

Abstract
Structural plasticity and dynamic protein-protein interactions are critical determinants of protein function within living systems. Quantitative chemical crosslinking with mass spectrometry (qXL-MS) is an emerging technology able to provide information on changes in protein conformations and interactions. Importantly, qXL-MS is applicable to complex biological systems, including living cells and tissues, thereby providing insights into proteins within their native environments. Here, we present an overview of recent technological developments and applications involving qXL-MS, including design and synthesis of isotope labeled crosslinkers, development of new LC-MS methodologies, and computational developments enabling interpretation of the data.
Keywords: quantitative crosslinking, protein interaction, protein conformation, mass spectrometry, quantitative interactomics
Graphical Abstract
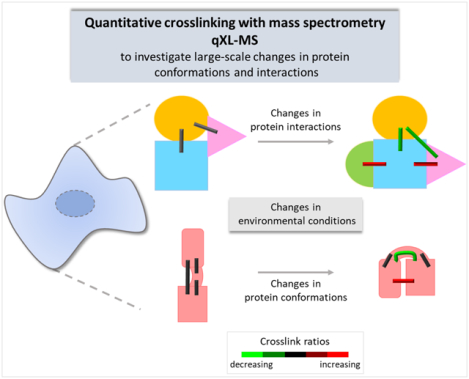
1. Introduction
Chemical crosslinking of proteins with mass spectrometry (XL-MS), combined with other structural methods, has become a powerful technique of increasingly utility for study of protein conformations and interactomics. This approach is based on the reactivity of crosslinkers to specific protein sites – usually primary amines, including side chains of lysine residues and protein N-termini, identifying proximal residues and yielding information on protein structures and interactions. Quantitative crosslinking with mass spectrometry (qXL-MS) brings another and yet more complex level of information, providing insights into large-scale changes in protein interactomes with varying biological states, including system perturbations such as drug treatment, age, phenotype, or disease state[1–*3]. More importantly, qXL-MS can be applied directly to complex biological systems, such as living cells, tissues, or organelles, therefore providing information on changes in protein structural dynamics and interactions as they occur within a native environment[4,5].
A variety of strategies have been used for qXL-MS. Label-free qXL-MS[6] includes extracting MS1 chromatographic peak areas, as well as the MS2-based quantitation employing parallel reaction monitoring (PRM)[7]. Isotopic labeling methods include stable isotope labeling by amino acids in cell culture (SILAC)[8] with XL-MS, and the use of light and heavy isotope-labeled crosslinkers[9], which enables large-scale qXL-MS studies. Isobaric labeling methods apply (i) isobaric reagents (e.g., TMT[10]) to label crosslinked peptides[11], or alternatively (ii) the isobaric quantitative protein interaction reporter (iqPIR) strategy, which includes isotope-encoded isobaric crosslinkers**[12] (Figure 1). Recently, a multiplexed version of the iqPIR has been developed, allowing for crosslinking and multiplexing of up to 6 different samples (Chavez et al., submitted). Moreover, recent developments in the publicly available XLinkDB platform enable the visualization and interpretation of qXL-MS results[13]. Software suitable and applied for qXL-MS include the MS1-based MassChroQ[14], MaxQuant[15,16], pQuant[17], xTract[5], and Skyline[18]. Here we review recent developments in qXL-MS methodologies and applications described in the following sections: (2) isotopic labeling, (3) label-free, including (3.2) PRM, and (4) isobaric labeling (Figure 1).
Figure 1. Quantitative crosslinking with mass spectrometry (qXL-MS).
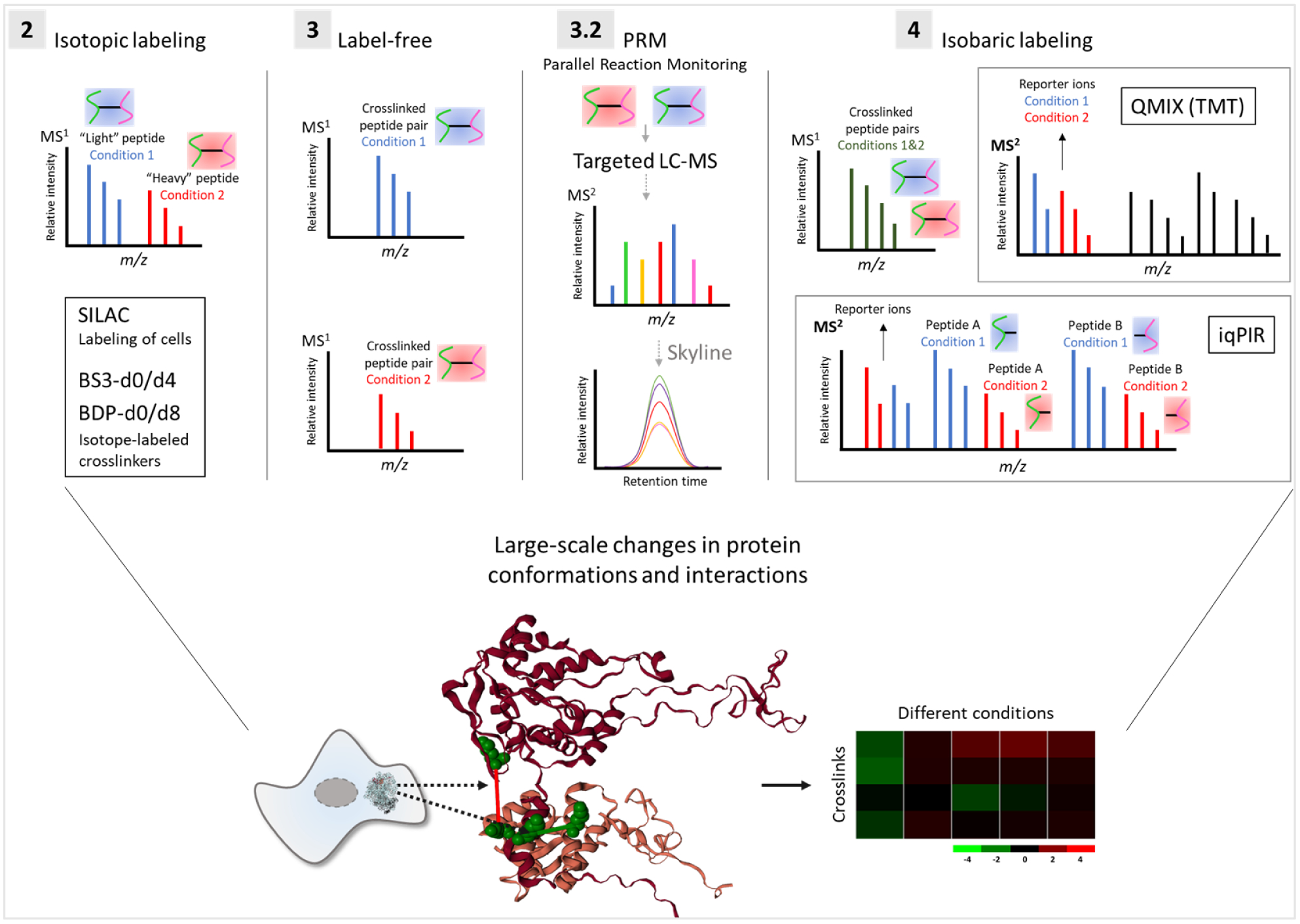
Isotope-labeled, label-free, PRM-based, and isobaric-labeled qXL-MS strategies are discussed in sections 2, 3, 3.2, and 4, respectively. iqPIR: isobaric quantitative protein interaction reporter.
