

Abstract
Foxp3+Tregs, CD4+Foxp3− and CD8+ T cells are composed of naïve (NP) and memory (MP) subsets. 10–20% of each MP T cell population are cycling (Ki-67+) in vivo. We investigated the contribution of co-stimulatory (CD28), and co-inhibitory (CTLA-4, PD-1) receptors on MP T cell homeostatic proliferation in vivo in the mouse. Blockade of CD28-CD80/86 signaling completely abolished MP Treg and profoundly inhibited MP CD4+Foxp3− T cell proliferation, but did not affect MP CD8+ T cell proliferation. Marked enhancement of homeostatic proliferation of MP Treg and MP CD4+Foxp3− T cells was seen after blocking CTLA4-CD80/CD86 interactions and PD-1-PD-L1/2 interactions and greater enhancement was seen with blockade of both pathways. The CD28 pathway also played an important role in the expansion of Treg and MP T cells after treatment of mice with agonistic antibodies to members of the TNF receptor superfamily which can act directly (anti-GITR, -OX40, -4-1BB) or indirectly (anti-CD40) on T cells. Induction of a cytokine storm by blocking the interaction of NK inhibitory receptors with MHC-Class I had no effect on Treg homeostasis, enhanced MP CD4+ proliferation and expansion in a CD28-dependent manner, but enhanced MP CD8+ T cell proliferation in a CD28-independent manner. As MP T cells exert potent biologic effects primarily before the induction of adaptive immune responses, these findings have important implications for the use of biologic agents designed to suppress autoimmune disease or enhance T effector function in cancer which may have negative effects on MP T cells.
Introduction
Conventional CD4+ (CD4+Foxp3−) T cells can be divided into naïve/resting phenotype (NP, CD44−CD62L+) and memory/effector (MP, CD44+CD62L−) populations. MP CD4+ T cells MP cells can be divided into pathogen-specific authentic memory cells and pathogen-independent MP cells (1). It is likely that most MP CD4+ cells develop in the absence of foreign antigen recognition and have distinct functions independent of antigenic stimulation. CD8+ T cells are more heterogeneous and are characterized as naïve (CD44− CD62L+), central memory (CM, CD44+CD62L+) and effector memory (EM, CD44+CD62L−) subtypes. A subpopulation of CD8+ MP T cells has been termed virtual memory T cells which express memory markers but are antigenically naïve (2). It has been proposed that this subpopulation of MP CD8+ T cells is generated by high affinity self-peptide interactions during thymic development (3). While the level of expression of CD44 can also distinguish NP and MP Treg, the GPI-linked surface marker Ly-6C has also proven to be useful to discriminate Tregs into NP (Ly-6C+, 20–30%) and MP (Ly-6C−,70–80%) subpopulations (4, 5) The Ly-6C− MP Treg cells are characterized by increased CD3ζ/TCR signaling, pAKT/mTOR, and NFAT/STAT5 signaling compared to NP Ly-6C+ Treg subset (4). Two studies in which the TCR was deleted from peripheral Treg demonstrated that MP Treg were specifically deleted while NP Treg maintained Foxp3 expression, and that deletion of MP Treg was accompanied by loss of Treg suppressor activity implying that MP Treg were responsible for Treg suppressor function in the steady state (6, 7).
One of the major characteristics of all MP T cells populations is that they are highly proliferative in vivo with ~10% dividing in a 24 h period based on Ki-67 staining and BrdU incorporation studies (8). Proliferation is balanced by an equivalent degree of cell death as the percentage and absolute numbers of the MP subpopulations remain constant over a period of weeks to months (9). The factors that drive and regulate the proliferation of MP T cells in the steady state in the absence of stimulation by exogenous antigen remain poorly characterized. .
While it is widely accepted that the proliferation of MP CD8+ T cells is cytokine driven (IL-7 and IL-15, (10)). MP CD4+ T cell proliferation was only modestly reduced when mice were treated with anti-IL-7 and not affected by treatment with anti-IL-15 (8). We and others have recently demonstrated (5, 11) that short-term treatment of mice with CTLA-4-Ig to block CD28/CD80-CD86 interactions reduced, but did not eliminate, MP CD4+ and Treg T cell cycling and total cell numbers, but had no effect on MP CD8+ T cell cycling or numbers. Administration of anti-CTLA-4 and anti-TCR mAbs induced proliferation of all three MP subsets. We concluded from these studies that CD28-driven signals are the main drivers of Treg and MP CD4+ T cells proliferation in vivo and that the CD28 signals are restrained in a complex manner by a combination of inhibitory signals mediated by engagement of CTLA-4 by CD80/CD86 and by MHC-II/TCR interactions (5).
In the present report, we re-examine the role of CD28/CD80-CD86 interactions in MP T cell homeostasis using more potent inhibitors of the interaction and demonstrate the MP Treg proliferation and accumulation are completely dependent on CD28-driven signals, MP CD4+ T cells are partially dependent, while MP CM and EM CD8+ T cells are completely independent. ICOS plays no role in MP T cell proliferation in the steady state, but in CD28 deficient (CD28−/− or CD80/86−/−) mice that have very few Treg, anti-ICOS substantially inhibits Treg and MP CD4+ proliferation. While CTLA-4 appears to be the main controller of MP Treg and CD4 MP proliferation and accumulation, anti-PD-1 treatment also enhanced MP Treg and MP CD4+ proliferation/accumulation and that combined treatment with anti-PD-1 and anti-CTLA-4 produced greater enhancement than either mAb alone suggesting that the pathways by which CTLA-4 and PD-1 restrain homeostasis are distinct. In addition to factors that directly mediate MP Treg and MP CD4+ and CD8+ T cell proliferation in the steady state, we also examined the role of the CD28 pathway in Treg and MP expansion observed after treatment of mice with IL-2 immune complexes and agonistic antibodies to members of the TNF receptor superfamily which are capable of acting directly (anti-GITR, -OX40, -4-1BB) or indirectly (anti-CD40) on T cells. Surprisingly, many of the effects of these agonistic mAbs were also dependent on CD28-driven co-stimulatory signals. Lastly, we evaluated the effects of a complex global activation of the immune system induced by blockade of MHC-I/Ly-49 interactions (12) which results in cytokine storm including the release of Th1 cytokines, APC-derived cytokines (IL-12, IL-15 and IL-18) and bystander proliferation of MP T cells. Curiously, under these conditions, Treg homeostasis was completely unaffected, while MP CD4+ proliferation was enhanced by CD28-driven signals, and MP CD8+ T cells proliferation enhanced by a CD28-independent pathway.
Taken together, these studies demonstrate that the homeostasis of MP T cells subsets is regulated by different pathways in the steady state. Furthermore, agonistic mAbs which have been used as biotherapeutics to enhance immune responses primarily for the treatment of malignancy can not only act directly on MP T cells, but their action can be potently modulated by CD28 driven signals. These findings have major implications for the use of these agents as biotherapeutics.
Materials and Methods
Mice
C57BL/6 mice were purchased from Charles River Laboratories. FcγR−/− mice obtained from Taconic farms. CD28−/−, CD80/CD86−/−, CD40−/− mice were procured from Jackson Laboratories. All mice were sex- and age-matched for experimentation and used between 7 and 12 wk of age. All animal protocols used in this study were approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee.
In vivo mAb treatment
C57BL/6 mice were injected with following mAbs as indicated in figure legends: 250 μg/dose mouse IgG1 (Clone: MOPC-21; BioXcell), mouse IgG2a (Clone: C1.18.4), Rat-IgG2a (Clone: 2A3; BioXcell), of human CD28 domain specific antibody (100 μg/dose; Bristol-Myers Squibb), 250 μg/dose of anti-CD80 (Clone: 1G10; BioXcell), anti-CD86 (Clone: GL-1; BioXcell) CTLA4-Ig (Abatacept, Bristol-Myers Squibb), Anti-ICOS (Clone: 7E.17G9; BioXcell), Anti-CTLA4 (Clone UC10-4F10-11; Bio X Cell); Anti-PD1(Clone: 29F.1A12; BioXcell), TIM-3(Clone: RMT3.23; BioXcell), TIGIT (Clone:1G9; BioXcell), BTLA (Clone:6A6, BioXcell), VISTA(Clone: 13F3; BioXcell), CD70(Clone: FR70; BioXcell), CD48(Clone: HM48.1; BioXcell), TNF-α (Clone: XT3.11; BioXcell), IFNγ (Clone: XMG1.2; BioXcell), IFNAR (Clone:MAR1–5A3; BioXcell), IFNγR (Clone:GR20), IL-7 (Clone:M25; BioXcell), IL-7Rα (Clone:A7R34; BioXcell), CD40 (50 μg/dose; Clone:FGK4.5; BioXcell, CD40L (MR-1; BioXcell) Clone:, GITR (Clone:DTA-1; BioXcell), OX40 (Clone:OX-86; BioXcell), 4-1BB (Clone:LOB12.3; BioXcell), and Anti-MHC-I (500 μg/dose; Clone: M1/42; BioXcell).
Lymphocyte isolation
Spleens were harvested from wildtype or knockout animals on d8 after antibody treatments. Intact spleens were homogenized using a cell strainer (70 μm, BD Falcon, USA). Red blood cells were lysed using sterile ACK lysing buffer (Gibco, Invitrogen). Lymphocytes were washed, suspended in sterile complete medium [RPMI medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine (2 mM), sodium pyruvate (1 mM), HEPES (1 mM), non-essential amino acids (0.1 mM), 2-mercaptoethanol (50 μM), and penicillin and streptomycin (100 U/ml)], and total live cells were counted by hemocytometer.
Flowcytometry
For surface staining, cells in staining buffer (PBS, 10% heat-inactivated FBS, and 0.05% sodium azide) were incubated with directly labeled mAbs for 30 mins at 4°C. For intracellular Foxp3 and Ki-67 staining, fixation and permeabilization were done according to the manufacturer’s guidelines (Foxp3 transcription factor buffer set, eBioscience). Phospho-flow staining was performed by adding fluorescence conjugated antibodies against pAKT, mTOR, STAT5 etc. (BD Bioscience and Cell Signaling Technology) according to manufacturer’s instructions (BD phosflow).
