

Abstract
Background 3F7 is a monoclonal antibody targeting the enzymatic pocket of activated factor XII (FXIIa), thereby inhibiting its catalytic activity. Given the emerging role of FXIIa in promoting thromboinflammation, along with its apparent redundancy for hemostasis, the selective inhibition of FXIIa represents a novel and highly attractive approach targeting pathogenic processes that cause thromboinflammation-driven cardiovascular diseases.
Methods The effects of FXIIa inhibition were investigated using three distinct mouse models of cardiovascular disease—angiotensin II-induced abdominal aortic aneurysm (AAA), an ApoE −/− model of atherosclerosis, and a tandem stenosis model of atherosclerotic plaque instability. 3F7 or its isotype control, BM4, was administered to mice (10 mg/kg) on alternate days for 4 to 8 weeks, depending on the experimental model. Mice were examined for the development and size of AAAs, or the burden and instability of atherosclerosis and associated markers of inflammation.
Results Inhibition of FXIIa resulted in a reduced incidence of larger AAAs, with less acute aortic ruptures and an associated fibro-protective phenotype. FXIIa inhibition also decreased stable atherosclerotic plaque burden and achieved plaque stabilization associated with increased deposition of fibrous structures, a >2-fold thicker fibrous cap, increased cap-to-core ratio, and reduction in localized and systemic inflammatory markers.
Conclusion Inhibition of FXIIa attenuates disease severity across three mouse models of thromboinflammation-driven cardiovascular diseases. Specifically, the FXIIa-inhibiting monoclonal antibody 3F7 reduces AAA severity, inhibits the development of atherosclerosis, and stabilizes vulnerable plaques. Ultimately, clinical trials in patients with cardiovascular diseases such as AAA and atherosclerosis are warranted to demonstrate the therapeutic potential of FXIIa inhibition.
Keywords: 3F7 monoclonal antibody, abdominal aortic aneurysm, atherosclerosis, coagulation factor XII, vulnerable plaques
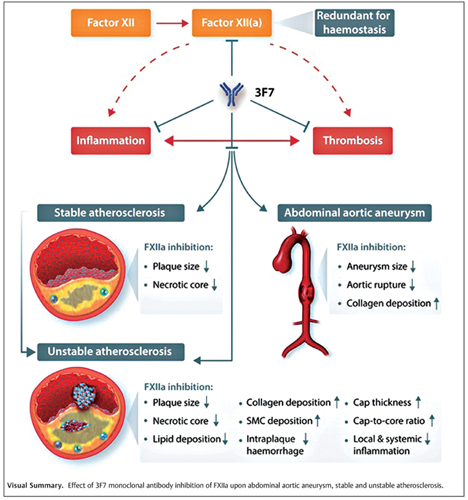
Introduction
The rupture of an atherosclerotic plaque and formation of an occlusive arterial thrombus are the inciting events leading to myocardial infarction and ischemic stroke, which represent the leading causes of mortality and morbidity worldwide. 1 Atherosclerosis, the leading pathology that causes these ischemic complications, is fuelled by chronic inflammatory processes. 2 While the role of the coagulation system initiating pathological thrombus formation upon plaque rupture is well appreciated, there is now a growing body of evidence that the coagulation system also directly contributes to atherosclerosis development, including plaque destabilization. 3 4 Indeed, multiple coagulation factors have been demonstrated to be present within atherosclerotic plaques. 5 Moreover, active coagulation factors such as tissue factor, thrombin, factor X (FX), and factor XII (FXII) have been shown to be present early on during the development of atherosclerotic plaques. 5 Supporting this notion, it has recently been demonstrated that genetic deficiency of FXII or FXI inhibits atherogenesis in a mouse model of atherosclerosis. 6 7 These findings support the concept of utilizing anticoagulants as a means to prevent atherosclerosis. 8 However, given that all clinically available anticoagulant therapeutics are associated with an inherent risk of bleeding, newer more advanced strategies avoiding bleeding are required. 8 9
FXII, the zymogen of the serine protease, FXIIa, is activated by several substances including polyphosphate, vascular collagen, misfolded proteins, and neutrophil extracellular traps. 10 11 12 13 The activation of FXII initiates the intrinsic coagulation pathway and liberates the formation of bradykinin (BK) thus promoting inflammation. 14 Significantly, recent data have demonstrated that FXII −/− mice are afforded protection from atherosclerosis. 6 Interestingly, the protective effects of FXII deficiency appear to occur independently of any systemic effects on coagulation, or BK production, thus suggesting an important proinflammatory role of FXII/FXIIa within the confines of the atherosclerotic plaque. In this regard, FXIIa induces the production of proinflammatory, proatherogenic cytokines, including interleukin (IL)-6, IL-1b, IL-12, and tumor necrosis factor-α from bone marrow-derived macrophages. 6 However, to date, the effects of pharmacological FXIIa inhibition as an antiatherosclerotic approach have not been investigated. FXIIa is a highly attractive therapeutic target given the fully recombinant human 3F7 monoclonal antibody (mAb), which binds to the catalytic site of activated FXII and potently inhibits both FXIIa and βFXIIa, and has been demonstrated to inhibit thrombosis without impeding hemostasis in preclinical animal models 15 16 and an affinity-improved version (CSL312) is currently in phase 3 clinical trials for the prevention of hereditary angioedema (ClinicalTrials.gov Identifier: NCT04656418). Therefore, given its key role in mediating pathological thrombosis and several inflammatory processes implicated in the development of atherosclerosis and AAA, we hypothesized that the therapeutic inhibition of FXIIa with the mAb 3F7 represents a novel approach to inhibit atherosclerosis and AAA by treating both coagulation and inflammation, while preserving hemostasis.
In this study, we demonstrate that inhibition of FXIIa with 3F7 has a protective effect in mouse models of AAA and atherosclerosis. Most importantly, using a unique mouse model of atherosclerotic plaque instability, 17 18 19 we show that FXIIa inhibition via 3F7 leads to the stabilization of vulnerable, unstable atherosclerotic plaques. Taken together, these data indicate that FXIIa inhibition suppresses thromboinflammation and thereby exerts beneficial effects in vascular diseases, such as AAA and atherosclerosis.