2. Isotopic labeling
2.1. Isotopic labeling of cells with SILAC for qXL-MS
Isotopic qXL-MS strategies include the labeling of living cells with SILAC[8] followed by crosslinking. This approach uses the MS1 information from light and heavy crosslinked peptide pairs to provide relative quantitation of crosslink levels between two samples. One advantage of SILAC over isotope-coded crosslinkers is that SILAC generates wider mass shifts in MS1 scans, that may provide higher quantitative accuracy. Optimized SILAC isotope combinations for quantitative crosslinking applications have not yet been explored, but could further improve quantitative capabilities.
A SILAC-based qXL-MS illustrated that differences related to acquired chemoresistance to the active agent in the anticancer therapy Irinotecan were detectable at the interactome level, and these differences could in part, help explain activity differences in cells that contributed to the Irinotecan resistance phenotype[1]. A significant aspect of those results was that they revealed for the first time that in vivo crosslinking with mass spectrometry could provide quantitative insight on the interactome inside cells. Following this, Chavez et al. employed SILAC and PIR crosslinking of cancer cells to probe altered conformations and interactions resultant from treatment of cells with multiple Hsp90 inhibitors[2]. These results revealed interactome changes that were both drug concentration-dependent and drug mechanism-specific. These studies revealed drug-dependent changes in Hsp90, indicating compact conformation enrichment from the cellular ensemble which appears specific to N-terminal ATP pocket-targeting Hsp90 inhibitors. Moreover, the study identified changes in Hsp90 interactome with co-chaperones STIP1, Hsp70, CHRD1, and CDC37. This approach has also been applied by the same group to investigate changes in protein conformations and interactions in cancer cells after treatment with the mitotic inhibitor paclitaxel (PTX), using different concentrations of the drug*[3]. The qXL-MS results provided insights into dose-dependent changes in cytoskeleton organization (Figure 2), which caused stabilization of microtubules and arrest of mitosis, consistent with the PTX mechanism of action [19,20].
Figure 2. Quantitative crosslinking reveals PTX stabilized microtubules (MT).
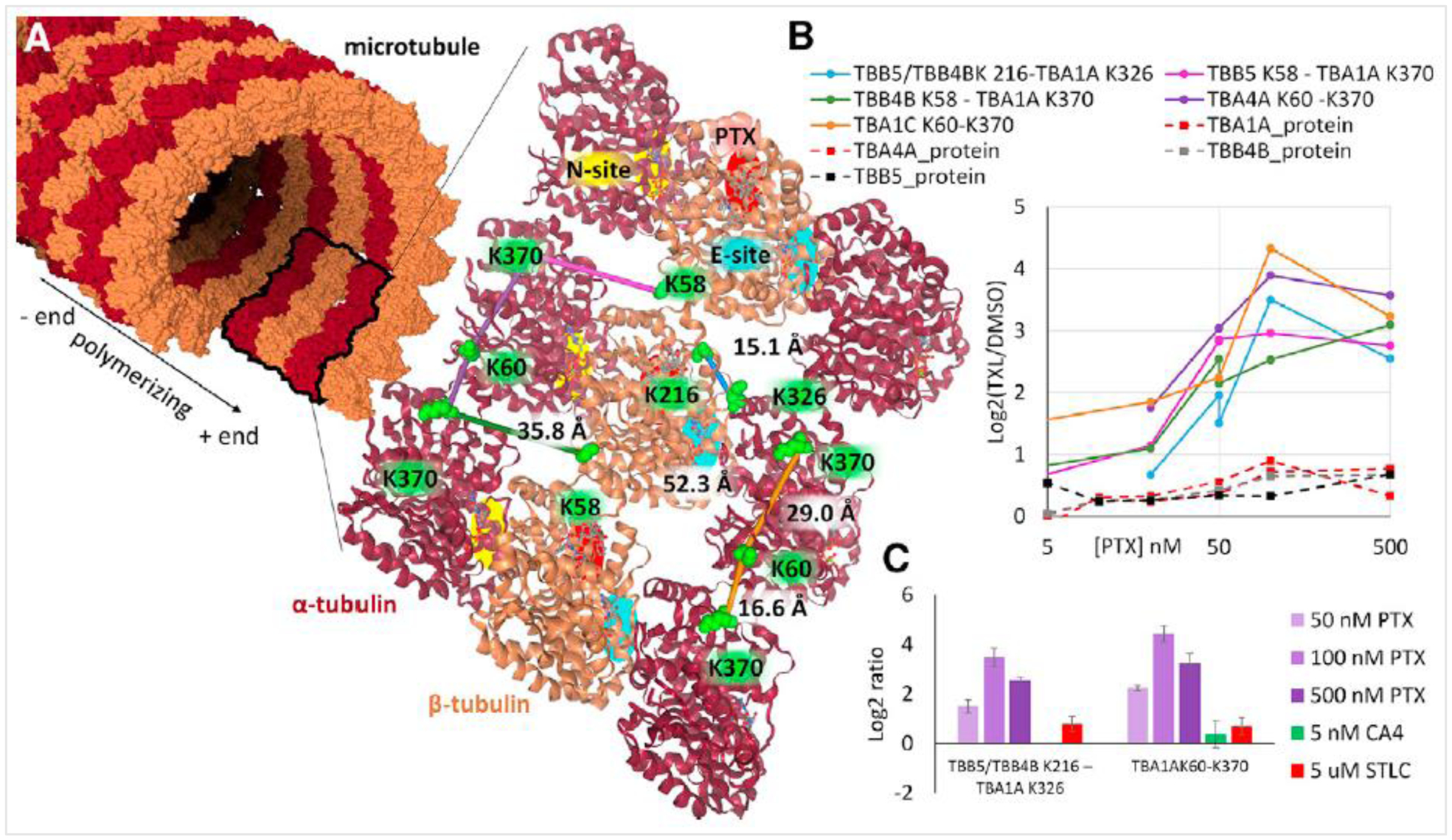
(A) MT structure (PDB 3EDL) displayed as a molecular surface with a ribbon structure inset, illustrating the a-tubulin (maroon) and b-tubulin (gold) subunits. Crosslinked Lys residues are shown as green space-filled residues, with crosslinks displayed as colored lines connecting them (TBA1A K60-K370, orange; TBA4A K60-K370, purple; TBB5/TBB4B K216-TBA1A K326, blue; TBB4B K58-TBA1A K370, green; TBB5 K58-TBA1A K370, magenta). The non-exchangeable GTP binding site (N-site) is indicated by a yellow-highlighted region on a-tubulin. The exchangeable GTP binding site (E-site) is indicated by a cyan-colored region on b-tubulin. The PTX binding site on b-tubulin is red. (B) Protein and crosslink levels measured by SILAC for four tubulin isoforms (TBA1A, dark red dashed line; TBA4A, red dashed line; TBB4B, gray dashed line; TBB5, black dashed line). Crosslinks are colored the same as in (a). (C) Quantified levels of TBB5/TBB4B K216-TBA1A K326 and TBA1A K60-K370 with PTX (50, 100, and 500 nM), CA4 (5 nM), and STLC (5 mM). Error bars represent 95% confidence intervals for n = 6 replicate injections of 2 biological samples. Reproduced with permission from Chavez et al., 2019[3].
