

ABSTRACT
Crocin (CRO) is feasible in alleviating atherosclerosis (AS), the mechanism of which was therefore explored in the study. High-fat diet (HFD)-induced apolipoprotein E-deficient (ApoE−/−) mice and lysophosphatidic acid (LPA)-treated macrophages received CRO treatment. Treated macrophage viability was determined via MTT assay. In both murine and macrophage, the lipid level and total Cholesterol/Cholesteryl l Ester (TC/CE) levels were quantified by oil-red-O staining and ELISA, respectively. Lipid droplet, aortic plaque formation and collagen deposition were detected via Oil-red-O staining, hematoxylin–eosin staining and Masson staining, respectively. Liver X Receptor-α (LXR-α), Peroxisome Proliferator-Activated Receptor γ (PPARγ), CD68, PCSK9, CD36, ATP Binding Cassette Subfamily A Member 1 (ABCA1), phosphorylated (p)-AKT, and AKT expressions were detected via Western blot, the former three also being detected using Immunohistochemistry and the first being measured by qRT-PCR. CRO decreased HFD-induced weight gain, ameliorated the abnormal serum lipid levels of HFD-treated mice, and inhibited aortic plaque formation and lipid deposition, and increased collagen fibers, with upregulated high-density lipoprotein-cholesterol (HDL-C) and downregulated TC and low-density lipoprotein-cholesterol (LDL-C). CRO alleviated the HFD-induced upregulations of CD68, PCSK9 and CD36 as well as downregulations of PPARγ/LXR-α, ABCA1 and AKT phosphorylation. In LPA-treated macrophages, CRO alone exerted no effect on the viability yet inhibited the lipid droplets formation and downregulated TC/CE levels. Silent LXR-α reversed the effect of CRO on the lipid droplets formation and levels of lipid metabolism-related factors. CRO ameliorated AS by inhibiting foam cells formation and promoting reverse cholesterol transport via PPARγ/LXR-α.
KEYWORDS: Crocin, atherosclerosis, macrophages, foam cell, lipid transporter, peroxisome proliferator-activated receptor γ, liver x receptor-α
Introduction
As the main pathological basis of the occurrence of most cardiovascular diseases (CVDs), atherosclerosis (AS) has been recognized as a metabolic-related disorder and a chronic and maladaptive inflammatory process due to passive lipid accumulation within the vessel wall, which threatens human health and imposes a heavy social burden[1, 2]. Nowadays, though anti-atherosclerotic and anti-thrombotic therapies have been developed and applied to the treatment of AS, the incidence of adverse clinical events in advanced AS remains high, making it urgent to hammer out a viable and effective therapeutic method for AS [3]. Therefore, delving into the pathological mechanism of AS formation and development is of great significance to prevent and treat AS.
Macrophages are heterogeneous cells that function in both inflammatory and tissue reparative responses, with accumulating evidence confirming their central role in the development of AS [4,5]. It was also well acknowledged that foam cell, which plays a major role in the early stage of atherogenesis, is the component of atherosclerotic plaques, and its formation from the macrophages is the main sign of early atherosclerotic lesions [6,7]. During the early stage of AS, monocytes are stimulated by inflammatory chemokines and differentiate into macrophages, which phagocytize loads of oxidatively modified low-density lipoprotein (ox-LDL) through the scavenger receptor (SR.) on their surface, leading to cholesterol influx and deposition and then foam cell formation [8]. Hence, promoting the intracellular cholesterol efflux, also known as reverse cholesterol transport (RCT), is a vital mechanism to prevent AS [9,10], whilst exploring avenues to promote RCT and inhibit the formation of foam cells may be of great help for the AS treatment.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) affects the RCT pathway, which promotes cholesterol efflux and plays an important role in AS formation and development; while cluster of differentiation 36 (CD36) affects the RCT process via regulating the liver X receptors (LXRs) expression [11]. The activated peroxisome proliferator-activated receptor γ (PPARγ)/LXRα/ATP-binding cassette transporter A1 (ABCA1) pathway can affect macrophage cholesterol efflux and play a critical role in the pathogenesis of AS [12]. In addition, it was found that macrophage lipid accumulation and atherosclerosis can be reduced by promoting the activation of the PI3K/AKT insulin pathway [13].
Many natural plant extracts, especially Chinese herbal medicine, have been revealed to have anti-atherosclerotic activity, including Paeonol, Sulforaphane, Luteolin and Quercetin [11,14–16]. Previous studies have reported that Shen-Hong-Tong-Luo pretreatment reduced lipid accumulation and inflammation of macrophages by activating the PPAR-γ/LXR-α/ABCA1 pathway [17]. Moreover, studies have suggested that Quercetin protects against AS by regulating the expressions of PPARγ, LXRα, ABCA1, PCSK9, and CD36 [12]. Besides, Danhong injection attenuates macrophage lipid accumulation and AS via regulating the PI3K/AKT insulin pathway [13]. Crocin (CRO) is one of the major active constituents of Saffron, which is the dried stigma of Crocus sativus L.and previously proved to possess anti-cancer, anti-oxidant, and anti-inflammation properties [18–21]. CRO can protect cardiomyocytes against lipopolysaccharide-induced inflammation [18], and amelioratecerulein-induced pancreatic inflammation and oxidative stress [19], with anti-cancer effect [21]. In addition, the previous study reported that CRO generated an ameliorative effect on experimental AS in quails, as well as inhibited lipid synthesis and induced M2 macrophage polarization in rat with AS, implicating that CRO can be used as a potential therapeutic strategy for AS [22,23]. However, the detailed mechanism of CRO on AS still needed more investigation.
Given the roles and effects of lysophosphatidic acid (LPA) and high-fat diet (HFD) on the onset of AS [24,25], this study therefore established LPA-induced macrophages to form foam cell as a cellular AS model in vitro and an HFD-induced AS mouse model in vivo. After treating both macrophages and mice with CRO, the effect and mechanism of CRO on foam cell formation, RCT in macrophages and the plaque formation in mice with AS were further explored.
Material and Methods
Ethics statement
All animal experiments were performed strictly based on the guidelines of China Council on Animal Care and Use, approved by the Committee of Experimental Animals of The First Hospital of Hebei Medical University (serial no. Z2019092123X) and performed in the First Hospital of Hebei Medical University. Every effort was made to minimize pain and discomfort for the animals.
Animal model construction
The animal experiments were performed according to the previous reports [11,12]. Apolipoprotein E (ApoE) as part of the high-density lipoprotein, has anti-inflammatory, antioxidant and antiatherogenic properties [26]. Established an animal model of HFD-induced AS using ApoE-deficient (ApoE−/−) mice was commonly used to study the progression of AS [12]. CRO (HY-N0697) used for our study was obtained from MedChemExpress (Princeton, NJ, USA), and 64 male Apolipoprotein E-deficient (ApoE−/−) C57BL/6 J mice (11-wk old were obtained from Cavens (Changzhou, China). All the mice were raised under a specific pathogen-free (SPF) environment and given free access to food and water with set conditions of light (a 12 h of light/dark cycle), temperature (22–24°C) and humidity (50–60%). All the mice were given adaptive feeding for a week prior to our study. After that, all mice (12-wk old, 28–30 g) were randomly divided into four groups: the Sham group, the HFD group, the HFD+CRO-L group, and the HFD+CRO-H group, 16 mice were included in each group (n = 16) and treated as mentioned subsequently. CRO was administered simultaneously with HFD in the animal model construction. The mice in the Sham group were fed on normal diet for 12 wk, while those in the remaining groups were fed on HFD with 21% fat and 0.15% cholesterol for 12 wk. Meanwhile, the mice in the HFD+CRO-L group and the HFD+CRO-H group, respectively, received a low dose of CRO (CRO-L; 4 mg/kg/d) and a high dose of CRO (CRO-H; 12.5 mg/kg/d) via gavage once a day for 12 wk, whilst those in the Sham group and the HFD group received equal volume of distilled water via oral administration once a day for 12 wk. The bodyweight of the mice was recorded by a body composition analyzer (LF50, Bruker Corporation, Billerica, MA, USA) every 2 wk following model construction (from 12-wk old to 24-wk old). After 12 wk of the experiment, the mice (24-wk old) were deeply anesthetized and sacrificed by inhalation of 5% isoflurane (R510-22-4, RWD Lifesciences, Shenzhen, China) and cervical dislocation. Then the blood of the mice was collected, and the aortic arch located 0.5 cm away from the aortic root of each mouse was removed for later use.