Anti-IL-2 and IL-2/Anti-IL-2 complex treatments
To prepare IL-2/anti–IL-2 mAb complexes, recombinant murine IL-2 (1 mg; PeproTech) was mixed with either S4B6 or JES6-1 (5 mg) at the optimal 1:2 molar ratio and incubated at 37°C for 10 min a room tempaerature. IL2:S4B6 or IL2:JES6 alone or together with anti-CD28 dAb were injected i.p. on day 0, 1, 2 and 4 and animals were sacrificed on day 5.
Cytometric Bead Array
Mouse whole blood was collected in a heparinized tube. After centrifugation at 2000g for 15 min, plasma was transferred to a new tube. Concentrations of cytokines were determined by Cytometric Bead Array Mouse Th1/Th2/Th17 kit (BD Biosciences) following the manufacturer’s instructions.
Statistics
Statistical analysis Comparisons between groups were tested by a two-tailed unpaired Student t test or one-way ANOVA using Prism 9 (GraphPad Software). A p value 0.05 was considered significant.
Results
CD28/CD80-CD86 signaling regulates cell cycling of MP Treg and MP CD4+, but not MP CD8+ T cells in vivo
We previously demonstrated that the cycling of MP Treg (Ly-6C−) and MP CD4+ T cells in vivo was largely dependent on CD28 driven stimulation as cycling was markedly inhibited by treatment of animals with human CTLA-4Ig (Abatacept)(5). However, CTLA-4 blockade only reduced proliferation to ~70% raising the possibility that other co-stimulatory pathways or cytokines could drive cell cycling in vivo. To more rigorously examine the role of the CD28 pathway in cell cycling in vivo, we blocked the CD28 pathway with an antagonistic anti-CD28 domain antibody, anti-CD28 dAb (13, 14) or high concentrations of a combination of anti-CD80/CD86 mAbs. In contrast to CTLA-4Ig which produced a partial inhibition of Treg cell cycling as defined by Ki-67 expression, treatment of mice with the anti-CD28 dAb or anti-CD80/CD86 completely abolished MP Treg proliferation (Fig. 1A, B) and markedly reduced the total number of Treg recovered on d8 (Fig. 1C). The effects of blocking CD28 signaling were seen in both lymphoid and non-lymphoid sites (Fig. 2, A–I). The effects of the mAb persisted for 21 d after cessation of mAb treatment, but all parameters returned to normal by d 36 consistent with the half-life of rat mAbs (Fig. 2, I–P). Taken together, these results strongly suggest that CD28 driven co-stimulation plays the primary role in mediating Treg homeostatic proliferation in vivo in a cytokine-independent manner. Ly-6C− Treg have been shown to be activated in vivo as manifest by elevated levels of pAKT, NFAT, NFκB, pSTAT5, and the mTOR pathway(4). Inhibition of CD28 signaling profoundly inhibited all of these pathway most likely secondary to a marked decrease in the total number of activated Ly-6C− Treg during the 8 day treatment period (Supplementary Fig. S1).
Figure 1.
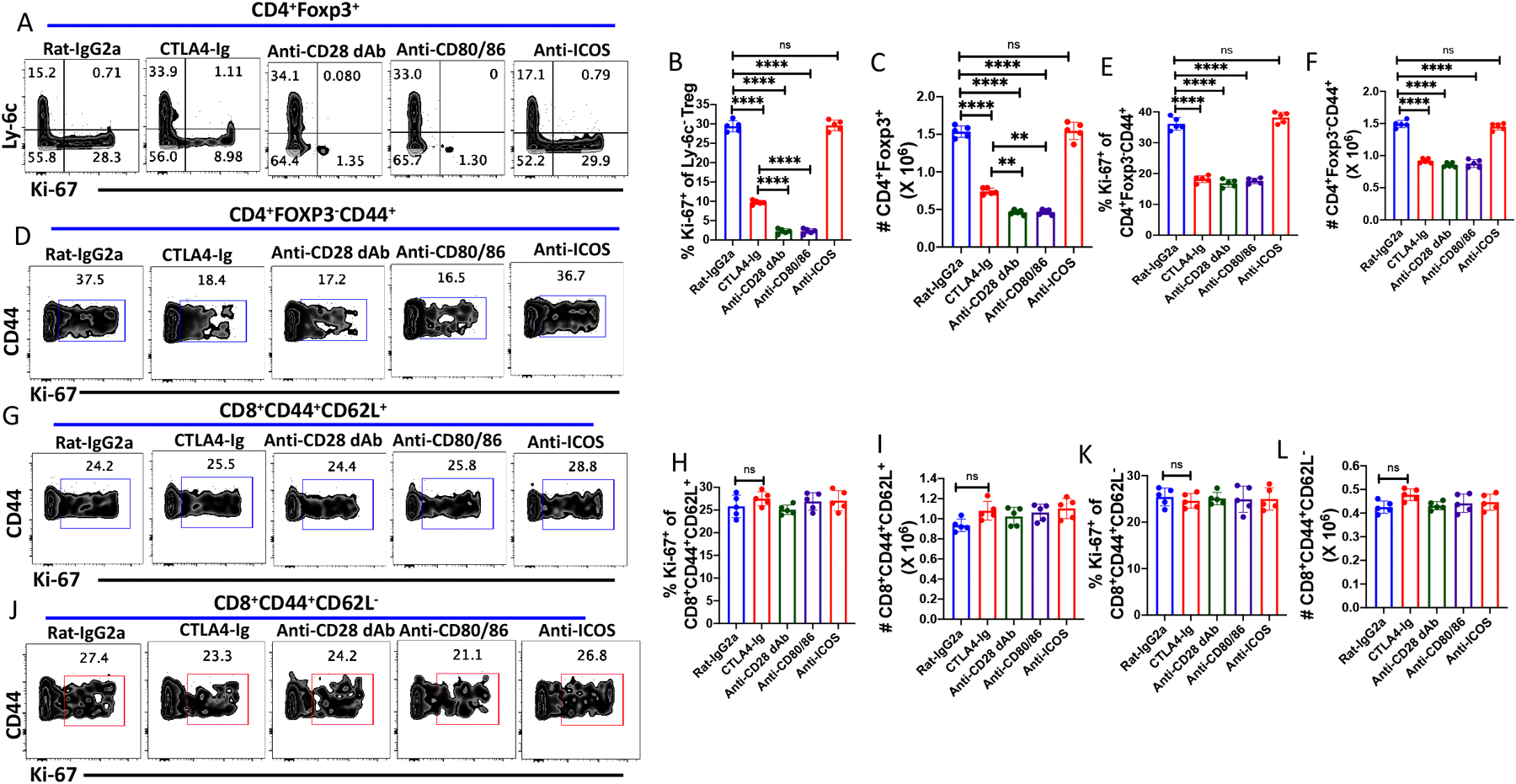
Steady state cycling of MP Treg and CD4+ MP T cells requires CD28 signaling in vivo. WT C57BL/6 mice were treated with Rat-IgG2a, anti-CD28 dAb, anti-CD80/86, CTLA4-Ig (abatacept) or anti-ICOS (250 μg/dose) on d 0, 2, 4 and 6 and spleens were harvested on day 8. (A) Representative plot of Ly-6C versus Ki-67 expression on splenic Tregs. (B) Percentage of Ly6C− Treg cells expressing Ki-67 after antibody treatments. (C) Absolute number of splenic CD4+Foxp3+ T cells after antibody treatments. (D) Representative plot of Ki-67 expression on CD4+Foxp3−CD44+ cells after antibody treatments. (E) Percentage of CD4+Foxp3−CD44+ MP T cells expressing Ki-67. (F) Absolute number of CD4+ MP T cells after antibody treatments. (G) Representative plot of Ki-67 expression on CD8+CM T cells. (H) Percentage of CD8+CD62L+ expressing Ki-67.(I) Absolute number of CD8+CM T cells after antibody treatments. (J) Representative plot of the percentage of CD8+EM cells expressing Ki-67. (K) Percentage of CD8+ EM cells expressing Ki-67 (L) Absolute number of CD8+ EM T cells after antibody treatments. (A-L) represent the results of one experiment using 2–5 mice per group.
Figure 2.