Methods
Mouse Experiments and 3F7 Administration
All animal procedures were approved by the Animal Ethics Committee of the Alfred Medical Research and Education Precinct (AMREP), Melbourne, Australia, under ethics application numbers E/1658/2016/B and E/1187/2012/B and conform to the current National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. ApoE −/− mice were generated from a C57BL/6 background, bred, and maintained at the AMREP Animal Centre.
Briefly, mouse models of AAA, stable, and unstable atherosclerosis were employed ( Supplementary Fig. S1 , available in the online version). For the investigation of atherosclerotic plaques, ApoE −/− and tandem stenosis (TS) mouse models were employed while an angiotensin II (AngII)-infusion mouse model was used for the AAA study. Animals were randomly assigned to receive either 3F7 or its isotype control BM4 every second day via intraperitoneal (i.p.) injection, with administration beginning directly after surgery or at 8-weeks old for the ApoE −/− study. Both 3F7 and BM4 (MuBM4-MuG1K) were supplied by CSL Limited as murine IgG1 antibodies.
Detailed information on the methods used in this publication can be found in the Supplementary Material (available in the online version).
Statistical Analysis
Unless otherwise specified, quantitative data are expressed as mean ± standard deviation. Comparisons of parameters between two groups were made using the unpaired Student's t -test after normal data distribution was confirmed. A p -value of <0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism software.
Results
FXIIa Is Localized within AAAs
Several negatively charged surfaces which can activate FXII have been described within the AAA microenvironment. 20 21 Therefore, we postulated that FXIIa is detectable in AAAs. Using immunofluorescence (IF), we confirmed the presence of FXIIa within AAAs ( Fig. 1A, B ).
Fig. 1.
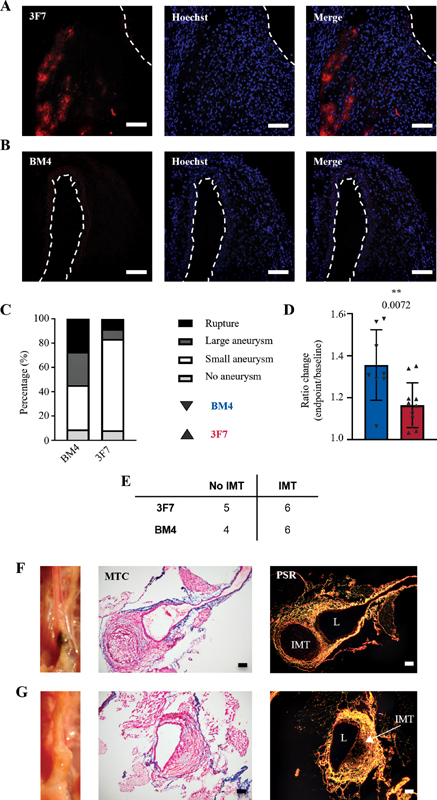
Presence of FXIIa in AAA and inhibition of FXIIa decrease severity and increase stability of AAA. ( A ) Representative IF images showing FXIIa (3F7-staining) deposition within the AAA. ( B ) BM4 was used as isotype control. The vessel lumen is marked by interrupted lines. ApoE −/− mice were infused with angiotensin-II via an implanted osmotic minipump and then treated with 3F7 or the control BM4. Ultrasound measurements of the dilatation of the abdominal aorta were taken. ( C ) Percentages of ruptured, large, small, and no aneurysm formation (BM4 n = 11; 3F7 n = 12). ( D ) Ratio change (endpoint/baseline) for mice that developed AAA and survived, i.e., did not experience rupture ( p = 0.0072; BM4 n = 7; 3F7 n = 10). ( E ) Table showing the incidence of IMT formation, determined by morphological assessment or necropsy (BM4 n = 10; 3F7 n = 11). Representative images showing the morphological analysis of collagen deposition using MTC and PSR for: ( F ) lower limit aneurysms from 3F7-treated animals ( n = 5) and ( G ) a lower limit aneurysm from BM4 control animals ( n = 1). The unpaired Student's t -test was used for comparison of BM4 and 3F7-treated mice. Scale bars = 200 µm. AAA, abdominal aortic aneurysm; FXII, factor 12; FXIIa, activated factor 12; IMT, intraluminal thrombosis; L, lumen; MTC, Masson's trichrome; PSR, Picro-Sirius Red.
Anti-FXIIa (3F7) Administration Results in Smaller AAAs and a More Stable Phenotype
We next investigated the efficacy of the anti-FXIIa mAb 3F7 to prevent the development of AAA. Mice received 3F7 or BM4 isotype control antibody every 48 hours for 28 days following the implantation of an osmotic minipump containing AngII to induce AAA. Although 3F7 did not significantly alter the incidence of aneurysm formation, it did result in less acute aortic ruptures and a higher proportion of small aneurysms on the lower limit of classification (lower-limit aneurysm 1.2–1.4 mm, aneurysm >1.5 mm; BM4 no aneurysm n = 1, lower-limit aneurysm n = 4, aneurysm n = 3, rupture n = 3; 3F7 no aneurysm n = 1, lower-limit aneurysm n = 9, aneurysm n = 1, rupture n = 1; Fig. 1C ). Ultimately, inhibition of FXIIa significantly impeded the progressive dilatation of the abdominal aorta throughout the 28-day experimental period, therefore forming smaller aneurysms as compared with the control animals receiving BM4 ( Fig. 1D ; p = 0.0072). Individual plots of the baseline and endpoint dilatation measurements can be found in Supplementary Fig. S2A and B , available in the online version.
Studies have suggested that the volatility of an AAA and its risk of acute rupture are correlated to the size and reactivity of the aneurysms' thrombus. 22 23 The AngII mouse model of AAA is characterized by the development of an intramural thrombus (IMT) in the absence of an intraluminal thrombus. 24 Although FXIIa inhibition did not impede IMT formation ( Fig. 1E ), morphological analysis of those aneurysms containing an IMT demonstrated a more stable phenotype in animals receiving 3F7 versus the control, BM4. Specifically, the IMT of 3F7-treated animals with lower-limit small aneurysms showed thick deposition of collagen between IMTs and the vessel–lumen interface ( Fig. 1F ), which was not seen in either lower-limit aneurysms ( Fig. 1G ) or aneurysms ( Supplementary Fig. S2C , available in the online version) from the BM4 control group. The formation of this protective layer of collagen may offer a potential mechanism by which inhibiting FXIIa might protect from AAA rupture.