Yu et al. combined the Quantitative analysis of Tandem Affinity-purified in vivo crosslinked (X) protein complexes (QTAX)[21] strategy with SILAC and disuccinimidyl sulfoxide (DSSO) crosslinker[22] for a qXL-MS study of the human 26S proteasome, assessing interactomic changes in response to H202-induced oxidative stress*[23]. Briefly, cells expressing histidine/biotin- (HB-) tagged proteasome were grown in SILAC media, crosslinked in vivo with formaldehyde, and on-bead purified proteasomes were crosslinked in vitro with DSSO. MS3 spectra were used for the identification of 746 crosslinks, and MS1 signal used for SILAC-based quantitation of 343 crosslinks (~46% of total links). While SILAC-based qXL-MS has enabled initial interactome dynamic measurements, in some cases even inside cells, the challenges associated with MS1 signal quantitation limit the efficiency of crosslink quantitation for complex systems. For instance, of the 3,323 crosslinked peptides detected in Chavez et al., 2016[2], only 2,582 were quantified (77,7%) and even fewer (559 crosslinks, ~17%) were quantified across all 5 different drug concentrations. This results from issues of co-eluting species that congest MS1 signals and contaminate extracted ion signals used for quantitation. Filtering of all forward and reverse SILAC-qXL-MS quantitation data for 95% confidence limits eliminates these spurious results, significantly reducing the depth of quantitative data. One strategy that could potentially improve upon the issue of MS1 signal congestion would be to further separate crosslinked peptides using gas phase fractionation techniques such as high-field asymmetric waveform ion mobility spectrometry (FAIMS)[24].
2.2. Isotopic crosslinkers
Isotope-labeled crosslinkers for qXL-MS include DSS-d0d12*[25], BDP-NHP-d0d8[26], CBDPS*[27], Leiker[28], DMDSSO-d0d10[29], BS3-d0d4[30] and BS3-d0d12[31,32], and DSBU*[33] (Table 1), which include variable numbers of deuterium atoms as the heavy labels. These isotope-encoded crosslinkers are useful for binary comparisons and most of them have been applied for in vitro studies, where extreme complexity in the MS1 signal is not likely to be such an issue [34–38]. The Rappsilber group applied BS3-d0d4 in solution to investigate conformational changes in the human complement protein C3 in comparison to its activated cleavage product C3b[34]. The same group developed a computational tool for analysis of isotope-coded crosslinking data, called XiQ[39]. BS3-d0d4 was also applied in vitro to study F1FO-ATPase isolated from chloroplasts[35], with conformational changes in the catalytic interface of the enzyme related to phosphorylation. Furthermore, Boelt et al. used BS3-d0d4 in solution to investigate conformational changes of calreticulin, a protein part of the endoplasmatic reticulum that regulates Ca2+ homeostasis[38]. More recently, BS3-d0d12 was applied to study the role of LEM2 in the reformation of nuclear envelope during cell division[40]. The crosslinking results indicated specific sites of interaction between CHMP7 and the WH domain of LEM2 that was proposed to form a macromolecular O-ring seal at the confluence between membranes, chromatin and the spindle. The qXL-MS results agreed with those from other experiments used in the study, thus providing insights on protein conformation and interactions of isolated complex samples. As with SILAC-based qXL-MS, quantitative measurements with isotopic-labeled crosslinkers also rely on MS1 information for crosslink quantitation and are subject to similar peak assignment challenges, particularly with complex large scale qXL-MS applications. Additionally, peak assignment can be further complicated by retention time shift between light and heavy deuterium-labeled peptides during LC separation. To date, all applications of isotope-labeled crosslinkers have utilized a binary comparison between a light and heavy pair. Conceptually, it should be possible to expand beyond binary situations, allowing for a greater number of comparisons to be made with the limitation of increasing the MS1 signal complexity by N as well as diluting the signal of any crosslinked product by 1/N (where N= number of isotopologue crosslinkers).
Table 1.