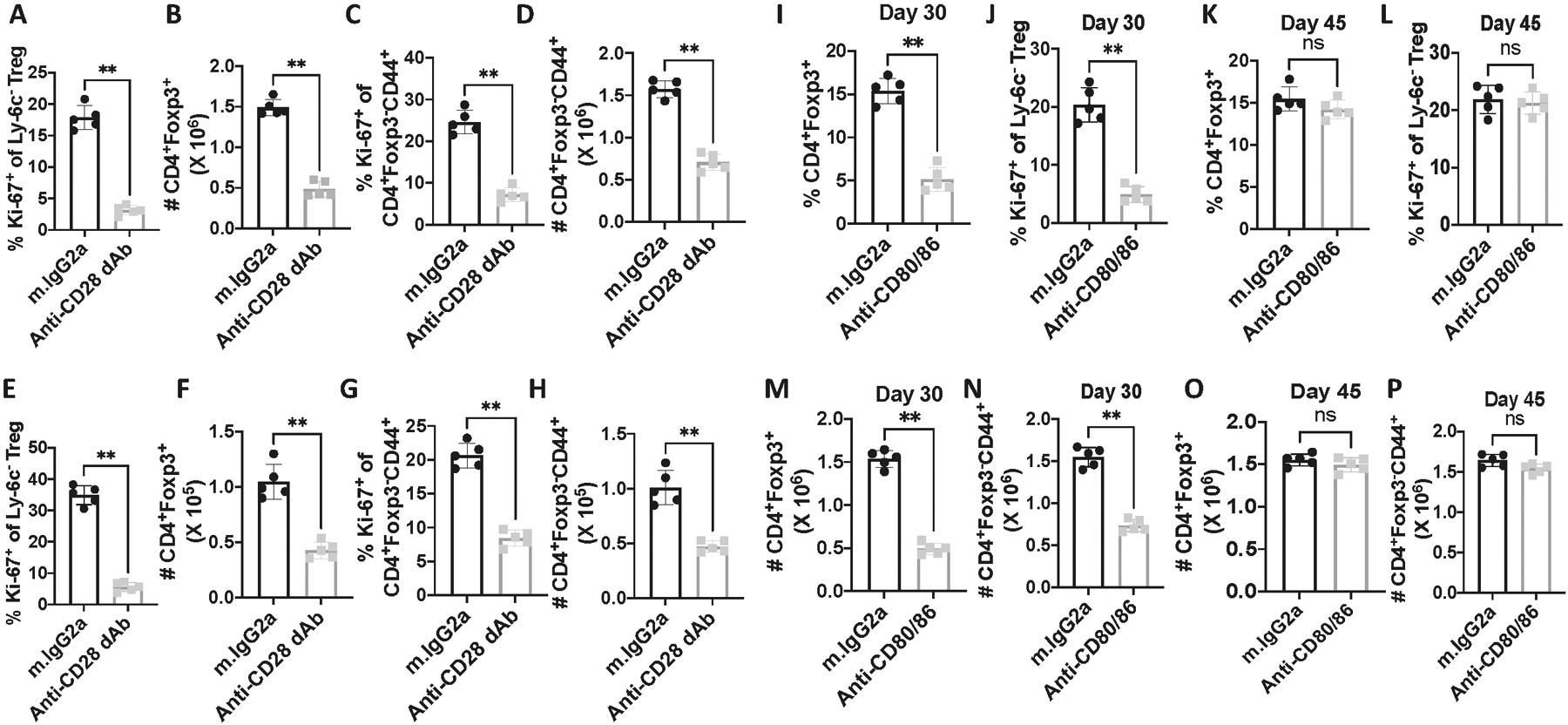
CD80/CD86 signaling is critical for CD4+ MP and Treg cell homeostasis in both lymphoid and non-lymphoid sites, but the effects of mAb treatment are reversible. WT C57BL/6 mice were injected with either Rat-IgG2a or anti-CD28 dAb every other d for 6d, and lymphocytes from mesenteric lymph nodes (A-D) and liver (E-H) were harvested on d8. (A) Ki-67 expression on Ly-6C−, (B) Absolute number of Tregs on d8, (C) Ki-67 expression on CD4+Foxp3−CD44+ T cells, (D) Absolute number of CD4+Foxp3−CD44+ T cells. (E) Ki-67 expression on Ly-6C− Treg, (F) Absolute number of Tregs, (G) Ki-67 expression on CD4+Foxp3−CD44+ T cells, (H) Absolute number of CD4+Foxp3−CD44+ T cells. (I-P) C57BL/6 mice were injected Rat IgG2a or anti-CD80/CD86 on d0, d2, d4, and d6. Splenic cells were harvested on d30 or d45. (I & J) Percentage of CD4+Foxp3+ Treg and Ki-67 expression on Ly-6C− Treg on d30. (K & L) Percentage of Treg and Ki-67 expression on Ly-6C− Tregs on d45. (M & N) Absolute number of Treg and CD4+Foxp3−CD44+ T cells on d30 (O & P) Absolute number of Treg and CD4+Foxp3−CD44+ T cells on d45. *p, 0.05; **p; 0.005; ***p, 0.0005, ****p, 0.00005.
In contrast to the results with MP Treg, blockade of CD28 signaling by all the reagents tested only partially inhibited the proliferation, and the absolute number of CD4+ MP T cells recovered (Fig. 1D–F). Notably, blocking CD80-CD86/CD28 interactions by any of the inhibitory reagents had minimal or no impact on MP CM (Fig.1G–I) or EM (Fig. 1J–L) CD8+ T cell proliferation or recovery. As some studies have suggested that ICOS/ICOS-L interactions may control T cell homeostasis in vivo (15), we also administered anti-ICOS to normal mice. Anti-ICOS had no effect on the proliferation or the recovery of Treg, MP CD4+, or MP CD8+ T cell subsets in CD28 sufficient WT animals (Fig. 1A–H). Curiously, although CD28−/− or CD80/CD86−/− mice have a 90% reduction of the total number of Treg, the percentage of cycling (Ki-67+) Treg cells and MP CD4+Foxp3− T cells is similar to that seen in WT mice. Anti-ICOS treatment significantly inhibited the proliferation of both MP Treg and MP CD4+ MP T cells in the absence of CD28 signaling, but had no effect on MP CD8+ T cell proliferation (Supplementary Fig. 1B–E).
Anti-CTLA-4 and Anti-PD-1 co-treatment enhance MP Treg and MP CD4+ T cell proliferation in a CD28-dependent manner
We have previously demonstrated that treatment of mice with anti-CTLA-4 resulted in a significant expansion in the frequency and absolute number of MP Tregs and MP CD4+ T cells, but minimal change in the absolute numbers of CD8+ T cells. We next compared the effects of anti-PD-1 on Treg and MP T cell compartments. Anti-PD-1 alone or in combination with anti-CTLA-4 were administrated to naïve mice every other d for 6 d, and mice were euthanized on d8. Both anti-PD-1 and anti-CTLA-4 enhanced the frequency of proliferating and the absolute numbers of Treg (Fig. 3A–B, Supplementary Fig. 2A), but the effects of anti-CTLA-4 treatment resulted in a greater stimulation of proliferation as assayed by Ki-67 expression of Ly-6C− Treg than anti-PD1 (Fig. 3A, Supplementary Fig. 2A). Simultaneous treatment with anti-CTLA-4 and anti-PD-1 enhanced the total number of Treg compared to a single treatment suggesting that CTLA-4 and PD1 inhibit Treg homeostasis by two distinct pathways (Fig. 3B). The enhanced proliferation and increase in absolute number of Treg cells after treatment with anti-CTLA-4, anti-PD1, singly or together, were completely abolished when the mice were co-treated with anti-CD28 dAb demonstrating that CD28 signaling drives Treg enhancement in the steady state as well as in the presence of co-inhibitory receptor blockade (Fig. 3A, B, Supplementary Fig. 2A).
Figure 3.
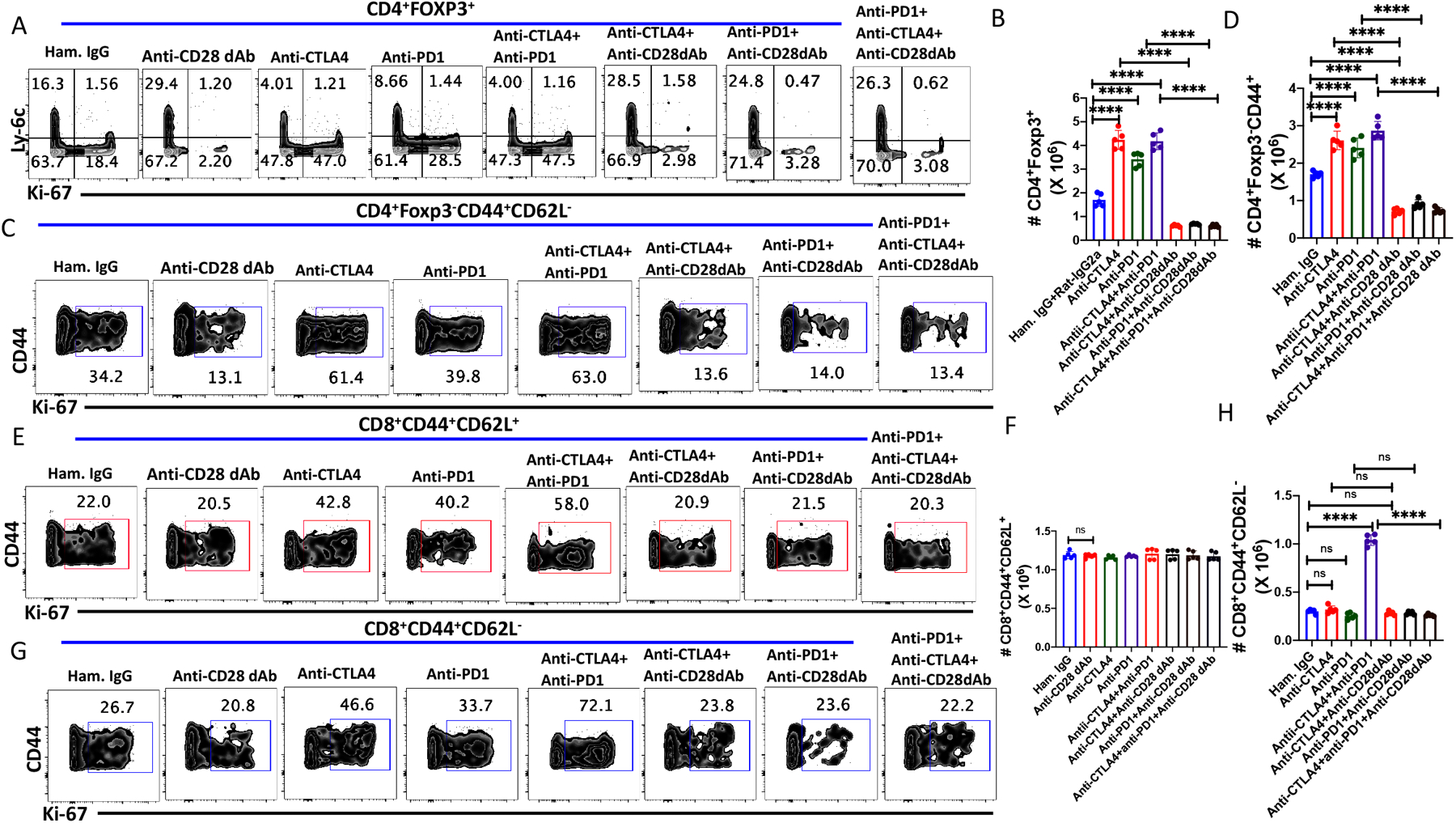
Co-treatment with anti-CTLA-4 and anti-PD-1 enhances CD28-dependent proliferation of MP Treg and CD4+ MP T cells in vivo. WT C57BL/6 mice were injected with either anti-CTLA4 (250 μg/dose), anti-PD1 (250 μg/dose) or both or cotreated with anti-CD28 dAb (100ug/dose) every other d for six d and splenocytes were harvested on d 8. (A) Representative plots of Ly-6C versus Ki-67 expression among splenic Tregs. (B) Absolute number of splenic CD4+Foxp3+ cells on d 8 after antibody treatments. (C) Representative plots of Ki-67 expression on CD4+Foxp3−CD44+ MP cells after antibody treatments on day 8. (D) Absolute number of CD4+ MP T cells after antibody treatments. (E) Representative plots of Ki-67 expression on CD8+ CM T cells. (F) Absolute number of CD8+CM T cells after antibody treatments. (G) Representative plot of Ki-67 expression on CD8+ EM T cells. (H) Absolute number of CD8+ EM T cells after antibody treatments. (A-H) represent the result of one experiment of two using five mice per group.
