

Abstract
Primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders (MCPH–SCKS) include a heterogeneous group of autosomal recessive inherited diseases characterized by primary (congenital) microcephaly, the absence of visceral abnormalities, and a variable degree of cognitive impairment, short stature and facial dysmorphism. Recently, biallelic variants in the nuclear pore complex (NPC) component nucleoporin 85 gene (NUP85) were reported to cause steroid-resistant nephrotic syndrome (SRNS). Here, we report biallelic variants in NUP85 in two pedigrees with an MCPH–SCKS phenotype spectrum without SRNS, thereby expanding the phenotypic spectrum of NUP85-linked diseases. Structural analysis predicts the identified NUP85 variants cause conformational changes that could have an effect on NPC architecture or on its interaction with other NUPs. We show that mutant NUP85 is, however, associated with a reduced number of NPCs but unaltered nucleocytoplasmic compartmentalization, abnormal mitotic spindle morphology, and decreased cell viability and proliferation in one patient’s cells. Our results also indicate the link of common cellular mechanisms involved in MCPH–SCKS spectrum disorders and NUP85-associated diseases. In addition to the previous studies, our results broaden the phenotypic spectrum of NUP85-linked human disease and propose a role for NUP85 in nervous system development.
Introduction
Primary autosomal recessive microcephaly (MCPH) and Seckel syndrome (SCKS) spectrum disorders (MCPH–SCKS) include a heterogeneous group of autosomal recessive inherited disorders characterized by primary (congenital) microcephaly and the absence of visceral abnormalities (1–4). Variable findings are the degree of cognitive impairment, short stature and facial dysmorphism. Cranial imaging demonstrates microcephaly but otherwise normal brain morphology in most cases. Although biallelic variants in some MCPH–SCKS-linked genes cause the MCPH or the SCKS phenotype only, others can cause both and/or intermediate phenotypes (Supplementary Material, Table S1). Further genetic heterogeneity exists given the high proportion of patients with MCPH–SCKS without pathogenic variants in known genes.
Here we report two families with MCPH–SCKS caused by biallelic variants in the nucleoporin 85 (NUP85) gene, a gene recently linked to childhood-onset steroid-resistant nephrotic syndrome [SRNS; MIM*618176; (5)]. SRNS was associated with intellectual disability (ID) without structural brain defects in one pedigree and additionally short stature and partial growth hormone deficiency in one of the two affected children. Microcephaly was not reported in these patients [Table 1; (5)].
Table 1.
Comparison of phenotype of index patients and other reported patients with NUP85 mutations
| Characteristics and Symptoms | Index patient (pedigree 1) | Index patient (pedigree 2) | Patient 1 (A5195-22A)1 | Patient (A3259–21)1 | Patient 3 (NCR3227)1 | Patient 4 (NCR3310)1 |
|---|---|---|---|---|---|---|
| NUP85 variant (NM_024844.5); (NP_079120.1) | c.932G > A; p.R311Q | c.1109A > G, c.1589 T > C; p.N370S, p.M530T |
c.1430C > T; p.A477V | c.1933C > T; p.R645W | c.405 + 1G > A; Donor splice site |
c.1741G > C; p.A581P |
| Parents consanguinity | + | − | + | − | − | − |
| Gender | Female | Female | Female | Male | Female | Male |
| Age at last assessment | 9 years | 27 GW | 8 years | 11 years | 7 years | 4 years |
| Age of onset | Birth | Prenatal | 8 | 11 | 7 | 4 |
| Primary microcephaly | + | + | − | − | − | − |
| Intrauterine growth retardation | + | − | − | − | − | − |
| Short stature | + | − | + | − | − | + |
| Dystrophy | + | − | − | − | − | |
| Upslanted palpebral fissures | + | − | − | − | − | − |
| Short philtrum | + | − | − | − | − | − |
| High nasal bridge | + | − | − | − | − | − |
| Reduced vision | + | unknown | − | − | − | − |
| Optic nerve atrophy | + | Unknown | − | − | − | − |
| Astigmatism | + | Unknown | − | − | − | − |
| Esophoria | + | Unknown | − | − | − | − |
| Long, skinny finger | − | − | − | − | − | |
| Syndactyly | + | − | − | − | − | − |
| Pes adductus | + | − | − | − | − | − |
| Epilepsy | + | N/A | − | − | − | − |
| ID, moderate | + | N/A | − | − | + | + |
| Delayed speech and language development | + | N/A | − | − | − | − |
| SRNS | − | N/A | + | + | + | + |
| Muscular hypotonia | + | N/A | − | − | − | − |
| Cranial MRI abnormalities | − | + | − | − | − | − |
| Abnormality of vision evoked potentials | − | N/A | − | − | − | − |
Notes: +, yes; −, no; N/A, not applicable.