Mice aortic arch dissection
All procedures were conducted as illustrated in a previous study [27]. For dissection of mice aortic arch, after the sacrifice of mice in each group, the skin of mice was cut from the base of the abdomen to the top of the thorax with blunt scissors and forceps, and laparotomy was performed below the rib cage. The sternum was lifted with forceps and the diaphragm was cut, and the bilateral rib cage was opened to expose the thymus and lungs. The esophagus and lungs were removed so as to expose the heart and gain access to the aorta. Two or three small incisions were made in the liver for drainage, and the blood from all the organs was washed out via perfusing phosphate buffered saline (PBS, P4417, Sigma-Aldrich, St Louis, MO, USA). All dissected areas were gently dried with paper towels. After gripping the segments on the diaphragm attached to the end of the thoracic aorta with scissors and forceps, the connective tissue between the aorta and the thoracic cavity muscle wall was cut to expose the area of the aortic arch. All the left and right carotid arteries and the left subclavian artery were removed to allow free access to both the heart and the aorta, which were dissected and placed in regular plastic dish with PBS. Under a stereo microscope (EZ4, Leica Microsystems, Wetzlar, Germany), the aorta was carefully removed by cutting at the point where it emerges from the heart and placed in a 1.5 mL Eppendorf tube, followed by being added with 1 mL of paraformaldehyde solution (P6148, Sigma-Aldrich, USA) at 4°C overnight. Then the aorta was embedded in pre-dissolved paraffin (1.07151, Supelco, Bellefonte, PA, USA) and subjected to both hematoxylin-eosin staining and Oil-red-O staining.
Hematoxylin-eosin staining
The histopathological changes of mouse aorta were observed by Hematoxylin-eosin staining. All procedures with hematoxylin-eosin staining were performed as previously described [27]. All the paraffin-embedded tissue samples were warmed in a dry oven at 55°C for 5 minutes and resected into 5-μm-thick sections. All samples were deparaffinized with xylene (214,736, Sigma-Aldrich, USA) twice (5 minutes for each time) and dehydrated with different concentrations of Ethanol (E7023, Sigma-Aldrich, USA) under the conditions as below: absolute Ethanol for 5 minutes, 95% Ethanol for 5 minutes and 70% Ethanol for 2 minutes (each procedure was performed twice). All samples were then washed with distilled water and firstly stained with hematoxylin from the Hematoxylin-eosin staining reagent (R20570, Shanghai Yuanye Biotechnology Co., Ltd, Shanghai, China) for 3 minutes. The tissues were successively rinsed in running tap water for 5 minutes and 95% Ethanol for 30 seconds. Then the samples were counterstained with eosin (diluted in 95% Ethanol) from the Hematoxylin-eosin staining reagent for 1 minute, washed with running tap water for 5 minutes and dehydrated using Ethanol. After being cleared by xylene for 2 minutes, the image on the aorta was observed and photographed using an automatic microscope (DMLA; Leica, Solms, Germany) at a magnification of × 8.
Masson staining
The collagen changes of mouse aorta were observed by Masson staining. All procedures with Masson staining were confirmed as described previously [28]. The aortic arch tissue was collected and used to prepare 4-μm-thick sections. All the tissues were stained with hematoxylin for 5 minutes, and treated with acid ponceau solution for 8 minutes, followed by treatment with aniline blue solution for another 5 minutes. Hematoxylin, acid ponceau fuchsin and aniline blue solutions were all provided from Masson’s trichrome staining reagent (HT15, Sigma-Aldrich, USA). Finally, the tissue was fixed in 1% acetic acid (A6283, Sigma-Aldrich, USA) for 2 minutes and rinsed in distilled water, dehydrated with 95% Ethanol and transparentized with xylene, followed by being sealed in neutral gum (G8590, Solarbio, Beijing, China). All tissues were finally observed and photographed using an automatic microscope at a magnification of × 8.
Immunohistochemistry
The levels of cluster of difference 68 (CD68), Liver X Receptor-α (LXR-α), and Peroxisome Proliferator-Activated Receptor γ (PPARγ) in mouse aorta were observed by immunohistochemistry. Immunohistochemistry was conducted as illustrated previously [29]. The aortic arch tissue was collected and used to prepare 4-μm-thick sections. Then, the samples were incubated with an antigen repair solution (P0081, Beyotime, Shanghai, China) at 100°C for 10 minutes, and blocked by 5% bovine serum albumin (BSA, A7030, Sigma-Aldrich, USA) for 1 hour, followed by incubation with the primary antibodies of atherosclerosis-related marker CD68 (1:200, ab125212, Abcam, Cambridge, UK), anti-LXR-α (1:500, ab106464, Abcam, UK) and anti-PPARγ (1:500, ab59256, Abcam, UK) antibodies overnight at 4°C. Then, the slices were incubated with the corresponding secondary antibodies: goat anti-rabbit IgG antibody (ab205718, Abcam, UK) or goat anti-mouse IgG antibody (ab205719, Abcam, UK) for 1 hour at room temperature and treated with the 3, 3ʹ-diaminobenzidine (DAB) reagent (DA1015, Solarbio, China) for 30 minutes. Next, the tissue was stained by hematoxylin for 10 minutes. Finally, the tissue was observed and photographed using a light microscope (SZX10, Olympus, Tokyo, Japan) at a magnification of × 40.
Enzyme-linked immunosorbent assay (ELISA)
All the processes with ELISA were performed as described in a prior study [30]. In detail, after sacrificing the mice, 2 mL blood samples from the retroorbital plexus of mice in each group were first harvested and kept at room temperature for 2 hours, followed by being centrifuged at 300 × g for 20 minutes at 4°C. The serum was subsequently collected and stored at −80°C until use.
Then, the levels of total cholesterol (TC, #76,211-700, VWR International, Radnor, PA, USA), triglyceride (TG, #KTE70365, Abbkine, Wuhan, China), high-density lipoprotein-cholesterol (HDL-C, #orb409066, Biorbyt LLC, Cambridge, UK), and LDL-cholesterol (LDL-C, #CSB-EQ027860MO, CusaBio Technology LLC, Houston, TX, USA) in the serum of mice were detected by ELISA using the corresponding kits as instructed by the manufacturers. OD values were finally measured with Crocodile 4-in-one assay miniWorkstation (Titertek-Berthold Detection Systems GmbH, Pforzheim, Germany), and their levels were presented as nmol/L.
Cell culture and treatment
Mouse RAW264.7 macrophages (CL-0190, Procell, Wuhan, China) were normally cultured in α-minimum essential medium (α-MEM, M8042, Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (FBS, F2442, Sigma-Aldrich, USA) and 1% penicillin-streptomycin (P4333, Sigma-Aldrich, USA) at 37°C with 5% CO2.
For the construction of in vitro cell models, crocin was administered simultaneously with LPA in the in vitro model. RAW264.7 cells were cultured in the α-MEM which have been co-treated with 200 μmol/L LPA (O832928, MREDA, Beijing, China) and different concentrations of CRO (1, 5, 10, 20 μmol/L) for 24 h. Then the cells were collected for later use. The concentrations of both LPA and CRO were selected according to those described previously but with slight modifications [31,32].
