associated genes whose average expression values fall within the top 2nd percentile of all genes expressed across all LECs’ RNA-seq datasets. i, Gene-sets obtained by functional enrichment of 25th percentile AD-associated genes expressed across the different LECs’ RNA-seq datasets. Data in a-i consists of n = 2 or 3 per group; individual RNA samples result from LECs pooled from 10 mice; genes used in a-i resulted from RNA-seq datasets obtained from LECs isolated from diaphragm, ear skin and meninges at 2–3 months (m.), from meninges at 2–3 or 20–24 months, from meninges at 20–24 months after injections with AAV1-CMV-eGFP (eGFP) or AAV1-CMV-mVEGF-C-WPRE (mVEGF-C, one month after i.c.m. injection − see methods for details) and from meninges of 6-month-old WT or 5xFAD mice (see Extended Data Fig. 1f–k for related data); in c, f and i the Benjamini-Hochberg correction was used to adjust the associated P-values (adj. P-values < 0.05) and the functional enrichment of differential expressed genes was determined with Fisher’s exact test. j, Pie charts showing the proportion of AD-associated genes, for which the average expression was in the top 2nd, 5th, 10th, or 25th percentile out of all genes in each cluster of brain blood endothelial cells (BECs): capillary BECs 1, capillary BECs 2, arterial BECs and venous BECs. k, Heatmap showing the expression values for AD-associated genes whose average expression values fall within the top 2nd percentile of all genes expressed in each cluster of BECs. l, The transcriptome of myeloid cells (live CD45+Ly6G−CD11b+ cells) sorted from the brain cortex of 5.5-month-old 5xFAD mice was analyzed by single-cell RNA-seq (scRNA-seq, see methods for more details). The graph shows the unsupervised clustering and tSNE representation of four distinct clusters of microglia (Mg). m, Pie charts showing the proportion of AD-associated genes, for which the average expression was in the top 2nd, 5th, 10th, or 25th percentile out of all genes in each Mg cluster. n, Heatmap showing the expression values for AD-associated genes whose average expression values fall within the top 2nd percentile of all genes expressed in each Mg cluster. o, Venn diagram showing the overlap between AD-associated genes in the top 10th percentile for meningeal LECs (mLECs), brain BECs (bBECs) and Mg. Data in j and k resulted from the analysis of a scRNA-seq dataset published by Vanlandewijck et al; data in l-n resulted from the scRNA-seq analysis of 651 microglia; in k and n, scale bars represent log2-normalized expression values.