As shown previously (5), both anti-CTLA-4 and anti-PD-1 enhanced MP CD4+ proliferation (Fig. 3C, Supplementary Fig. 2B) and absolute numbers (Fig. 3D), but the magnitude of the enhancement was less marked than that seen on Treg. Co-administration of anti-CTLA-4 and anti-PD-1 resulted in an increase in the absolute number of MP CD4+ T cells greater than that seen with either mAb alone (Fig. 3D). The enhancement of MP CD4+ T cell homeostasis by blocking CTLA-4 and PD-1 was reversed by anti-CD28 dAb, but the magnitude of inhibition was less than that seen with similarly treated Treg. Anti-PD-1, anti-CTLA-4, or the combination of anti-CTLA-4 and anti-PD1 enhanced CM and EM CD8+ T cell proliferation (Fig. 3 E, G). While the absolute number of CM CD8+ cells remained unchanged (Fig. 3F), co-treatment with anti-CTLA-4 and anti-PD-1 markedly enhanced the absolute numbers of EM CD8+ T cell recovered (Fig. 3H). The enhancement, but not the homeostatic, proliferation of CM and EM CD8+ proliferation and was inhibited by anti-CD28 dAb.
Taken togther, these studies demonstrate that CTLA-4 and to a somewhat lesser extent PD-1 play a major role in restraining CD28 driven Treg, MP CD4+ and MP CD8+ homeostasis. To examine the potential contribution of other co-inhibitory receptors in MP T cell homeostasis, we treated mice with anti-BTLA, anti-TIM3, anti-TIGIT (agonistic), anti-VISTA, anti-CD48, anti-CD70. In general, none of these reagents had any effects on MP Treg and MP CD4+ and MP CD8+ T cell homeostasis as assayed by Ki-67 expression or absolute numbers (Supplementary Fig. 2-E–H).
Effects of IL-2 on MP T Cell homeostasis
Cytokines are crucial factors in T cell differentiation and effector function. However, the role of cytokines in MP T cell homeostasis remains poorly defined. IL-2 is an essential cytokine for Th1 and Th2 differentiation (16), the optimal function of Treg cells (17) and for facilitating the induction of Treg in the presence of TGFβ (18). Targeted manipulation of IL-2 signaling on Treg and MP CD4+ and MP CD8+ T cells is an active area of study for immunotherapy of autoimmunity and enhancement of immune responses to tumors (19, 20);. The primary effects of IL-2 deprivation in vivo were manifest as a decrease in the absolute numbers of resting or naïve Ly-6C+ (5) or CD25hi Treg (15), while the homeostatic cycling of the MP (or effector) Treg (Ly-6C−) was only slightly decreased. Here, we have examined the effects of enhancement of IL-2 signalling on Treg and MP T cell phenotype in vivo. Given that our studies have shown that CD28 signaling plays a major role in Treg and MP CD4+ T cells homeostasis in vivo (Fig. 1), we have also addressed whether IL-2 activation of Treg, MP CD4+, and MP CD8+ T cells operates independently, or whether CD28 costimulation is crucial for optimal IL-2 function in vivo. Mice were treated (d0, 1, 2, and 4 and euthanized on d5) with anti-IL-2 or IL-2/anti-IL-2 immune complexes that target the IL-2R β-chain signaling (IL-2/S4B6) or IL-2R α-chain signaling (IL-2/JES6) alone or in the presence of anti-CD80/CD86.
The administration of IL-2/S4B6 complex enhanced the proliferation of Treg, but had only a modest effect on the total number of Treg recovered (Fig 4A, B. Supplementary Fig. 3A). The IL-2/JES6 complex had a more potent effect than IL-2/S4B6 on Treg proliferation, but markedly expanded the total number of Treg recovered on d5 (Fig. 4B). While the co-administration of anti-CD80/CD86 had slight inhibitory effects on the proliferation of Treg induced by IL-2/S4B6 (Supplementary Fig. 3A), the enhancement of total Treg absolute numbers by either IL-2/anti-IL-2 immune complex was not changed by co-administration of anti-CD80/CD86 (Fig. 4B). Both immune complexes equally enhanced the proliferation and total numbers of MP CD4+ T cells and this enhancement was independent of CD28-driven co-stimulation (Fig. 4 C, D, Supplementary Fig. 3B). As shown previously, the effects of IL-2/S4B6 complexes on the proliferation Fig. 4 E, F and supplementary Fig. 4 C, D) and total yield of either CM or EM CD8+ T cells (Fig. 4 F, H) were substantially greater than the effects of IL-2/JES6 complexes. Co-administration of anti-CD80/CD86 had no effects on any parameters of either IL-2 immune complex induced activation of CM or EM CD8+ T cells (Fig. 4E–H, supplementary Fig. 4 C, D).
Figure 4.
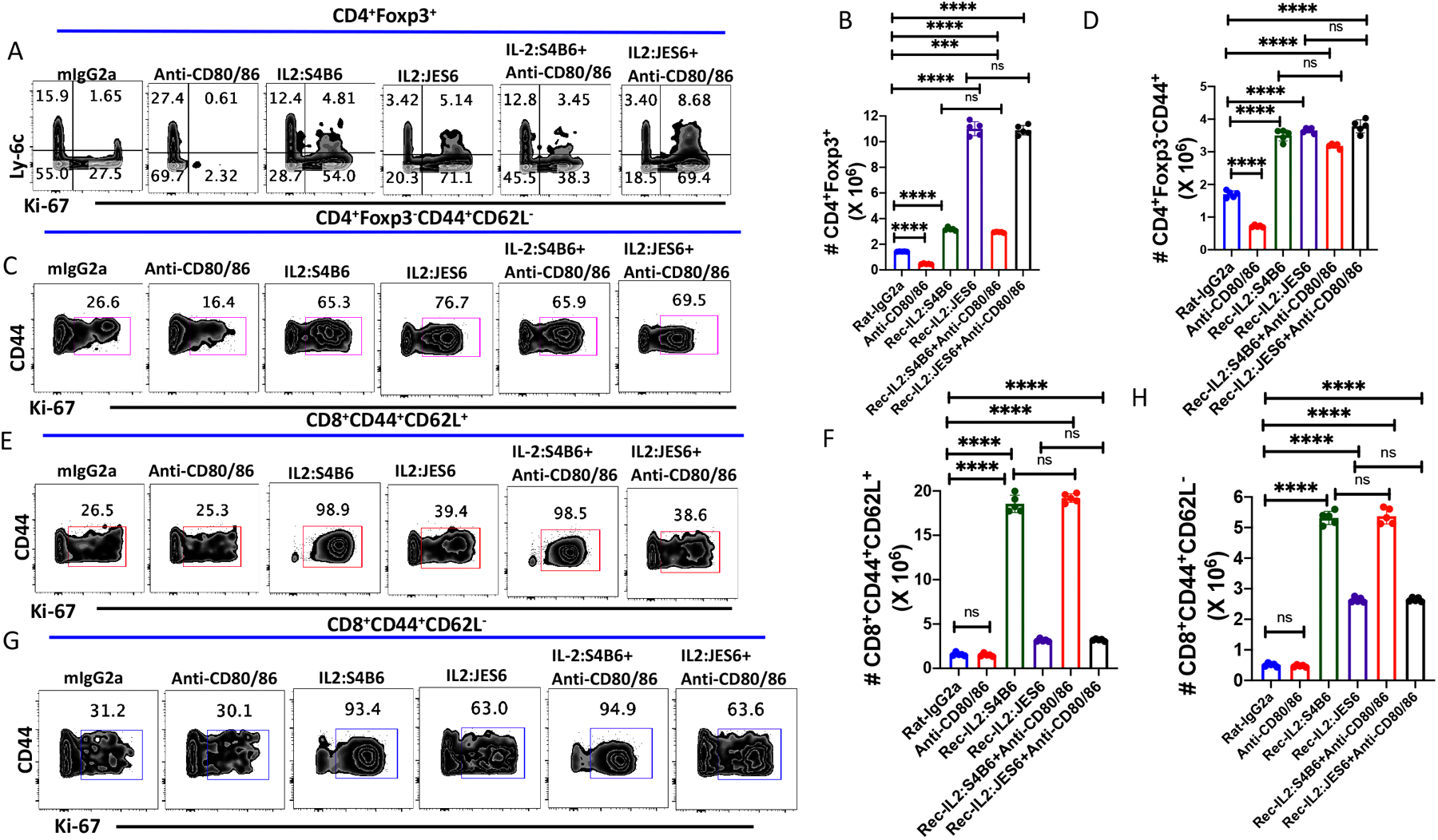
Impact of IL-2 signaling on MP T cell homeostasis. WT C57BL/6 mice were injected with either recombinant IL-2, IL-2 anti-IL-2 complexes (S4B6 or JES6) either alone or in combination with anti-CD80/86 antibodies on d 0, 1, 2 and 4. Immunophenotyping of splenic lymphocytes was performed by flowcytometry on d 5. (A) Representative plots of Ly-6C versus Ki-67 expression on CD4+Foxp3+ Tregs. (B) Absolute number of splenic CD4+Foxp3+ T cells on d 5 after antibody treatment. (C) Representative plot of Ki-67 expression ong CD4+Foxp3−CD44+ MP cells after antibody treatment on d 5. (D) Absolute number of CD4+ MP T cells after antibody treatment. (E) Representative plots of Ki-67 expression on CD8+ CM T cells. (F) Absolute number of CD8+ CM T cells after antibody treatment. (G) Representative plot of Ki-67 expression on CD8+ EM T cells. (H) Absolute number of CD8+ EM T cells after antibody treatment. (A-H) represent the result of one experiment of two using five mice per group.
To rule out the contribution of a number of other cytokines, we examined the effects of anti-type I IFN, anti-type II IFN, anti-TNFα, anti-IL-7 anti-IL-7Ra and anti-IL-33 on the homeostatic proliferation of MP Treg, and MP CD4+ and CD8+ T cells, but observed no effects on Ki-67 incorporation (Supplementary Fig. 3E–H).