Isotope-coded crosslinkers for qXL-MS. Dashed line indicates the cleavage site.
| Crosslinker | Structure | Feature | Reference |
|---|---|---|---|
| DSS |
X indicates H or D isotope for d0-DSS (X = H) or d12-DSS (X = D) |
Vendor: Creative Molecules | Khakzad et al.1 |
| Formula (X = H): C16H20N2O6 | |||
| MW (X = H): 368.34 | |||
| BS3 |
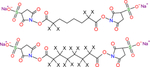 X indicates H or D isotope for d0-BS3 (X = H) or d4-BS3/d12-BS3 (X = D) |
Vendor: ThermoFisher Creative Molecules | Schmidt et al.2 Linden et al.3 Appen et al.4 |
| Formula: C16H18N2Na2O14S2 | |||
| MW: 572.43 | |||
| DMDSSO |
X indicates H or D isotope for d0-DMDSSO (X = H) or d10-DMDSSO (X = D) |
MS cleavable | Yu et al.5 |
| Formula: C14H16N2O9S | |||
| MW: 388.35 | |||
| DSBU |
X indicates H or D isotope for d0-DSBU (X = H) or d12-DSBU (X = D) |
MS cleavable | Ihling et al.6 |
| Formula: C17H22N4O9 | |||
| MW: 426.38 | |||
| CBDPS |
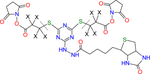 X indicates H or D isotope for d0-CBDPS (X = H) or d8-CBDPS (X = D) |
MS cleavable | Makepeace et al.7 |
| Biotin tagged | |||
| Formula: C27H33N9O10S3 | |||
| MW: 739.80 | |||
| Leiker (bAL2) |
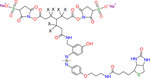 X indicates H or D isotope for d0-bAL2 (X = H) or d6-bAL2 (X = D) |
Photo cleavable | Tan et al.8 |
| Biotin tagged | |||
| Formula: C43H52N8O13S | |||
| MW: 920.98 | |||
| PIR BDP-NHP |
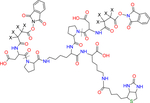 X indicates H or D isotope for d0-PIR (X = H) or d8-PIR (X = D) |
MS cleavable | Zhong et al.9 |
| Biotin tagged | |||
| Peptide synthesis | |||
| Formula: C64H79N13O22S | |||
| MW: 1414.45 | |||
| 2-plex iqPIR BDP-NHP |
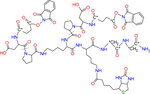 * indicates the position can be isotope-coded with C13 |
MS cleavable | Chavez et al.10 |
| Biotin tagged | |||
| Peptide synthesis | |||
| Formula: C68H85N15O24S | |||
| MW: 1532.55 | |||
| 6-plex iqPIR BDP-NHP |
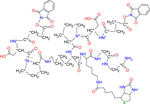 * indicates the position can be isotope-coded with C13 or N15 |
MS cleavable | Chavez et al. (submitted) |
| Biotin tagged | |||
| Peptide synthesis | |||
| Formula: C68H85N15O24S | |||
| MW: 1546.42 |
3. Label-free qXL-MS
As with traditional quantitative proteomics, another strategy for qXL-MS is to utilize label-free quantitation (LFQ). Benefits of LFQ include its compatibility with nearly any crosslinking reagent, avoiding costly heavy isotope labeled reagents, and allowing for higher level comparisons. A consequence of LFQ is that each sample needs to be prepared and analyzed separately, requiring increased and reproducible sample preparation and LC-MS instrument time and being subject to experimental variabilities introduced at each step along the process. Müller et al. evaluated the quantitative reproducibility of a MS1-based LFQ qXL-MS approach using BS3 crosslinked human serum albumin (HSA) samples and found more variability resulted from comparison across different crosslinking reactions than from LC-MS injections[6]. The qXL-MS strategies for label-free and isotope labeled BS3 from the Rappsilber laboratory were summarized in a recent protocol[42]. MS1-based LFQ of crosslinks and acetylated peptides revealed that acetylation disrupts dimer formation and decreases the activity of the muscle isoform of creatine kinase [43]. MS1-based LFQ was used to reveal the conformational changes to the peroxisome proliferator-activated receptors gamma (PPARγ) upon binding the antagonist SR11023[44]. To group multiple protein conformations detected in different biological samples, Kurt et al. developed a software that clusters crosslink identifications according to their MS1-based quantitative profile across multiple samples, the QUIN-XL[45].
3.1. DIA-QCLMS
The majority of qXL-MS studies have used a DDA approach. However, data-independent acquisition (DIA) qXL-MS methods have also been explored. Müller et al. demonstrated a LFQ DIA qXL-MS approach at the MS1 and MS2 levels using samples consisting of a mixture of 7 proteins crosslinked with BS3*[46]. The authors discussed that, despite the ratio compression in MS1/MS2 spectra observed for complex mixtures, the Spectronaut-based (Biognosis) quantitation of crosslinks showed increased accuracy and reproducibility. The Rappsilber laboratory further demonstrated use of the DIA qXL-MS protocol[47] to detect pH-dependent conformational changes in photo-crosslinked HSA and cytochrome c[48].
3.2. Targeted PRM
Targeted quantitation offers multiple benefits to qXL-MS including increased reproducibility, sensitivity, precision and accuracy. Furthermore, targeted analysis is applicable with both LFQ approaches or those using isotope labeling. A limitation of targeted analysis is that it often requires some a priori knowledge of the crosslinked analytes of interest, requiring development of special LC-MS methods devoted to a select number of analytes, making it a relatively low throughput strategy. The first application of parallel reaction monitoring (PRM) qXL-MS was applied in a cross-laboratory study which utilized Skyline[18] for method sharing and data analysis[7]. PRM qXL-MS was also used to help elucidate important conformational changes in heat shock protein 90 (Hsp90) that occur with nucleotide binding, interaction with co-chaperone Aha1, and with a phosphomimetic mutant Y313E [49]. Gutierrez et al. utilized PRM qXL-MS with three different MS-cleavable crosslinkers to detect structural dynamics of the COP9 signalosome*[50]. Mehnert et al. used PRM qXL-MS as part of a multilayered proteomic workflow to detect topological changes associated with cancer mutations on kinase complexes[51]. As with traditional quantitative proteomics, when using DIA and PRM for quantitation of crosslinked peptides it is important to implement good practices to ensure accurate quantitation of the correct analyte[52,53]. These include use of proteotypic peptides, quantifying multiple high mass accuracy matching fragment ions, ideally originating from both peptides, and ensuring accurate retention time matching. Spectral library searching can also be useful to confirm identification of quantified species[54].