Braun et al. (5).
NUP85 is one of nine members (NUP160, NUP133, NUP107, NUP96, NUP85, NUP43, NUP37, SEH1 and SEC13) of the NUP107–160 subcomplex (synonym Y complex) within the nuclear pore complex [NPC; (6)]. NPCs are key for nucleocytoplasmic transport, but also regulate the mitotic machinery, transcription and chromatin organization through transport-independent mechanisms (7–9). The NUP107–160 complex is acknowledged to contribute to the assembly and maintenance of the NPC structure (10). Members of this largest subcomplex of the NPC associate with kinetochores, mitotic spindles, centrosomes and mitotic checkpoint regulators for proper completion of the cell cycle (7,8,11–13). Downregulation of NUP107–160 subcomplex members resulted in defective cytokinesis, compromised microtubule structures, altered cytoskeletal dynamics, impaired chromosome segregation and differentiation (5,8,14). Variants in several genes encoding NUP components have been linked to human disease [Supplementary Material, Table S2; (15)]. Of these, the following have been implicated in neurologic disorders with microcephaly and/or ID: NUP214 in encephalopathies (16,17), NUP62 in striatonigral degeneration (18), NUP93 in congenital ataxia (19), NUP188 in Sandestig–Stefanova syndrome (20), NUP107 and NUP37 in SRNS with microcephaly and ID (5), NUP107 and NUP133 in Galloway-Mowat syndrome [GMS (21)] and AAAS in triple-A syndrome (22).
In this study, we extend the human phenotype spectrum of NUP85 variants, proposing its role in brain development and add NUP85 to the list of genes associated with MCPH–SCKS.
Results
Clinical presentation
We report two individuals of unrelated pedigrees of Caucasian descent with MCPH–SCKS and biallelic variants in the NUP85 gene (Fig. 1A).
Figure 1 .
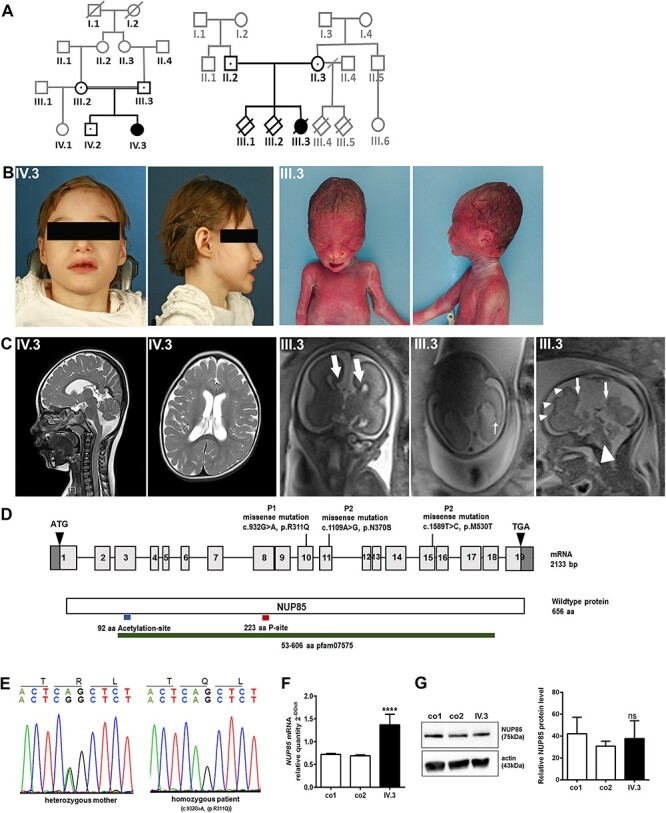
Phenotype and genotype of index patients with NUP85 mutation. (A) Pedigree of index family 1 and family 2 indicating affected patients (IV.3; III.3). (B) Photomicrographs of P1 (IV.3). AP and lateral photos of the fetus demonstrate sloping forehead, high nasal bridge and micrognathia (P2, III.3). (C) MRI of the head and sagittal and axial T2 images revealed reduced brain volume and delayed myelination in P1 at 6 years (IV.3). In P2 (III.3), bull-horn appearance of the frontal horns of the lateral ventricles (thick arrows) due to agenesis of the corpus callosum, irregularity and focal puckering of the lateral ventricular contour with heterotopia in the trigone of the hemisphere on the left (thin arrow), and abnormal sulcation in the left frontal lobe (arrows) and absence of the corpus callosum (arrowhead) were observed in fetal head MRI images. (D) Pictogram representing the NUP85 cDNA with exons 1–19 and the NUP85 wild-type protein with the identified variants. Through WES, the homozygous missense c.932G > A (p.R311Q) variant was identified in exon10 of NUP85 gene (NM_024844.5) in IV.3. In III.3, WES identified compound heterozygous variants c.1109A > G (maternally inherited; p.N370S) and c.1589T > C (paternally inherited; p.M530T) in exon 11 and exon 15, respectively. (E) Electropherogram traces depicting single