Cell transfection
Small interfering RNA (siRNA) for LXR-α (siLXR-α, SR414579) and siRNA negative control (siNC) for LXR-α (SR30002) were purchased from Origene (Rockville, MD, USA). Before transfection, 2 mL of α-MEM medium containing 10% FBS and RAW264.7 cells at a density of 1.0 × 106 cells/well were placed into a 6-well plate and cultured until the cell confluence reached about 80%. Then, the siRNAs including siLXR-α and siNC were transfected into the cells using 3 μL of lipofectamine 3000 reagent (L3000-015, Invitrogen, Carlsbad, CA, USA) at 37°C and cells were further cultured. All cells were collected 48 hours later for subsequent studies.
MTT assay
RAW264.7 cells at a density of 1 × 105 cells/well were cultured in a 96-well plate at 37°C with 5% CO2 and treated with different concentrations of CRO (1, 5, 10 and 20 μM) or not for 24 hours. 10 μL MTT reagent (R20228, Shanghai Yuanye Biotechnology Co., Ltd, China) was added at 37°C for additional 4 hours. 100 μL dimethyl sulfoxide (DMSO, S24295, Shanghai Yuanye Biotechnology Co., Ltd, China) was added to make those visible formazan salts dissolve. Optical density (OD) value at 570 nm was determined with iMark microplate reader (Bio-Rad, Hercules, CA, USA).
Oil-red-O staining
Two schemes of Oil-red-O staining were used to evaluate the formation of the lipid deposition in mice aorta and lipid droplets in RAW264.7 cells in line with the previous publications [27,33]. For visualization on lipid deposition in the mice aorta, all processes with Oil-red-O staining were conducted at room temperature as described previously [27]. After cleaning the aorta by removal of any adventitial fat under a stereo microscope and cutting the aorta into 5-μm-thick sections, 1 mL of 78% Methanol (322,415, Sigma-Aldrich, USA) was added into the aorta within the tubes, which were placed on a tilted roller with gentle movement for 5 minutes twice. The methanol was discarded and 1 mL of fresh Oil-red-O solution (O0625, Sigma-Aldrich, USA) was added. The tubes were incubated on the tilted roller for 1 hour, and the aorta was transferred to a new tube and washed twice with 1 mL of 78% methanol for 5 minutes. The methanol was finally discarded again and the tubes were refilled with 1 mL PBS. The aorta was observed and photographed using an automatic microscope at a magnification of × 8.
For the RAW264.7 cells, after transfection and LPA or CRO treatment, the cells were fixed with formaldehyde (F8775, Sigma-Aldrich, USA) for 10 minutes and immersed with 60% isopropanol (W292907, Sigma-Aldrich, USA). Then cells were collected and incubated with Oil Red O at 25°C for 30 minutes. Next, the cells were washed with 75% ethanol. After washing the cells with PBS, the cells were observed under an automatic microscope at a magnification of × 200.
Cholesteryl l Ester/Total Cholesterol (CE/TC) quantitation
All procedures with quantifying CE/TC were calculated by TC and CE quantitation assay kit (K603, BioVision, Milpitas, CA, USA) as described previously [34]. For analysis on the TC and CE, CRO- and LPA-treated RAW264.7 cells were first lysed with RIPA lysis buffer (P0013 C, Beyotime, China) and 50 μL of the lysates were diluted in 1 × reaction buffer provided from the quantitation kit and then diluted in either TC reaction mixture (for quantifying TC) or CT reaction mixture (for quantifying CE) (both were set at 50 μL) for an hour at 37°C in the dark. The OD value at 570 nm was recorded with Crocodile 4-in-one assay miniWorkstation (Titertek-Berthold Detection Systems GmbH, Germany).
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
For RNA extraction, after transfection and treatment with LPA and CRO, the RAW264.7 cells were collected and lysed by TRIzol (15,596–018, Invitrogen, USA), and the extracted RNA was subsequently stored in an −80°C refrigerator. A NanoDrop 2000 c spectrometer (ND-2000 c, ThermoFisher Scientific, Waltham, MA, USA) was then used to measure the concentration. In regard to qRT-PCR, cDNA was synthesized by a First Strand cDNA Synthesis Kit (K1651, ThermoFisher Scientific, USA). All processes of qRT-PCR were performed in triplicate using standard SYBR green PremixEx Taq II kit (RR820L, TaKaRa, Shiga, Japan) on CFX384 real-time PCR system (Bio-Rad, USA). β-actin was used as the internal control. The expression was quantified using the 2−ΔΔCT method [35]. The primers used in this experiment were listed as follows: LXR-α-Forward: 5ʹ-CAAGGGAGCACGCTATGTCTG-3ʹ, LXR-α-Reverse: 5ʹ-GGACACCGAAGTGGCTTGAG-3ʹ; β-actin-Forward: 5ʹ-CCTAGAAGCATTTGCGGTGG-3ʹ, and β-actin-Reverse: 5ʹ-GAGCTACGAGCTGCCTGACG-3ʹ.
Western blot
All protein expressions were measured via Western blot as expounded previously [12]. The total protein in the RAW264.7 cells and the mice aorta root were extracted by RIPA lysis buffer and the concentration was determined by the bicinchoninic acid (BCA) protein kit (P0012S, Beyotime, China). After denaturation, the protein (20 μg) was electrophoresed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (P0012A, Beyotime, China), and transferred into polyvinylidene fluoride (PVDF) membranes (FFP33, Beyotime, China). Then, the membrane was incubated with following primary antibodies at 4°C overnight, including those against PPARγ (1:1000, ab272718, Abcam, UK), LXR-α (1:2000, ab176323, Abcam, UK), PCSK9 (1:2000, ab31762, Abcam, UK), CD36 (1:1000, ab133625, Abcam, UK), ABCA1 (1:500, ab18180, Abcam, UK), phosphorylated (p)-AKT (1:1000, ab38449, Abcam, UK), AKT (1:10,000, ab179463, Abcam, UK), and internal control β-actin (1:1000, ab8226, Abcam, UK). Following being incubated with secondary horseradish peroxidase (HRP)-conjugated antibodies: goat anti-rabbit IgG H&L (ab205718, 1:2000, Abcam, UK) or goat anti-mouse IgG H&L (ab205719, 1:2000, Abcam, UK) at room temperature for 1 hour and washed using tris-buffer saline tween (TBST, T196393, Aladdin, China) for three times, protein bands were visualized by a Pierce ECL Western blotting substrate (32,109, ThermoFisher Scientific, USA). Image J (version 5.0, National Institutes of Health, Bethesda, MD, USA) was used to capture the chemiluminescent signals and the data were analyzed with iBright CL750 Imaging System (A44116, ThermoFisher Scientific, USA).
Statistical analysis
Every experiment was performed in at least three times independently. Data were expressed as mean ± standard deviation (SD). In the animal experiment, samples were collected from all 16 animals for each assay/measurement presented. All the data involved were analyzed using Graphpad prism software (version 8.0, GraphPad, Inc., La Jolla, CA, USA). Statistical significance was determined using one-way ANOVA followed by Bonferroni post hoc test. P < 0.05 indicated that the data were statistically significant.
Results
CRO decreased HFD-induced body weight gain and ameliorated the abnormal serum lipid levels of HFD-treated mice
To prove the inhibitory effect of CRO on AS in vivo, the AS mice (12-wk old) model was established by HFD feeding, and then the body weight of mice in each group was measured every 2 wk since model construction. When the experiment starts, the body weight of the mice (12-wk old) had no difference (Fig. 1A). However, after the HFD feeding for 12 wk (i.e. the week 24), the body weight of the mice in the HFD group was dramatically increased (Fig. 1A, P < 0.05), whilst, by contrast, the body weight of the mice induced by HFD and further treated with CRO was reduced compared with HFD group (Fig. 1A, P < 0.05), which indicated that CRO decreased the body weight of HFD-treated mice.
Figure 1.
