

Abstract
The inteleukin-36 (IL-36) family, including IL-36α, IL-36β, IL-36γ, and IL-36 receptor antagonist (IL-36Ra), belong to the IL-1 super-family. It was reported that IL-36 plays a role in immune diseases. However, it remains unclear how IL-36 regulates inflammation. To determine the role of IL-36/IL-36R signaling pathways, we established an acute hepatitis mouse model (C57BL/6) by intravenous injection of the plant lectin concanavalin A (Con A). We found that the levels of IL-36 were increased in the liver following Con A injection. Our results demonstrated the infiltrated neutrophils, but not the hepatocytes were the main source of IL-36 in the liver. Using the IL-36R−/− mouse model (H-2b), we surprisingly found that the absence of IL-36 signals led to aggravated liver injury, as evidenced by increased mortality, elevated serum ALT and AST levels and severe liver pathological changes. Further investigations demonstrated that a lack of IL-36 signaling induced intrahepatic activation of CD4+ and CD8+ T lymphocytes and increased the production of inflammatory cytokines. In addition, IL-36R−/− mice had reduced Treg cell numbers and chemokines in the liver. Together, our results from the mouse model suggested a vital role of IL-36 in regulating T cell function and homeostasis during liver inflammation.
Keywords: IL-36, acute hepatitis, Con A, T cells, Foxp3
INTRODUCTION
Interleukin (IL)-36 is a novel member of the IL-1 family and is involved in innate and acquired immune responses. It includes three agonist ligands (IL-36α, β and γ, previously known as IL-1F6, IL-1F8, IL-1F9, respectively) that can bind to heterodimeric receptor complexes, including the IL-36 receptor (IL-36R, also known as IL-1RL2) and co-receptor IL-1 receptor accessory protein (IL-1RAcP) (1–3), which induces inflammatory mediators through MyD88-, MAPK- and NF-κB-dependent signaling pathways (4–6). IL-36 receptor antagonist (IL-36Ra), is an antagonist ligand that can bind to IL-36R. It prevents the recruitment of co-receptor IL-1 receptor accessory protein (IL-1RAcP) (1–3) and doesn’t induce any pro-inflammatory responses (7). In addition, IL-38 (also called IL-1F10) can also bind to IL-36R and exert antagonistic effects similar to those of IL-36Ra (8). Immune cells, including neutrophils, monocytes, macrophages, dendritic cells (DCs), and T cells, can express IL-36R and respond to the stimulation of IL-36 ligands to play immune regulatory functions (9–13).
IL-36 plays a critical role in autoimmune diseases. IL-36 and its receptors can be detected in the skin, lung, gastrointestinal tract, brain and other tissues (14–17). It is well known that IL-36 is highly expressed in psoriatic lesions (14, 18), while IL-36R knockout (IL-36R−/−) mice are resistant to psoriasis-like dermatitis (19). Specific mutations in the gene encoding the IL-36Ra is closely associated with generalized pustular psoriasis (20, 21). Inhibition of the IL-36 signaling pathway has been proved to be an effective therapeutic way for psoriasis (22). In addition, IL-36α and IL-36γ were elevated in mucosal biopsies from patients with inflammatory bowel disease (IBD) (16, 23, 24) and promoted pro-inflammatory cell infiltration into the inflamed colon lamina propria (23) by inducing the expression of CXC chemokines in intestinal epithelial cells (16). IL-36R−/− mice showed less severe clinical manifestations in dextran sodium sulfate (DSS)-induced colitis (23). However, their recovery and wound healing of the intestinal epithelium following DSS-induced damage were defective as compared to wild-type (WT) mice (24, 25). IL-36R signaling can stimulate fibroblast activation and promote epithelial cell proliferation through the MyD88 signaling pathway in chronic intestinal inflammation, which can be reversed when IL-36R is inhibited or knocked out (25, 26). Hence, the IL-36/IL-36R signaling pathway showed an additional protective mechanism in intestinal diseases.
The role of IL-36 in liver diseases is not entirely understood. In an analgesic acetaminophen (APAP)-induced hepatitis mouse model, hepatic IL-36γ and its primary downstream chemokine target CCL20 were detectable. The application of IL-36Ra decreased CCL20 expression but increased liver damage in the late phases of liver injury (27). Lower IL-36α expression was found in poorly differentiated hepatocellular carcinoma, and correlated with tumor size, histological differentiation, tumor stage, vascular invasion, and survival rates (28). Despite a wealth of evidence on IL-36/IL-36R signaling in immune regulation and autoimmune diseases, their roles in Con A-induced acute hepatitis and hepatic T cell homeostasis are less well understood.
Using a Con A-induced murine model of acute hepatitis, we found increased IL-36 cytokine levels in the murine liver. We demonstrated that leukocytes, but not hepatocytes, were the main source of IL-36 in the murine liver. Among the different immune cell subsets, intrahepatic neutrophils were the main source of IL-36. Importantly, deficiency of IL-36 signaling resulted in lower survival rates, higher serum ALT and AST levels and increased hepatic necrosis. Moreover, IL-36R−/− animals exhibited exaggerated T cell responses as evidenced by increased hepatic infiltration of effector T cells and enhanced inflammatory cytokine production in the liver. Interestingly, the numbers of Treg cells were lower in the liver of IL-36R−/− mice compared with those of WT mice, suggesting that IL-36 may play a role in T cell function and homeostasis in Con A-induced hepatitis.
MATERIALS AND METHODS
Animals and treatment
C57BL/6 (B6) mice were purchased from Jackson Laboratory. The IL-36R−/− mouse breeding pairs were obtained from Amgen under a Material Transfer Agreement and housed at the animal facility of the University of Texas Medical Branch (UTMB), Galveston, TX. A single i.v. Con A injection (12 mg/kg body weight; Sigma-Aldrich, St. Louis, MO) was performed to induce T cell-mediated hepatitis (29, 30). All mice were maintained and bred under specific pathogen-free conditions. Seven- to ten-week-old male mice were used in our study. All experiments were reviewed and approved by the UTMB Institutional Animal Care and Use Committees.
Antibodies and reagents
All fluorochrome-labeled monoclonal antibodies (mAbs) and their corresponding isotype controls were purchased from Thermo Fisher Scientific (San Diego, CA) and Biolegend (San Diego, CA): Allophycocyanin-conjugated anti-IFN-γ (XMG1.2), PE-Cy7-conjugated anti-CD3 (17A2), Alexa Flour700-conjugated anti-CD3 (17A2), Pacific Blue-conjugated anti-CD4 (RM4–5), FITC-conjugated anti-CD4 (GK1.5), Allophycocyanin-eFlour780-conjugated anti-CD4 (RM4–5), PerCp-Cy5.5-conjugated anti-CD8 (53–6.7), Allophycocyanin-eFlour780-conjugated anti-CD8 (53–6.7), PerCp-eFlour710-conjugated anti-TNF-α (MP6-XT22), BV711-conjugated anti-CD44 (IM7), Alexa Fluor700-conjugated anti-CD11b (M1/70), PE-conjugated anti-CD69 (H1.2F3), PE-conjugated anti-CD45R/B220 (RA3–6B2), PE-CF594-conjugated anti-NK1.1 (PK136), BV421-conjugated anti-F4/80 (T45–2342), Alexa-Fluor488-conjugated anti-Ly-6G (1A8), PE-conjugated Foxp3 (FJK-16s), Allophycocyanin-conjugated anti-CD62L (MEL-14), APC anti-mouse IgG1 antibody (RMG1–1) and purified anti-CD16/32 (2.4G2). Fixable Viability Dye efluor 506 was used to exclude dead cells. Mouse IL-36α antibody (AF2297) was purchased from R&D system. Mouse IL-36γ antibody (PA5–99822) was purchased from Thermo Fisher Scientific. Anti-β-actin (#4970) was purchased from Cell Signaling Technology. Mouse TGF-β, IL-2, anti-IFN-γ, recombinant mouse IL-36α, IL-36β and IL-36γ were purchased from Biolegend.
H&E and serum biochemical analysis
Livers of Con A-administrated mice were collected, immediately fixed with 10% formalin, embedded in paraffin, and cut into 5-μm sections. Paraffin-embedded sections were stained with H&E for histological evaluation of necrotic areas. Images were obtained with a digital microscope (Olympus Microscopes, Tokyo, Japan), and the surface area of the necrotic regions was measured using Image J software. Necrotic regions were counted according to a previous method (31). Briefly, the area of whole tissue section was marked as A, and the area of each of the necrotic areas was marked as B. The sum of areas of the whole tissue section is designated as At (A total) (At = A1 + A2 +… An). The sum of necrotic regions in the whole tissue section is designated as Bt (B total) (Bt = B1 + B2 +… Bm). Hepatic necrosis was assessed in each section as the percentage of liver parenchyma with necrotic damage using the following formula: N = (Bt / At) × 100. N represents the percentage (%) of necrotic area in the whole tissue section. Serum AST and ALT were measured by an automatic biochemistry analyzer (Beckman Coulter) in the Department of Clinical Chemistry, UTMB.