base exchange of G to A at position 932 of the NUP85 gene present in heterozygous state in both parents and homozygous state in P1 of pedigree 1. (F) Quantitative real-time PCR revealed an increased level of NUP85 mRNA in P1’s (IV.3) fibroblasts compared with controls (n = 3, ****P < 0.0001. Error bars represent ± SD). (G) Unaltered levels of NUP85 protein was observed in P1’s (IV.3) fibroblasts (NUP85 75 kDa, actin 43 kDa; n = 3, one-way ANOVA, ns = non-significant, Error bars represent ± SD).
Proband 1 (P1) is a 10-year-old girl born at term as the second child of healthy consanguineous German parents (Fig. 1A and B; pedigree 1, IV.3). Intrauterine growth restriction was noticed during the sixth month of pregnancy and resulted in induced but otherwise uncomplicated birth in the 37th week of gestation (GW). At birth, microcephaly was observed with an occipitofrontal head circumference (OFC) of 31 cm [< 3rd centile, < −3.6 standard deviations (SD)]. In addition, hypotrophy [weight 1970 g (−2.8 SD), length 45 cm (−2.7 SD), both <3rd centile], facial dysmorphism (upslanted palpebral fissures, short philtrum, high nasal bridge and beaked nose appearance), syndactyly, long and thin fingers, and bilateral pes adductus were also noted (Table 1). Dystrophy remained throughout life despite feeding high-calorie meals through a percutaneous endoscopic gastrostomy tube. Severe developmental delay was apparent early on with strongly delayed motor milestones and absent speech (Supplementary Material, Table S3). During the last neurological examination at the age of 9 years, she could sit and bottom-shuffle, use a wheelchair in daily life for locomotion and walk a few steps with pronounced assistance. She had global muscular hypotonia, hypotrophy and reduced muscle strength. All reflexes other than the patellar reflex were absent. Speech was limited to rudimentary sounds indicating unhappiness or pleasure, and she could not follow simple instructions. Ophthalmological assessment revealed bilateral hyperopia, astigmatism, pale papillae as an indication of bilateral optic atrophy and unilateral esophoria. The patient was treated with lamotrigine (previously levetiracetam and valproic acid) for drug-resistant, genetic epilepsy with focal-onset seizures that had manifested in the first year of life. At the last presentation she still had repetitive episodes of hypoxia (minimal oxygen saturation 68%) and bradycardia (minimal 56/min) at night as revealed by a home monitor. Electroencephalography showed multifocal epileptic potentials, and repeated cranial magnetic resonance imaging (MRI) had detected reduced brain volume and delayed myelination (Fig. 1C, IV.3). A muscle biopsy performed at 1 year-of-life did not revealed any specific histopathologic or electron microscopic pathologies (Supplementary Material, Fig. S1). She had no clinical, laboratory or radiological findings indicative of kidney dysfunction. Sonography results of the abdomen, kidneys, urinary tract and bladder were normal. Echocardiography showed a persistent ductus arteriosus without clinical significance, electrocardiography results were normal.