CD28 co-stimulatory signals regulate the effects of agonistic anti-TNFRSF mAbs
GITR, OX40, and 4-1BB are members of the TNFRSF and are expressed primarily on the cell surface of Treg and MP CD4+ and CD8+ T cells (21). Ligation of these TNFRSF members by their ligands (GITRL, OX40L, and 4-1BBL), which are primarily expressed on on antigen-presenting cells results in activation of intracellular TRAF1 and TRAF2 signaling ultimately leading to activation of multiple interlinked signaling pathways (AKT, NFκB, Jun, p38MAPK, pERK, etc.). Agonistic mAbs to these TNFRSF members have been generated and been shown to frequently mimic the effects of the TNFRSF ligands (22). Several of these mAbs are now being developed as therapeutic agents to enhance tumor immunity and immunity to infectious agents. While it is widely assumed that these agents act directly on their target cells, we asked whether an inter-relationship exists between CD28-driven signaling and anti-TNFRSF signaling by administrering anti-GITR, anti-OX40, and anti-4-1BB alone or together with anti-CD28 dAb on alternative d for six days to unmanipulated wild-type B6 mice and analyzed the splenocytes on Day 8. Adminstration of all 3 reagents markedly enhanced the proliferation of MP Treg, but an increase in the absolute number of Treg was only seen with anti-OX40 and anti-4-1BB suggesting that anti-GITR stimulation did not promote Treg survival. While cell death is undoubtedly occurring, apoptotic cells are rapidly removed precluding detection. (Fig. 5A, B, Supplementary Fig. 4A). Surprisingly, the Treg proliferative responses to all 3 mAbs and the increase in the absolute number of Treg were all completely abolished by co-treatment with anti-CD28 dAb to levels below that seen with the control IgG2a mAb (Fig 5A, B, Supplementary Fig. 4A). This result indicates that the steady state Treg proliferation as well and the enhancement induced by the anti-TNFRSF mAbs were absolutely dependent on CD28-driven costimulation.
Figure 5.
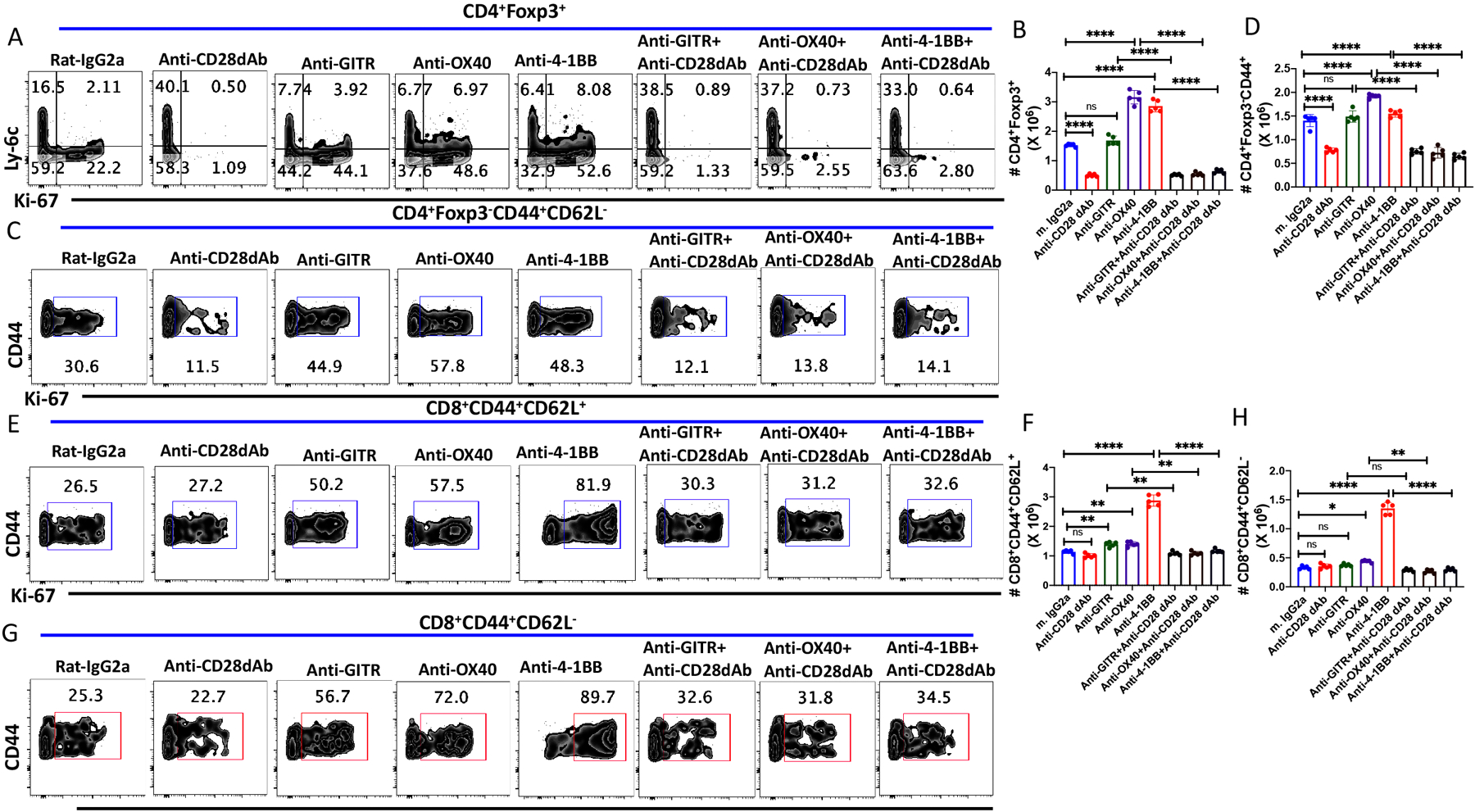
CD28 co-stimulatory signals modulate the effects of agonistic anti-TNFRSF mAbs in vivo. WT C57BL/6 mice were treated with either agonistic anti-GITR, anti-OX40, anti-4-1BB alone (250 μg/dose; i.p.) or together with anti-CD28 dAb (100 μg/dose; i.p.) every other d for six d and splenocytes were harvested on d 8. (A) Representative plots of Ly-6C versus Ki-67 expression on CD4+Foxp3+ Tregs. (B) Absolute number of splenic CD4+Foxp3+ T cells on d 8 after antibody treatment. (C) Representative plots of Ki-67 expression on CD4+Foxp3−CD44+CD62L− MP T cells after antibody treatment on d 8. (D) Absolute number of CD4+ MP T cells after antibody treatment. (E) Representative plot of Ki-67 expression on CD8+ CM T cells. (F) Absolute number of CD8+ CM T cells after antibody treatments. (G) Representative plot of Ki-67+ expresson on CD8+ EM T cells. (H) Absolute number of CD8+ EM T cells after antibody treatment. (A-H) represent the result of one experiment of two using five mice per group.
Very similar results were seen when we analyzed the effects of the 3 mAbs on the proliferation and expansion of MP CD4+ T cells. All 3 mAbs markedly enhanced the proliferation of MP CD4+ T cells and anti-OX40 and anti-4-1BB, but not anti-GITR treatment, modestly increased the absolute number of MP CD4+ T cells (Fig 5C, D, Supplementary Fig. 4B). Again, co-treatment with anti-CD28 dAb reduced MP CD4+ T cell proliferation and total numbers recovered to the baseline proliferation seen with the control IgG2a mAb (Fig. 5C, D, Supplementary Fig. 4B). CM (Fig. 5E, F and supplementary Fig. 4C) and EM (Fig. 5G, H Supplementary Fig. 4D) MP CD8+ T cells behaved similarly to stimulation by all 3 stimuli. All 3 mAbs induced proliferation of both subsets, but substantial enhancement of the numbers of CM and EM CD8+ T cells was only seen when the mice were treated with anti-4-1BB. Curiously, the enhanced proliferation induced by all 3 mAbs was abolished by co-treatment with anti-CD28 dAb, while the steady state proliferation was resistant to anti-CD28 dAb. Furthermore, the increase in absolute cells numbers seen with anti-4-1BB were completely CD28-dependent. This result is consistent with the data in Figure 1 demonstrating that the steady state homeostatic proliferation of MP CD8+ T cells is independent of CD28 signaling.
CD28 Co-stimulation regulates CD40-induced Treg and MP CD4+ proliferation, but not MP CD8+ T cell proliferation
While the targets of anti-GITR, anti-OX40, and anti-4-1BB are primarily expressed on T cells, CD40 is expressed on antigen presenting cells and B cells and interacts with CD40 ligand (CD40L) expressed primarily by activated T lymphocytes. Ligation of CD40 with CD40L induces cellular maturation and enhances multiple proliferation and signaling pathways, including increased surface expression of CD80/CD86 and MHC class II, and the production of proinflammatory cytokines (IL-12, IL-15, IL-18, etc.). As the therapeutic use of agonistic anti-CD40 mAbs has been proposed to augment immune responses to tumors and infectious agents (23), we also examined the effects of agonistic anti-CD40 treatment on MP Treg, MP CD4+ amd MP CD8+ T cells homeostasis. Treatment of mice with anti-CD40 mAb doubled MP Treg cell proliferation and increased the total number of Treg almost 3-fold. All the effects of the CD40 agonistic mAb were reversed by co-treatment with anti-CD28 dAb and are likely secondary to increased CD80/CD86 expression on B cells and APC (Fig. 6A–C). Curiously, treatment of mice with anti-CD40L decreased the baseline homeostatic proliferation (Ki-67 expression) and the total number of Treg by 50% suggesting that CD40L/CD40 interactions may be occurring to a certain extent in the steady state in the absence of exogenous stimuli (Fig. 6A–C).
Figure 6.