Isolation of intrahepatic lymphocytes (IHLs)
Mouse IHLs were isolated, according to our previous method with slight modifications (32). In brief, the inferior vena cava was cut above the diaphragm, and the liver was perfused with cold PBS through the portal vein. Liver tissues were collected and digested in RPMI 1640 containing collagenase IV (0.05%; Roche Applied Science, Indianapolis, IN) at 37°C for 30 min. After digestion, cell suspensions were passed through 70-μm nylon cell strainers to remove aggregates and yield single-cell suspensions. IHLs were purified by centrifugation (400 g) at room temperature for 30 min over a 30/70% discontinuous Percoll gradient (Sigma-Aldrich St. Louis, MO). The IHLs were collected from the interphase, thoroughly washed, and resuspended in complete RPMI 1640 containing 10% FBS (Hyclone, Logan, UT). The absolute numbers of IHLs per liver were counted by a light microscopy. The relative percentages of cell populations were analyzed by flow cytometry, and the absolute numbers of these lymphocyte subpopulations were calculated by their percentages and IHL numbers.
Flow cytometry analysis
For surface staining, cells were first incubated with FcγR blocker (CD16/32) at room temperature for 5 min, followed by staining with fluorochrome-labeled Abs at 4°C for 30 min in the dark. For intracellular staining, cells were incubated with PMA (50 ng/mL), ionomycin (750 ng/mL), and Brefeldin A (BD Biosciences) for 5 h. After incubation, cells were collected for surface staining, followed by fixation and permeabilization using a fixation/permeabilization kit (Thermo Fisher Scientific). Incubation of intracellular Abs was performed at 4°C for 60 min in the dark. Samples were processed on a LSRII FACS Fortessa (Becton Dickinson, San Jose, CA, USA), and data were analyzed using FlowJo X software (Tree Star, Ashland, OR).
Hepatocyte isolation
Hepatocytes were isolated by the collagenase perfusion technique (33). Mouse livers were perfused through the vena cava with warm HBSS, followed by perfusion with 30 mL of warm collagenase IV (0.05%; Roche Applied Science, Indianapolis, IN). During perfusion by portal vein, the vena cava was periodically clamped for 10 s. The liver was gently placed in a petri dish on ice and opened with a forceps. Liver cells were resuspended in DMEM, and spun down at 50 g × 3 times, and again with 25% Percoll solution (Sigma) at 400 g for 10 min. Cells were washed and resuspended with DMEM. Cell viability was determined by trypan blue dye exclusion.
Quantitative reverse transcriptase-PCR (qRT-PCR)
Total RNA was extracted using a RNeasy Mini Kit, according to the instructions (Qiagen, Valencia, CA). The synthesis of cDNA was performed using an iScript Reverse Transcription Kit (Bio-Rad, Hercules, CA). cDNA was amplified in a 10-μl reaction mixture containing 5 μl of iTaq SYBR Green Supermix (Bio-Rad) and 5 μM each of gene-specific forward and reverse primers. The PCR assays were denatured for 30 s at 95°C, followed by 40 cycles of 15 s at 95°C, and 60 s at 60°C, utilizing the CFX96 Touch real-time PCR detection system (Bio-Rad). Relative quantitation of mRNA expression was calculated using the 2−ΔΔCt method. The primers are listed in Table S1.
Western blot analysis and ELISA assay
Total protein was extracted using a RIPA lysis buffer (Cell Signaling Technology) in the presence of a phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentrations were measured using a BCA Protein Assay Kit (Pierce). Protein samples were separated by SDS-PAGE (4–15%) and electro-transferred onto a PVDF membrane (Bio-Rad), which was then blocked with 5% BSA for 60 min. The membrane was incubated with primary Abs overnight at 4°C. After incubation, the membrane was washed 3 times with TBST, incubated with secondary Abs for 60 min at room temperature and developed using the ECL western blotting substrate reagent (Thermo Fisher Scientific). The signal intensity was analyzed by Image J and normalized to β-actin. Hepatic cytokine levels were assayed using specific ELISA kits according to the manufacturer’s instructions. ELISA kits specific to mouse TNF-α, IFN-γ, IL-10, IL-4, and IL-17A were purchased from Biolegend. The IL-1β ELISA kit was purchased from Thermo Fisher Scientific, and the IL-6 ELISA kit was purchased from R&D Systems.
Chemotaxis of Treg cells
Naive CD4+ T cells (CD4+ CD44−) were purified from the spleens by magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany), and cultured in anti-CD3/CD28 coated plates under the Treg cell culture condition (10 ng/mL of IL-2, 1000 ng/mL of anti-IFN-γ, and 0.5 ng/mL of TGF-β) for 5 days. A total of 3×106 differentiated Treg cells in 200 μL was placed in each Transwell insert (5.0-μm pore, 24-well format, Corning Costar). Transwell inserts were placed in 24-well plates containing 500 μL of RPMI 1640 medium without FBS. Recombinant IL-36 (200 ng/mL) were added in the lower chamber. Cells were allowed to migrate for 3, 6 and 24 h in a 5% CO2 incubator at 37°C. The numbers of migrated cells to the lower chambers were counted at indicated time-points.
PrimeFlow RNA assay
PrimeFlow RNA assay (Thermo Fisher Scientific, San Diego, CA) was performed on IHLs. The specific target probe set for mouse IL-36γ (Assay ID # VB1–3046123-PF, type 1) was designed by and purchased from Thermo Fisher Scientific. When applicable, cell surface staining was performed for 30 min at 4°C with fluorochrome-conjugated antibodies. Cells were then fixed for 30 min at 4°C. After permeabilization, cells were fixed a second time for 1 h at RT with Fixation buffer 2. A hybridization step was performed by incubating cells with the appropriate target probe sets for 2 h at 40°C. Samples were stored over night at 4°C in dark. Next day, pre-amplification and amplification of the hybridization were performed by two consecutive incubations of 1.5 h at 40°C with the pre-Amplification mix and subsequently the Amplification mix. Finally, cells were incubated with the label probe sets for 1 h at 40°C. Samples were processed on a LSRII FACS Fortessa (Becton Dickinson, San Jose, CA, USA), and data were analyzed using FlowJo X software (Tree Star, Ashland, OR).
Proliferation of Treg cells
Total CD4+ T cells were purified from spleens by magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany), and stained with CFSE (5 μM, Biolegend, San Diego, CA) for 10 min at room temperature. Ten volumes of ice-cold culture medium with 10% FBS were added to quench any dye remaining in the buffer, and cells were washed twice with culture medium. Cells were then cultured in an anti-mouse CD3/CD28 antibody coated plate in the presence of IL-2 (20 ng/mL), with or without IL-36 (200 ng/mL). After 3 days of incubation, cell proliferation was determined by measuring CFSE fluorescence using flow cytometry.
Statistical analysis
All data were analyzed with GraphPad Prism software. The data are shown as the mean ± SEM. A two-tailed Student’s T-test is used for comparisons between two groups. Significance of the survival rates was assessed by Chi-square test. *, **, ***, and **** represents P-value<0.05, < 0.01, < 0.001 or < 0.0001, respectively.
RESULTS
Hepatic IL-36 expression was upregulated in Con A-induced hepatitis
It has been reported that IL-36 levels are elevated in several inflammatory diseases (14, 18, 23, 27); however, the pattern of IL-36 expression in acute hepatitis remains unclear (15, 18). We used a Con A-induced liver injury murine model, and observed a significant upregulation of hepatic IL-36α, IL-36γ, and IL-38 at 12 and 24 h post-Con A injection, with a modest increase in IL-36β at 12 h (Fig. 1A). The levels of IL-36Ra didn’t increase until 24 h post-Con A injection (Fig. 1A). No significant changes in IL-36R were detected at either 12 or 24 h (Fig. 1A). We further noted increased protein levels of IL-36α and IL-36γ in the livers of Con A-injected mice compared with those of control animals at 12 and 24 h (Fig. 1B). Hence, those results showed that IL-36 cytokines were elevated in the murine model of acute hepatitis.
Figure 1. Hepatic IL-36 expression was upregulated in Con A-induced hepatitis.