Inhibition of FXIIa Promotes Stable Atherosclerosis
Following the observation of reduced dilatation and rupture, and evidence of increased collagen content in AAA, we next explored the potential for 3F7 to promote stable atherosclerosis. We investigated the effect of FXIIa inhibition on the development of atherosclerosis in ApoE −/− mice fed a high-fat diet. We found that 3F7 decreased total atherosclerotic plaque size and necrotic core area ( Fig. 2A, B ; p = 0.0031; 0.0489) and increased total collagen content ( Fig. 2C ; p = 0.0169), as compared with those mice receiving isotype control antibody BM4. No significant changes in lipid profile were observed ( Supplementary Fig. S3 , available in the online version). This demonstrates the protective effect of blocking FXIIa, a finding consistent with a previous study demonstrating that genetic deficiency of FXII in FXII −/− ApoE −/− double-knockout mice reduced the development of atherosclerosis. 6
Fig. 2.
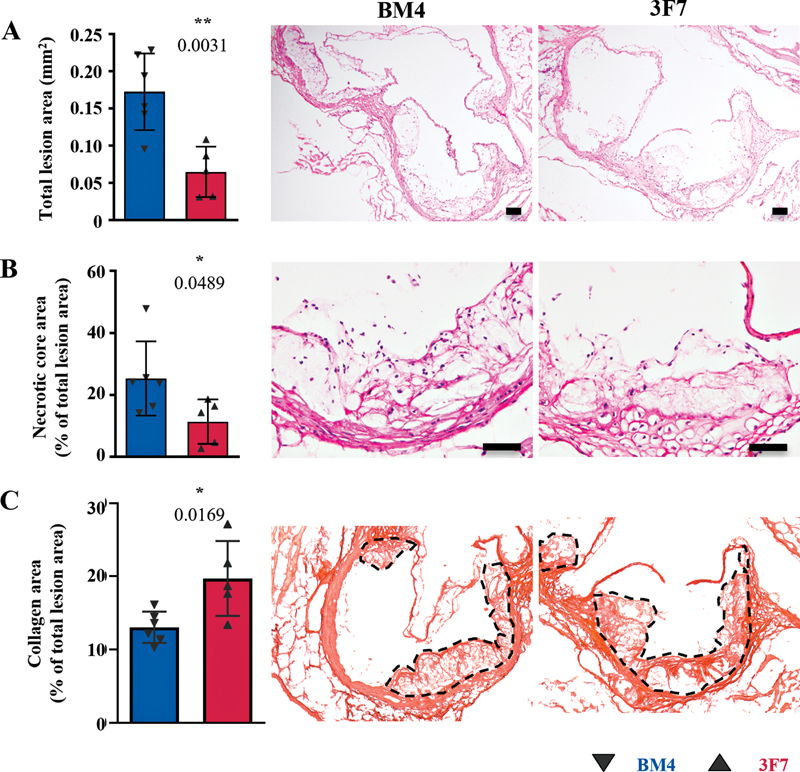
FXIIa inhibition attenuated the development of stable atherosclerosis. ApoE −/− mice were placed on an HFD for 8 weeks to develop stable atherosclerosis. ( A ) H&E was used to morphologically assess plaque size ( p = 0.0031), ( B ) necrotic core area ( p = 0.0489), and ( C ) the collagen content ( p = 0.0169) in stable plaques of the aortic sinus, as compared with the BM4 control–treated animals (BM4 n = 6; 3F7 n = 5). Values are mean ± SD. Scale bars = 200 µm. Assays were assessed using unpaired Student's t -tests. Interrupted lines indicate total lesion area used for collagen content analysis. Representative images show one valve of the aortic sinus only. All valves were included in the analysis. H&E, hematoxylin and eosin; HFD, high-fat diet; SD, standard deviation.
FXIIa Is Localized within the Microenvironment of Mouse-Unstable Atherosclerotic Plaque
To assess the efficacy of 3F7 in a more translationally relevant animal model of atherosclerosis, we utilized a TS mouse model, which is characterized by thin-capped, rupture-prone, unstable carotid artery plaques with a phenotype similar to the culprit lesions often responsible for ischemic events in patients. 17 Anatomical information regarding the location of the TS and the predefined area of plaque instability within this model can be found in the Methods section and Chen et al. 17
Prior to investigating the therapeutic capacity of 3F7 in the TS mouse model, we confirmed the accumulation of FXIIa within the unstable atherosclerotic plaques. Fluorescence (AF546)-labeled 3F7 was detected inside the unstable atherosclerotic plaques using both IF ( Fig. 3A, B ) and in vivo imaging system ( Fig. 3C, D ). This is an important finding both in regard to demonstrating a pathological activation of FXII in unstable plaque and also in regard to the suitability of FXIIa targeting as a selective therapeutic approach.
Fig. 3.