4. Isobaric labeling
4.1. TMT labeling for qXL-MS
A qXL-MS multiplexed method based on labeling of crosslinked peptides with isobaric mass tags, i.e., TMT, was developed by Yu et al., called Quantitation of Multiplexed Isobaric-labeled cross (X)-linked peptides (QMIX)[11]. The authors used DSSO to crosslink cytochrome c, followed by proteolytic digestion of separate samples. Digested samples were then labeled with a binary set of TMT reagents (126 or 127 reporter) followed by multiplexing samples, and LC-MS analysis of crosslinked peptides. To eliminate the signal interference at the MS1 level and compression of measured TMT ratios due to co-isolation of precursor ions[55], the quantitation of crosslinks was done at the MS3 level. Hence, the QMIX approach benefits from advanced mass spectrometers with higher sensitivity MS3 capabilities.
4.2. Isobaric quantitative PIR (iqPIR)
Recently, isobaric quantitative Protein Interaction Reporter (iqPIR) crosslinking technology was developed and demonstrated in our laboratory**[12]. iqPIR crosslinkers are synthesized using 13C and 15N isotope labels, enabling the quantitation of crosslinked peptide pairs using the relative abundance of multiple isotope fragment ions unique to each crosslink in the MS2 spectra. This strategy eliminates any retention time shifts – as it happens with deuterated crosslinkers (Section 2.2), and ratio compression of the reporter ion signal by quantifying fragment ions that retain an isotope encoded portion of the tag, analogous to the use of complement reporter ions with TMT[55] or the EASY-tag approach[56] used in traditional quantitative proteomics. Fragmentation of iqPIR crosslinked peptides generates a number of quantifiable ions, including the released intact peptides, as well as b-type and y-type backbone fragment ions containing the crosslinked residue. The iqPIR, as well as other PIR-based crosslinkers[57], contain MS-cleavable bonds, affinity tags, and membrane permeability for the crosslinking of live cells, tissues, and isolated organelles. These features enable isotope-encoded crosslinking of proteins in complex biological systems with one single labeling step, thus eliminating the need for additional chemical labeling reactions and cleanup steps. Moreover, the biotin affinity tag enables crosslink enrichment, which combined with the quantitative features, allows complex cellular applications.
iqPIR was pursued to enable multiplex interactome quantitation but was initially demonstrated with binary comparisons with 2 different isotope-coded crosslinkers**[12]. Recently, a multiplexed strategy was developed with 6 different iqPIR crosslinkers, called 6-plex iqPIR (Chavez et al., submitted). Because the 6-plex iqPIR crosslinkers are isobaric, the precursor ion signal from a crosslinked peptide pair originating from 6 different multiplexed samples overlap in MS1 but generate unique fragments in MS2 (Figure 3), which are further used for crosslink quantitation (Chavez et al., submitted). The 6-plex iqPIR strategy was demonstrated in vivo on MCF-7 cells treated with five different Hsp90 inhibitors, revealing drug class specific interactome dynamics from qXL-MS analysis of a multiplexed sample (Wippel et al., submitted). From 1,756 crosslinks identified in this study, 1,650 were quantified (~94%), and a total of 1,257 crosslinks were quantified across all 6 channels (~71%). These results represent an improvement in crosslinking quantitation if compared with the values discussed above from Chavez et al.[2], where 77,7% of crosslinked peptide pairs were quantified, and 17% quantified across all drug concentrations.
Figure 3. 6-plex iqPIR for multiplexed qXL-MS.
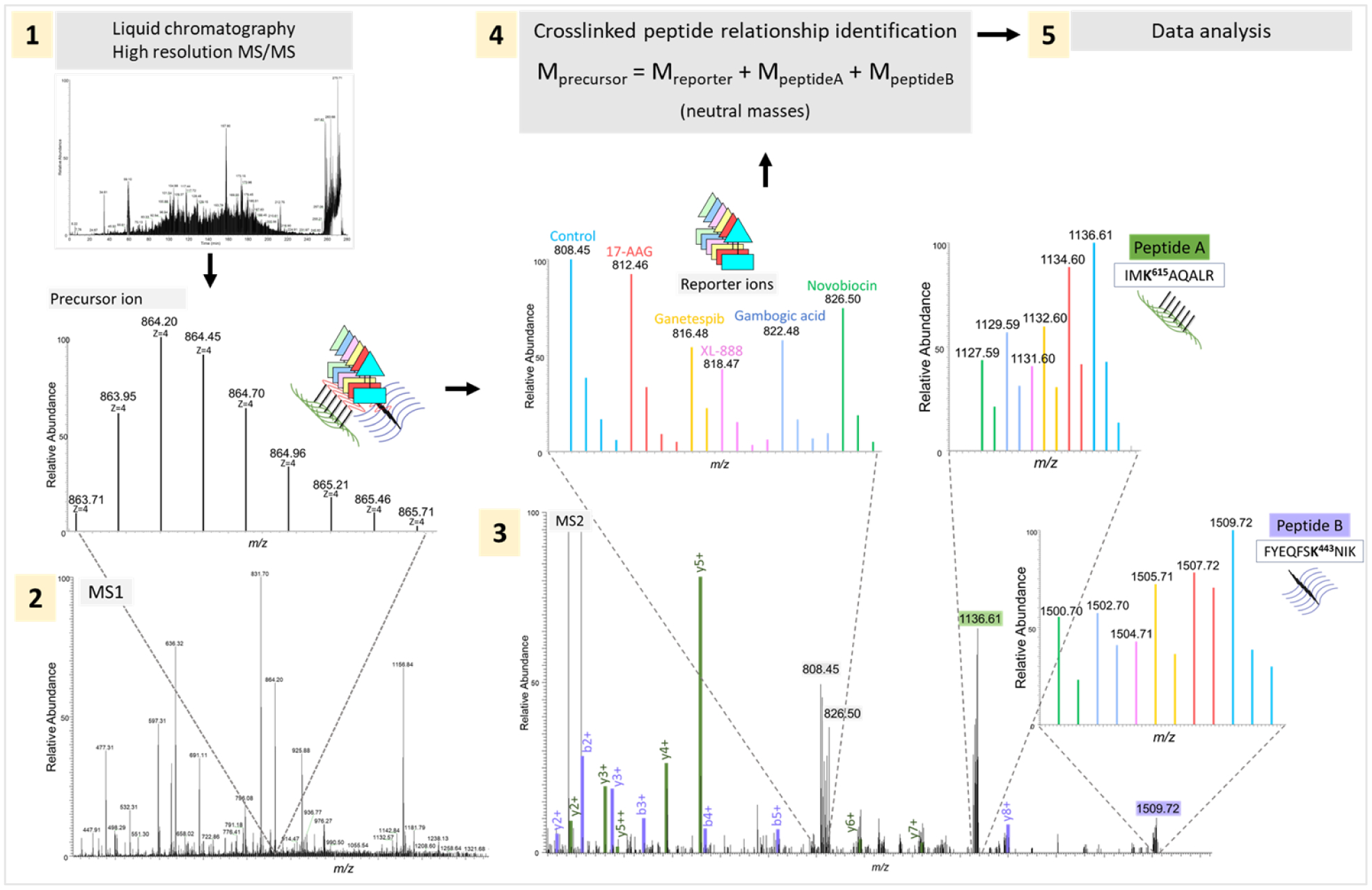