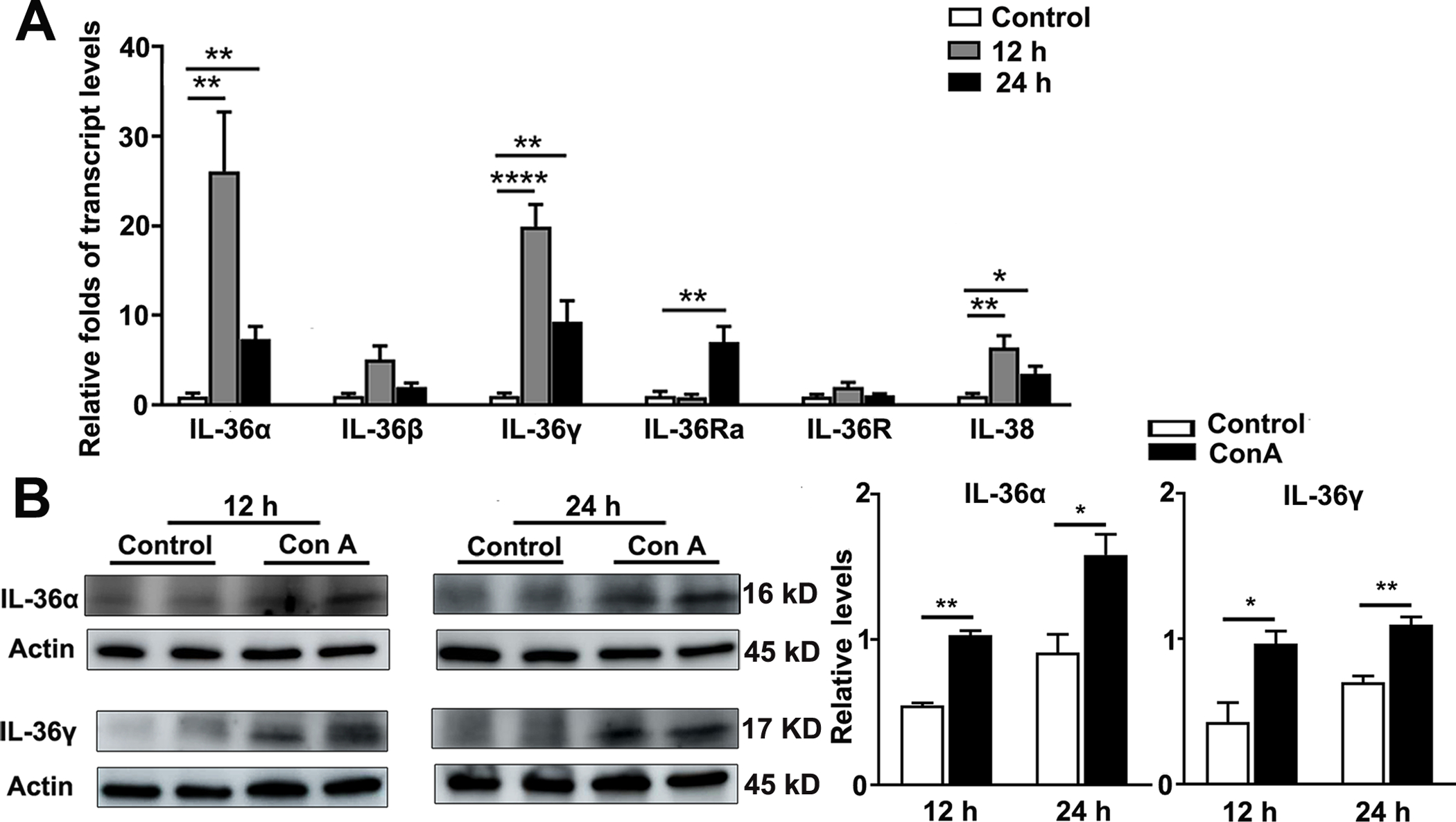
C57BL/6 mice were i. v. injected with Con A (12 mg/kg). PBS-injected mice were used as controls. Liver tissues were collected at 12 and 24 h following Con A injection. (A) Transcript and (B) protein levels of IL-36 family members in the livers were analyzed by qRT-PCR and western blot, respectively. n= 3–6 samples/group from single animal experiments. The data are shown as mean ± SEM of each group from single experiments and are representative of three animal experiments performed. Two-tailed unpaired T test was used for statistical analysis. *p < 0.05; **p < 0.01; **** p < 0.0001.
Infiltrated neutrophils were the main source of IL-36 in the liver following Con A injection
IL-36 proteins are predominately expressed in skin keratinocytes and bronchial epithelial cells (18, 34). Besides this, several kinds of immune cells, including T cells, plasma cells and macrophages are considered the source of IL-36 in the colonic mucosa in IBD (16, 25). To investigate the source of IL-36 in the liver, we perfused the liver with PBS and isolated hepatocytes and intrahepatic leukocytes from control and Con A-injected mice. We found that leukocytes expressed markedly higher levels of IL-36 genes, including IL-36α, IL-36β and IL-36γ compared with those of hepatocytes (Fig. 2A). Furthermore, the transcript levels of IL-36α, IL-36β and IL-36γ were elevated in intrahepatic leukocytes at 12 h after Con A injection compared with the control group (Fig. 2A). To investigate what types of immune cells are the main sources of IL-36, we used the PrimeFlow RNA assay and identified that neutrophils were the main source of IL-36γ in the liver (Fig. 2B). Therefore, our data indicated that intrahepatic neutrophils were the main source of IL-36 in the liver following Con A injection.
Figure 2. Infiltrated neutrophils were the main source of IL-36 in the liver following Con A injection.
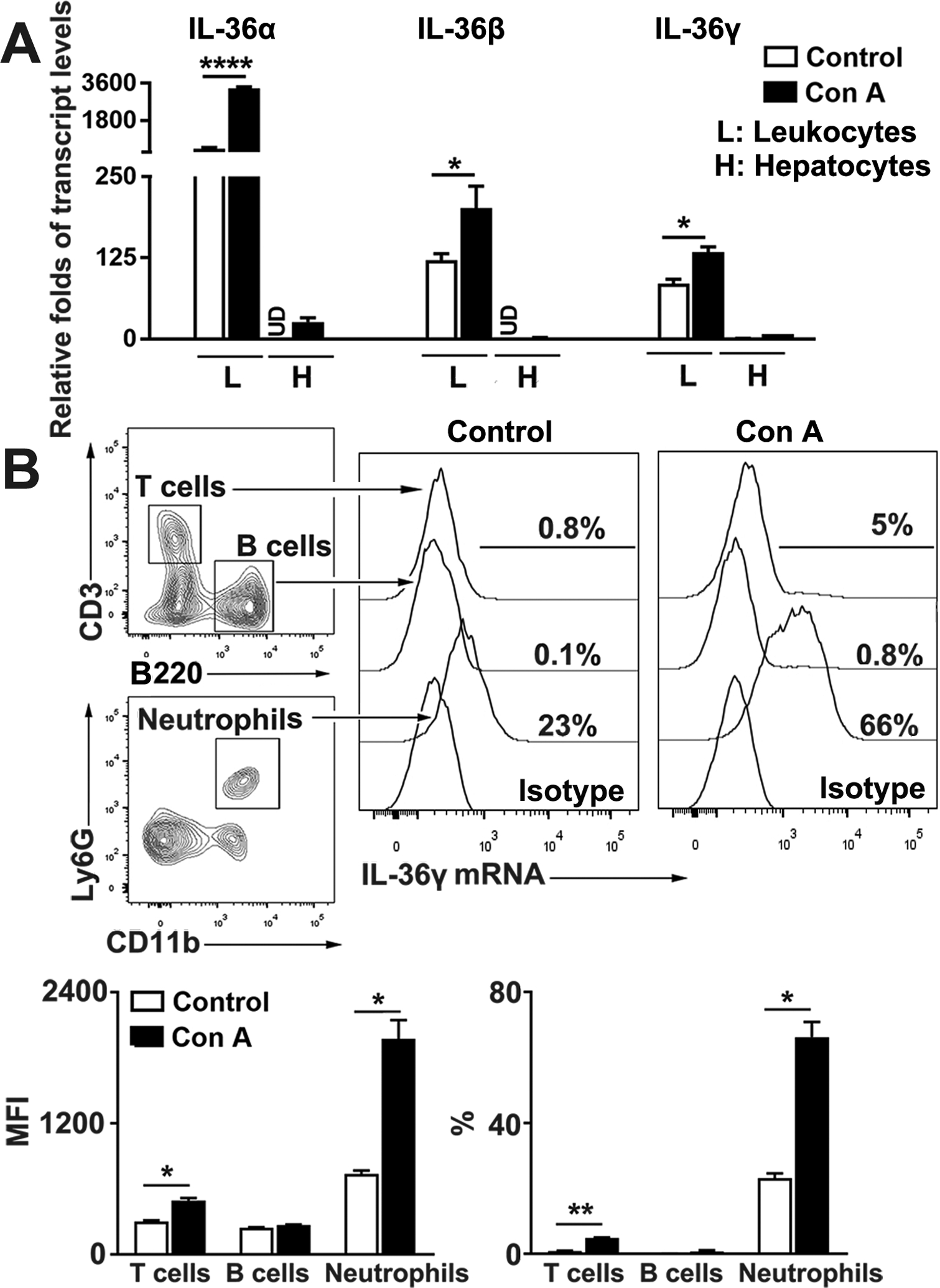
C57BL/6 mice were i. v. injected with Con A (12 mg/kg). PBS-injected mice were used as controls. After 12 h, leukocytes and hepatocytes were isolated from liver tissues. (A) Transcript levels of IL-36α, β and γ were analyzed by qRT-PCR. (B) Leukocytes were stained with surface flow cytometry antibodies for T cells, B cells and neutrophils, followed by the hybridization of the specific probe to IL-36γ RNA transcript (PrimeFlow RNA assay). The transcript levels of IL-36γ were measured in different types of immune cells by flow cytometry. n = 3–5 samples/group from single experiments. The data are shown as mean ± SEM of each group from single experiments and are representative of three experiments performed. Two-tailed unpaired T test was used for statistical analysis. * p < 0.05; **p < 0.01; **** p < 0.0001; UD, undetectable.