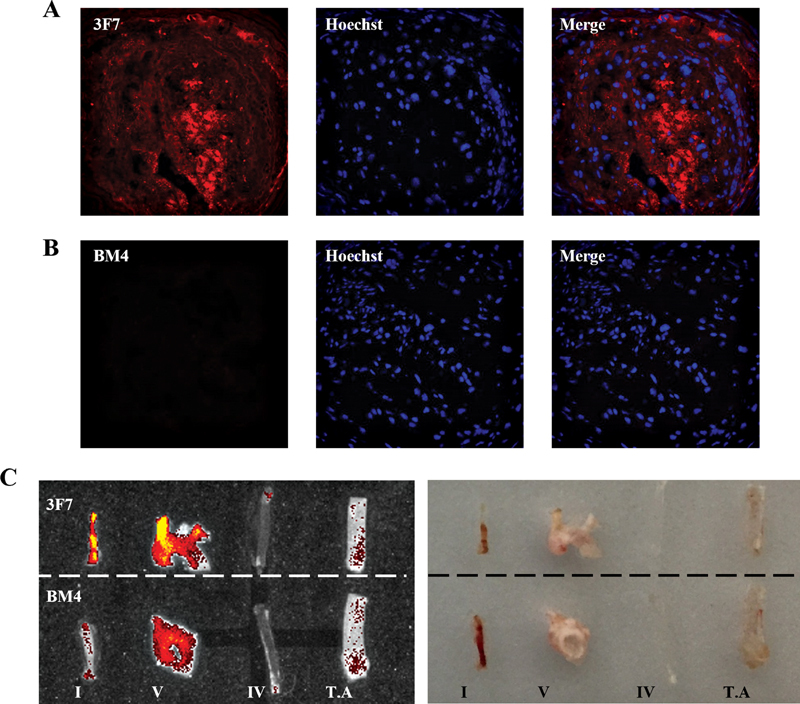
FXIIa accumulates abundantly in unstable atherosclerotic plaques as compared with stable plaques. TS mice were injected with fluorescently labeled (AF546) 3F7 or BM4 and the presence of FXIIa was detected in unstable and stable atherosclerotic plaques using IF microscopy and IVIS. Representative IF images show: ( A ) the presence of FXIIa detected by in vivo labeling within the unstable plaque of a TS mouse; ( B ) a section of unstable plaque from an animal injected with BM4 isotype control. The atherosclerotic vessels, both stable (segment V & TA) and unstable plaques (segment I), as well as healthy vessels (segment IV), were excised and imaged using IVIS ( left ) and brightfield ( right ). ( C ) Segment I shows strong accumulation of fluorescently labeled 3F7 but not the isotype control fluorescence antibody. Areas of stable atherosclerosis also reveal some FXIIa binding, but to a lesser extent. FXIIa, activated factor 12; IVIS, in vivo imaging system; TA, thoracic aorta; TS, tandem stenosis.
3F7 Stabilizes Vulnerable Atherosclerotic Plaques
Inhibition of FXIIa via administration of 3F7 began 24 hours after TS surgery. We assessed various markers of plaque stability. At the endpoint of this study, there were no differences in the body weight, spleen weight, or serum lipids between animals receiving 3F7 and BM4 ( Supplementary Fig. S4 , available in the online version). 3F7 administration decreased the total plaque area, necrotic core size, and lipid deposition within the vulnerable plaques, as compared with animals receiving BM4 ( Fig. 4A – C ; p = 0.0101; 0.0391; 0.0429). Furthermore, there was a marked increase in total collagen deposition within the vulnerable plaques of those animals receiving 3F7, as well as increases in cap thickness and cap-to-core ratio, two important measurements of plaque instability directly translatable to clinical measurements of plaque instability in humans ( Fig. 4D – F ; p = 0.0004; 0.0197; 0.0015). This increase in collagen deposition was mirrored by an increase in intimal smooth-muscle cells, another structural element supporting atherosclerotic plaque stability ( Fig. 4G ; p < 0.0001). Another important sign of plaque instability is intraplaque hemorrhage. The antibody TER119 specifically binds to the glycophorin A-associated protein (Ly-76) expressed on erythrocytes and so can be used to detect intraplaque hemorrhage. Mice treated with 3F7 showed markedly reduced staining of TER119 in segment I of the TS model as compared with those mice receiving the isotype control antibody BM4 ( Fig. 4H ; p = 0.0029), again strongly indicating the plaque-stabilizing effect of 3F7.
Fig. 4.
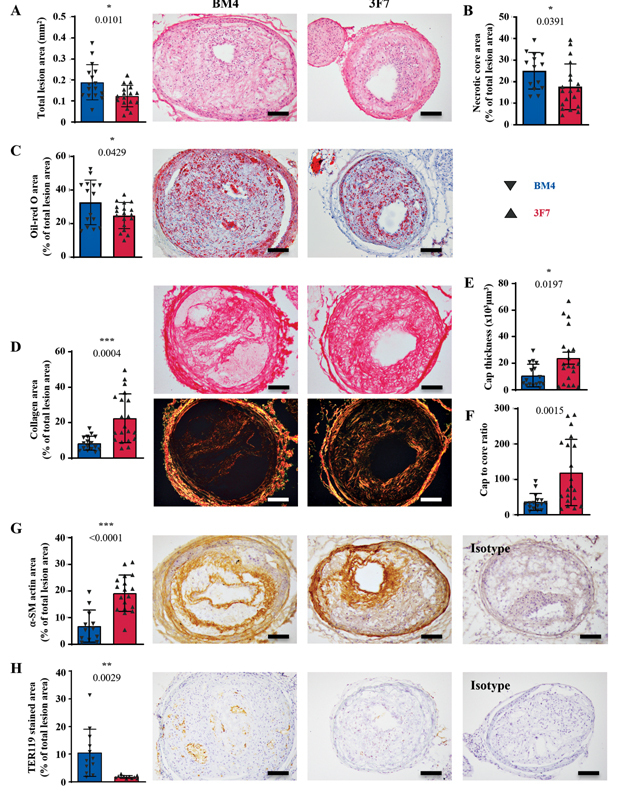
Inhibition of FXIIa via antibody 3F7 stabilizes vulnerable atherosclerotic plaques. ApoE −/− mice were placed on an HFD for 6 weeks prior to TS surgery. Following surgery, animals remained on the HFD for 7 weeks and 3F7 or BM4 isotype control was administered. ( A ) H&E was used to morphologically assess plaque size ( p = 0.0101; BM4 n = 16; 3F7 n = 17) and ( B ) necrotic core area ( p = 0.0391; BM4 n = 14; 3F7 n = 19) in unstable plaques, as compared with the BM4-treated animals. ( C ) Oil-red O staining was used to assess the lipid deposition within the plaques ( p = 0.0429; BM4 n = 15; 3F7 n = 18). Picro-Sirius Red was used to assess the degree of protective collagen deposition within the plaques, with histological examination performed using BF and polarized light. ( D ) The total intimal collagen deposition, ( E ) fibrous cap thickness, and ( F ) cap-to-core ratio, as compared with BM4-treated animals ( p -values = 0.0004; 0.0197; 0.0015; BM4 n = 16; 3F7 n = 20). ( G ) Intimal smooth-muscle cell deposition ( p = < 0.0001; BM4 n = 12; 3F7 n = 18) and ( H ) intra-plaque hemorrhage, as indicated by erythrocyte/TER119 staining ( p = 0.0029; BM4 n = 12; 3F7 n = 11). Isotype refers to the isotype control for the antibody used in the respective immunohistochemical stain. Assays were assessed using unpaired Student's t -tests. Values are mean ± SD. Scale bars = 200 µm. α-SM, α-smooth muscle; BF, brightfield; FXII, factor 12; FXIIa, activated factor 12; H&E, hematoxylin and eosin; HFD, high-fat diet; SD, standard deviation.