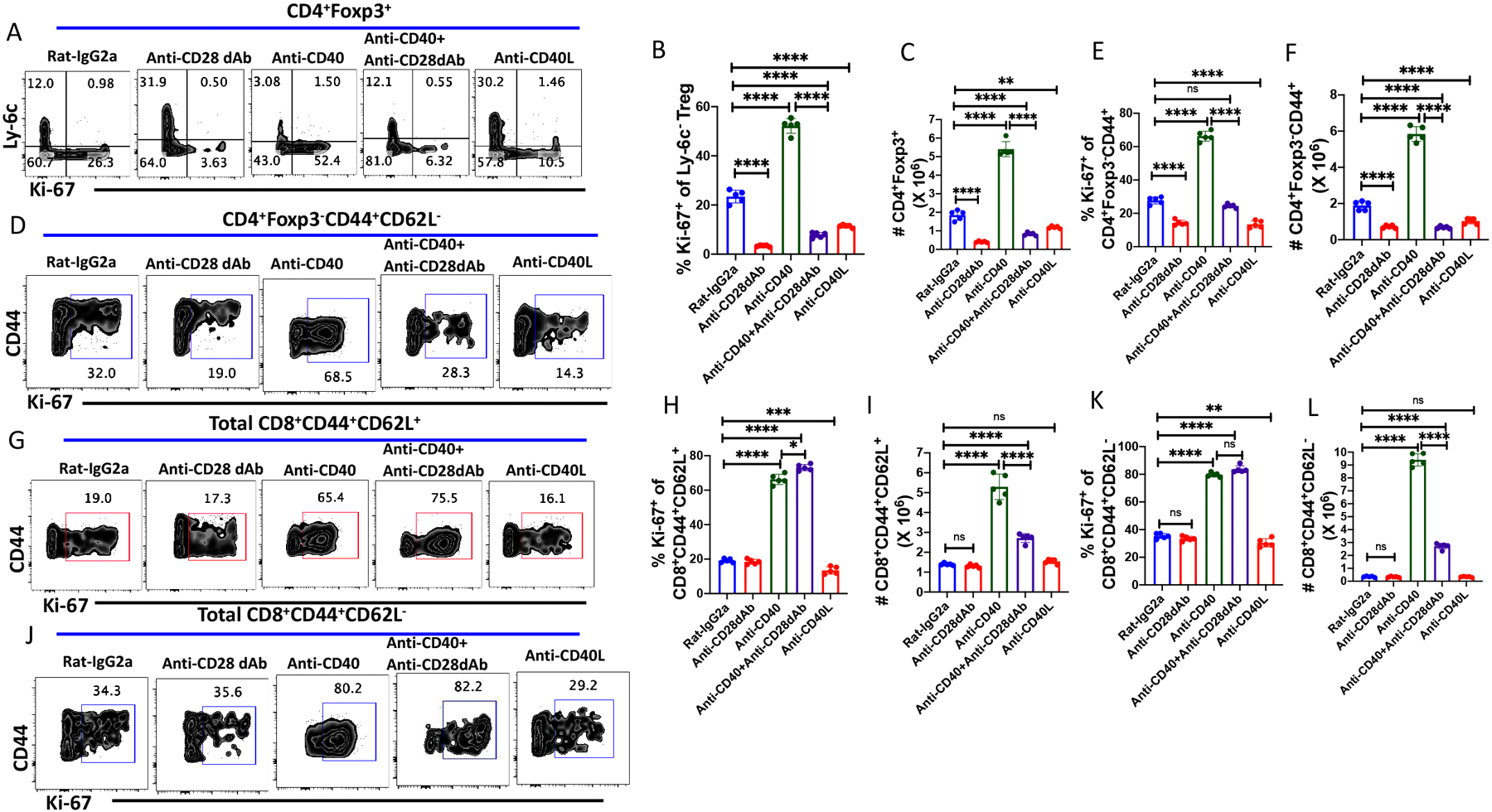
CD28 signaling modulates Treg and CD4+MP proliferation after treatment with agonistic anti-CD40. WT C57BL/6 mice were treated with rat-IgG2a, anti-CD28 dAb (100 μg/dose; i.p.), anti-CD40 (50 μg/dose; i.p.), anti-CD40L (250 μg/dose) or anti-CD40 together with anti-CD28 dAb on d 0, 2, 4 and 6 and spleens were harvested on d 8. (A) Representative plots of Ly-6C versus Ki-67 expression on CD4+Foxp3+ Tregs. (B) Ki-67 expression on Ly6C− Treg cells after antibody treatment. (C) Absolute number of splenic CD4+Foxp3+ T cells after antibody treatment. (D) Expression of Ki-67 on CD4+Foxp3−CD44+ MP T cells after antibody treatment. (E) Percentage of Ki-67 expression on CD4+ MP T cells. (F) Absolute number of CD4+ MP T cells after antibody treatment. (G) Representative plot of the epression of Ki-67 on CD8+ central CM T cells. (H) Ki-67 expression on CD8+ CM T cells. (I) Absolute number of CD8+ CM T cells after antibody treatment. (J) Representative plot of the expression of Ki-67 on CD8+ EM T cells. (K) Ki-67 expression on CD8+ EM T cells. (L) Absolute number of CD8+ EM T cells after antibody treatment. (A-L) represent the result of one experiment of two using five mice per group.
The effects of anti-CD40 treatment on MP CD4+ T cells were similar to those seen with Treg. Anti-CD40 induced a doubling of MP CD4+ proliferation and about a 4-fold increase in the absolute number (Fig. 6D–F). Anti-CD40L treatment slightly decreased Treg cell proliferation and absolute numbers. Anti-CD40 treatment profoundly enhanced the proliferation and the absolute numbers of both CM and EM MP CD8+ T cells (Fig. 6G–K). The effects of anti-CD40 on the proliferation of MP CD8+ T cells were completely independent of CD28 signaling, while the enhancement of the total numbers of both MP CD8+ subpopulations were markedly reduced by co-administration of anti-CD28 dAb. This result raises the possibility that the induction of MP CD8+ T cell proliferation may be cytokine driven (e.g, IL-15), but survival may be CD28-dependent. Anti-CD40L had minimal effects on the proliferation or recovery of either MP CD8+ T cell subpopulation (Fig. 6G–K).
Induction of cytokine production in vivo by blocking the interaction of NK inhibitory receptors with MHC I disrupts MP T cells, but not Treg, homeostasis
We have previously demonstrated (12) that administration of a pan-anti-MHC-I mab, M1/42, to normal mice for six days resulted in marked global activation of NK cells accompanied by induction of a Th1 pro-inflammatoryphenotype (Fig. 7), enhanced IL-15 production, proliferation of APC, activation of T cells, and enhanced anti-viral and anti-tumor responses It was therefore of in interest to examine whether this global disruption of immune homeostasis resembling a cytokine storm had a bystander effect on Treg and MP homeostasis. Surprisingly, this global induction of cytokine production had no effect on MP Treg proliferation or absolute numbers and both proliferation and enhancement of total numbers remained susceptible to inhibition by blocking CD28/CD80-CD86 interaction (Fig. 8A–C). In marked contrast, M1/42 treatment significantly enhanced MP CD4+ proliferation and total numbers and both enhancements were inhibited by blocking CD28 signaling (Fig. 8D–F). M1/42 treatment remarkably enhanced both CM (Fig. 8 G–I) and EM CD8+ (Fig. 8J–L) proliferation and absolute numbers. Blockade of CD28 signaling had no effect on MP CD8+ proliferation and absolute numbers which is consistent with our previous observation (12) that MP CD8+ expansion was mediated by IL-15 trans-presentation. Thus, in this complex model of disruption of systemic immune homeostasis, Treg were completely unaffected, while MP CD4+ were activated in a CD28 driven manner, and MP CD8+ T cells were activated by a pathway independent of CD28-driven co-stimulation.
Figure 7.
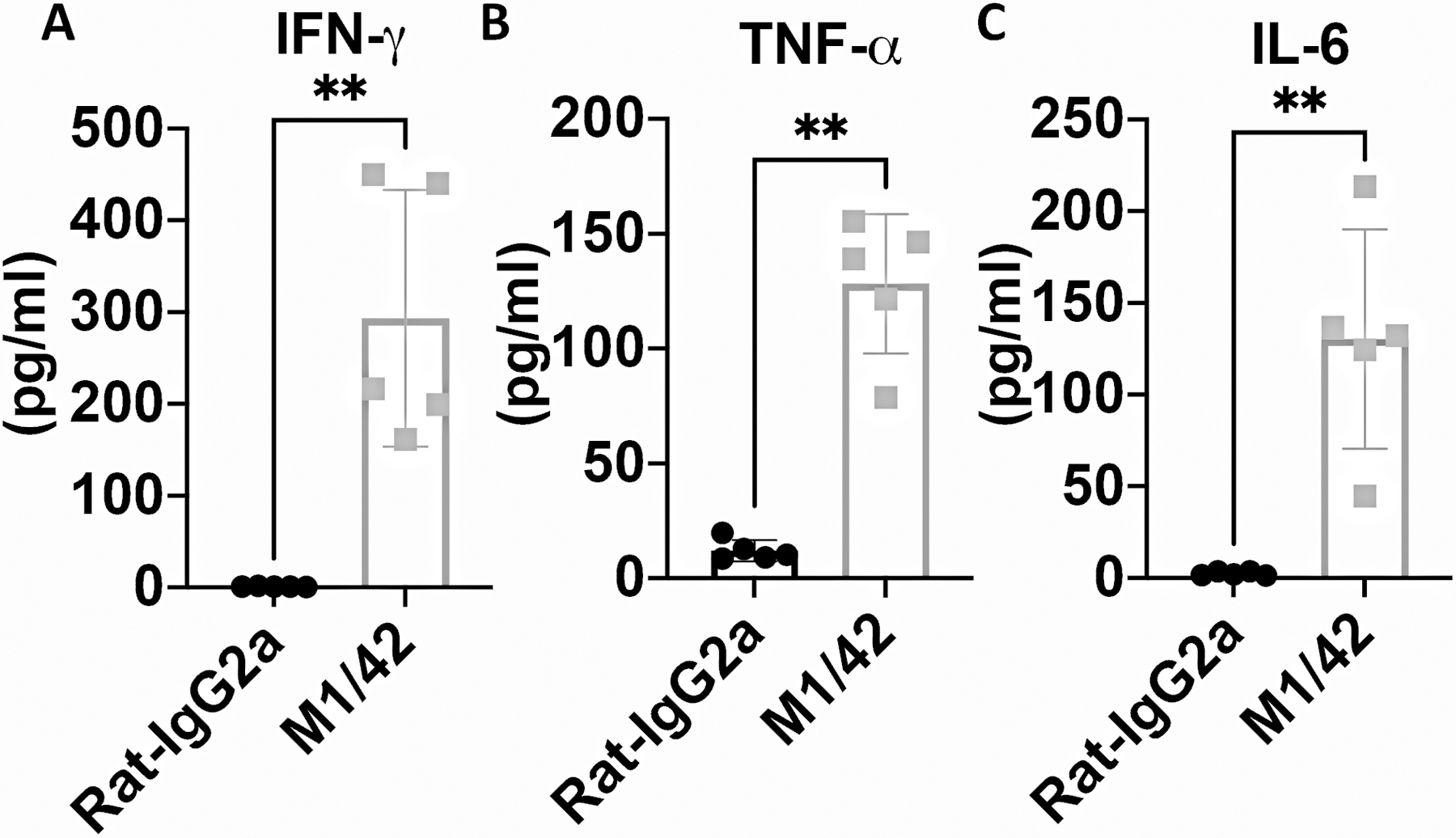
M1/42 administration enhances systemic pro-inflammatory cytokine production. WT C57BL/6 mice were injected with Rat-IgG2a or M1/42 (pan-anti-MHC-I) on day 0-, 2-, 4- and 6. Plasma was collected on d8 to measure IFN-γ, TNF-α and IL-6 levels by cytometric bead array. *p, 0.05; **p; 0.005; ***p, 0.0005, ****p, 0.00005.