IL-36R-deficiency resulted in exacerbated liver damage
It has been reported that administering IL-36Ra can aggravate hepatic damage and hinder recovery in APAP-induced liver injury (27). To explore whether IL-36 plays a role in T cell-mediated acute hepatitis, WT and IL-36R−/− mice were i.v. injected with Con A. While all animals survived in the first 12 h, IL-36R−/− mice exhibited increased mortality compared with WT controls at 24 h post-Con A administration (Fig. 3A), indicating exacerbated liver damage in the absence of IL-36/IL-36R signaling (Fig. 3B). Liver histopathological evaluation revealed a widespread necrosis with extensive infiltration of mononuclear cells in IL-36R−/− mice at 24 h, suggesting uncontrolled inflammation compared with those in the control group (Fig. 3C–D). No differences in survival rates, serum enzymes or necrotic areas were observed between WT and IL-36R−/− mice at 12 h (Fig. 3). These results demonstrated that IL-36R signaling is protective in T cell-mediated acute hepatitis.
Figure 3. IL-36R-deficiency resulted in exacerbated liver damage.
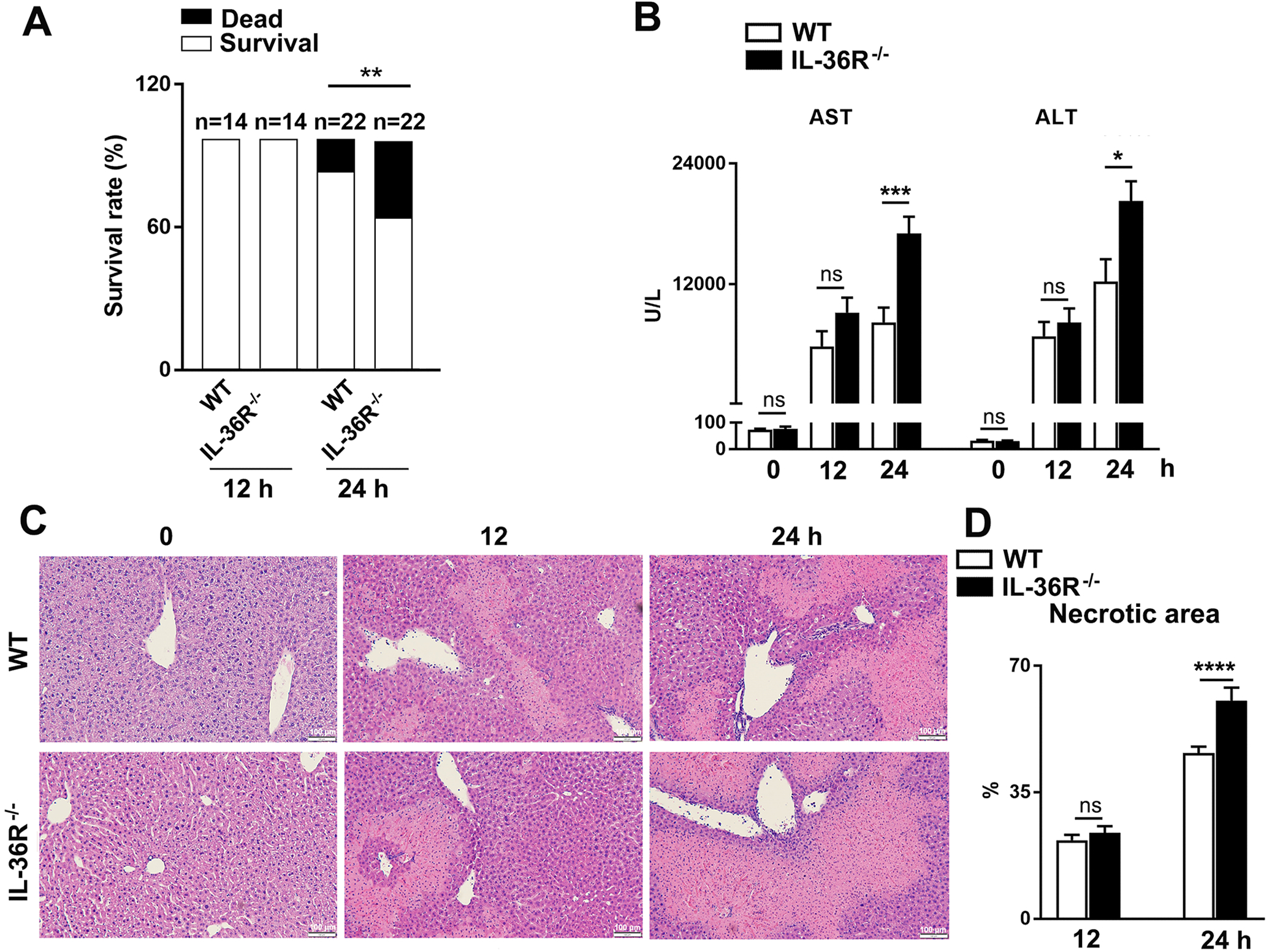
WT and IL-36R−/− mice were i.v. treated with 12 mg/kg of Con A. PBS-injected mice were used as controls (0 h). Animals were euthanatized at 12 and 24 h after Con A treatment. (A) Survival rates of WT and IL-36R−/− mice. (B) Serum ALT and AST levels. (C) Representative images of H&E staining for livers. (D) Statistical analysis of necrotic areas. n= 4–6 samples/ control group. n = 14–22 samples/ Con A-treated group from pooled experiments. The data are shown as mean ± SEM of each group from three independent experiments. Two-tailed unpaired T test was used for statistical analysis. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; ns, no significant difference.
IL-36R-deficiency resulted in exaggerated T cell activities in hepatitis
T cells play a central role in disease progression and pathogenesis in Con A-induced hepatitis (35–37). To investigate whether IL-36/IL-36R play a role in T cell responses in hepatitis, we analyzed intrahepatic lymphocytes (IHLs) in WT and IL-36R−/− mice by flow cytometry. The numbers of CD4+ and CD8+ T cells were initially comparable at 12 h post-Con A injection, but expanded quickly at 24 h in the liver and spleen of IL-36R−/− mice (Fig. 4A and Fig. S1A–B). Increased numbers of CD4+, CD8+ effector T cells and CD69+ T cells were also observed in the liver of IL-36R−/− mice at 24 h following Con A treatment (Fig. S2A–B). Since IFN-γ and TNF-α are critical for inducing hepatocyte apoptosis and hepatic fibrosis in T cell-dependent liver injury (38, 39), we asked whether IFN-γ and TNF-α contributed to Con A-induced hepatitis. We found that IL-36R−/− mice had increased percentages and numbers of IFN-γ+ CD4+ T cells at both 12 and 24 h following Con A treatment (Fig. 4B). Cytokine-producing CD8+ T cells were also increased at 24 h in the liver of IL-36R−/− mice (Fig. 4B). We also noted that effector, TNF-α+, IFN-γ+TNF-α+ and IFN-γ+ CD4+ and CD8+ T cells were increased greatly in IL-36R−/− mice when compared to WT mice in the spleen (Fig. S1C–D). We also evaluated innate cell populations, including NKT cells and macrophages. Our results revealed that NKT cells had no obvious changes, but macrophages were decreased in the liver of IL-36R−/− mice at 12 and 24 h following Con A treatment (Fig. S2C). Together, IL-36R-deficiency resulted in enhanced T cell activities in Con A-induced hepatitis.
Figure 4. IL-36R-deficiency resulted in exaggerated T cell activities in hepatitis.