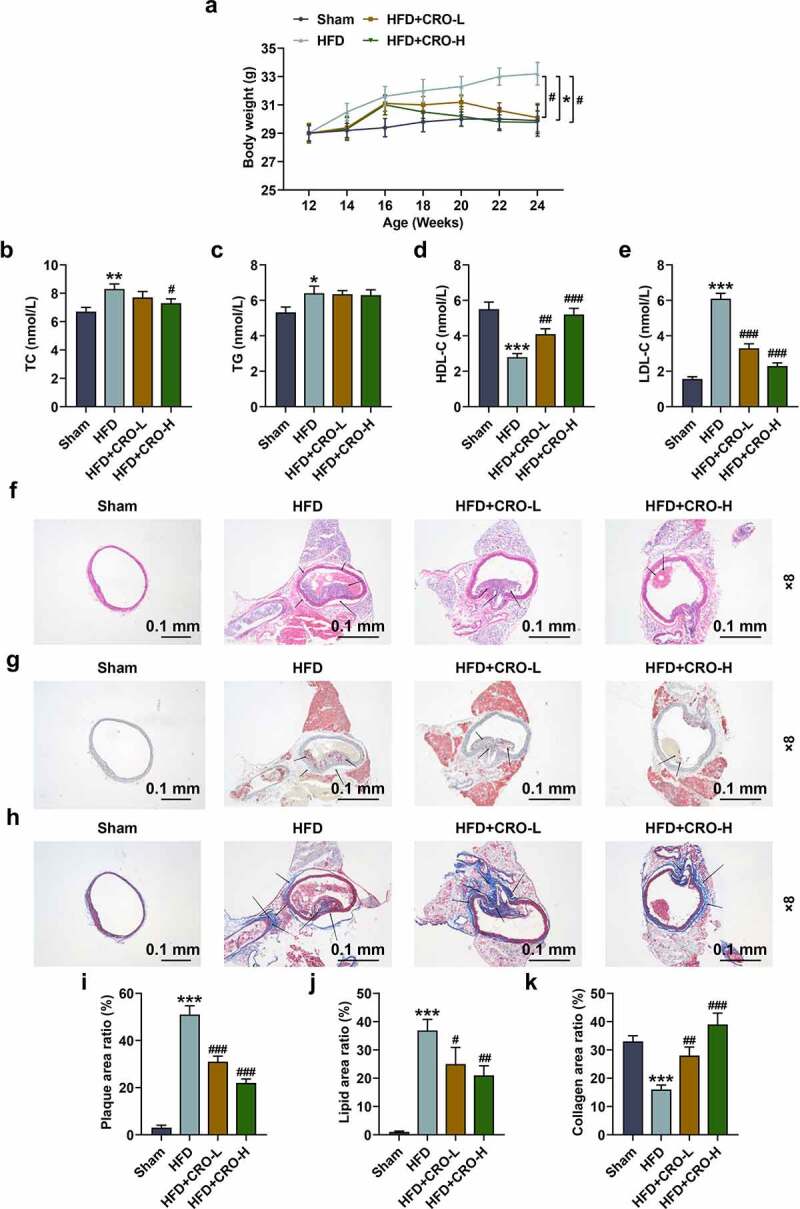
CRO decreased the weight gain, ameliorated the abnormal serum lipid levels, and inhibited the aortic plaque formation and the deposition of lipid but increased the collagen fibers in the plaque of mice with AS.
(A) The body weight of mice after being fed with HFD and CRO treatment or not was recorded. (B-E) The serum levels of TC (B), TG (C), HDL-C (D), and LDL-C (E) after mice being fed with HFD and CRO treatment or not were determined by ELISA. (F, I) The aortic plaque formation of the mice after being fed with HFD and CRO treatment or not was detected by hematoxylin-eosin staining (magnification: ×8). (G, J) The deposition of lipid in the aortic plaque of the mice after being fed with HFD and CRO treatment or not was evaluated by Oil-red-O staining (magnification: ×8). (H, K) The deposition of collagen in the aortic plaque of the mice after being fed with HFD and CRO treatment or not was evaluated by Masson staining (magnification: ×8). All experiments have been performed independently in triplicate and data were expressed as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001, vs. Sham; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. HFD. AS: Atherosclerosis, CRO-L: low dose of Crocin, CRO-H: high dose of Crocin, HFD: high-fat diet, TC: total cholesterol, TG: triglyceride, HDL-C: high-density lipoprotein-cholesterol, LDL-C: low-density lipoprotein-cholesterol; ELISA: enzyme-linked immunosorbent assay.
Then, the serum lipid levels of the mice were determined by evaluating the serum level of lipid-related factors. In HFD group, the mice had higher levels of TC, TG, and LDL-C yet a lower level of HDL-C than those in normal mice (the Sham group) (Fig. 1B–1E,P < 0.05). On the contrary, in the HFD+CRO-L and the HFD+CRO-H groups, the mice had lower levels of TC and LDL-C and high level of HDL-C level compared with those in mice of the HFD group. Besides, no difference of TG level was observed in mice among the three groups (HFD+CRO-L, HFD+CRO-H and HFD groups) (Fig. 1B–1E,P < 0.05). All this evidence suggested that CRO had an improving effect on abnormal serum lipid levels of HFD-treated mice.
CRO inhibited the aortic plaque formation and the deposition of lipid but increased the collagen in the plaque of HFD-treated mice
Subsequently, the aortic plaque in the mice with or without AS was determined through hematoxylin-eosin staining. As delineated in Fig. 1F, there was no aortic plaque in mice of the Sham group, while certain aortic plaques existed in the HFD-treated mice, the number of which was decreased after treatment with CRO (Fig. 1F, 1I, P < 0.001). Besides, the levels of lipid and collagen in the plaque of HFD-treated mice were further determined by the Oil-red-O staining (Fig. 1G) and Masson staining (Fig. 1H). As depicted, in terms of lipid deposition and collagen contents, the area of lipid was low in the mice of Sham group (Fig. 1G,1J) but were high in the mice of HFD group, and collagen content was low in the mice of HFD group (Fig. 1H, 1K), while the two constituents in the aortic plaque of HFD-treated mice were ameliorated after treatment with CRO (Fig. 1G-1 H, 1 J-1 K, P < 0.001). These results further verified that CRO inhibited the aortic plaque formation and the deposition of lipid but increased collagen in the plaque of HFD-treated mice.
CRO regulated CD68, PPARγ/LXR-α, lipid metabolism-related factors and AKT phosphorylation in the aorta of HFD-treated mice
We examined the levels of PPARγ/LXR-α and CD68 in the aorta of AS mice treated with or without CRO by immunohistochemistry. It turned out that CD68 level was upregulated yet PPARγ/LXR-α levels were downregulated in mice of the HFD group, while the opposite trends were observed after CRO treatment (Fig. 2A-2 F, P < 0.01).
Figure 2.
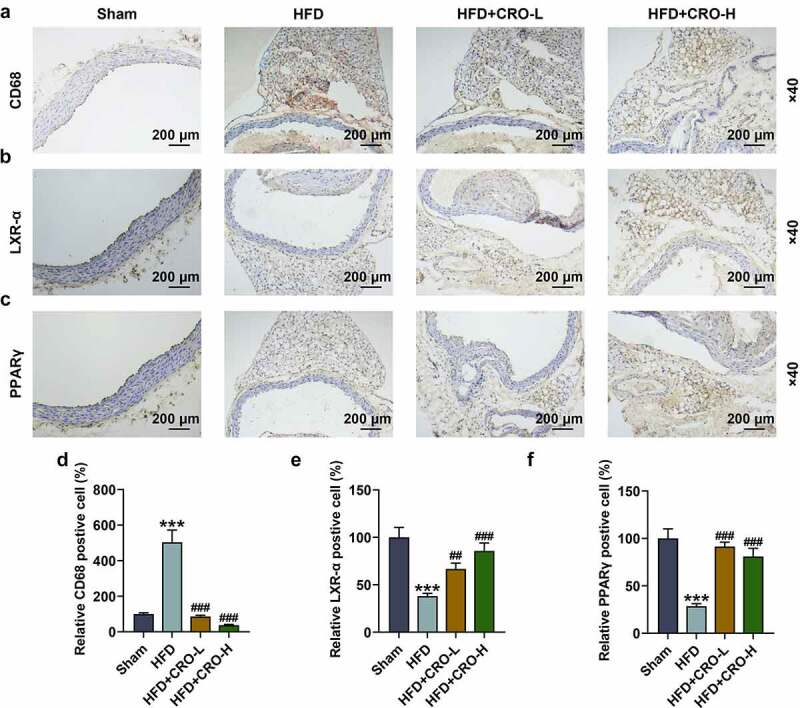
CRO regulated the abnormal CD68, LXR-α, and PPARγ levels in the aorta of mice with AS.