Figure 8.
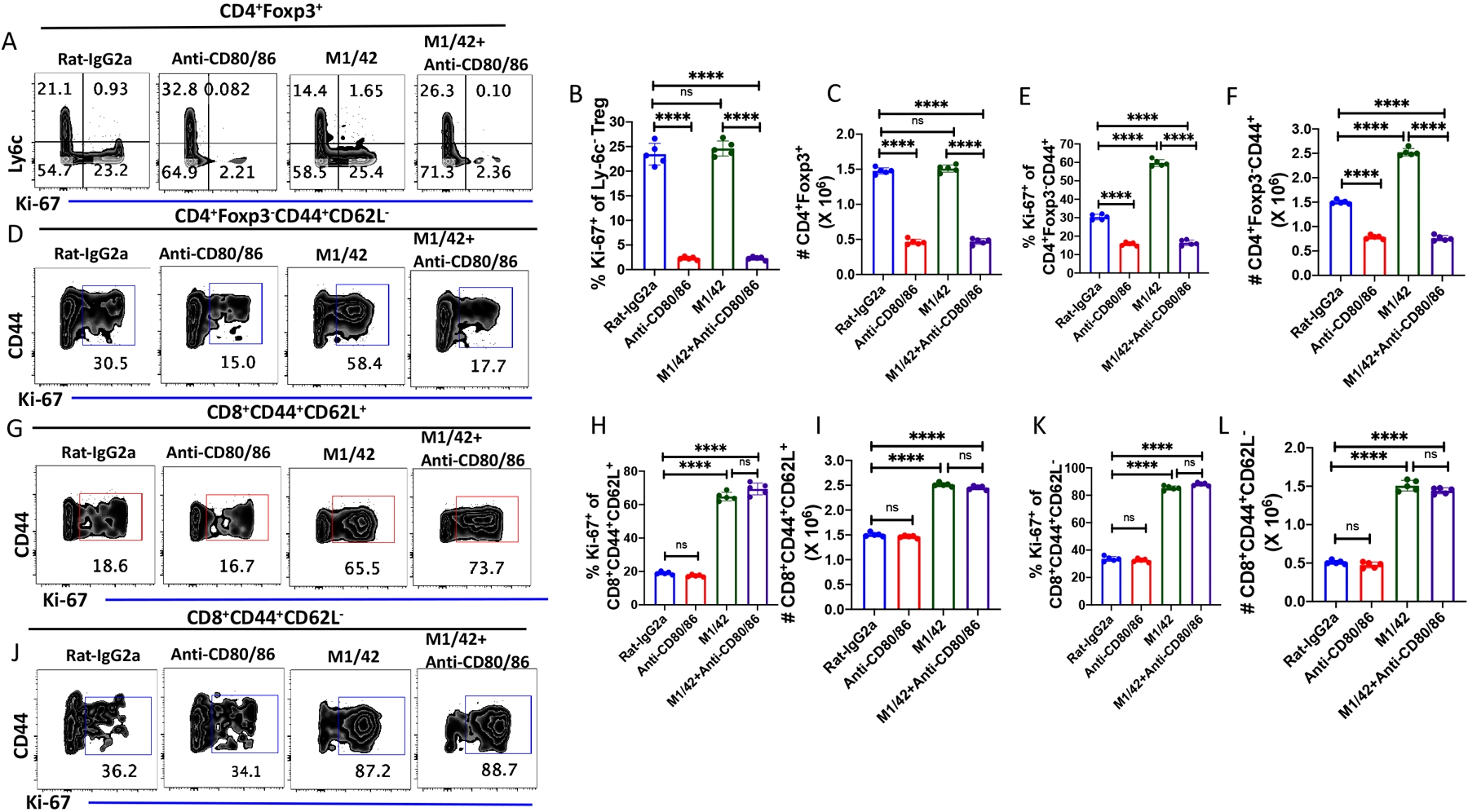
CD28 signaling regulates NK-dependent enhancement of CD4+ MP T cell, but not CD8+MP T cell, proliferation. C57BL/6 mice were treated with rat-IgG2a, M1/42 (500 μg/dose), anti-CD80/86 (250 μg/dose) and M1/42 together with anti-CD80/86 on d 0, 2, 4 and 6 and spleens were harvested on d 8. (A) Representative plots of Ly-6C versus Ki-67 expression on CD4+Foxp3+ splenic Tregs. (B) Expression of Ki-67 on among Ly6C− Treg cell after antibody treatments. (C) Absolute number of splenic CD4+Foxp3+ T cells after antibody treatment. (D) Representative plots of the expression of Ki-67 on CD4+Foxp3−CD44+MP T cells after antibody treatment. (E) Expression of Ki-67 on CD4+ MP T cells. (F) Absolute number of CD4+ MP T cells after antibody treatment. (G) Representative plots of the expression of Ki-67 on CD8+ CM T cells. (H) Percentage of Ki-67+ CD8+ CM T cells. (I) Absolute number of CD8+ CM T cells after antibody treatment. (J) Representative plots of Ki-67 expression on CD8+ EM T cells. (K) Percentage of Ki-67+ CD8+CM T cells. (L) Absolute number of CD8+ EM T cells after antibody treatment. (A-L) represent the result of one experiment of two using five mice per group.
Discussion
MP cells can be indentified as a subpopulation of CD4+, CD8+ and Treg by high expression of CD44 and in the case of Treg, low expression of Ly-6C. Each of these MP subsets has several unique properties when compared to the their resting NP counterparts. Most prominently, they are highly proliferative in the steady state in vivo with about 10–12% of each subset proliferating in a 24h period and an equivalent percentage dying as the percentages of each subset remain constant. MP Treg appear to be the major subset mediating suppressor function under homeostatic condition (6, 7). In the case of CD4+ T cells, the MP subset has innate-like properties and can respond to IL-12 in a pathogen-independent fashion to enhance antigen-specific effector CD4+ T cells involved in resistance to infection (11, 24). Similarly, MP CD8+ T cells can rapidly respond to infectious challenges by rapidly producing effector cytokines (25). The rapid production of cytokines is also a property of CD8+ virtual memory cells (26). The focus of this study was an examination of the factors that drive and can potentially influence the proliferation and accumulation of MP cells over a short-term period (5–7d) in vivo.
The major driver of Treg and MP CD4+ proliferation was CD80/CD86-CD28 stimulation. MP Treg were completely dependent on this pathway as Treg proliferation in vivo could be almost completely blocked by treatment of mice with a potent anti-CD28 dAb. While MP CD4+ T cell proliferation was also substantially reduced by this treatment, a certain component was reproducibly CD28-independent and is likely cytokine driven potentially by IL-7 (8). In addition to splenic Treg and MP CD4+ T cells, similar results were observed in mesenteric lymph nodes and liver. Although it is likely that both CD28 and TCR signals are involved in MP Treg and MP CD4+ T cell proliferation, the precise contribution of TCR signals to this process is difficult to assess as blocking TCR signaling in vivo paradoxically resulted in CD28-dependent expansion of both MP Treg, MP CD4+ and MP CD8+ cell populations (5). We hypothesized that this result indicated that TCR signals also restrained MP Treg homeostasis and that blocking TCR signals together with unrestrained CD28 signals led to an abrogation of TCR suppression and expansion of CD4+ and particularly CD8+ MP T cells. In contrast to the results with Treg and MP CD4+ T cells, the homeostatic proliferation of both CM and EM CD8+ T cells was completely CD28-independent which is consistent with the prevailing view that homeostatic proliferation of this subset is primarily cytokine-driven.
Both anti-CTLA-4 and anti-PD-1 treatment enhanced Treg proliferation and MP CD4+ T cell proliferation. Co-treatment produced greater augmentation than either mAb alone suggesting that the effects of the two agents were mediated by different pathways. This view is consistent with the proposed mechanisms for the two inhibitory molecules. While CTLA-4 inhibits T cell activation by removing CD80/CD86 from APC, followed by transendocytosis and degradation (27), PD-1 mediates its effects by inhibiting signaling via the interaction of SHP-2 with an ITIM or an ITSM in its cytoplasmic domain (28, 29). Surprisingly, the enhancement produced by either CTLA-4 or PD-1 blockade could be overcome by simultaneous treatment of the mice with the anti-CD28 dAb confirming the direct relationship between CD28 and CTLA-4 and suggesting that the primary inhibitory target for PD-1 is CD28. While anti-CD28 had no effect of the baseline homeostatic proliferation of EM CD8+ T cells, combined treatment of mice with anti-CTLA-4 and anti-PD-1 markedly enhanced EM CD8+ T cell proliferation and this enhancement, but not the basal, proliferation of EM CD8+ EM cells was blocked by the anti-CD28 dAb. One question which remains unresolved is whether the effects of anti-CTLA-4 are mediated directly on MP CD4+ and CD8+ T cells which normally do not express CTLA-4 or indirectly by acting on Treg with resultant inhibition of Treg suppressor function as proposed for blockade of TCR-MHC II interactions (5). It should also be noted that blocking mAbs to a number of other inhibitory receptors had no effect of MP proliferation.