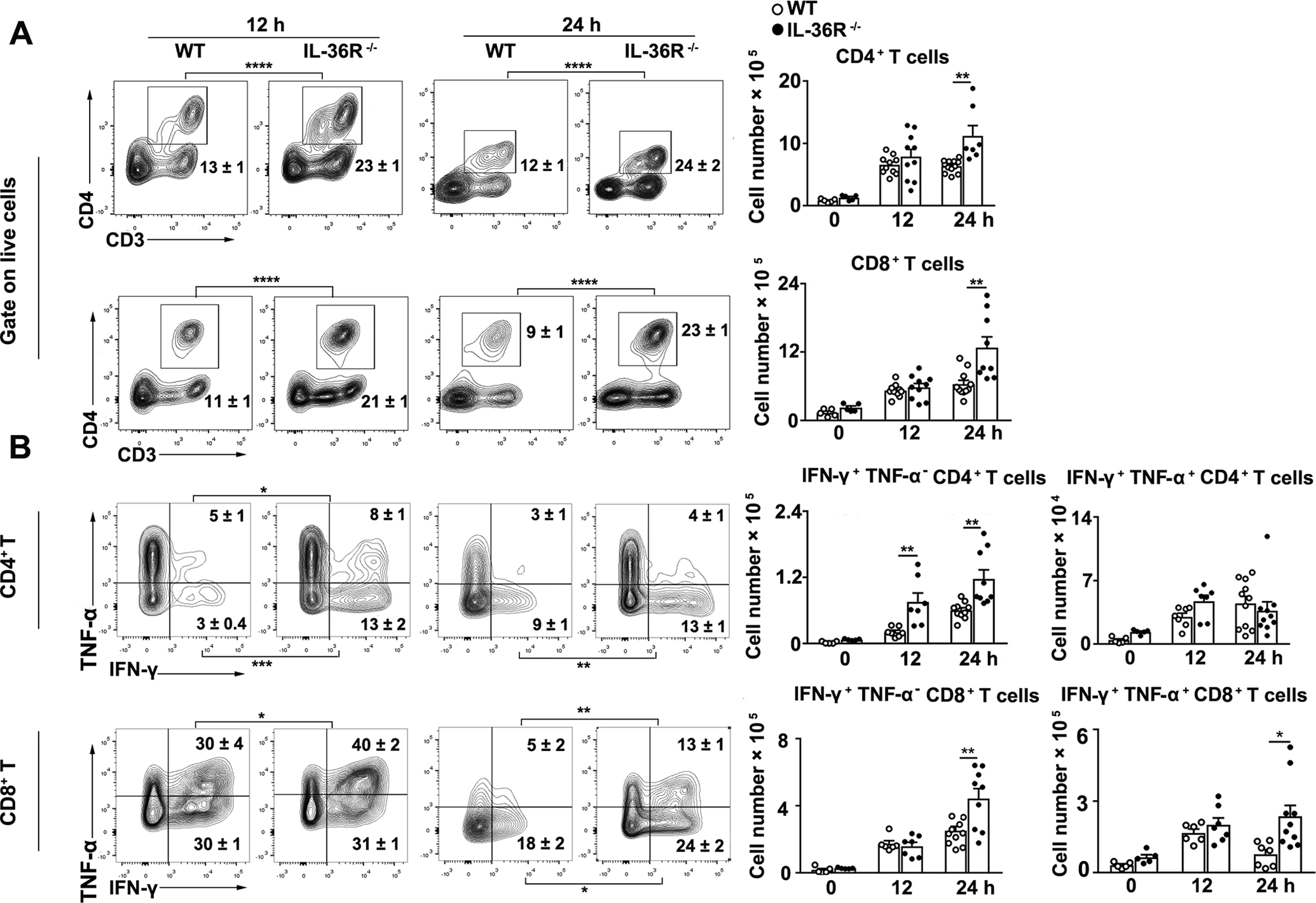
WT and IL-36R−/− mice were i.v. treated with 12 mg/kg of Con A. PBS-injected mice were used as controls (0 h). Intrahepatic lymphocytes were isolated at 12 and 24 h after Con A treatment for flow cytometry analysis. (A) Percentages and numbers of CD4+ and CD8+ T cells in livers. (B) Percentages and numbers of cytokine-producing CD4+ and CD8+ T cells in livers. n= 3–4 samples/ control group. n = 7–12 samples/ Con A-treated group from single experiments. The data are shown as mean ± SEM of each group from single experiments and are representative of three experiments performed. Two-tailed unpaired T test was used for statistical analysis. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
IL-36R-deficiency resulted in increased liver inflammatory responses
Studies with Con A-induced hepatitis have shown that inflammatory cytokines produced by liver infiltrating immune cells cause hepatocyte apoptosis and lead to liver damage, and that Th1, Th2, and Th17 responses contribute to this process (40–42). To determine the role of IL-36 in regulating liver inflammation in T cell-mediated hepatitis, we analyzed several key cytokines in the mouse liver by ELISA. We found that IL-36R−/− mice had increased levels of Th1 (IFN-γ and TNF-α), Th2 (IL-4) and Th17 (IL-17) cytokines at 12 and 24 h, respectively (Fig. 5A). Furthermore, IL-36R-deficiency resulted in higher IL-6, IL-1β, and IL-10 in the liver at 24 h post-Con A injection (Fig. 5A). Since chemokines are crucial for inflammatory cell recruitment in the liver and can subsequently lead to liver damage (43–45), we examined chemokine levels and found that IL-36R−/− mice had higher transcript levels of CXCL9 and CXCL10, which can recruit CXCR3+ effector T cells into the liver. CCL2, a key chemokine for T cell and monocyte migration, was also increased at 24 h in IL-36R−/− animals (Fig. 5B). Our results demonstrated excessive hepatic inflammation in the absence of IL-36R signaling in Con A-induced hepatitis.
Figure 5. IL-36R-deficiency resulted in increased liver inflammatory responses.
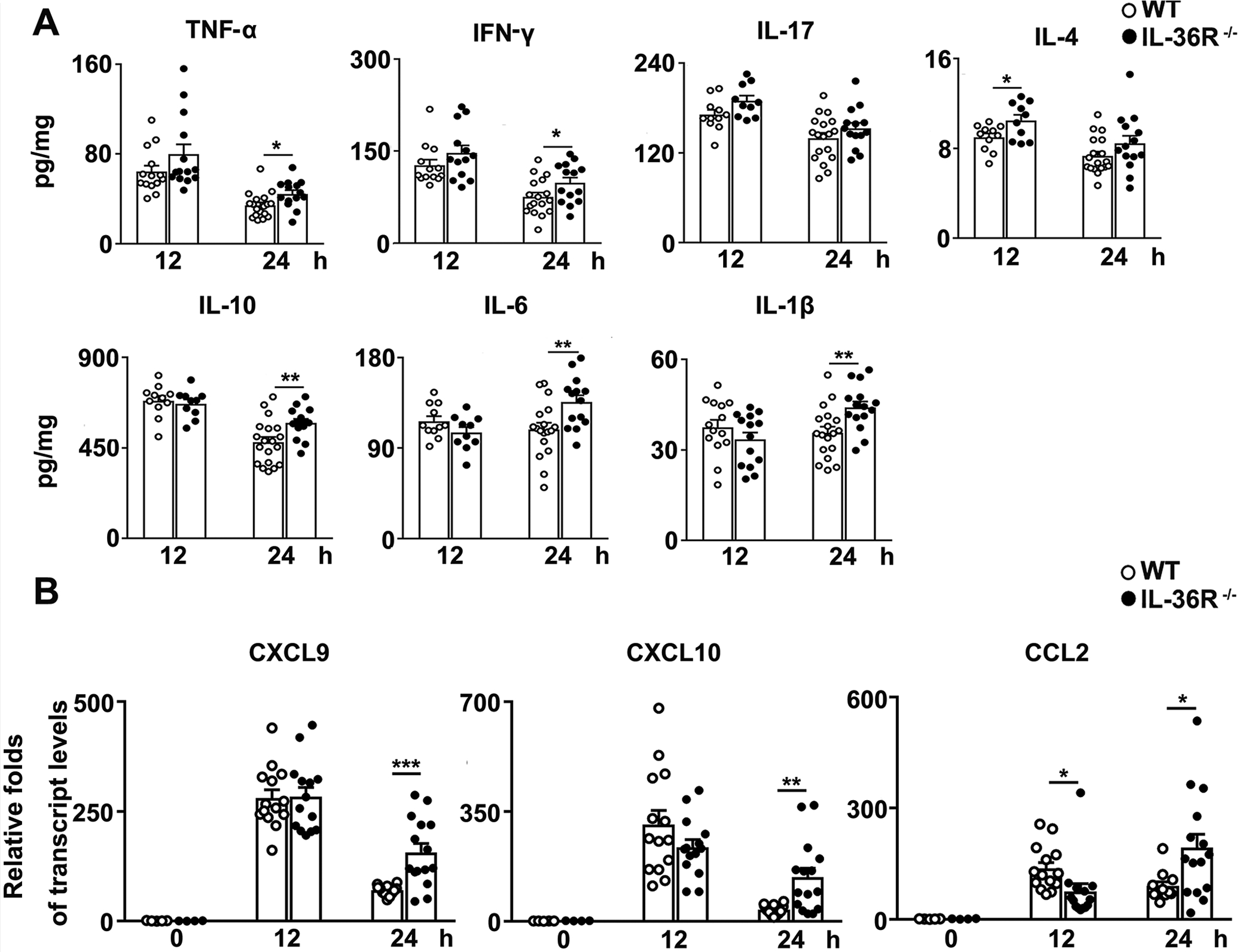
WT and IL-36R−/− mice were i.v. treated with 12 mg/kg of Con A. PBS-injected mice were used as controls (0 h). Protein and RNA were extracted from the livers at 12 and 24 h after Con A treatment. (A) IFN-γ, TNF-α, IL-17, IL-4, IL-10, IL-6 and IL-1β levels in the livers were analyzed by ELISA. (B) Expressions of CXCL9, CXCL10 and CCL2 mRNA in the livers were determined by qRT-PCR. n= 4–5 samples/ control group. n = 10–19 samples/ Con A-treated group from pooled experiments. The data are shown as mean ± SEM of each group from three independent experiments. Two-tailed unpaired T test was used for statistical analysis. * p < 0.05; ** p < 0.01; *** p < 0.001.
Absence of IL-36R resulted in reduced regulatory T cells in the liver following Con A injection
IL-36R can be detected on Treg cells and may play a role in their immunoregulatory functions (46, 47). To investigate whether IL-36 plays a role in regulating Treg cells in T cell-mediated hepatitis, we measured Treg cells in the liver of WT and IL-36R−/− mice following Con A injection. Although the numbers of Treg cells were comparable at 12 h, IL-36R-deficient mice had considerably lower numbers of Treg cells in the liver at 24 h compared with WT mice (Fig. 6A). To confirm these findings, we analyzed chemokines that are associated with Treg cell recruitment and function in the liver (48, 49). We observed decreased transcript levels of CCL5, CCL17, and CXCL12 in IL-36R−/− mice at 24 h (Fig. 6B). To determine whether IL-36 can affect chemotaxis of Treg cells, we performed a transwell migration assay. We found that IL-36 didn’t directly affect chemotaxis of Treg cells (Fig. S3A). However, we found that IL-36 can promote Treg cell proliferation (Fig. S3B). These results suggest that IL-36 signaling may play a role in Treg cell hemostasis. The repressed Treg cell responses in IL-36R-deficient mice may lead to overzealous T cell effector functions in the liver microenvironment.