Proband 2 (P2) is a female, naturally conceived fetus from healthy non-consanguineous Australian parents (Fig. 1A and B, pedigree 2, III.3). The pregnancy was preceded by two early miscarriages in the sixth GW. Although the result of an ultrasound scan at 19 GW was noted to be normal, repeat ultrasound at 24 GW revealed mild ventriculomegaly, absent septum pellucidum and corpus callosum agenesis. MRI of the fetal brain at 24 + 2 GW indicated complete agenesis of the corpus callosum, abnormal sulcation in the left frontal lobe, nodularity of the frontal horn and trigone with focal puckering of the left lateral ventricle (Fig. 1C, III.3). Considering the poor prognosis associated with these neuronal abnormalities, the couple elected to terminate the pregnancy at 27 GW. At autopsy, body weight was 1120 g (971 ± 188 g; ~90th centile), crown-heel length was 399 mm (358 ± 21 mm; >90th centile), head circumference was 265 mm (245 ± 15 mm; ~50th centile) and brain weight of 157.4 g [133 ± 22 g; (23)]. Complete agenesis of the corpus callosum was confirmed, with investigations also showing grey matter heterotopia and cortical malformation of the left frontal lobe. Macro- and microscopic analysis of the fetal brain confirmed the absence of the cingulate gyrus, and suggested maturational delay, as indicated by the micro-columnar architecture of the cortex. No other organ malformations were detected. The fetus was mildly macerated but had some facial findings that could be consistent with MCPH–SCKS such as sloping forehead, high nasal bridge and micrognathia (Table 1 and Fig. 1B, III.3).
Molecular genetic studies
To identify the genetic cause of the disease phenotypes, we performed whole exome sequencing (WES) followed by bioinformatic analysis in both index families. In pedigree 1, we identified the homozygous exchange of a single base G to A in exon 10 of the NUP85 gene in P1 (c.932G > A; NM_024844.5, rs199764435; Fig. 1D). Sanger sequencing confirmed the identified missense mutation that segregates with the phenotype in the pedigree (Fig. 1E). The variant is reported six times in heterozygosity in gnomAD v2.1. The mutation lies in a highly conserved region and leads, at the protein level, to an exchange of the polar basic amino acid arginine to the polar amino acid glutamine (p.R311Q, NP_079120.1/UniProt ID, Q9BW27). To analyze the effect of the identified mutation (c.932G > A; p.R311Q) in NUP85 mRNA and protein levels, fibroblasts and Epstein–Barr virus-transformed, immortalized lymphocytes (LCLs) were cultured from P1 and control individuals. Although quantitative PCR analysis revealed significantly increased levels of NUP85 mRNA in P1 compared with control cells (Fig. 1F), NUP85 protein levels were unchanged between P1 and control samples by Western blot analysis (Fig. 1G). This indicates the presence of mutant NUP85 protein in the system.
WES analysis in pedigree 2 identified compound heterozygous missense variants in NUP85 (NM_024844.5): a maternally inherited c.1109A > G (p.N370S) variant and a paternally inherited c.1589T > C (p.M530T) variant (Fig. 1D). The maternal variant is absent from population databases and the paternal variant is reported twice in heterozygosity in gnomAD v2.1. Both variants are highly conserved with GERP scores of 5.83 (maternal) and 5.71 (paternal) and are predicted to be deleterious by CADD, which scored the variants with 27 (maternal) and 26 (paternal). In line with these findings, no homozygous loss-of-function variants are reported in gnomAD (24), suggesting that loss of both alleles has a deleterious effect.
Structural implications of the NUP85 mutations
In order to gain insight into the possible impact of the identified mutations that affect residues R311, N370 and M530 of the NUP85 protein as well as the consequences for the NUP107–160 complex and the overall NPC (Fig. 2A and B), we performed a structure analysis of NUP85 using known structures and protein structure prediction tools. According to protein data bank (PDB)-based structural alignment using PDB structures from Saccharomyces cerevisiae, Myceliophthora thermophila, Xenopus laevis and Homo sapiens, and a newly generated model of NUP85 (25,26), the identified variants are found in three different regions of NUP85 (Fig. 2C). The amino acid exchange R311Q is located in a structurally conserved region. This position is located in helix 12 and directed into a lumen formed by a triangular-shaped 3 helix bundle of helix 12 with helices 11 and 13 (Fig. 2D). Variants that affect this region may alter the overall conformation of this NUP85 three-helix bundle and, thus, may have an indirect effect on the overall structure of the NUP107–160 complex by reorientation of the tip of the short arm. The putative effects of the other two amino acid exchanges, N370S and M530T, are more obvious. They are located at positions in the vicinity to the β-propeller proteins SEH1 and NUP43, respectively (Fig. 2E and F). N370 is located in helix 16, directly neighboring the blade in SEH1 provided by NUP85 (Fig. 2E). The predictions and the structural information on cytoplasmic NUP85 suggest a location for M530 within a region of NUP85 that is located at the periphery of blade one of the 7 bladed β-propeller protein NUP43. This blade harbors two extended loops. One, shown in blue, is located between β-strand 2 and 3 (14 residues) and one located between ß-strand 4 and 5 (15 residues in length of which 10 are missing; in the structure indicated by a dashed line; Fig. 2F). Due to their close proximity to SEH1 and NUP43, respectively, both mutations, N370S and M530T, could be predicted to interfere with the binding properties and/or the orientation of the β-propeller proteins to NUP85.