Agonistic mAbs to several member of the TNFRSF (GITR, OX-40, 4-1BB) can induce Treg and MP T cell proliferation and activation in vitro and in vivo (30). Anti-CD40 activation of different APC populations can also result in indirect effects on MP T cells. Agonistic mAbs to GITR, OX-40 and 4-1BB had similar effects on MP Treg and MP CD4+ and CD8+ T cells in that they enhanced to varying degrees proliferation and expansion of all MP subsets. The effects on Treg were completely reversed by anti-CD28 blockade while the effects on MP CD4+ and CD8+ T cells were a reduction to basal proliferation levels. Similar results were observed with Treg and MP CD4+ T cells during anti-CD40 stimulation. The effects of anti-CD40 treatment on MP CD8+ T cells were different from the agonistic mAbs to the other TNFRSF members, as it induced proliferation that was not blocked by anti-CD28, while accumulation of MP CD8+ T cells was inhibited. It remains possible that proliferation of MP CD8+ T cells was cytokine driven and not affected by anti-CD28, while survival was CD28-dependent. Importantly, it also remains unclear why the effects of agonistc mAbs to TNFRSF members would be dependent on CD28 co-stimulation as the effects of these mAbs may be independent of TCR signaling (31). One possibility which has been proposed for the role of CD28 on TCR signaling is that CD28 lowers the threshold for TCR mediated activation which results in enhanced proliferation, cytokine production, and cell survival. Our results are consistent with a model in which CD28 engagement lowers the signal for cellular activation for other stimulatory ligands including membrs of the TNFRSF.
IL-2 signaling via STAT5 is critical for maintaining mature Treg cell fitness, Foxp3 expression, and suppressive function in vitro and in vivo (32, 33). However, the administration of anti-IL-2 (S4B6) had minimal effects on MP Treg homeostasis and Treg suppressor function (20). A modest decrease in percentage NP Treg was observed which is consistent with the higher levels of CD25 expressed by this subpopulation and their greater requirement for IL-2 for survival (15). When IL-2 is complexed with either mAbs S4B6 or JES6, different populations of T cells are stimulated depending on the mAb used to generate the complex (34). We examined the potential requirements for CD28 co-stimulation for activation of MP Treg by IL-2/anti-IL-2 complexes. In contrast to the results observed with the agonistic mAbs to the TNFRSF, the potent expansion of all 3 MP subsets was unaffected by blocking CD28 driven co-stimulation.
Lastly, we examined the requirements for co-stimulatory signals when a generalized cytokine storm was induced in vivo by blocking MHC-I/NK inhibitory receptor interactions with a pan anti-MHC-mAb (12). In this model, NK cells are initially activated to secrete IFNγ which drives a forward-feedback loop resulting in activation Th1 cytokine production, and activation of different APC subpopulations to production of IL-15. In contrast to what we have observed previously, in this model, MP CD4+ T cells proliferation was augmented in a CD28-dependent manner, while no effects were observed on MP Treg homeostasis although basal homeostatic proliferation remained susceptible to inhibition by anti-CD28 dAb. MP CD8+ T cell proliferation was enhanced and was completely CD28-independent which is consistent with it being cytokine (IL-15)-driven (12). While the homeostatic proliferation of both MP Treg and MP CD4+ T cells was for the most part CD28-dependent, the reason for the differential susceptibility of MP CD4+ T cells versus Treg cells to expansion in this model in contrast to all the other models is unclear. We did not observe enhanced expression of CD80/CD86 on APC although other both MHC-I and MHC-II were upregulated. It remains possible that enhancement of MP CD4+ T cell proliferation requires lower level of CD80/86 than Treg require or that other factors preferentially act on MP CD4+ T cells in this model.
In summary, one major characteristic of MP phenotype T cells is their rapid proliferation in vivo in the absence of immunization. We have demonstrated that MP Treg, MP CD4+ and MP CD8+ T cells have distinct factors responsible for driving their homeostatic proliferation. In addition, the responses of the three MP T cell subsets to other stimuli in vivo including both cytokines, stimululation via members of the TNFRSF, and inhibitory receptor blockade are markedly different. Many of the agents used for the treatment of autoimmune diseases or for cancer immunotherapy may have unwanted effects on MP T cells. For example, our studies suggest that treatment of autoimmune diseases by blocking CD28 signaling on T effector cells may also have deleterious effects on Treg, while enhancement of effector cell function in cancer by treatment with agonistic mAbs to members of the TNFRSF may inadvertently enhance MP Treg function and suppresson of anti-tumor immunity.
Supplementary Material
Key points:
Homeostatic proliferation of MP Treg is absolutely CD28-dependent.
Both anti-CTLA-4 and anti-PD-1 enhanced Treg, CD4 and CD8 homeostatic proliferation.
Stimulation of MP T cell proliferation by agonistic anti-TNFRS mAbs is CD28-dependent.
Acknowledgments
We thank members of the Shevach laboratory, Ke Weng, and Dr. Jeff Zhu for helpful insights and suggestions to this project.
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Abbreviations used in this article:
- CM
central memory
- dAb
domain antibody
- EM
effector memory
- MP
memory phenotype
- NP
naïve phenotype
- TNFRSF
TNF receptor superfamily
- Treg
T regulatory cells
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Sprent J, and Surh CD. 2011. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol 12: 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JY, Hamilton SE, Akue AD, Hogquist KA, and Jameson SC. 2013. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci U S A 110: 13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller CH, Klawon DEJ, Zeng S, Lee V, Socci ND, and Savage PA. 2020. Eomes identifies thymic precursors of self-specific memory-phenotype CD8(+) T cells. Nat Immunol 21: 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delpoux A, Yakonowsky P, Durand A, Charvet C, Valente M, Pommier A, Bonilla N, Martin B, Auffray C, and Lucas B. 2014. TCR signaling events are required for maintaining CD4 regulatory T cell numbers and suppressive capacities in the periphery. J Immunol 193: 5914–5923. [DOI] [PubMed] [Google Scholar]
- 5.Holt MP, Punkosdy GA, Glass DD, and Shevach EM. 2017. TCR Signaling and CD28/CTLA-4 Signaling Cooperatively Modulate T Regulatory Cell Homeostasis. J Immunol 198: 1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine AG, Arvey A, Jin W, and Rudensky AY. 2014. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, Rieß D, Hein MY, Buch T, Polic B, Schönle A, Zeiser R, Schmitt-Gräff A, Kretschmer K, Klein L, Korn T, Sakaguchi S, and Schmidt-Supprian M. 2014. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 41: 722–736. [DOI] [PubMed] [Google Scholar]
- 8.Younes SA, Punkosdy G, Caucheteux S, Chen T, Grossman Z, and Paul WE. 2011. Memory phenotype CD4 T cells undergoing rapid, nonburst-like, cytokine-driven proliferation can be distinguished from antigen-experienced memory cells. PLoS Biol 9: e1001171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surh CD, and Sprent J. 2008. Homeostasis of naive and memory T cells. Immunity 29: 848–862. [DOI] [PubMed] [Google Scholar]
- 10.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, and Surh CD. 2002. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med 195: 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawabe T, Jankovic D, Kawabe S, Huang Y, Lee PH, Yamane H, Zhu J, Sher A, Germain RN, and Paul WE. 2017. Memory-phenotype CD4(+) T cells spontaneously generated under steady-state conditions exert innate T(H)1-like effector function. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panda AK, Gangaplara A, Buszko M, Natarajan K, Boyd LF, Sharma S, Margulies DH, and Shevach EM. 2020. Cutting Edge: Inhibition of the Interaction of NK Inhibitory Receptors with MHC Class I Augments Antiviral and Antitumor Immunity. J Immunol 205: 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang AL, Teijaro JR, Njau MN, Chandran SS, Azimzadeh A, Nadler SG, Rothstein DM, and Farber DL. 2008. CTLA4 expression is an indicator and regulator of steady-state CD4+ FoxP3+ T cell homeostasis. J Immunol 181: 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Badell IR, and Ford ML. 2018. Selective CD28 blockade attenuates CTLA-4-dependent CD8+ memory T cell effector function and prolongs graft survival. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ. 2014. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 211: 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, and Paul WE. 2004. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A 101: 3880–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY. 2005. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 6: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 18.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, and Wahl SM. 2003. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198: 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyman O, and Sprent J. 2012. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12: 180–190. [DOI] [PubMed] [Google Scholar]
- 20.Hayes ET, Hagan CE, Khoryati L, Gavin MA, and Campbell DJ. 2020. Regulatory T Cells Maintain Selective Access to IL-2 and Immune Homeostasis despite Substantially Reduced CD25 Function. J Immunol 205: 2667–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward-Kavanagh LK, Lin WW, Šedý JR, and Ware CF. 2016. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 44: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bullock TN 2017. TNF-receptor superfamily agonists as molecular adjuvants for cancer vaccines. Curr Opin Immunol 47: 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li DK, and Wang W. 2020. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol Lett 20: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krueger PD, Osum KC, and Jenkins MK. 2021. CD4(+) Memory T-Cell Formation during Type 1 Immune Responses. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laidlaw BJ, Craft JE, and Kaech SM. 2016. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haluszczak C, Akue AD, Hamilton SE, Johnson LD, Pujanauski L, Teodorovic L, Jameson SC, and Kedl RM. 2009. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med 206: 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, and Sansom DM. 2011. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332: 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, Sica GL, Sharpe AH, Freeman GJ, Blazar BR, Turka LA, Owonikoko TK, Pillai RN, Ramalingam SS, Araki K, and Ahmed R. 2017. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355: 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, and Okazaki T. 2019. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front Immunol 10: 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edner NM, Carlesso G, Rush JS, and Walker LSK. 2020. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov 19: 860–883. [DOI] [PubMed] [Google Scholar]
- 31.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, and Byrne MC. 2002. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16: 311–323. [DOI] [PubMed] [Google Scholar]
- 32.Malek TR 2008. The biology of interleukin-2. Annu Rev Immunol 26: 453–479. [DOI] [PubMed] [Google Scholar]
- 33.Stolley JM, and Campbell DJ. 2016. A 33D1+ Dendritic Cell/Autoreactive CD4+ T Cell Circuit Maintains IL-2-Dependent Regulatory T Cells in the Spleen. J Immunol 197: 2635–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, and Sprent J. 2009. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 206: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.