Extended Data Figure 10 |. Integrative analyses of gene expression profiles of microglia from the brain of 5xFAD mice with intact or ablated meningeal lymphatics and from the human brain.
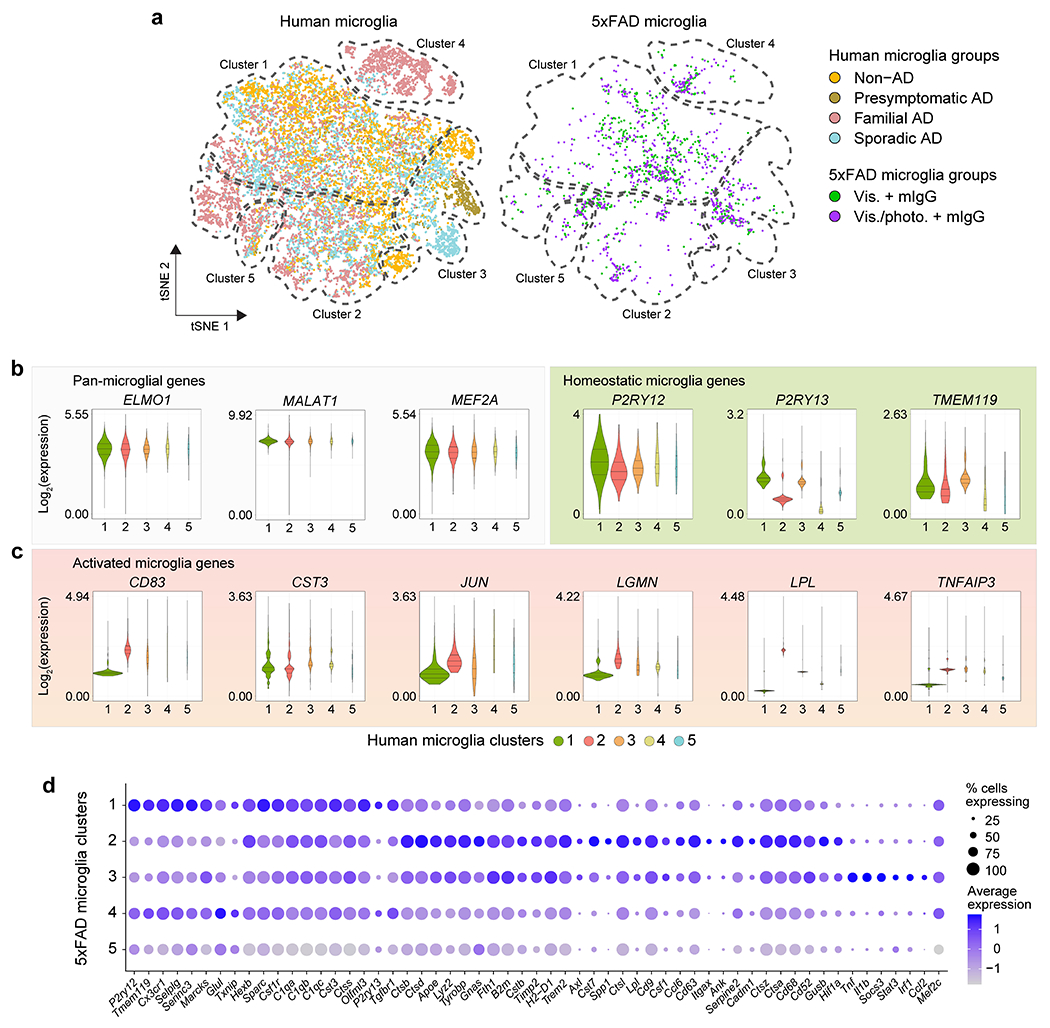
a, Representation of the tSNE plots showing the segregation of microglia from human donors or 5xFAD mice by groups (dashed line represents the approximate boundaries between microglial clusters) upon cross-species RNA-seq data integration and analysis (see also Fig. 3b). b, c, Violin plots with expression levels of the b) pan-microglial genes ELMO1, MALAT1 and MEF2A, homeostatic genes P2RY12, P2RY13 and TMEM119, and c) activation genes CD83, CST3, JUN, LGMN, LPL and TNFAIP3 in the different human microglial clusters. d, Dot plot depicting the average scaled expression levels of specific genes (in the x axis), as well as the percentage of gene-expressing 5xFAD microglia within each cluster. Data in a-d are related to Fig. 3b–g and were obtained from the integrated analysis of a total of 5,462 non-AD, 618 presymptomatic AD, 4,548 familial AD and 6,461 sporadic AD microglia (single-nucleus RNA-seq data) from human brain parietal lobes, and from a total of 781 and 770 microglia (scRNA-seq data) from the brains of 5xFAD mice of the Vis. plus mIgG and the Vis./photo. plus mIgG groups, respectively.
Supplementary Material
Acknowledgements.
We thank S. Smith for editing the manuscript and Susan Blackburn and N. Al-Hamadani for animal colony maintenance. This work was supported by grants from the National Institutes of Health/National Institute on Aging (AG034113 and AG057496), PureTech Health, the Cure Alzheimer’s Fund and the Ed Owens Family Foundation to J. K.; RF1AG053303 and RF1AG058501 to C.C.; AG057777 and AG067764 to O.H.; and AG062734 to C.M.K. O.H. is an Archer Foundation Research Scientist. We thank all the members of the Kipnis laboratory for their valuable comments during numerous discussions of this work. We acknowledge the staff of the Neuropathology Cores and other personnel of the Charles F. and Joanne Knight Alzheimer Disease Research Center (P30 AG066444, P01AG026276, P01AG03991). Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer’s Network (DIAN, UF1AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI). This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the Knight ADRC and DIAN (see full list of DIAN consortium members in Supplementary Notes) research and support staff at each of the participating sites for their contributions to this study.