The levels of CD68, LXR-α, and PPARγ in the aorta of mice after being fed with HFD and CRO treatment or not were determined by immunohistochemistry (magnification × 40). ***P < 0.001, vs. Sham; ##P < 0.01, ###P < 0.001, vs. HFD. AS: Atherosclerosis, CRO-L: low dose of Crocin, CRO-H: high dose of Crocin, HFD: high-fat diet; CD68: cluster of difference 68; LXR-α: Liver X Receptor-α; PPARγ: Peroxisome Proliferator-Activated Receptor γ.
Furthermore, the expressions of PPARγ/LXR-α were measured using the Western blot, which were found to be down-regulated following HFD treatment but were up-regulated following CRO treatment (Fig. 3A–3C, P < 0.001). Since PCSK-9, CD36 and ABCA1 have been suggested to be related to lipid metabolism [36–38], their expressions following HFD and CRO treatment were measured as well, validating that PCSK9 and CD36 expressions were up-regulated yet that of ABCA1 was down-regulated in mice of the HFD group, while the down-regulated PCSK9 and CD36 expressions and up-regulated ABCA1 expression in mice were induced by CRO treatment (Fig. 3D-3 F, P < 0.001). Since AKT phosphorylation has been proven to be implicated in AS of mice [39], the expressions of AKT and its phosphorylated form (p-AKT) were subsequently measured, the latter of which was decreased in mice of the HFD group, but was increased by CRO treatment (Fig. 3G-3 H, P < 0.01). Although there is no difference of AKT expression among these groups, the ratio of p-AKT to AKT shared the same tendency with p-AKT among these groups (Fig. 3I, P < 0.01). All these results above indicated that the therapeutic effect of CRO was realized by regulating the expressions of PPARγ/LXR-α, lipid-metabolism-related factors and the phosphorylation of AKT.
Figure 3.
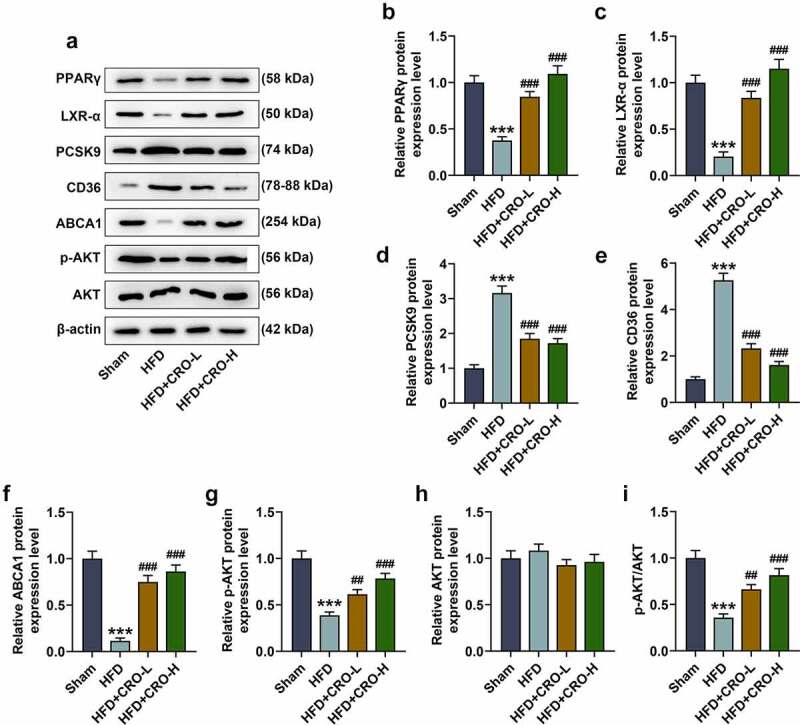
CRO regulated CD68, PPARγ/LXR-α, the lipid metabolism-related factors and AKT phosphorylation in the aorta of mice with AS.
(A-H) The protein/β-actin expressions of PPARγ/LXR-α, PCSK9, CD36, ABCA1, p-AKT, and AKT in the aorta of mice with AS were detected by Western blot. β-actin was used as an internal control. (I) The ratio of p-AKT to AKT was calculated based on the data of Western blot. All experiments have been performed independently in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001, vs. Sham; ##P < 0.01, ###P < 0.001, vs. HFD. AS: Atherosclerosis, CRO-L: low dose of Crocin, CRO-H: high dose of Crocin, HFD: high-fat diet; PCSK9: Proprotein convertase subtilisin/kexin type 9; CD68: cluster of difference 68; ABCA1: ATP Binding Cassette Subfamily A Member 1; p-AKT: phosphorylated-AKT.
CRO inhibited lipid droplets formation and regulated lipid metabolism-related factors in LPA-treated macrophages
The effect of CRO on the viability of RAW264.7 cells was first evaluated after treating macrophages with different concentrations of CRO (1, 5, 10 and 20 μmol/L), uncovering that CRO treatment alone generated no effect on the viability of RAW264.7 cells (Fig. 4A).
Figure 4.
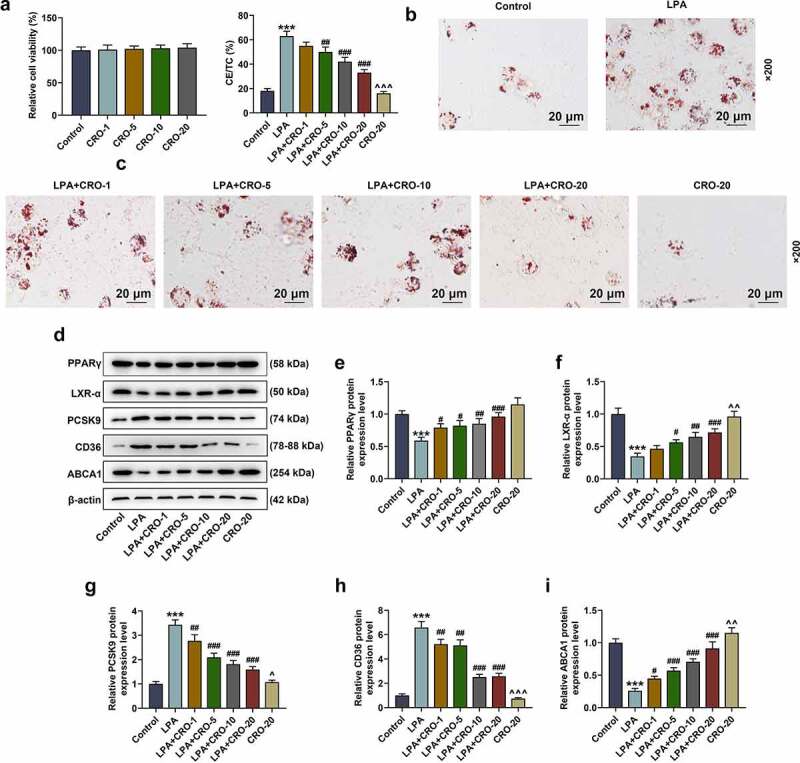
CRO inhibited the lipid droplets formation and regulated the levels of lipid metabolism-related factors in LPA-induced macrophages
(A) The viability of RAW264.7 macrophages after being treated with different concentrations of CRO was detected by MTT assay. (B) The formation of lipid droplets in RAW264.7 macrophages after being treated with LPA or CRO was detected by Oil-red-O staining (magnification × 200). (C) The levels of CE/TC in RAW264.7 macrophages after being treated with LPA or CRO were detected and calculated. (D-I) The protein/β-actin expressions of PPARγ/LXR-α, PCSK9, CD36, and ABCA1 of RAW264.7 macrophages after treated with LPA or CRO were detected by Western blot. β-actin was used as an internal control. ***P < 0.001, vs. Control; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. LPA; ^P< 0.05, ^^P< 0.01, ^^^P< 0.001, vs. LPA + CRO-20. CRO: Crocin, LPA: lysophosphatidic acid, CE: cholesteryl ester, TC: total cholesterol.
