
Figure 1.
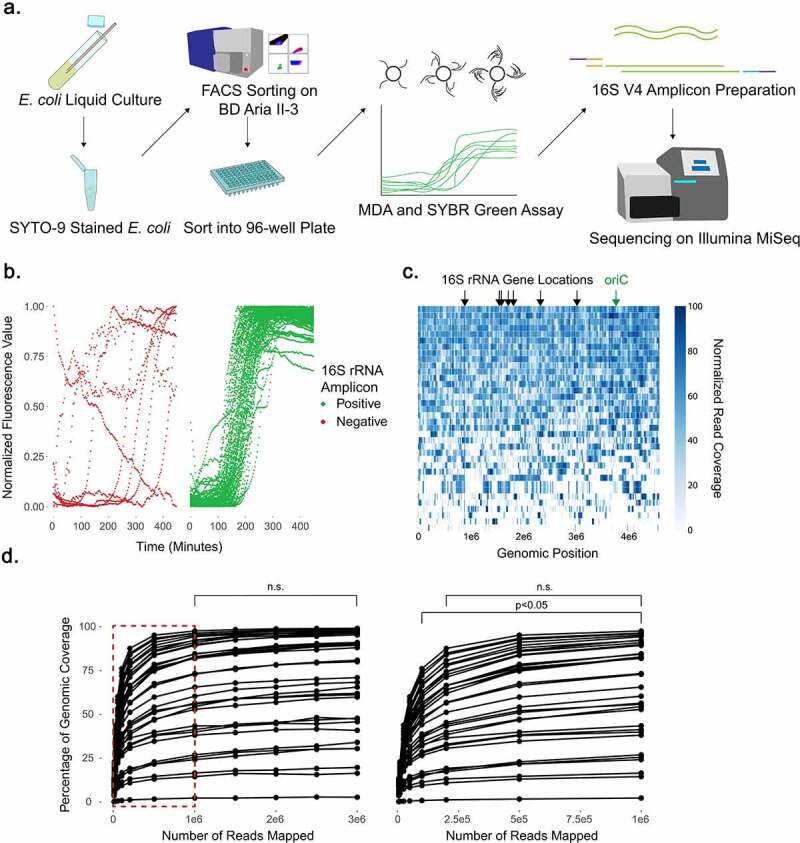
Near-complete E. coli genomes can be obtained from single cells, though increasing sequencing depth fails to resolve gaps in coverage. (a) Schematic for single-cell sorting and analysis of E. coli. Cells were stained with SYTO-9 nucleic acid stain, sorted, and then amplified by multiple displacement amplification (MDA) in a reaction tracked with SYBR Green nucleic acid stain. The V4 region of the 16S rRNA gene was amplified, and 16S V4 amplicon-positive samples were further shotgun sequenced on the Illumina MiSeq platform. (b) SYBR Green fluorescence values during MDA, grouped by subsequent positive or negative 16S V4 PCR result. (c) Read coverage of 36 individual cells mapped against the reference genome by bowtie2. Read coverage displayed as a heatmap with each bp represented. Overall genomic coverage ranged from 4.1% to 99.6%. Locations of seven 16S rRNA genes and origin of replication (oriC) on the genome are indicated. (d) Comparison of genomic coverage to number of reads used for mapping. Reads were subsampled from the original read set using bbtools and re-mapped by bowtie2 to the reference genome. Subsampling and re-mapping were performed 10 times for each sample to a maximum of 3e6 reads per cell. Statistical comparison at all depths was performed by ANOVA and Tukey’s test; n.s. = non-significant. The boxed region in the left plot is shown expanded on the right.