Figure 6. Absence of IL-36R resulted in reduced regulatory T cells in the liver following Con A injection.
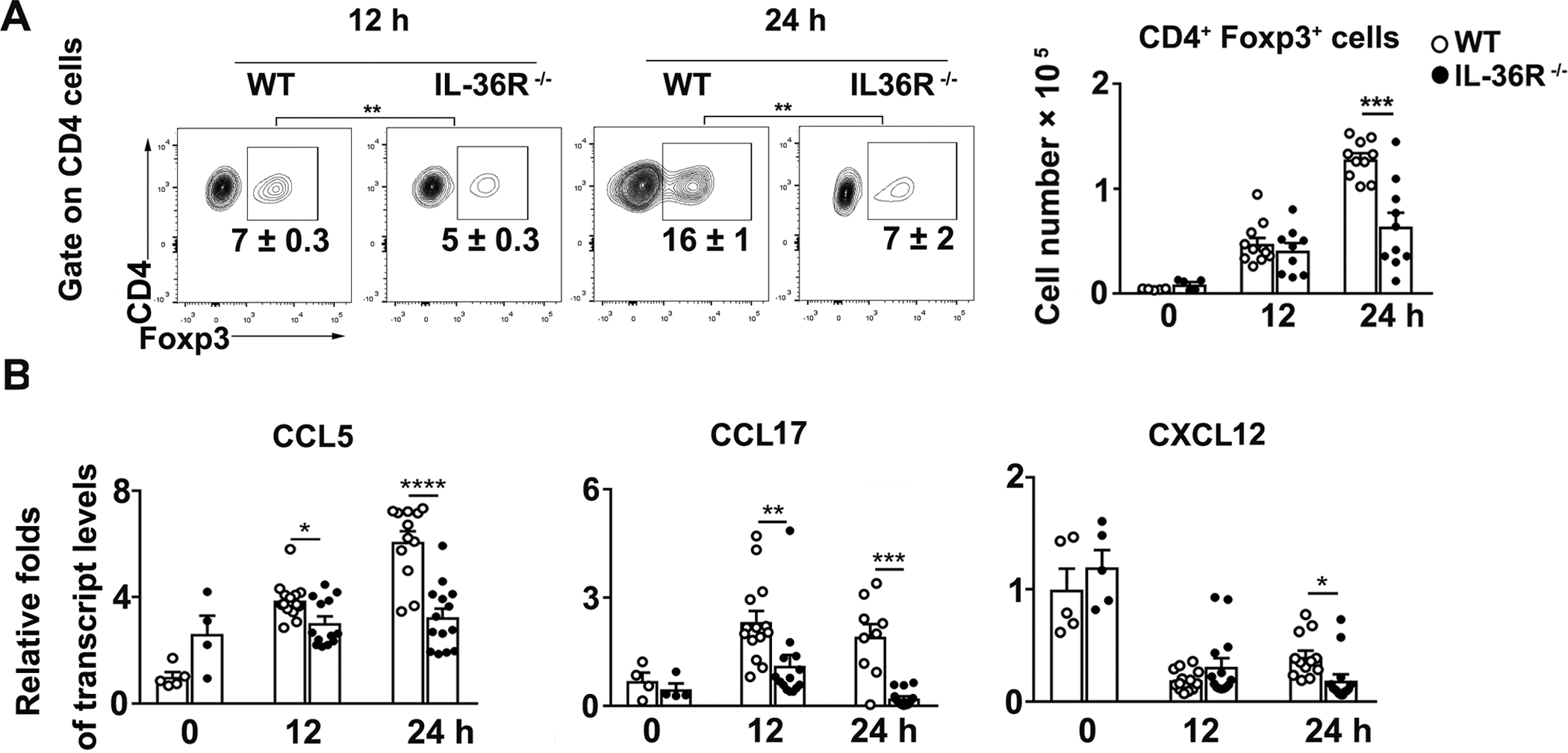
WT and IL-36R−/− mice were i.v. treated with 12 mg/kg of Con A. PBS-injected mice were used as controls (0 h). Intrahepatic lymphocytes and RNA were obtained from the livers at 12 and 24 h after Con A treatment. (A) Percentages and numbers of Foxp3+ CD4+ Treg cells were analyzed in the livers by flow cytometry. (B) Total RNAs were extracted from the liver at 12 and 24 h after Con A treatment. Expressions of CCL5, CCL17 and CXCL12 mRNA in the liver were determined by qRT-PCR. n= 4–5 samples/ control group. n = 9–12 samples/ Con A-group from pooled experiments. The data are shown as mean ± SEM of each group from three independent experiments. Two-tailed unpaired T test was used for statistical analysis. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
DISCUSSION
Being a member of the IL-1 superfamily, IL-36 is widely studied in different types of autoimmune diseases, including psoriasis, systemic lupus erythematosus and IBD (16, 18). IL-36 was increased and acted as an inflammatory factor in the pathogenesis of psoriasis lesions (18, 50). In phase Ⅰ clinical trials, a single dose of an anti-IL-36R mAb inhibited the IL-36 pathway and greatly alleviated the severity of generalized pustular psoriasis in patients (22, 51). In addition, increased IL-36 was detected in mucosa biopsies from IBD patients and dextran sulfate sodium-induced IBD animal models (23, 52), resulting in enhanced Th1 and reduced Th17 responses (23). In a recent study, Scheibe and co-worker reported that inhibition of IL-36R using the mAb ameliorated inflammation and reversed the fibrosis in experimental animals (25). However, blockage of IL-36R can also exacerbate DSS-induced colitis and impair wound healing (24, 25), raising great interest in the multifunctional IL-36 signaling pathway in inflammatory diseases.
Despite these findings, the putative role that IL-36 plays in liver diseases is not clear. Treatment with IL-36 receptor antagonist (IL-36Ra) reduced Con A-induced pro-inflammatory cytokine production and alleviated liver injury in BALB/c mice (53). However, IL-36γ expression was enhanced in APAP-induced liver injury, and application of IL-36Ra aggravated liver damage and impaired recovery in the late phase of liver damage (27). In our study, we found elevated IL-36 expression in T cell-mediated acute hepatitis in mice (Fig. 1). IL-36 cytokines were mainly produced by leukocytes, and its expression elevated in the liver following Con A treatment (Fig. 2A) (54, 55). Our study also demonstrated that IL-36 is mainly expressed by neutrophils among different immune cell subsets (Fig. 2B), as reported previously (54, 55). More importantly, we demonstrated that IL-36R−/− mice (H-2b) exhibited a higher mortality rate, more severe liver damage with expanded areas of necrosis (Fig. 3). These findings suggested that IL-36 signaling is protective in T cell-mediated hepatitis. Peng et al. reported that treatment with IL-36Ra plasmid decreased liver inflammation and serum aminotransferase levels in Con A-induced hepatitis of BALB/c (H-2d) mice. IL-36Ra can bind to IL-36R, and may trigger potential signaling pathways (5). However, IL-36 failed to activate any downstream signaling pathway in our IL-36R−/− mouse model. The application of different strategies may account for the discrepancy between our and other’s studies in Con A-induced hepatitis.
Activation of T cells is a critical process in the pathophysiology of Con A-induced liver diseases (56, 57). IL-36 can amplify T cell proliferation and activation in vitro, resulting in increased pro-inflammatory cytokine production (46, 58). Here, we reported that the immune responses of CD4+ and CD8+ T cells were enhanced in the liver and spleen in IL-36R−/− mice (Fig. 4 and Fig. S1), leading to severe liver injury. In addition, effector T cells were also significantly elevated in the liver of IL-36R−/− mice (Fig. S2). T cell-related chemokines play critical roles in regulating the accumulation and infiltration of T cells in inflamed tissues (59). The balance between various T cell subgroups determines the outcome of local inflammation and is central to the pathogenesis of inflammatory diseases (59). CXCL9, CXCL10 and CCL2 can direct pro-inflammatory T cell trafficking and positioning within tissues, thus regulating tissue-specific immune responses and inflammation (44, 45, 59). Increased T cell activities can result in the elevated production of proinflammatory cytokines, leading to the destruction of the balance between protection and injury. Our data demonstrated that the accumulation and activation of T cells in the liver resulted in the elevation of T cell associated proinflammatory factors, including Th1 (TNF-α and IFN-γ), Th17 (IL-17), and other pro-inflammatory factors (such as IL-6, IL-1β, etc.), and contributed to serious liver damage in IL-36R−/− mice (Fig. 5). A decrease in macrophages was also found in IL-36R−/− mice (Fig. S2). IL-36 is a critical upstream amplifier of macrophage activities and has been found to correlate with the number of infiltrating macrophages (10, 55, 60). This result suggests that IL-36 may also regulate macrophage activities in Con A-induced hepatitis. Overall, these results demonstrated that the lack of IL-36/IL-36R signaling contributed to severe liver injury and dysregulated T cells responses in T cell-mediated hepatitis.