Representation of the 6-plex iqPIR qXL-MS LC-MS pipeline. 1) Crosslinked peptide pairs are separated by liquid chromatography and analyzed with high resolution MS/MS; 2) MS1 spectrum highlighting the isotope envelope of the precursor ion, embedding all 6 crosslinked peptides; 3) MS2 spectrum with highlighted fragment ions from peptides A (b/y ions in green) and B (b/y ions in purple), and isotope envelopes from released reporter ions and backbone peptides A and B, with the peaks colored according to the corresponding channel/Hsp90 inhibitor (shown as an example of environmental conditions here); 4) Relationship check between reporter ion and peptides A and B, comparing the monoisotopic neutral masses with the experimental masses observed (with maximum 10 ppm error allowed)[58]. After Mango, in silico analysis continues with Comet[59] for peptide search of 6-plexed fragment ions, followed by validation of identified crosslinks, and quantitation of 6-plex crosslinks (Chavez et al., in preparation). Reproduced with permission from Wippel et al., submitted.
CONCLUSIONS
A challenge of systems-level biology is to better understand how molecules act and interact to confer function within a crowded cellular environment. XL-MS is an emerging tool that can probe protein conformations and interactions to gain unique insight on molecular mechanisms and biological pathways. Just as seeing how the components of complex machines or instruments work together to function can improve fundamental understanding, visualizing how protein conformations and interactions change within a system can help better understand large-scale protein biological function. Thus, perturbations in the system are frequently necessary to allow comparative analysis among different conditions, where qXL-MS can uniquely reveal protein and interactome dynamics in response to environmental changes. While many successful qXL-MS studies have used MS1 signal levels for crosslink quantitation or from targeted MS2 spectra, isobaric approaches based on MS2 spectral quantitation offer increased quantitation efficiency as well as multiplexed quantitation. Recent developments have allowed for the crosslinking, multiplexing, and quantitation of up to 6 different biological samples, with mixing at the protein level. In parallel, computational improvements are needed to allow the visualization of qXL-MS data. Although far from comprehensive, quantitative XL-MS studies and the advancements made so far are providing unique insight on protein, complex and interactome dynamics.
HIGHLIGHTS.
Quantitative chemical crosslinking provides information on changes in protein interactomics;
Overview of recent technological developments involving qXL-MS;
Applications of qXL-MS in biological systems with varying conditions.
ACKNOWLEDGEMENTS
The authors acknowledge and thank all members of the Bruce Lab for helpful comments and suggestions during the course of preparation of this manuscript.
FUNDING
This work was supported by the US national Institutes of Health through grants: R35GM136255, R01HL144778, and R01GM086688.
ABBREVIATIONS
- BS3
bis(sulfosuccinimidyl)suberate
- CBDPS
CyanurBiotinDimercaptoPropionylSuccinimide
- DSBU
disuccinimidyl dibutryic urea
- DSS
disuccinimidyl suberate
- iqPIR
isobaric quantitative protein interaction reporter
- LC-MS
liquid chromatography – mass spectrometry
- MS
mass spectrometry
- NHP
N-hyrdoxyphthalimide
- PIR
protein interaction reporter
- PRM
parallel reaction monitoring
- PTX
paclitaxel
- SILAC
stable isotope labeling by amino acids in cell culture
- TMT
tandem mass tags
- qXL-MS
quantitative chemical crosslinking with mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
Papers of particular interest, published within the period of review, have been highlighted as:
(*) of special interest
(**) of outstanding interest
- 1.Chavez JD, Schweppe DK, Eng JK, Zheng C, Taipale A, Zhang Y, Takara K, Bruce JE: Quantitative interactome analysis reveals a chemoresistant edgotype. Nat Commun 2015, 6:7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chavez JD, Schweppe DK, Eng JK, Bruce JE: In Vivo Conformational Dynamics of Hsp90 and Its Interactors. Cell Chem Biol 2016, 23:716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *3.Chavez JD, Keller A, Zhou B, Tian R, Bruce JE: Cellular Interactome Dynamics during Paclitaxel Treatment. Cell Rep 2019, 29:2371–2383.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was an in vivo application of SILAC-based qXL-MS in HeLa cells treated with mitotic inhibitor paclitaxel.