3F7 Administration Decreases Plaque Macrophage Content and Circulating Proinflammatory Markers, as well as Increasing Markers Associated with Fibrosis in Vulnerable Plaques
Dual inhibition of prothrombotic and inflammatory pathways is a unique feature of 3F7. The effect of FXIIa inhibition via 3F7 on inflammation was assessed locally using immunohistochemistry and systemically using a mouse cytokine/chemokine multiplex array.
Assessment of localized inflammation showed a marked decrease in VCAM-1 expression following 3F7 treatment ( Fig. 5A ; p = 0.0002), which correlated to a reduction in macrophages infiltrating into the plaque ( Fig. 5B ; p ≤ 0.0001). Multiplexing identified significant downregulation of circulating chemoattractants: monocyte chemoattractant protein-1 (MCP-1, CCL2), eotaxin-1 (CCL11), keratinocyte-derived chemokine (CXCL1), and lipopolysaccharide-induced CXC chemokine (CXCL5), in the plasma of animals treated with 3F7 ( Fig. 5C ; p = 0.0133; 0.0350; 0.0493; 0.0421). In addition, circulating proinflammatory markers IL-1α, IL-1β, IL-12p40, and IL-17 ( Fig. 5D ; p = 0.0335; 0.0499; 0.0485; 0.0347), and stimulating factors granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor were downregulated ( Fig. 5E ; p = 0.0459; 0.0234), while factors associated with wound healing and fibro-protection, IL-4, IL-13, and IL-5, were all significantly upregulated following treatment with 3F7 ( Fig. 5F ; p = 0.0491; 0.0410; 0.0055). Interestingly, the level of circulating BK as determined by liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS; Supplementary Fig. S5 , available in the online version) was not influenced by 3F7 administration.
Fig. 5.
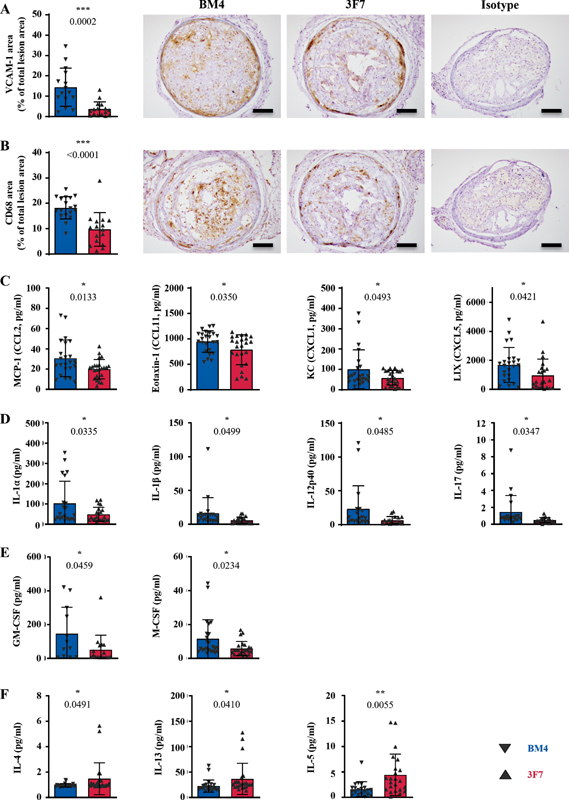
Inhibition of FXIIa via 3F7 reduces plaque-localized and systemic inflammation. Immunohistochemistry of unstable plaques shows: ( A ) reduction in VCAM-1 expression and ( B ) CD68+ macrophage infiltration by 3F7 administration ( p = 0.002; <0.0001; BM4 n = 15–18; 3F7 n = 16). Isotype refers to the isotype control for the antibody used in the respective immunohistochemical stain. In addition, further insights into the degree of inflammatory markers circulating in the sera were obtained: ( C ) the chemoattractants MCP-1 (CCL2), eotaxin-1 (CCL11), KC (CXCL1), and LIX (CXCL5) are significantly reduced ( p -values = 0.0133; 0.0350; 0.0493; 0.0421), ( D ) the proinflammatory markers IL-1α, IL-1β, IL-12p40, and IL-17 are significantly reduced ( p -values = 0.0335; 0.0499; 0.0485; 0.0347), ( E ) the stimulating factors GM-CSF and M-CSF ( p -values = 0.0459; 0.0234) and ( F ) the factors associated with wound healing and fibrosis, IL-4, IL-13, and IL-5 ( p -values = 0.0491; 0.0410; 0.0055), are also significantly reduced (BM4 n = 12–24; 3F7 n = 17–24). Values are mean ± SD. Assays were assessed using unpaired Student's t -tests. Scale bars = 200 µm. FXIIa, activated factor 12; GM-CSF, granulocyte-macrophage colony-stimulating factor; KC, keratinocyte-derived chemokine; LIX, LPS-induced CXC chemokine; MCP-1, monocyte chemoattractant protein-1; M-CSF, macrophage colony-stimulating factor; SD, standard deviation.
Discussion
Cardiovascular disease (CVD) remains a leading cause of mortality and morbidity globally. With the rising diabetes and obesity epidemic in the developed world, in addition to the significant risk of recurrent events for patients with CVD, there remains a pressing need for novel drugs for CVD treatment and prevention. Moreover, therapeutic options for patients with AAA remain limited to surgical options, which are often inappropriate for a significant proportion of patients with AAA and are also associated with high mortality rates. Therefore, given this unmet clinical need, nonsurgical preventive and treatment options for AAA are required. 25 The availability of therapies that have the potential to inhibit the development of atherosclerosis and to stabilize unstable atherosclerotic plaques, as well as rupture-prone AAAs, would represent a major clinical need. Atherosclerosis and AAA are both characterized by chronic inflammation, with single-cell RNA-sequencing studies beginning to highlight the significant diversity of immune cells in both diseases. 26 27 28 Similarly, thrombosis is also a central feature of both pathologies. 29 30 As a result, therapies targeting both inflammation and thrombosis are of key interest in cardiovascular research. Accordingly, recent evidence has highlighted that FXIIa plays an important role in mediating pathological thrombosis, in addition to possessing several previously unappreciated roles in regulating inflammation. Therefore, given the importance of the thromboinflammatory process in contributing to atherogenesis and AAA development, we investigated the efficacy of FXIIa inhibition as a potential approach for prevention and treatment of atherosclerosis and AAA.