In this study, we found that IL-36R−/− mice had markedly lower numbers of hepatic Treg cells (Fig. 6), which can play an important role in the regulation of immune responses in Con A-induced hepatitis (61). It is reported recently that an increased proliferation of Treg cells were observed in the presence of IL-36 in vitro (47, 62). Our data suggested that IL-36 may promote proliferation of Treg cells (Fig. S3). Moreover, Treg cells were functional in suppressing T-cell proliferation and immune responses in vitro and in vivo (63). Our data suggested that in the absence of IL-36 signaling, reduced Treg cell activities may result in overzealous T effector responses and subsequent liver injury. Unlike T effector cells, Treg cells have a significant positive association with the levels of CCL5, CCL17 and CXCL12 (63–65). In this study, we observed higher effector T cell-associated chemokine levels, but lower Treg cell-associated ones in IL-36R−/− mice (Fig. 5–6). Moreover, although IL-36 didn’t directly drive Treg cell migration (Fig. S3), it may orchestrate various chemokines, leading to an imbalance of pro-inflammatory T cells and immune suppressive cells in the liver.
In summary, our results showed that Con A treatment induced expression of liver IL-36 cytokines, which were predominantly produced by neutrophils. Further analysis showed that deficiency of IL-36R resulted in increased liver inflammation and exacerbated tissue damage. Moreover, IL-36R-deficiency resulted in increased effector T cell responses and decreased Treg cell activities. In all, our study revealed a protective role of IL-36 in acute hepatitis.
Supplementary Material
Key points:
Increased IL-36 levels were found in Con A-injected mice.
Deficiency of IL-36R led to exacerbated liver injury and increased mortality.
A lack of IL-36 signaling exaggerated T cell responses and inflammation in the liver.
ACKNOWLEDGMENTS
We thank Dr. Sherry Haller for her assistance with the manuscript preparation.
This work was supported partly by grants from the National Natural Science Foundation of China 81800506 and Natural Science Foundation of Hunan Province China 2019JJ40494 to PY, the National Institutes of Health grants (EY028773 to JC and JS, AI132674 to LS, and AI153586 to YL), as well as the UTMB Institute of Human Infections & Immunity Pilot grant (to LS and YL).
Abbreviations:
- Con A
concanavalin A
- IBD
inflammatory bowel disease
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- IHL
intrahepatic lymphocyte
- IL-1RAcP
IL-1 receptor accessory protein
- DSS
dextran sodium sulfate
Footnotes
DISCLOSURES
The authors have no financial conflicts of interest.
REFERENCES
- 1.Towne JE, Garka KE, Renshaw BR, Virca GD, and Sims JE. 2004. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem 279: 13677–13688. [DOI] [PubMed] [Google Scholar]
- 2.Palomo J, Mastelic-Gavillet B, Woldt E, Troccaz S, Rodriguez E, Palmer G, Siegrist CA, and Gabay C. 2016. IL-36-Induced Toxicity in Neonatal Mice Involves TNF-alpha Production by Liver Myeloid Cells. J Immunol 197: 2239–2249. [DOI] [PubMed] [Google Scholar]
- 3.Gunther S, and Sundberg EJ. 2014. Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J Immunol 193: 921–930. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen TT, Niyonsaba F, Ushio H, Akiyama T, Kiatsurayanon C, Smithrithee R, Ikeda S, Okumura K, and Ogawa H. 2012. Interleukin-36 cytokines enhance the production of host defense peptides psoriasin and LL-37 by human keratinocytes through activation of MAPKs and NF-kappaB. J Dermatol Sci 68: 63–66. [DOI] [PubMed] [Google Scholar]
- 5.Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, Wagner J, Edwards G, Clifford T, Menon S, Bazan JF, and Kastelein RA. 2001. Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J Immunol 167: 1440–1446. [DOI] [PubMed] [Google Scholar]
- 6.Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, and Kramer D. 2018. IkappaBzeta is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci U S A 115: 10088–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, and Sims JE. 2011. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem 286: 42594–42602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, Joosten LA, van der Meer JW, Hao R, Kalabokis V, and Dinarello CA. 2012. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci U S A 109: 3001–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Li ZY, Jiang WX, Liao B, Zhai GT, Wang N, Zhen Z, Ruan JW, Long XB, Wang H, Liu WH, Liang GT, Xu WM, Kato A, and Liu Z. 2018. The activation and function of IL-36gamma in neutrophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol 141: 1646–1658. [DOI] [PubMed] [Google Scholar]
- 10.Dietrich D, Martin P, Flacher V, Sun Y, Jarrossay D, Brembilla N, Mueller C, Arnett HA, Palmer G, Towne J, and Gabay C. 2016. Interleukin-36 potently stimulates human M2 macrophages, Langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 84: 88–98. [DOI] [PubMed] [Google Scholar]
- 11.Gresnigt MS, Rosler B, Jacobs CW, Becker KL, Joosten LA, van der Meer JW, Netea MG, Dinarello CA, and van de Veerdonk FL. 2013. The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur J Immunol 43: 416–426. [DOI] [PubMed] [Google Scholar]
- 12.Sims JE, and Smith DE. 2010. The IL-1 family: regulators of immunity. Nat Rev Immunol 10: 89–102. [DOI] [PubMed] [Google Scholar]
- 13.Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE, and Gabay C. 2011. IL-36R ligands are potent regulators of dendritic and T cells. Blood 118: 5813–5823. [DOI] [PubMed] [Google Scholar]
- 14.Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, Wohn C, Prens EP, Wang F, Maier LE, Kang S, Voorhees JJ, Elder JT, and Gudjonsson JE. 2011. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 186: 2613–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoyagi T, Newstead MW, Zeng X, Kunkel SL, Kaku M, and Standiford TJ. 2017. IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol 10: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishida A, Hidaka K, Kanda T, Imaeda H, Shioya M, Inatomi O, Bamba S, Kitoh K, Sugimoto M, and Andoh A. 2016. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflamm Bowel Dis 22: 303–314. [DOI] [PubMed] [Google Scholar]
- 17.Berglof E, Andre R, Renshaw BR, Allan SM, Lawrence CB, Rothwell NJ, and Pinteaux E. 2003. IL-1Rrp2 expression and IL-1F9 (IL-1H1) actions in brain cells. J Neuroimmunol 139: 36–43. [DOI] [PubMed] [Google Scholar]
- 18.Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, Morel F, Lecron JC, Rolli-Derkinderen M, Bourreille A, Vigne S, Gabay C, Palmer G, Le Goff B, and Blanchard F. 2016. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol 184: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, Renauld JC, Werner S, Kisielow J, and Kopf M. 2012. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest 122: 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, Towne JE, Douangpanya J, Pons C, Mansour S, Serre V, Makni H, Mahfoudh N, Fakhfakh F, Bodemer C, Feingold J, Hadj-Rabia S, Favre M, Genin E, Sahbatou M, Munnich A, Casanova JL, Sims JE, Turki H, Bachelez H, and Smahi A. 2011. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med 365: 620–628. [DOI] [PubMed] [Google Scholar]
- 21.Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO, Burden AD, Capon F, Trembath RC, and Barker JN. 2011. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 89: 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, Turki H, Hall DB, Shear M, Baum P, Padula SJ, and Thoma C. 2019. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N Engl J Med 380: 981–983. [DOI] [PubMed] [Google Scholar]
- 23.Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, Statovci D, Moran T, Fallon PG, Shanahan F, Brint EK, Melgar S, Hussey S, and Walsh PT. 2016. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol 9: 1193–1204. [DOI] [PubMed] [Google Scholar]
- 24.Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, Neumann PA, Geem D, Lili LN, Ramadas RA, Chassaing B, Gewirtz AT, Kohlmeier JE, Parkos CA, Towne JE, Nusrat A, and Denning TL. 2016. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J Immunol 196: 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, Probst HC, Bopp T, Neurath MF, and Neufert C. 2017. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 66: 823–838. [DOI] [PubMed] [Google Scholar]
- 26.Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carle B, Knieling F, Claussen J, Klimowicz AC, Zheng J, Baum P, Meyer S, Schurmann S, Friedrich O, Waldner MJ, Rath T, Wirtz S, Kollias G, Ekici AB, Atreya R, Raymond EL, Mbow ML, Neurath MF, and Neufert C. 2019. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation. Gastroenterology 156: 1082–1097 e1011. [DOI] [PubMed] [Google Scholar]