Figure 2 .
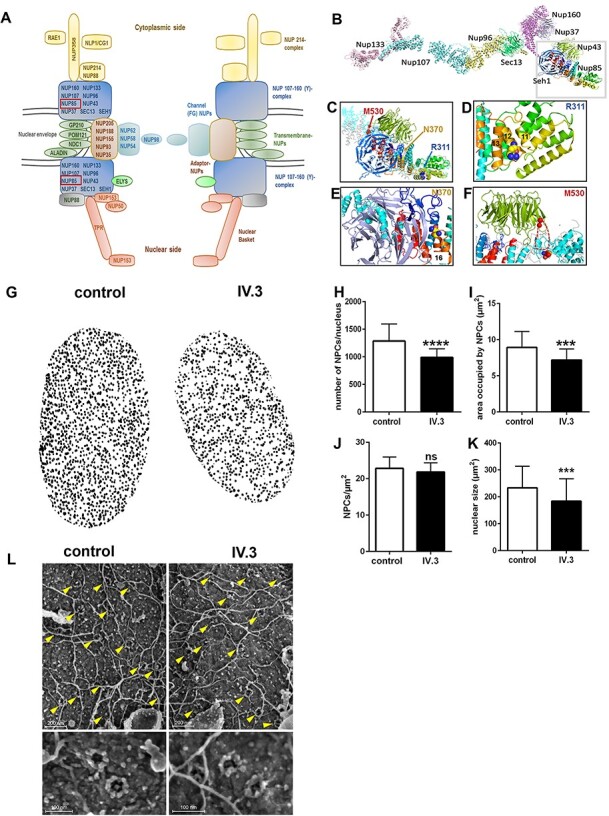
Effect of identified variants on NUP85 and NPCs. (A) Schematic representation of the NPC. NUP85 is an essential member of the NUP107–160 complex (Y-complex), the latter representing the scaffold unit of a NPC. (B) Model NUP107–160 complex derived from PBDid: 5a9q with the individual components indicated. (C) Structure of human NUP85 based on PDBid 5a9q and an ITasser model (cyan lines) and the structure from M. thermophila (grey lines). Rainbow-coloring from N- (blue) to C-terminus (red) within the trans blade in the N-terminal region. The remaining part is all helical with some helices indicated by numbers. The first helix (dark blue) marks the beginning of a U-shaped helical arrangement with helix 8 at the turning point, and then heading back to helices 12 with the site of mutation as well as helices nearby, 11 and 13. (D) Magnification of G to highlight the region comprising helices 11–12–13. The mutated residue is depicted as spheres with the carbon in yellow and nitrogen and oxygen colored in blue and red, respectively. (E) Magnification of the region comprising the N370 mutation. The blade inserted into SEH1 (light purple) by NUP85 is depicted in blue. The site of mutation is represented as spheres and colored as in (D). (F) Blow up of a slightly rotated NUP43 and its binding region of NUP85. Coloring as in G. The dashed red lines indicate a missing loop and the site of mutation is shown as spheres (colored as in H). (G) Graphical representative mAb414-stained nucleus of control and patient fibroblasts. Patient fibroblasts showed a significant reduction in the (H) total number of NPCs per nucleus and (I) the area occupied by NPCs in each nucleus (J), however, there was no significant difference in the NPCs per area between control and P1-derived cells. (K) The nuclear size was reduced in patient cells. (n = 30–33 cells, Student’s t-test, ***P < 0.001, ****P < 0.0001, ns-non-significant, Error bars represent ± SEM). (L) Direct surface imaging of NPCs by scanning electron microscopy of control and patient-derived fibroblasts. Cells were subjected to mild hypotonic treatment to expose intact nuclei and further processed for high-resolution imaging. Representative flat areas of the exposed outer nuclear surface are shown in the upper panels with yellow arrowheads marking the position of NPCs (scale bar 200 nm). Higher magnification images depicting individual NPCs appear in the lower panels (scale bar 100 nm).