For AAA, there is increasing evidence that the thrombotic burden is associated with the risk of aneurysm rupture. The highly proteolytic microenvironment of the thrombus is proposed to decrease wall strength and thereby increase the risk of aneurysm rupture. 22 23 31 In our study, although similar numbers of animals developed aneurysms and IMTs, the 3F7-treated cohort experienced less acute ruptures and developed smaller aneurysms in comparison to the control mice. Upon histological examination of collagen deposition within AAAs, an indicator of structural stability, we observed a thick, fibrous layer of collagen deposited at the IMT–vessel interface in animals treated with 3F7. Recently, a study by Moran et al 32 investigated the gene and protein profiles of both an ApoE −/− FXII −/− double-knockout and an ApoE −/− 3F7-treated mouse model. The authors reported decreases in ADAM-17, endothelial growth factor receptor, and the matrix metalloproteinases MMP-2 and MMP-9 expression, 32 correlating to reduced local inflammation and remodeling, as well as to the preservation of extracellular matrix integrity. As the luminal portion of the IMT is the most inflamed area of an aneurysm, containing the highest proportion of proinflammatory infiltrating cells, 33 the increased deposition of collagen at this interface is an exciting finding and might contribute to the increased rate of survival associated with 3F7 administration.
3F7 significantly attenuated the development of atherosclerosis in ApoE −/− mice. Most strikingly, pharmacological inhibition of FXIIa prevented the development of vulnerable atherosclerotic plaques in the TS model of plaque instability. Mice treated with 3F7 exhibited significantly decreased necrotic core size and lipid deposition. In accordance with our AAA data, we also observed marked increases in collagen deposition, cap thickness, and the cap-to-core ratio. These are particularly important findings, since it is well accepted that the extent of cross-linked collagen deposited within the cap region is proportional to the stability of the plaque and increases in cap thickness confer a more stable atherosclerotic phenotype. 34 Moreover, inhibition of FXIIa resulted in increases in intimal smooth-muscle cell content and almost eliminated intraplaque hemorrhage, further highlighting the compelling plaque-stabilizing effects of FXIIa inhibition. The remarkable magnitude of this plaque-stabilizing effect achieved by 3F7 can also be seen in comparison to the effects seen with the use of a myeloperoxidase inhibitor in the TS model, which despite reporting on only small plaque-stabilizing effects attracted broad interest. 35
Recent experimental data have highlighted the role of FXII/FXIIa in regulating inflammation, including direct regulation of neutrophil function and wound healing 36 in addition to a central role in the pathogenesis of experimental autoimmune encephalomyelitis. 37 These findings have served to highlight that pharmacological inhibition of FXIIa can achieve significant anti-inflammatory effects. In this regard, our findings that 3F7 administration significantly decreases VCAM-1 expression and CD68+ macrophage infiltration point to a potential role of FXIIa inhibition as an anti-inflammatory strategy for the prevention and potential treatment of atherosclerosis.
This concept is further supported by the fact that we observed downregulation of circulating cytokines IL-1β, IL-12p40, and MCP-1. These cytokines and chemoattractants play important roles in mediating monocyte/macrophage recruitment and differentiation into proinflammatory M1 macrophages, and, specifically for IL-1β, foam cell apoptosis, 38 and likely help explain the reduction in macrophage infiltration and overall disease attenuation. While the present study did not specifically examine the downstream signaling effects of FXIIa, our data are consistent with a recent report demonstrating that genetic deficiency of FXII afforded ApoE −/− mice protection from atherosclerosis. 6 Here, FXIIa was shown to stimulate the secretion of proinflammatory cytokines, including IL-1β and IL-12, from bone marrow–derived macrophages and antigen-presenting cells, which likely explains the beneficial anti-inflammatory effects of FXIIa inhibition. It is important to note that another important function of FXIIa relates to its ability to activate the kallikrein/kinin system (KKS) to yield the proinflammatory oligopeptide BK. However, we found no detectable differences in plasma BK levels between 3F7 and BM4-treated mice, suggesting that the proinflammatory effects of FXIIa in the context of atherosclerosis are either largely localized to the site of the lesion and any BK generated not detectable systemically, or independent of the KKS.
An outstanding issue pertaining to the role of FXII in mediating atherosclerosis relates to what activates FXII within the confines of an atherosclerotic plaque. While this was not a focus of our current study, it is likely that the activation of FXII in atherosclerotic plaques is linked to the multitude of physiological FXII activators that have been previously demonstrated to be abundant within atherosclerotic plaques. Indeed, extracellular traps, misfolded protein aggregates, and activated platelets have all been previously demonstrated to be present within plaques and are well-described physiological activators of FXII. 39 Further emphasizing the central role of FXII in mediating atherothrombosis, deficiency of FXII or FXIIa inhibition has been demonstrated to diminish thrombus formation on atherosclerotic plaque material ex vivo. 40 Moreover, a recent study investigating pharmacological inhibition of FXI not only implicated FXI/FXIa in the development of atherosclerosis and thereby indirectly supporting the pathological role of FXIIa within the plaque confines, 41 but also provides further evidence that therapeutic targeting of coagulation factors holds immense potential to impact cardiovascular clinical outcomes.
The TS mouse model representing unstable plaques as seen in patients is a unique preclinical tool both to develop diagnostic approaches for the detection of unstable plaques and for the development and testing of drugs for plaque-stabilizing effects. 42 For the latter, the promising effects of FXIIa inhibition hold great promise for clinical translation, particularly as first clinical applications, utilizing the anti-inflammatory and antithrombotic effects of FXIIa inhibition are currently trialed. However, our data also indicate that FXIIa targeting can be used for diagnostic approaches. Anti-FXIIa imaging was shown to be a potential means to identify unstable atherosclerotic plaques, which is a long sought-after diagnostic approach with the strong translational perspective of identifying patients at risk and ultimately preventing myocardial infarctions.