- 27.Scheiermann P, Bachmann M, Hardle L, Pleli T, Piiper A, Zwissler B, Pfeilschifter J, and Muhl H. 2015. Application of IL-36 receptor antagonist weakens CCL20 expression and impairs recovery in the late phase of murine acetaminophen-induced liver injury. Sci Rep 5: 8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan QZ, Pan K, Zhao JJ, Chen JG, Li JJ, Lv L, Wang DD, Zheng HX, Jiang SS, Zhang XF, and Xia JC. 2013. Decreased expression of interleukin-36alpha correlates with poor prognosis in hepatocellular carcinoma. Cancer Immunol Immunother 62: 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bi Y, Li J, Yang Y, Wang Q, Wang Q, Zhang X, Dong G, Wang Y, Duan Z, Shu Z, Liu T, Chen Y, Zhang K, and Hong F. 2019. Human liver stem cells attenuate concanavalin A-induced acute liver injury by modulating myeloid-derived suppressor cells and CD4(+) T cells in mice. Stem Cell Res Ther 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiegs G, Hentschel J, and Wendel A. 1992. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 90: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, Pejnovic N, Arsenijevic N, and Lukic ML. 2012. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J Hepatol 56: 26–33. [DOI] [PubMed] [Google Scholar]
- 32.Liang Y, Yi P, Wang X, Zhang B, Jie Z, Soong L, and Sun J. 2020. Retinoic Acid Modulates Hyperactive T Cell Responses and Protects Vitamin A-Deficient Mice against Persistent Lymphocytic Choriomeningitis Virus Infection. J Immunol 204: 2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldstein I, Paakinaho V, Baek S, Sung MH, and Hager GL. 2017. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat Commun 8: 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Li H, Wang Y, Li H, Du S, Zou X, Zhang X, and Cao B. 2020. High Expression of IL-36gamma in Influenza Patients Regulates Interferon Signaling Pathway and Causes Programmed Cell Death During Influenza Virus Infection. Front Immunol 11: 552606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ando Y, Yasuoka C, Mishima T, Ikematsu T, Uede T, Matsunaga T, and Inobe M. 2014. Concanavalin A-mediated T cell proliferation is regulated by herpes virus entry mediator costimulatory molecule. In Vitro Cell Dev Biol Anim 50: 313–320. [DOI] [PubMed] [Google Scholar]
- 36.Palacios R 1982. Concanavalin A triggers T lymphocytes by directly interacting with their receptors for activation. J Immunol 128: 337–342. [PubMed] [Google Scholar]
- 37.Boilard E, and Surette ME. 2001. Anti-CD3 and concanavalin A-induced human T cell proliferation is associated with an increased rate of arachidonate-phospholipid remodeling. Lack of involvement of group IV and group VI phospholipase A2 in remodeling and increased susceptibility of proliferating T cells to CoA-independent transacyclase inhibitor-induced apoptosis. J Biol Chem 276: 17568–17575. [DOI] [PubMed] [Google Scholar]
- 38.Gantner F, Leist M, Lohse AW, Germann PG, and Tiegs G. 1995. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology 21: 190–198. [DOI] [PubMed] [Google Scholar]
- 39.Kimura K, Ando K, Ohnishi H, Ishikawa T, Kakumu S, Takemura M, Muto Y, and Moriwaki H. 1999. Immunopathogenesis of hepatic fibrosis in chronic liver injury induced by repeatedly administered concanavalin A. Int Immunol 11: 1491–1500. [DOI] [PubMed] [Google Scholar]
- 40.Huang J, Yuan Q, Zhu H, Yin L, Hong S, Dong Z, Jin W, and Dong C. 2017. IL-17C/IL-17RE Augments T Cell Function in Autoimmune Hepatitis. J Immunol 198: 669–680. [DOI] [PubMed] [Google Scholar]
- 41.Feng XX, Chi G, Wang H, Gao Y, Chen Q, Ru YX, Luo ZL, Yan W, Li PY, Liu M, Feng ZH, and Tian DA. 2019. IL-37 suppresses the sustained hepatic IFN-gamma/TNF-alpha production and T cell-dependent liver injury. Int Immunopharmacol 69: 184–193. [DOI] [PubMed] [Google Scholar]
- 42.Sternak M, Jakubowski A, Czarnowska E, Slominska EM, Smolenski RT, Szafarz M, Walczak M, Sitek B, Wojcik T, Jasztal A, Kaminski K, and Chlopicki S. 2015. Differential involvement of IL-6 in the early and late phase of 1-methylnicotinamide (MNA) release in Concanavalin A-induced hepatitis. Int Immunopharmacol 28: 105–114. [DOI] [PubMed] [Google Scholar]
- 43.Cedile O, Wlodarczyk A, and Owens T. 2017. CCL2 recruits T cells into the brain in a CCR2-independent manner. APMIS 125: 945–956. [DOI] [PubMed] [Google Scholar]
- 44.Shinde P, Liu W, Menoret A, Luster AD, and Vella AT. 2017. Optimal CD4 T cell priming after LPS-based adjuvanticity with CD134 costimulation relies on CXCL9 production. J Leukoc Biol 102: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brownell J, and Polyak SJ. 2013. Molecular pathways: hepatitis C virus, CXCL10, and the inflammatory road to liver cancer. Clin Cancer Res 19: 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E, Olleros ML, Vesin D, Garcia I, Ronchi F, Sallusto F, Sims JE, and Gabay C. 2012. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 120: 3478–3487. [DOI] [PubMed] [Google Scholar]
- 47.Qu Q, Zhai Z, Xu J, Li S, Chen C, and Lu B. 2020. IL36 Cooperates With Anti-CTLA-4 mAbs to Facilitate Antitumor Immune Responses. Front Immunol 11: 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, and Fujii H. 2008. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer 122: 2286–2293. [DOI] [PubMed] [Google Scholar]
- 49.Lim HW, Broxmeyer HE, and Kim CH. 2006. Regulation of trafficking receptor expression in human forkhead box P3+ regulatory T cells. J Immunol 177: 840–851. [DOI] [PubMed] [Google Scholar]
- 50.He Q, Chen HX, Li W, Wu Y, Chen SJ, Yue Q, Xiao M, and Li JW. 2013. IL-36 cytokine expression and its relationship with p38 MAPK and NF-kappaB pathways in psoriasis vulgaris skin lesions. J Huazhong Univ Sci Technolog Med Sci 33: 594–599. [DOI] [PubMed] [Google Scholar]
- 51.Todorovic V, Su Z, Putman CB, Kakavas SJ, Salte KM, McDonald HA, Wetter JB, Paulsboe SE, Sun Q, Gerstein CE, Medina L, Sielaff B, Sadhukhan R, Stockmann H, Richardson PL, Qiu W, Argiriadi MA, Henry RF, Herold JM, Shotwell JB, McGaraughty SP, Honore P, Gopalakrishnan SM, Sun CC, and Scott VE. 2019. Small Molecule IL-36gamma Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci Rep 9: 9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fonseca-Camarillo G, Furuzawa-Carballeda J, Iturriaga-Goyon E, and Yamamoto-Furusho JK. 2018. Differential Expression of IL-36 Family Members and IL-38 by Immune and Nonimmune Cells in Patients with Active Inflammatory Bowel Disease. Biomed Res Int 2018: 5140691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng X, Pan X, Tan J, Li Y, and Li M. 2020. Protective effect of interleukin-36 receptor antagonist on liver injury induced by concanavalin A in mice. Iran J Basic Med Sci 23: 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bozoyan L, Dumas A, Patenaude A, and Vallieres L. 2015. Interleukin-36gamma is expressed by neutrophils and can activate microglia, but has no role in experimental autoimmune encephalomyelitis. J Neuroinflammation 12: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koss CK, Wohnhaas CT, Baker JR, Tilp C, Przibilla M, Lerner C, Frey S, Keck M, Williams CMM, Peter D, Ramanujam M, Fine J, Gantner F, Thomas M, Barnes PJ, Donnelly LE, and El Kasmi KC. 2021. IL36 is a critical upstream amplifier of neutrophilic lung inflammation in mice. Commun Biol 4: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sucher E, Sucher R, Gradistanac T, Brandacher G, Schneeberger S, and Berg T. 2019. Autoimmune Hepatitis-Immunologically Triggered Liver Pathogenesis-Diagnostic and Therapeutic Strategies. J Immunol Res 2019: 9437043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heymann F, Hamesch K, Weiskirchen R, and Tacke F. 2015. The concanavalin A model of acute hepatitis in mice. Lab Anim 49: 12–20. [DOI] [PubMed] [Google Scholar]
- 58.Arakawa A, Vollmer S, Besgen P, Galinski A, Summer B, Kawakami Y, Wollenberg A, Dornmair K, Spannagl M, Ruzicka T, Thomas P, and Prinz JC. 2018. Unopposed IL-36 Activity Promotes Clonal CD4(+) T-Cell Responses with IL-17A Production in Generalized Pustular Psoriasis. J Invest Dermatol 138: 1338–1347. [DOI] [PubMed] [Google Scholar]
- 59.Oo YH, Shetty S, and Adams DH. 2010. The role of chemokines in the recruitment of lymphocytes to the liver. Dig Dis 28: 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wein AN, Dunbar PR, McMaster SR, Li ZT, Denning TL, and Kohlmeier JE. 2018. IL-36gamma Protects against Severe Influenza Infection by Promoting Lung Alveolar Macrophage Survival and Limiting Viral Replication. J Immunol 201: 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erhardt A, Biburger M, Papadopoulos T, and Tiegs G. 2007. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 45: 475–485. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Zhao X, Feng C, Weinstein A, Xia R, Wen W, Lv Q, Zuo S, Tang P, Yang X, Chen X, Wang H, Zang S, Stollings L, Denning TL, Jiang J, Fan J, Zhang G, Zhang X, Zhu Y, Storkus W, and Lu B. 2015. IL-36gamma Transforms the Tumor Microenvironment and Promotes Type 1 Lymphocyte-Mediated Antitumor Immune Responses. Cancer Cell 28: 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, Blazar BR, and Serody JS. 2005. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood 106: 3300–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, Shetty S, Harki J, Shaw JC, Eksteen B, Hubscher SG, Walker LS, and Adams DH. 2010. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol 184: 2886–2898. [DOI] [PubMed] [Google Scholar]
- 65.Zhang B, Wu C, Zhang Z, Yan K, Li C, Li Y, and Li L. 2019. CXCL12 is associated with FoxP3(+) tumor-infiltrating lymphocytes and affects the survival of patients with oral squamous cell carcinoma. Oncol Lett 18: 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.