To investigate the effect of CRO on AS in vitro, LPA was used to induce the RAW264.7 macrophages to establish the foam cells model. As depicted in Fig. 4B-4 C, the lipid droplet formation and the level of CE/TC in the LPA-induced RAW264.7 macrophages were obviously increased (Fig. 4B-4 C, P < 0.001), which indicated that the foam cells model was successfully established. In addition, the LPA-induced increment on lipid droplets and CE/TC of macrophages was notably decreased by CRO treatment (Fig. 4B-4 C, P < 0.01), affirming that CRO inhibited the formation of foam cells. Besides, the expression of lipid-metabolism-related factors and PPARγ/LXR-α were evaluated to further verify the discovery above. As illustrated in Fig. 4D–4I, LPA down-regulated the expressions of PPARγ/LXR-α and ABCA1 but up-regulated those of PCSK9 and CD36 in RAW264.7 cells (Fig. 4D–4I, P < 0.001), while CRO treatment reversed the effect of LPA above (Fig. 4D–4I, P < 0.05), which further corroborated that CRO inhibited the lipid droplets formation and exerted regulatory effects on the expressions of lipid metabolism-related factors in macrophages.
Silent LXR-α reversed the effect of CRO on the lipid droplets formation and lipid metabolism-related factors levels in LPA-treated macrophages
We subsequently transfected siLXR-α into LPA- and CRO-treated macrophages, and the mRNA and protein expressions of LXR-α were evaluated. It could be noted that the expression of LXR-α in LPA-treated macrophages was up-regulated by CRO treatment (Fig. 5A-5 C, P < 0.001), which was overturned by silent LXR-α (Fig. 5A-5 C, P < 0.001).
Figure 5.

LXR-α silencing reversed the effect of CRO on the lipid droplet formation, and the levels of lipid metabolism-related factors in LPA-induced macrophages
(A-B) The protein/β-actin expression of LXR-α in RAW264.7 macrophages after transfection or LPA or CRO treatment was detected by Western blot. β-actin was used as an internal control. (C) The mRNA expression of LXR-α in RAW264.7 macrophages after transfection or LPA or CRO treatment was detected by qRT-PCR. β-action was used as an internal control. (D) The formation of lipid droplets in RAW264.7 macrophages after transfection or LPA or CRO treatment was detected by Oil-red-O staining (magnification × 200). (E) The levels of CE/TC in RAW264.7 macrophages after transfection or LPA or CRO treatment were detected. (F-K) The protein/β-actin expressions of PCSK9, CD36, ABCA1, p-AKT, and AKT in RAW264.7 macrophages after transfection or LPA or CRO treatment were detected by Western blot. β-action was used as an internal control. (L) The ratio of p-AKT to AKT was calculated based on the data of Western blot. ***P < 0.001, vs. LPA; ##P < 0.01, ###P < 0.001, vs. LPA+CRO-20+ siNC. siNC: small interfering RNA for negative control; siLXR-α: small interfering RNA targeting LXR-α.
Also, as depicted in Fig. 5D–5E, CRO treatment suppressed the lipid droplets formation and downregulated CE/TC level in LPA-treated macrophages, while silent LXR-α reversed the inhibitory effect of CRO (Fig. 5D–5E,P < 0.001). Furthermore, data in Fig. 5F-5 L disclosed that CRO inhibited the expressions of PCSK9 and CD36 and promoted the levels of ABCA1, p-AKT and p-AKT/AKT ratio in the LPA-induced cells (Fig. 5F-5 L, P < 0.001), while silent LXR-α inverted the effect of CRO on these factors (Fig. 5F-5 L, P < 0.01). All these phenomena evidenced that silent LXR-α could reverse the effects of CRO on the lipid droplets formation and the expressions of lipid metabolism-related factors in LPA-treated macrophages.
Discussion
AS is a progressive disease starting with the accumulation of immune cells, lipoproteins, and lipids in the arterial wall [40]. Despite the continuing efforts that have been made to explore the pathogenesis and progression of AS, our understanding of AS mechanisms remains limited [41]. Established an animal model of HFD-induced AS using ApoE-deficient (ApoE−/−) mice was commonly used to study the progression of AS [12]. Previous studies have reported that the body weight of HFD-induced AS model mice was significantly higher than that of control mice, and the mice in the model group had higher serum TC and LDL‑C levels [12]. In this work, we also found that after the inducement of HFD, the bodyweight of the mice in the HFD group was significantly increased. Interestingly, early studies pointed out that CRO inhibits lipid synthesis and induced M2 macrophage polarization in rat with AS so that it can be used as a promising therapeutic strategy for AS [23]. Nevertheless, the detailed mechanism awaited to be additionally elucidated. Similarly, in our study, we found that CRO decreased HFD-induced body weight increase, ameliorated the abnormal serum lipid levels, inhibited the aortic plaque formation, the deposition of lipid in the plaque of HFD-treated mice, which proved that CRO had an anti-AS effect and its mechanism still needed more exploration.
The formation of macrophage foam cells in the intima is a major marker of early atherosclerotic lesions [34]. To verify the effect of CRO on AS, the macrophage foam cells model was established by inducing macrophages with LPA, which is widely used in basic research [34]. Also, such formation is thought to be the result of an imbalance between cholesterol influx and efflux [6]. During the development of AS, monocytes differentiate into macrophages in the vascular intima with uncontrolled uptake of more ox-LDL and impaired intracellular cholesterol excretion through the SR (such as CD36 and SR-1), causing the formation of excess cholesterol esterification and storage in cells, and finally contributing to the formation of foam cells [37,42]. In this study, after being treated with LPA, the lipid droplets formation and the levels of CE/TC in the macrophages were remarkably enhanced, indicating that the foam cell model was successfully established. In accordance with the results from both in vivo and in vitro research, we come to conclude that the AS model was successfully constructed to support our follow-up research.
RCT, a vital mechanism to prevent AS, refers to the process of transporting cholesterol from the surrounding tissues, including the arterial wall to the liver for recirculation or excretion in the form of bile acid [43,44]. With respect to the findings above that the CRO inhibited the deposition of lipid and increased the collagen fibers in the foam cells and the plaque of mice with AS, we then conjectured that the anti-AS effect of CRO was achieved by promoting the RCT progression, with the efflux of cholesterol being deemed to be the critical step [45]. ABCA1, as an integral membrane protein and a member of ATP-binding cassette transporter, is indispensable for mediating the lipid efflux [11]. The expression of ABCA1 is regulated by LXR-α, a nuclear receptor that regulates cholesterol metabolism [46]. PPARγ, a ligand-activated nuclear receptor, has regulatory effects on glucose and lipid metabolism, endothelial function and inflammation, as evidenced in AS [47,48]. In addition, PCSK9 is a kind of subtilisin that regulates lipid metabolism and directly affects the vessel wall. PCSK9 is expressed in both AS aortic plaque of humans and in the liver of ApoE−/− mice [11,12,34]. Similarly, in this study, we discovered that the expressions of PPARγ, LXR-α and ABCA1 were downregulated and that of PCSK9 was upregulated in both LPA-induced foam cells and HFD-induced mice with AS, while the CRO treatment reversed the tendency of PPARγ, LXR-α, and PCSK9 expressions. Previous studies have reported that CRO promoted macrophage polarization to the M2 phenotype via elevating M2 markers (CD206 and CD68) and reducing M1 markers (CD40 and CD11c), while we manifested that the CRO decreased the upregulated level of CD68, a marker of M2 phenotype macrophage [23], in AS mice, which was contrary from the previous study [23] and further validated that CRO inhibited the formation of foam cells. All the evidence of the changes of PPARγ, LXR-α, PCSK9, and CD68 expressions confirmed that the anti-AS effect of CRO was implemented by promoting the RCT and inhibiting the formation of foam cells.