Together, our data demonstrate that inhibition of FXIIa impedes the development of atherosclerosis and stabilizes vulnerable atherosclerotic plaques, in addition to preventing AAA rupture in a process linked to its anti-inflammatory and stabilizing effects. These data, coupled with the antithrombotic benefits of FXIIa inhibition without interference with normal hemostasis, identify FXIIa inhibition as a potential novel preventative and therapeutic strategy for unstable atherosclerosis and AAA.
However, our study does have some limitations. While we show for the first time that the specific pharmacological targeting of FXIIa with 3F7 attenuated disease severity, any contributions of zymogen FXII to the observed pathology cannot be ascertained given the specificity of 3F7 for activated forms of FXIIa. As zymogen FXII is reported to directly influence innate immune functions and exerts mitogenic activity in endothelial and smooth muscle cells, 14 further studies are required to dissect the contributions of zymogen and activated forms of FXII. In addition, we used only male mice for our investigations which, although these diseases disproportionally affect males, may have led us to miss potential sex-specific effects of FXIIa inhibition. Follow-up studies will need to include female mice to exclude sex differences. Finally, our experimental models are designed to test for prophylactic benefits of 3F7 in developing AAA and atherosclerosis. Further investigations are required to establish the efficacy of 3F7 as a truly therapeutic approach. This includes ascertaining the optimal dose and administration interval.
Conclusion
We demonstrate that 3F7 administration results in the stabilization of both AAAs and vulnerable atherosclerotic plaques. 3F7 restricts the development of large aneurysms and results in a lower incidence of acute aortic rupture, potentially a result of the demonstrated increased collagen deposition between the IMT and the vessel lumen. In unstable atherosclerosis, we observed less unstable plaque development with 3F7 administration, including increased collagen and smooth-muscle cell density. This finding was accompanied by significant reductions in both systemic and local proinflammatory markers. The ability of 3F7 to prevent pathological thrombosis and reduce inflammation without impeding hemostasis is an additional unique and supportive feature of this potential therapeutic approach. Our preclinical data indicate that FXIIa inhibition has the potential to stabilize and prevent the rupture of AAAs and of vulnerable atherosclerotic plaques. Clinical trials are warranted to demonstrate the translatability of our preclinical data to clinically meaningful benefits in patients with AAA and atherosclerosis.
Acknowledgments
We thank the Monash Micro Imaging Platform, particularly Irena Carmichael and Betty Kouscousis, for their technical advice and support. Amy Searle received the 2020 Monash Faculty Indigenous Award for Doctoral Thesis Excellence.
Funding Statement
Funding This work was supported by the National Heart Foundation of Australia for A.K.S., Y.-C.C., J.D.M., and X.W.; by the German Research Foundation for M.W.; and by the National Health and Medical Research Council for J.D.M. and K.P. CSL Limited supported this research with an unrestricted research fund.
Conflict of Interest H.L., M.B., and C.P. are employees of CSL Limited. M.W.N. and P.R. are employees of CSL Behring Innovation GmbH. Y.-C.C., C.P., M.W.N., H.H., and K.P. are inventors on patent applications describing antibody-mediated anti-FXIIa therapies.
Author Contributions
Y.-C.C., C.P., X.W., H.H., and K.P. designed the study. A.K.S., Y.-C.C., M.W., J.D.M., A.C.M., J.N., P.K., M.T.K.Z., A.H., H.L., M.B., M.W.N., P.R., X.W., and H.H. were involved in the acquisition and interpretation or analysis of data. A.K.S. drafted the manuscript. Y.-C.C., M.W., J.D.M., J.N., A.B., C.P., X.W., and K.P. provided critical review of the intellectual content of the manuscript. All authors approved the final version of the manuscript prior to submission.
Equally contributing first authors.
Equally contributing senior authors.
What is known about the topic?
Coronary atherosclerosis and abdominal aortic aneurysm and their resulting complications of myocardial infarction and aortic rupture, respectively, are globally dominant causes of mortality and morbidity.
Medical therapies that can prevent and stabilize vulnerable atherosclerotic plaques, and rupture-prone AAAs are highly sought-after.
Activated coagulation factor XII (FXIIa) sits at the interface of both coagulation and inflammation.
What does this paper add?
Inhibition of FXIIa via a monoclonal antibody reduces the size and increases the stability of atherosclerotic plaques and abdominal aortic aneurysms.
FXII inhibition via 3F7 decreases markers of local and systemic inflammation in a mouse model of vulnerable plaque.
3F7 mAb and its derivatives warrant further testing as potential drug candidates for the prevention of myocardial infarction and the development and rupture of abdominal aortic aneurysms.