Reduced number of NPCs in mutant fibroblasts
All functional studies described below have been performed using patient derived cells from P1, as patient derived cells from P2 were not available for functional assessment. To understand the effect of the identified homozygous p.R311Q NUP85 variant in P1 on the nuclear envelope and NPC integrity, we performed immunofluorescence staining on control and patient-derived fibroblasts using the mAB414 antibody, specific for NPC proteins. We identified a significant decrease in the total number of NPCs per nucleus in the patient cells (Fig. 2G and H). In addition, the area occupied by NPCs in patient cells was significantly reduced compared with control cells (Fig. 2I), whereas the NPCs per area was not significantly different (Fig. 2J). The nuclear size was also significantly reduced in patient cells compared with the control (Fig. 2K; n = 30–33 cells, Student’s t-test). Hypotonic treatment and exposure of intact fibroblast nuclei for direct surface imaging via a scanning electron microscope revealed a similar distribution pattern of NPCs in patient-derived and control nuclei (Fig. 2L, yellow arrowheads). Higher magnifications focusing on individual NPCs (Fig. 2L, lower panels) showed no overt change in NPC architecture, which is characterized by an eightfold rotational symmetry around the central pore channel with the occasional occurrence of central channel particles or glimpses of the internal nuclear basket structure. Additionally, immunofluorescence data on NPCs using various nucleoporin-specific antibodies (NUP107, NDC1, NUP133, NUP88, NUP153, NUP155, NUP98, NUP160, ALADIN, NUP214, POM121, NUP358, SEC13, NUP62, TPR, NUP50 and NUP93) did not show any striking difference between control and patient fibroblasts (Supplementary Material, Fig. S2). Taken together, these results indicate that there are no discernible changes in the distribution or architecture of NPCs in patient-derived fibroblasts.
Mutant NUP85 does not affect nucleocytoplasmic compartmentalization
Disturbances in the compartmentalization of nucleus and cytosol due to loss of NPC integrity could lead to dysfunction of various cellular processes. In order to understand the effect of the identified p.R311Q mutation on nucleocytoplasmic compartmentalization, we performed label-free shotgun proteomic analysis of the nuclear and cytosolic proteome of control and P1-derived fibroblasts. For that, cells were fractionated into cytosolic and nuclear fractions and protein fold changes calculated from label-free quantified intensities. Whole cell lysates were also analyzed in parallel. In total, 6850 proteins were identified in the experiment and 5262 were quantified in at least two replicates and in both experimental groups. Analysis of whole cell lysates indicate major changes in the proteome of P1-derived fibroblasts compared with control (Fig. 3A, middle panel, n = 6, moderated t-test with Benjamini–Hochberg false discovery rate [FDR] correction). Proteome changes observed in cytosolic and nuclear fractions were largely explained by whole proteome (Fig. 3B), suggesting that compartmentalization of nucleo-cytoplasm is not strongly affected in P1-derived fibroblasts. For functional investigation of the 116 up- and 77 downregulated proteins in whole cell lysate of P1-derived fibroblasts, biological process GO enrichment analysis was performed (Fig. 3C). We observed a significant enrichment of terms related to cell adhesion and cytoskeletal dynamics in the group of proteins downregulated in the P1 fibroblasts (Fig. 3C, lower panel).
Figure 3 .