Competing interests.
J.K. is a member of a scientific advisory group for PureTech. J.K., S.D.M. and A.L. are holding patents and patent applications related to the findings described herein. D.M.H. is an inventor on a patent for anti-Aβ antibodies licensed to Eli Lilly by Washington University. D.M.H. and H.J. are inventors on a patent for anti-apoE antibodies licensed to NextCure. D.M.H. and H.J. are listed as inventors on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies. D.M.H. co-founded and is on the scientific advisory board of C2N Diagnostics. C2N Diagnostics has licensed certain anti-tau antibodies to AbbVie for therapeutic development. D.M.H. is on the scientific advisory board of Denali and consults for Genentech, Merck, and Cajal Neuroscience. C.C. receives research support from Biogen, EISAI, Alector and Parabon. The funders of the study had no role in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to submit the paper for publication. C.C. is a member of the advisory board of Vivid genetics, Halia Therapeutics and ADx Healthcare. J.P.C. has served on a medical advisory board for Otsuka Pharmaceuticals. The authors declare no other competing interests.
Footnotes
Extended data is linked to the online version of the paper at www.nature.com/nature.
Supplementary information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Jack CR Jr. et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562, doi: 10.1016/j.jalz.2018.02.018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mawuenyega KG et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774, doi: 10.1126/science.1197623 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarasoff-Conway JM et al. Clearance systems in the brain--implications for Alzheimer diseaser. Nat Rev Neurol 12, 248, doi: 10.1038/nrneurol.2016.36 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Sweeney MD, Sagare AP & Zlokovic BV Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14, 133–150, doi: 10.1038/nrneurol.2017.188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Da Mesquita S et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560, 185–191, doi: 10.1038/s41586-018-0368-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Da Mesquita S, Fu Z & Kipnis J The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron 100, 375–388, doi: 10.1016/j.neuron.2018.09.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker S et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis 43, 575–588, doi: 10.3233/JAD-140741 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kastanenka KV et al. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J Neurosci 36, 12549–12558, doi: 10.1523/JNEUROSCI.2080-16.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logovinsky V et al. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther 8, 14, doi: 10.1186/s13195-016-0181-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sevigny J et al. Addendum: The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 546, 564, doi: 10.1038/nature22809 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Howard R & Liu KY Questions EMERGE as Biogen claims aducanumab turnaround. Nat Rev Neurol, doi: 10.1038/s41582-019-0295-9 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Louveau A et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 21, 1380–1391, doi: 10.1038/s41593-018-0227-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rustenhoven J et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016 e1027, doi: 10.1016/j.cell.2020.12.040 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruchaga C et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505, 550–554, doi: 10.1038/nature12825 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamazaki Y, Zhao N, Caulfield TR, Liu CC & Bu G Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15, 501–518, doi: 10.1038/s41582-019-0228-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong M et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci Transl Med 13, doi: 10.1126/scitranslmed.abd7522 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbasi J Promising Results in 18-Month Analysis of Alzheimer Drug Candidate. JAMA 320, 965, doi: 10.1001/jama.2018.13027 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341, doi: 10.1038/nature14432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu JK et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol 19, 1212–1223, doi: 10.1038/s41590-018-0232-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L et al. Deep cervical lymph node ligation aggravates AD-like pathology of APP/PS1 mice. Brain Pathol 29, 176–192, doi: 10.1111/bpa.12656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217, doi: 10.1016/j.cell.2017.05.018 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569, doi: 10.1016/j.immuni.2017.08.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong S et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716, doi: 10.1126/science.aad8373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montagne A et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76, doi: 10.1038/s41586-020-2247-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480, doi: 10.1038/nature25739 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142, doi: 10.1038/s41591-019-0695-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078, doi: 10.1016/j.cell.2015.10.067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uhlmann RE et al. Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat Neurosci, doi: 10.1038/s41593-020-00737-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naj AC et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436–441, doi: 10.1038/ng.801 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012, doi: 10.1093/nar/gky1120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Yan Q et al. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry 26, 309–321, doi: 10.1038/s41380-018-0246-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deming Y et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med 11, doi: 10.1126/scitranslmed.aau2291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deming Y et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 133, 839–856, doi: 10.1007/s00401-017-1685-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang KL et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20, 1052–1061, doi: 10.1038/nn.4587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430, doi: 10.1038/s41588-019-0358-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del-Aguila JL et al. Assessment of the Genetic Architecture of Alzheimer’s Disease Risk in Rate of Memory Decline. J Alzheimers Dis 62, 745–756, doi: 10.3233/JAD-170834 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merlini M et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 101, 1099–1108 e1096, doi: 10.1016/j.neuron.2019.01.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spangenberg EE et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 139, 1265–1281, doi: 10.1093/brain/aww016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216, 2546–2561, doi: 10.1084/jem.20190980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tinevez JY et al. TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90, doi: 10.1016/j.ymeth.2016.09.016 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Finck R et al. Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494, doi: 10.1002/cyto.a.22271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine JH et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197, doi: 10.1016/j.cell.2015.05.047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419, doi: 10.1038/nmeth.4197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774, doi: 10.1101/gr.135350.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soneson C, Love MI & Robinson MD Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521, doi: 10.12688/f1000research.7563.2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287, doi: 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu G, Wang LG, Yan GR & He QY DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31, 608–609, doi: 10.1093/bioinformatics/btu684 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hong G, Zhang W, Li H, Shen X & Guo Z Separate enrichment analysis of pathways for up- and downregulated genes. J R Soc Interface 11, 20130950, doi: 10.1098/rsif.2013.0950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCarthy DJ, Campbell KR, Lun AT & Wills QF Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186, doi: 10.1093/bioinformatics/btw777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lun AT, Bach K & Marioni JC Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol 17, 75, doi: 10.1186/s13059-016-0947-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van den Berge K et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol 19, 24, doi: 10.1186/s13059-018-1406-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durinck S et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440, doi: 10.1093/bioinformatics/bti525 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Durinck S, Spellman PT, Birney E & Huber W Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191, doi: 10.1038/nprot.2009.97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Leeuw C, Sey NYA, Posthuma D & Won H A response to Yurko et al: H-MAGMA, inheriting a shaky statistical foundation, yields excess false positives. bioRxiv, 2020.2009.2025.310722, doi: 10.1101/2020.09.25.310722 (2020). [DOI] [Google Scholar]
- 61.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337, doi: 10.1038/s41586-019-1195-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324, doi: 10.1016/j.cels.2019.03.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296, doi: 10.1186/s13059-019-1874-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Del-Aguila JL et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 11, 71, doi: 10.1186/s13195-019-0524-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pliner HA, Shendure J & Trapnell C Supervised classification enables rapid annotation of cell atlases. Nat Methods 16, 983–986, doi: 10.1038/s41592-019-0535-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sul JH et al. Accurate and fast multiple-testing correction in eQTL studies. Am J Hum Genet 96, 857–868, doi: 10.1016/j.ajhg.2015.04.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duong D et al. Using genomic annotations increases statistical power to detect eGenes. Bioinformatics 32, i156–i163, doi: 10.1093/bioinformatics/btw272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Darnell G, Duong D, Han B & Eskin E Incorporating prior information into association studies. Bioinformatics 28, i147–153, doi: 10.1093/bioinformatics/bts235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eskin E Increasing power in association studies by using linkage disequilibrium structure and molecular function as prior information. Genome Res 18, 653–660, doi: 10.1101/gr.072785.107 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nettleton D & Doerge RW Accounting for variability in the use of permutation testing to detect quantitative trait loci. Biometrics 56, 52–58, doi: 10.1111/j.0006-341x.2000.00052.x (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.