Considering that PPARγ is a key protein during the RCT and the formation of foam cells, and its interaction with LXR-α has been evidenced in AS [49–51], we wondered that whether the effect of CRO on RCT and foam cells formation was mediated directly via modulating PPARγ/LXR-α. Therefore, we transfected siLXR-α into LPA-treated macrophages to verify our hypothesis, unveiling that after the foam cells were treated with CRO and silent LXR-α, the effect of CRO on the macrophages was all reversed, which verified that the effect of CRO on AS was mediated by modulating LXR-α. Furthermore, we also discovered that the level of p-AKT in the foam cells was downregulated whilst CRO treatment increased p-AKT level. AKT is a key protein in much signal transduction pathway including P38/MAPK and PI3K/AKT pathway. During the transduction of these pathways, the phosphorylation level of AKT (p-AKT) can be elevated, which also determined both the viability of macrophages and their resistance against those pro-apoptotic stimuli [39,52,53]. The increased p-AKT in CRO-treated foam cells indicated that the signaling pathway such as P38/MAPK and PI3K/AKT pathways might be involved in the development of AS and the effect of CRO on AS.
Nevertheless, some limitations in our study are waited to be addressed. We have confirmed the protective role of CRO on AS-induced lesion both in vivo and in vitro, but the detailed mechanism and effect of CRO on AS in vivo remained vague. Besides, several studies have documented that vasa vasorum angiogenesis, mitochondrial dysfunction, and ion-channels are involved in the progression of AS [54–56]. Interestingly, several studies have identified genes involved in the progression of neurodegenerative diseases by gene analysis techniques such as RNA-Seq, which are involved in the progression of retinitis pigmentosa or retinal degeneration by regulating angiogenesis, mitochondrial dysfunction, and ion channels [57–59]. In future research, we will also further explore the specific mechanism of action of CRO in AS by using the RNA-Seq.
Conclusion
To conclude, we provided another evidence on the efficacy of CRO on AS in the light of in vivo and in vitro studies. It was discovered and summarized in our research that CRO inhibited the aortic plaque formation and the deposition of lipid by alleviating the lipid metabolism in HFD-treated mice, promoting the RCT and inhibiting the foam cell formation in LPA-treated macrophages, which authenticated that CRO could be developed as a candidate drug for AS prevention and treatment.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Authors’ contributions
Substantial contributions to conception and design: Feng Zhang, Peng Liu
Data acquisition, data analysis and interpretation: Zhaopeng He, Like Zhang, Xinqi He, Feng Liu, Jinsheng Qi
Drafting the article or critically revising it for important intellectual content: Feng Zhang, Peng Liu
Final approval of the version to be published: Feng Zhang, Peng Liu, Zhaopeng He, Like Zhang, Xinqi He, Feng Liu, Jinsheng Qi
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved: Feng Zhang, Peng Liu, Zhaopeng He, Like Zhang, Xinqi He, Feng Liu, Jinsheng Qi
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Xu H, Jiang J, Chen W, et al. Vascular Macrophages in Atherosclerosis. J Immunol Res. 2019;2019:4354786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schaftenaar F, Frodermann V, Kuiper J, et al. Atherosclerosis: the interplay between lipids and immune cells. Curr Opin Lipidol. 2016;27:209–215. [DOI] [PubMed] [Google Scholar]
- [3].Sun C, Fu Y, Gu X, et al. Macrophage-Enriched lncRNA RAPIA: a Novel Therapeutic Target for Atherosclerosis. Arterioscler Thromb Vasc Biol. 2020;40:1464–1478. [DOI] [PubMed] [Google Scholar]
- [4].Artyomov MN, Sergushichev A, Schilling JD.. Integrating immunometabolism and macrophage diversity. Semin Immunol. 2016;28:417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barrett TJ. Macrophages in Atherosclerosis Regression. Arterioscler Thromb Vasc Biol . 2020;40:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li L, Du Z, Rong B, et al. Foam cells promote atherosclerosis progression by releasing CXCL12. Biosci Rep. 2020;40:BSR20193267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. [DOI] [PubMed] [Google Scholar]
- [8].Cao H, Jia Q, Yan L, et al. Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells. Int J Mol Sci. 2019;20: 6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peng Y, Xu J, Zeng Y, et al. Polydatin attenuates atherosclerosis in apolipoprotein E-deficient mice: role of reverse cholesterol transport. Phytomedicine. 2019;62:152935. [DOI] [PubMed] [Google Scholar]
- [10].Rohatgi A. Reverse Cholesterol Transport and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li SS, Cao H, Shen DZ, et al. Effect of Quercetin on Atherosclerosis Based on Expressions of ABCA1, LXR-α and PCSK9 in ApoE(-/-) Mice. Chin J Integr Med. 2020;26:114–121. [DOI] [PubMed] [Google Scholar]
- [12].Jia Q, Cao H, Shen D, et al. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARgamma, LXRalpha and ABCA1. Int J Mol Med. 2019;44:893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou M, Ren P, Li S, et al. Danhong Injection Attenuates High-Fat-Induced Atherosclerosis and Macrophage Lipid Accumulation by Regulating the PI3K/AKT Insulin Pathway. J Cardiovasc Pharmacol. 2019;74:152–161. [DOI] [PubMed] [Google Scholar]
- [14].Lu L, Qin Y, Chen C, et al. Beneficial Effects Exerted by Paeonol in the Management of Atherosclerosis. Oxid Med Cell Longev. 2018;2018 :1098617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shehatou GS, Suddek GM. Sulforaphane attenuates the development of atherosclerosis and improves endothelial dysfunction in hypercholesterolemic rabbits. Exp Biol Med (Maywood). 2016;241:426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ding X, Zheng L, Yang B, et al. Luteolin Attenuates Atherosclerosis Via Modulating Signal Transducer And Activator Of Transcription 3-Mediated Inflammatory Response. Drug Des Devel Ther. 2019;13:3899–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Z, Zhai L, Lu J, et al. Shen-Hong-Tong-Luo Formula Attenuates Macrophage Inflammation and Lipid Accumulation through the Activation of the PPAR-gamma/LXR-alpha/ABCA1 Pathway. Oxid Med Cell Longev. 2020;2020:3426925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baradaran Rahim V, Khammar MT, Rakhshandeh H, et al. Crocin protects cardiomyocytes against LPS-Induced inflammation. Pharmacol Rep. 2019;71:1228–1234. [DOI] [PubMed] [Google Scholar]
- [19].Godugu C, Pasari LP, Khurana A, et al. Crocin, an active constituent of Crocus sativus ameliorates cerulein induced pancreatic inflammation and oxidative stress. Phytother Res. 2020;34:825–835. [DOI] [PubMed] [Google Scholar]
- [20].Korani S, Korani M, Sathyapalan T, et al. Therapeutic effects of Crocin in autoimmune diseases: a review. Vol. 45. England): BioFactors (Oxford; 2019. p. 835–843. [DOI] [PubMed] [Google Scholar]
- [21].Zhou Y, Xu Q, Shang J, et al. Crocin inhibits the migration, invasion, and epithelial-mesenchymal transition of gastric cancer cells via miR-320/KLF5/HIF-1α signaling. J Cell Physiol. 2019;234:17876–17885. [DOI] [PubMed] [Google Scholar]