- 4.Chen ZA, Rappsilber J: Protein Dynamics in Solution by Quantitative Crosslinking/Mass Spectrometry. Trends in Biochemical Sciences 2018, 43:908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walzthoeni T, Joachimiak LA, Rosenberger G, Röst HL, Malmström L, Leitner A, Frydman J, Aebersold R: xTract: software for characterizing conformational changes of protein complexes by quantitative cross-linking mass spectrometry. Nat Methods 2015, 12:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller F, Fischer L, Chen ZA, Auchynnikava T, Rappsilber J: On the Reproducibility of Label-Free Quantitative Cross-Linking/Mass Spectrometry. J Am Soc Mass Spectrom 2018, 29:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.havez JD, Eng JK, Schweppe DK, Cilia M, Rivera K, Zhong X, Wu X, Allen T, Khurgel M, Kumar A, et al. : A General Method for Targeted Quantitative Cross-Linking Mass Spectrometry. PLoS ONE 2016, 11:e0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M: Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol Cell Proteomics 2002, 1:376–386. [DOI] [PubMed] [Google Scholar]
- 9.Müller DR, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S, Steinmetz MO: Isotope-Tagged Cross-Linking Reagents. A New Tool in Mass Spectrometric Protein Interaction Analysis. Anal Chem 2001, 73:1927–1934. [DOI] [PubMed] [Google Scholar]
- 10.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C: Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal Chem 2003, 75:1895–1904. [DOI] [PubMed] [Google Scholar]
- 11.Yu C, Huszagh A, Viner R, Novitsky EJ, Rychnovsky SD, Huang L: Developing a Multiplexed Quantitative Cross-Linking Mass Spectrometry Platform for Comparative Structural Analysis of Protein Complexes. Anal Chem 2016, 88:10301–10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **12.Chavez JD, Keller A, Mohr JP, Bruce JE: Isobaric Quantitative Protein Interaction Reporter Technology for Comparative Interactome Studies. Anal Chem 2020, doi: 10.1021/acs.analchem.0c03128. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents the first multiplex qXL-MS strategy for large-scale in vivo interactome studies using isobaric crosslinkers.
- 13.Keller A, Chavez JD, Eng JK, Thornton Z, Bruce JE: Tools for 3D Interactome Visualization. J Proteome Res 2019, 18:753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valot B, Langella O, Nano E, Zivy M: MassChroQ: A versatile tool for mass spectrometry quantification. Proteomics 2011, 11:3572–3577. [DOI] [PubMed] [Google Scholar]
- 15.Cox J, Mann M: MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 2008, 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 16.Chen ZA, Fischer L, Cox J, Rappsilber J: Quantitative Cross-linking/Mass Spectrometry Using Isotope-labeled Cross-linkers and MaxQuant. Molecular & Cellular Proteomics 2016, 15:2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, Song C-Q, Yuan Z-F, Fu Y, Chi H, Wang L-H, Fan S-B, Zhang K, Zeng W-F, He S-M, et al. : pQuant Improves Quantitation by Keeping out Interfering Signals and Evaluating the Accuracy of Calculated Ratios. Anal Chem 2014, 86:5286–5294. [DOI] [PubMed] [Google Scholar]
- 18.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ: Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T: Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 2011, 8:244–250. [DOI] [PubMed] [Google Scholar]
- 20.Belotti D, Vergani V, Drudis T, Borsotti P, Pitelli MR, Viale G, Giavazzi R, Taraboletti G: The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res 1996, 2:1843–1849. [PubMed] [Google Scholar]
- 21.Guerrero C, Tagwerker C, Kaiser P, Huang L: An Integrated Mass Spectrometry-based Proteomic Approach. Molecular & Cellular Proteomics 2006, 5:366–378. [DOI] [PubMed] [Google Scholar]
- 22.Kao A, Chiu C, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L: Development of a Novel Cross-linking Strategy for Fast and Accurate Identification of Cross-linked Peptides of Protein Complexes. Mol Cell Proteomics 2011, 10:M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Yu C, Wang X, Huszagh AS, Viner R, Novitsky E, Rychnovsky SD, Huang L: Probing H2O2-mediated Structural Dynamics of the Human 26S Proteasome Using Quantitative Cross-linking Mass Spectrometry (QXL-MS). Mol Cell Proteomics 2019, 18:954–967. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents a new strategy for the qXL-MS study of HB-tagged proteasome in vivo, using SILAC-labeling of cells and a 2-step crosslinking reaction with formaldehyde and DSSO.
- 24.Pfammatter S, Bonneil E, McManus FP, Prasad S, Bailey DJ, Belford M, Dunyach J-J, Thibault P: A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Molecular & Cellular Proteomics 2018, 17:2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25.Khakzad H, Happonen L, Tran Van Nhieu G, Malmström J, Malmström L: In vivo Cross-Linking MS of the Complement System MAC Assembled on Live Gram-Positive Bacteria . Front Genet 2020, 11:612475. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is an in vivo application (in living bacteria) of the isotope-coded crosslinker DSS-d0d12 for qXL-MS.
- 26.Zhong X, Navare AT, Chavez JD, Eng JK, Schweppe DK, Bruce JE: Large-Scale and Targeted Quantitative Cross-Linking MS Using Isotope-Labeled Protein Interaction Reporter (PIR) Cross-Linkers. J Proteome Res 2017, 16:720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *27.Makepeace KAT, Mohammed Y, Rudashevskaya EL, Petrotchenko EV, Vögtle F-N, Meisinger C, Sickmann A, Borchers CH: Improving Identification of In-organello Protein-Protein Interactions Using an Affinity-enrichable, Isotopically Coded, and Mass Spectrometry-cleavable Chemical Crosslinker. Molecular & Cellular Proteomics 2020, 19:624–639. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows an application of MS-cleavable crosslinker CBDPS-dod8 in intact yeast mitochondria for qXL-MS.
- 28.Tan D, Li Q, Zhang M-J, Liu C, Ma C, Zhang P, Ding Y-H, Fan S-B, Tao L, Yang B, et al. : Trifunctional cross-linker for mapping protein-protein interaction networks and comparing protein conformational states. eLife 2016, 5:e12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C, Kandur W, Kao A, Rychnovsky S, Huang L: Developing New Isotope-Coded Mass Spectrometry-Cleavable Cross-Linkers for Elucidating Protein Structures. Anal Chem 2014, 86:2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt C, Robinson CV: A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat Protoc 2014, 9:2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schilling B, Row RH, Gibson BW, Guo X, Young MM: MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. J Am Soc Mass Spectrom 2003, 14:834–850. [DOI] [PubMed] [Google Scholar]
- 32.Seebacher J, Mallick P, Zhang N, Eddes JS, Aebersold R, Gelb MH: Protein Cross-Linking Analysis Using Mass Spectrometry, Isotope-Coded Cross-Linkers, and Integrated Computational Data Processing. J Proteome Res 2006, 5:2270–2282. [DOI] [PubMed] [Google Scholar]
- *33.Ihling CH, Springorum P, Iacobucci C, Hage C, Götze M, Schäfer M, Sinz A: The Isotope-Labeled, MS-Cleavable Cross-Linker Disuccinimidyl Dibutyric Urea for Improved Cross-Linking/Mass Spectrometry Studies. J Am Soc Mass Spectrom 2020, 31:183–189. [DOI] [PubMed] [Google Scholar]; This paper describes a new isotope encoded MS-cleavable DBSU-d0d12 crosslinker for qXL-MS.