Supplementary Material
References
- 1.The World Health Organisation The top 10 causes of death worldwide 2016World Health Organization Fact Sheets. Published May 24, 2018. Accessed October 27, 2021 at:http://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death
- 2.Libby P, Buring J E, Badimon L. Atherosclerosis. Nat Rev Dis Primers. 2019;5(01):56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 3.Koch M, Zernecke A. The hemostatic system as a regulator of inflammation in atherosclerosis. IUBMB Life. 2014;66(11):735–744. doi: 10.1002/iub.1333. [DOI] [PubMed] [Google Scholar]
- 4.Borissoff J I, Spronk H MH, ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364(18):1746–1760. doi: 10.1056/NEJMra1011670. [DOI] [PubMed] [Google Scholar]
- 5.Borissoff J I, Heeneman S, Kilinç E. Early atherosclerosis exhibits an enhanced procoagulant state. Circulation. 2010;122(08):821–830. doi: 10.1161/CIRCULATIONAHA.109.907121. [DOI] [PubMed] [Google Scholar]
- 6.Vorlova S, Koch M, Manthey H D. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost. 2017;117(01):176–187. doi: 10.1160/TH16-06-0466. [DOI] [PubMed] [Google Scholar]
- 7.Shnerb Ganor R, Harats D, Schiby G. Factor XI deficiency protects against atherogenesis in apolipoprotein E/factor XI double knockout mice. Arterioscler Thromb Vasc Biol. 2016;36(03):475–481. doi: 10.1161/ATVBAHA.115.306954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olie R H, van der Meijden P EJ, Ten Cate H. The coagulation system in atherothrombosis: implications for new therapeutic strategies. Res Pract Thromb Haemost. 2018;2(02):188–198. doi: 10.1002/rth2.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran C S, Seto S-W, Krishna S M. Parenteral administration of factor Xa/IIa inhibitors limits experimental aortic aneurysm and atherosclerosis. Sci Rep. 2017;7:43079. doi: 10.1038/srep43079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller F, Mutch N J, Schenk W A. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(06):1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maas C, Govers-Riemslag J WP, Bouma B. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118(09):3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Brühl M-L, Stark K, Steinhart A. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(04):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Meijden P EJ, Munnix I CA, Auger J M. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114(04):881–890. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 14.Renné T, Stavrou E X. Roles of factor XII in Innate Immunity. Front Immunol. 2019;10:2011. doi: 10.3389/fimmu.2019.02011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Worm M, Köhler E C, Panda R. The factor XIIa blocking antibody 3F7: a safe anticoagulant with anti-inflammatory activities. Ann Transl Med. 2015;3(17):247. doi: 10.3978/j.issn.2305-5839.2015.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsson M, Rayzman V, Nolte M W. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6(222):222ra17. doi: 10.1126/scitranslmed.3006804. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y-C, Bui A V, Diesch J. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res. 2013;113(03):252–265. doi: 10.1161/CIRCRESAHA.113.301562. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y C, Rivera J, Peter K. Tandem stenosis to induce atherosclerotic plaque instability in the mouse. Methods Mol Biol. 2015;1339:333–338. doi: 10.1007/978-1-4939-2929-0_23. [DOI] [PubMed] [Google Scholar]
- 19.Koay Y C, Chen Y-C, Wali J A. Plasma levels of TMAO can be increased with “healthy” and “unhealthy” diets and do not correlate with the extent of atherosclerosis but with plaque instability. Cardiovasc Res. 2021;117(02):435–449. doi: 10.1093/cvr/cvaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinosa M, Su G, Salmon M D.Resolvin D1 decreases abdominal aortic aneurysm formation by inhibiting NETosis in a mouse model J Vasc Surg 201868(6S):93S–103S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meher A K, Spinosa M, Davis J P. A novel role of IL-1β in NETosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2018;38(04):843–853. doi: 10.1161/ATVBAHA.117.309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haller S J, Crawford J D, Courchaine K M. Intraluminal thrombus is associated with early rupture of abdominal aortic aneurysm. J Vasc Surg. 2018;67(04):10510–10580. doi: 10.1016/j.jvs.2017.08.069. [DOI] [PubMed] [Google Scholar]
- 23.Koole D, Zandvoort H JA, Schoneveld A. Intraluminal abdominal aortic aneurysm thrombus is associated with disruption of wall integrity. J Vasc Surg. 2013;57(01):77–83. doi: 10.1016/j.jvs.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Sénémaud J, Caligiuri G, Etienne H, Delbosc S, Michel J-B, Coscas R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2017;37(03):401–410. doi: 10.1161/ATVBAHA.116.308534. [DOI] [PubMed] [Google Scholar]
- 25.Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019;16(04):225–242. doi: 10.1038/s41569-018-0114-9. [DOI] [PubMed] [Google Scholar]
- 26.Zernecke A, Winkels H, Cochain C. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127(03):402–426. doi: 10.1161/CIRCRESAHA.120.316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez D M, Rahman A H, Fernandez N F. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25(10):1576–1588. doi: 10.1038/s41591-019-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao G, Lu H, Chang Z. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. 2021;117(05):1402–1416. doi: 10.1093/cvr/cvaa214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuivaniemi H, Ryer E J, Elmore J R, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13(09):975–987. doi: 10.1586/14779072.2015.1074861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cameron S J, Russell H M, Owens A P., III Antithrombotic therapy in abdominal aortic aneurysm: beneficial or detrimental? Blood. 2018;132(25):2619–2628. doi: 10.1182/blood-2017-08-743237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vorp D A, Lee P C, Wang D HJ. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J Vasc Surg. 2001;34(02):291–299. doi: 10.1067/mva.2001.114813. [DOI] [PubMed] [Google Scholar]
- 32.Moran C S, Seto S-W, Biros E. Factor XII blockade inhibits aortic dilatation in angiotensin II-infused apolipoprotein E-deficient mice. Clin Sci (Lond) 2020;134(09):1049–1061. doi: 10.1042/CS20191020. [DOI] [PubMed] [Google Scholar]
- 33.Wilson J S, Virag L, Di Achille P, Karšaj I, Humphrey J D. Biochemomechanics of intraluminal thrombus in abdominal aortic aneurysms. J Biomech Eng. 2013;135(02):21011. doi: 10.1115/1.4023437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansson G K, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278(05):483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rashid I, Maghzal G J, Chen Y-C. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur Heart J. 2018;39(35):3301–3310. doi: 10.1093/eurheartj/ehy419. [DOI] [PubMed] [Google Scholar]
- 36.Stavrou E X, Fang C, Bane K L. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. 2018;128(03):944–959. doi: 10.1172/JCI92880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Göbel K, Pankratz S, Asaridou C-M. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun. 2016;7:11626. doi: 10.1038/ncomms11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramji D P, Davies T S. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26(06):673–685. doi: 10.1016/j.cytogfr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII contact activation. Semin Thromb Hemost. 2017;43(08):814–826. doi: 10.1055/s-0036-1598003. [DOI] [PubMed] [Google Scholar]
- 40.Kuijpers M JE, van der Meijden P EJ, Feijge M AH. Factor XII regulates the pathological process of thrombus formation on ruptured plaques. Arterioscler Thromb Vasc Biol. 2014;34(08):1674–1680. doi: 10.1161/ATVBAHA.114.303315. [DOI] [PubMed] [Google Scholar]
- 41.Ngo A TP, Jordan K R, Mueller P A. Pharmacological targeting of coagulation factor XI mitigates the development of experimental atherosclerosis in low-density lipoprotein receptor-deficient mice. J Thromb Haemost. 2021;19(04):1001–1017. doi: 10.1111/jth.15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noonan J, Bobik A, Peter K. The tandem stenosis mouse model: towards understanding, imaging, and preventing atherosclerotic plaque instability and rupture. Br J Pharmacol. 2021:1–19. doi: 10.1111/bph.15356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.