- [22].He SY, Qian ZY, Tang FT, et al. Effect of crocin on experimental atherosclerosis in quails and its mechanisms. Life Sci. 2005;77:907–921. [DOI] [PubMed] [Google Scholar]
- [23].Li J, Lei HT, Cao L, et al. Crocin alleviates coronary atherosclerosis via inhibiting lipid synthesis and inducing M2 macrophage polarization. Int Immunopharmacol. 2018;55:120–127. [DOI] [PubMed] [Google Scholar]
- [24].Cui MZ. Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin Lipidol. 2011;6:413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Retterstøl K, Svendsen M, Narverud I, et al. Effect of low carbohydrate high fat diet on LDL cholesterol and gene expression in normal-weight, young adults: a randomized controlled study. Atherosclerosis. 2018;279:52–61. [DOI] [PubMed] [Google Scholar]
- [26].Kacperczyk M, Kmieciak A, Kratz EM. The Role of ApoE Expression and Variability of Its Glycosylation in Human Reproductive Health in the Light of Current Information. Int J Mol Sci. 2021;22:7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Andres-Manzano MJ, Andres V, Dorado B. Oil Red O and Hematoxylin and Eosin Staining for Quantification of Atherosclerosis Burden in Mouse Aorta and Aortic Root. Methods Mol Biol. 2015;1339:85–99. [DOI] [PubMed] [Google Scholar]
- [28].Zhang X, Liu H, Hao Y, et al. Coenzyme Q10 protects against hyperlipidemia-induced cardiac damage in apolipoprotein E-deficient mice. Lipids Health Dis. 2018;17:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nasiri-Ansari Ν, Dimitriadis GK, Agrogiannis G, et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol. 2018;17:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhan Y, Yu J, Ding R, et al. Triglyceride to high density lipoprotein cholesterol ratio, total cholesterol to high density lipoprotein cholesterol ratio and low ankle brachial index in an elderly population. Vasa. 2014;43:189–197. [DOI] [PubMed] [Google Scholar]
- [31].Yang L, Andrews DA, Low PS. Lysophosphatidic acid opens a Ca(++) channel in human erythrocytes. Blood. 2000;95:2420–2425. [PubMed] [Google Scholar]
- [32].Hashemzaei M, Mamoulakis C, Tsarouhas K, et al. Crocin: a fighter against inflammation and pain. Food Chem Toxicol. 2020;143:111521. [DOI] [PubMed] [Google Scholar]
- [33].Pang JL, Wang JW, Hu PY, et al. HOTAIR alleviates ox-LDL-induced inflammatory response in Raw264.7 cells via inhibiting NF-kappaB pathway. Eur Rev Med Pharmacol Sci. 2018;22:6991–6998. [DOI] [PubMed] [Google Scholar]
- [34].Shen D, Zhao D, Yang X, et al. Geniposide against atherosclerosis by inhibiting the formation of foam cell and lowering reverse lipid transport via p38/MAPK signaling pathways. Eur J Pharmacol. 2019;864:172728. [DOI] [PubMed] [Google Scholar]
- [35].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego. Calif). 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [36].Lin XL, Xiao LL, Tang ZH, et al. Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed Pharmacother. 2018;104:36–44. [DOI] [PubMed] [Google Scholar]
- [37].Zhao L, Varghese Z, Moorhead JF, et al. CD36 and lipid metabolism in the evolution of atherosclerosis. Br Med Bull. 2018;126:101–112. [DOI] [PubMed] [Google Scholar]
- [38].Delgado-Lista J, Perez-Martinez P, Perez-Jimenez F, et al. ABCA1 gene variants regulate postprandial lipid metabolism in healthy men. Arterioscler Thromb Vasc Biol. 2010;30:1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Linton MF, Moslehi JJ, Babaev VR. Akt Signaling in Macrophage Polarization, Survival, and Atherosclerosis. Int J Mol Sci. 2019;20:2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lin J, Kakkar V, Lu X. Impact of MCP-1 in atherosclerosis. Curr Pharm Des. 2014;20:4580–4588. [DOI] [PubMed] [Google Scholar]
- [41].Poznyak AV, Silaeva YY, Orekhov AN, et al. Animal models of human atherosclerosis: current progress. Braz J Med Biol Res. 2020;53:e9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chistiakov DA, Melnichenko AA, Myasoedova VA. Mechanisms of foam cell formation in atherosclerosis. J Mol Med. Vol. 95; 2017. p. 1153–1165. [DOI] [PubMed] [Google Scholar]
- [43].Wang HH, Garruti G, Liu M, et al. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: recent Advances in Reverse Cholesterol Transport. Ann Hepatol. 2017;16 Suppl 1:S27–s42. [DOI] [PubMed] [Google Scholar]
- [44].Zou TB, Zhu SS, Luo F, et al. Effects of Astaxanthin on Reverse Cholesterol Transport and Atherosclerosis in Mice. Biomed Res Int. 2017;2017:4625932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Favari E, Chroni A, Tietge UJ, et al. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. 2015;224:181–206. [DOI] [PubMed] [Google Scholar]
- [46].Lu J, Chen X, Xu X, et al. Active polypeptides from Hirudo inhibit endothelial cell inflammation and macrophage foam cell formation by regulating the LOX-1/LXR-α/ABCA1 pathway. Biomed Pharmacother. 2019;115:108840. [DOI] [PubMed] [Google Scholar]
- [47].Wang S, Dougherty EJ, Danner RL. PPARγ signaling and emerging opportunities for improved therapeutics. Pharmacol Res. 2016;111:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xiong W, Zhao X, Villacorta L, et al. Brown Adipocyte-Specific PPARγ (Peroxisome Proliferator-Activated Receptor γ) Deletion Impairs Perivascular Adipose Tissue Development and Enhances Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol. 2018;38:1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Toh SA, Millar JS, Billheimer J, et al. PPARγ activation redirects macrophage cholesterol from fecal excretion to adipose tissue uptake in mice via SR-BI. Biochem Pharmacol. 2011;81:934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gao Q, Wei A, Chen F, et al. Enhancing PPARγ by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol Res. 2020;160:105059. [DOI] [PubMed] [Google Scholar]
- [51].Gwon MH, Im YS, Seo AR, et al. Phenethyl Isothiocyanate Protects against High Fat/Cholesterol Diet-Induced Obesity and Atherosclerosis in C57BL/6 Mice. Nutrients. 2020;12:3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. 2015;74:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Song M, Bode AM, Dong Z, et al. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019;79:1019–1031. [DOI] [PubMed] [Google Scholar]
- [54].Sedding DG, Boyle EC, Demandt JAF, et al. Vasa Vasorum Angiogenesis: key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol. 2018;9:706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peng W, Cai G, Xia Y, et al. Mitochondrial Dysfunction in Atherosclerosis. DNA Cell Biol. 2019;38:597–606. [DOI] [PubMed] [Google Scholar]
- [56].Zhang RJ, Yin YF, Xie XJ, et al. Acid-sensing ion channels: linking extracellular acidification with atherosclerosis. Clin Chim Acta. 2020;502:183–190. [DOI] [PubMed] [Google Scholar]
- [57].Donato L, Scimone C, Alibrandi S, et al. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants (Basel). 2020;9:1158 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Scimone C, Alibrandi S, Scalinci SZ, et al. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants (Basel). 2020;9:1154 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Donato L, Scimone C, Alibrandi S, et al. New Omics-Derived Perspectives on Retinal Dystrophies: could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype? Int J Mol Sci. 2020;22:70. [DOI] [PMC free article] [PubMed] [Google Scholar]