- 34.Chen Z, Fischer L, Tahir S, Bukowski-Wills J-C, Barlow P, Rappsilber J: Quantitative cross-linking/mass spectrometry reveals subtle protein conformational changes. Wellcome Open Res 2016, 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt C, Zhou M, Marriott H, Morgner N, Politis A, Robinson CV: Comparative cross-linking and mass spectrometry of an intact F-type ATPase suggest a role for phosphorylation. Nat Commun 2013, 4:1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomko RJ, Taylor DW, Chen ZA, Wang H-W, Rappsilber J, Hochstrasser M: A Single α Helix Drives Extensive Remodeling of the Proteasome Lid and Completion of Regulatory Particle Assembly. Cell 2015, 163:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beilsten-Edmands V, Gordiyenko Y, Kung JC, Mohammed S, Schmidt C, Robinson CV: eIF2 interactions with initiator tRNA and eIF2B are regulated by post-translational modifications and conformational dynamics. Cell Discov 2015, 1:15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boelt SG, Norn C, Rasmussen MI, André I, Čiplys E, Slibinskas R, Houen G, Højrup P: Mapping the Ca2+ induced structural change in calreticulin. Journal of Proteomics 2016, 142:138–148. [DOI] [PubMed] [Google Scholar]
- 39.Fischer L, Chen ZA, Rappsilber J: Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J Proteomics 2013, 88:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Appen A, LaJoie D, Johnson IE, Trnka MJ, Pick SM, Burlingame AL, Ullman KS, Frost A: LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature 2020, 582:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linden A, Deckers M, Parfentev I, Pflanz R, Homberg B, Neumann P, Ficner R, Rehling P, Urlaub H: A Cross-linking Mass Spectrometry Approach Defines Protein Interactions in Yeast Mitochondria. Mol Cell Proteomics 2020, 19:1161–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen ZA, Rappsilber J: Quantitative cross-linking/mass spectrometry to elucidate structural changes in proteins and their complexes. Nat Protoc 2019, 14:171–201. [DOI] [PubMed] [Google Scholar]
- 43.Walker MA, Chavez J, Villet O, Tang X, Keller A, Bruce JE, Tian R: Acetylation of muscle creatine kinase negatively impacts high-energy phosphotransfer in heart failure. JCI Insight 2021, 6:e144301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng J, Corzo C, Chang MR, Shang J, Lam VQ, Brust R, Blayo A-L, Bruning JB, Kamenecka TM, Kojetin DJ, et al. : Chemical Crosslinking Mass Spectrometry Reveals the Conformational Landscape of the Activation Helix of PPARγ; a Model for Ligand-Dependent Antagonism. Structure 2018, 26:1431–1439.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurt LU, Clasen MA, Santos MDM, Lyra ESB, Santos LO, Ramos CHI, Lima DB, Gozzo FC, Carvalho PC: Characterizing protein conformers by cross-linking mass spectrometry and pattern recognition. Bioinformatics 2021, doi: 10.1093/bioinformatics/btab149. [DOI] [PubMed] [Google Scholar]
- *46.Müller F, Kolbowski L, Bernhardt OM, Reiter L, Rappsilber J: Data-independent Acquisition Improves Quantitative Cross-linking Mass Spectrometry. Mol Cell Proteomics 2019, 18:786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study brings a DIA-MS method for label-free qXL-MS.
- 47.Müller F, Rappsilber J: A protocol for studying structural dynamics of proteins by quantitative crosslinking mass spectrometry and data-independent acquisition. Journal of Proteomics 2020, 218:103721. [DOI] [PubMed] [Google Scholar]
- 48.Müller F, Graziadei A, Rappsilber J: Quantitative Photo-crosslinking Mass Spectrometry Revealing Protein Structure Response to Environmental Changes. Anal Chem 2019, 91:9041–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu W, Beebe K, Chavez JD, Boysen M, Lu Y, Zuehlke AD, Keramisanou D, Trepel JB, Prodromou C, Mayer MP, et al. : Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat Commun 2019, 10:2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *50.Gutierrez C, Chemmama IE, Mao H, Yu C, Echeverria I, Block SA, Rychnovsky SD, Zheng N, Sali A, Huang L: Structural dynamics of the human COP9 signalosome revealed by cross-linking mass spectrometry and integrative modeling. Proc Natl Acad Sci USA 2020, 117:4088–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper brings an application of PRM-based qXL-MS to study COP9 signalosome. The authors used 3 different MS-cleavable crosslinkers in the study.
- 51.Mehnert M, Ciuffa R, Frommelt F, Uliana F, van Drogen A, Ruminski K, Gstaiger M, Aebersold R: Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat Commun 2020, 11:3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Hüttenhain R, Koomen JM, et al. : Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics 2014, 13:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ: The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev 2020, 39:229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schweppe DK, Chavez JD, Navare AT, Wu X, Ruiz B, Eng JK, Lam H, Bruce JE: Spectral Library Searching To Identify Cross-Linked Peptides. J Proteome Res 2016, 15:1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wühr M, Haas W, McAlister GC, Peshkin L, Rad R, Kirschner MW, Gygi SP: Accurate Multiplexed Proteomics at the MS2 Level Using the Complement Reporter Ion Cluster. Anal Chem 2012, 84:9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Virreira Winter S, Meier F, Wichmann C, Cox J, Mann M, Meissner F: EASI-tag enables accurate multiplexed and interference-free MS2-based proteome quantification. Nat Methods 2018, 15:527–530. [DOI] [PubMed] [Google Scholar]
- 57.Chavez JD, Mohr JP, Mathay M, Zhong X, Keller A, Bruce JE: Systems structural biology measurements by in vivo cross-linking with mass spectrometry. Nat Protoc 2019, 14:2318–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mohr JP, Perumalla P, Chavez JD, Eng JK, Bruce JE: Mango: A General Tool for Collision Induced Dissociation-Cleavable Cross-Linked Peptide Identification. Anal Chem 2018, 90:6028–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eng JK, Jahan TA, Hoopmann MR: Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13:22–24. [DOI] [PubMed] [Google Scholar]