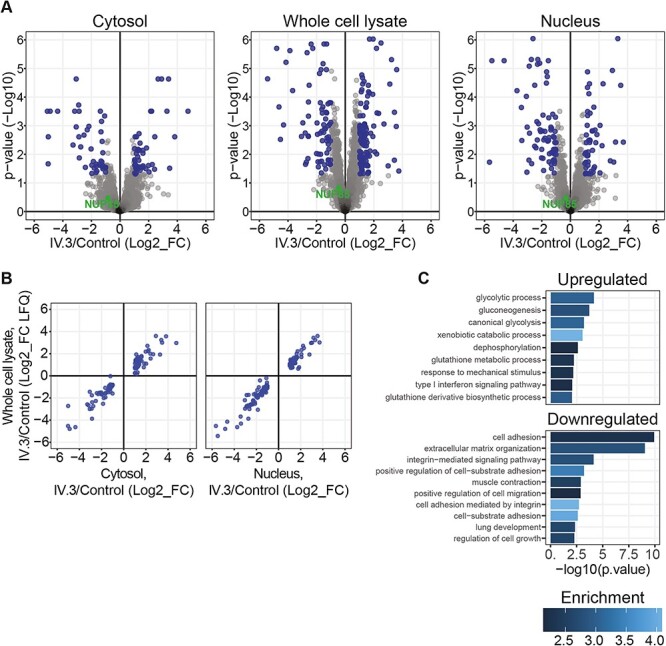
Unaffected nucleocytoplasmic compartmentalization in mutant-NUP85 (p.R311Q) fibroblasts. Proteomic analysis was performed in the nuclear and cytosolic fractions, and whole cell lysates of the control and patient-derived fibroblasts to understand the effect of mutant-NUP85 on nucleocytoplasmic compartmentalization. (A) Volcano plot shows changes in protein levels between control and patient (IV.3) cells in nuclear fraction (right), cytosolic fraction (left) and whole cell lysate (middle). Fold changes were calculated from protein LFQ intensities provided by MaxQuant. Moderated t-test was used and corrected for multiple tests with the Benjamini-Hochberg method. Fold changes higher than two and adjusted P-values smaller than 0.05 were considered differential (blue). (B) Scatter plot depicting protein fold changes between patient (IV.3) and control fibroblasts in whole cell lysates against cytosolic (left) and nuclear fractions (right). Only differentially expressed proteins in fractions are plotted (blue). (C) GO term enrichment analysis of up- and downregulated processes in the whole cell lysate from patient cells (IV.3).
Altogether, our results indicate that the NUP85 mutation (c.932G > A; p.R311Q) leads to proteomic alterations in cytoskeletal dynamics regulators, with no significant impact on nucleocytoplasmic compartmentalization.
Abnormal mitotic spindles and cytoskeletal structures in patient cells
Since NUP85 has been shown to play a role in mitosis by promoting spindle assembly, we investigated the effect of the identified NUP85 mutation (c.932G > A) on the mitotic machinery of P1-derived LCLs. We identified a significant increase of distorted mitotic spindle apparatus (broader and unfocused spindle poles) of metaphase cells in P1-derived (25%) versus control LCLs (2–5%). Whereas, the centrosome morphology and spindle pole distance appeared normal (Fig. 4A and B, n = 100 cells, one-way ANOVA). Cell viability and proliferation rate of P1-derived fibroblasts was also significantly reduced compared with control cells (Fig. 4C and D n = 8, one-way ANOVA). The abnormal mitotic spindle apparatus and the imbalance in expression levels of cytoskeletal proteins in patient fibroblasts led us to further analyze the effect of mutant NUP85 on cytoskeletal structures and cellular morphology. Analysis on cellular morphology of cultured fibroblasts using actin/phalloidin staining revealed a striking difference in the morphology of P1-derived cells compared with the controls. Control fibroblasts were elongated and spindle-shaped, whereas patient fibroblasts seemed more compact and polygonal-shaped (Fig. 4E). To better understand the spatial organization of the actin cytoskeleton, we plated P1-derived and control fibroblasts on crossbow-shaped micropatterns and stained for actin filaments. When cells are plated on engineered micropatterns the actin cytoskeleton develops a stereotypical architecture (27). The analysis revealed that the actin architecture was generally unchanged, but that ventral and dorsal actin stress fibers and contractile actin arcs were diminished in P1-derived fibroblasts (Fig. 4F).
Figure 4 .
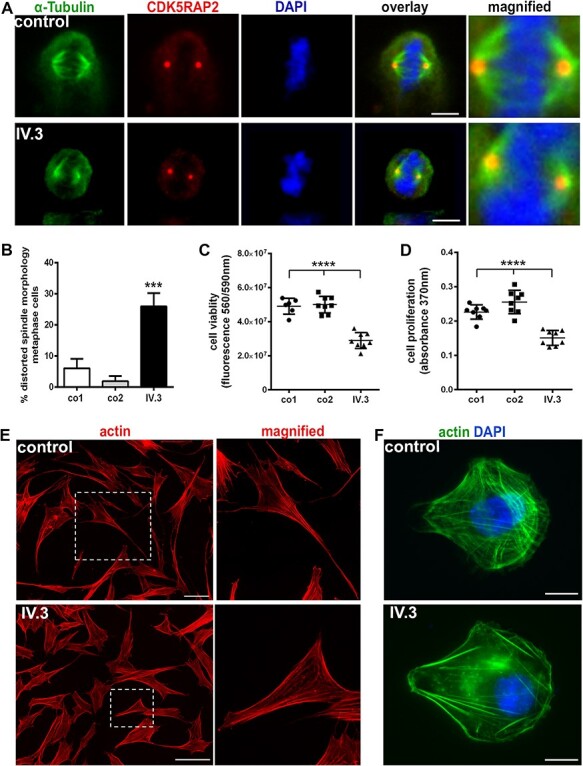
Abnormal mitotic spindles, reduced cell viability and altered cytoskeletal structures in P1-derived cells (IV.3). (A) Distorted mitotic spindle morphology in immortalized P1-derived lymphoblastoid cells (LCLs; metaphase cells, spindle marker α-tubulin (green), centrosome marker CDK5RAP2 (red), and DNA was stained with DAPI (blue), scale bar 5 μm). Abnormal spindles are shown in the magnified image of patient cells in comparison to control. (B) Quantification of metaphase cells revealed an increased percentage of P1-derived LCLs with abnormal mitotic spindles compared with the controls (n = 100, one-way ANOVA). (C, D) Reduced cell viability and proliferation of P1-derived fibroblasts compared with the controls (n = 8, one-way ANOVA). (E) Analysis of the cellular morphology of P1’s fibroblasts through actin (red) staining showed a pattern of compact and polygonal-shaped compared with elongated and spindle-shaped control cells (magnified image showing a representative cell of control and index patient), (scale bar 100 μm). (F) Spatial organization of the cytoskeleton was examined by plating the control and P1-derived fibroblasts on crossbow-shaped micropatterns and stained for actin (Alexa Fluor 488 phalloidin, green). The staining pattern reveals unchanged actin architecture, but ventral and dorsal actin stress fibers and contractile actin arcs were diminished in P1-derived fibroblasts. Nuclei were stained using DAPI (blue), (scale bar 10 μm). ***P < 0.001, ****P < 0.0001. Error bars represent ± SD.
Discussion
In this study, we report two unrelated individuals with MCPH–SCKS caused by biallelic mutation in the NUP85 gene. Although some of the clinical features of the probands from pedigree 1 and 2 appear to differ, both probands had findings consistent with MCPH–SCKS. Proband 1 had primary microcephaly, small stature, developmental delay, epilepsy and dysmorphic facial findings consistent with MCPH–SCKS without neuronal migration or corpus callosum abnormalities. Proband 2 was deceased at a GW when microcephaly may not have manifested in MCPH–SCKS. Agenesis of the corpus callosum and neuronal migration defects have been described in some cases of MCPH–SCKS (1–4). Our study also extends the phenotype spectrum of NUP85-associated disease, which has been previously linked to SRNS without neurodevelopmental disorder (5).
Mutations in the NUPs could have a major impact on the structural and functional aspects of NPC that has been implicated in various human diseases (28). We discussed here that for all three identified variants the suggested conformational changes could in turn interfere with proteins interacting with NUP85 resulting in a reorientation or loss of interaction partners. In analogy of the overall arrangement of NPC, one could speculate that at its cytoplasmic location a disturbance of the NUP85 structure could interfere with the shuttling process. Here the NUP214 complex is a central player of the export platform, which harbors a coiled-coil bundle that is interacting with the NUP85–SEH1 arm and the hub region of the Y-complex (29). On the nuclear side, the NUP85 interacts with TPR, which could be altered as well if NUP85 alters shape due to mutations. Our data suggests that the mutant NUP85 does not majorly affect the nucleocytoplasmic compartmentalization and do not exclude alterations of nucleocytoplasmic shuttling in general.
In line with previous findings on the effect of number of NPCs by mutations in various NUPs (28), our data on reduced number of NPCs in our index patient also suggests the role of nucleoporins in the formation and maintenance of NPCs. Several studies on mice have shown the link between reduced NPC and serious health problems (14,28). Future research on underlying mechanism is necessary; nevertheless, we postulate that the reduced NPC numbers might be due to partial perturbance in the NPC assembly process, alternatively cell cycle defects or defective nucleocytoplasmic transport.
Our data on abnormal cytoskeletal structures and distorted mitotic spindle morphology in patient cells are in line with existing research showing the regulatory role of NUP107–160 in mitotic machinery. It is known that the NUP107–160 complex localizes to kinetochore during early stages of mitosis and is key for the assembly of bipolar spindles (7,8,11,12). Indeed, CENP-F (Centromere Protein F) also plays role in the localization of NUP107–160 complex to kinetochores, and mutations in CENP-F have been reported in humans with microcephaly and a ciliopathic phenotype (30). Moreover, genes involved in the regulation of mitotic spindle and centrosome function have been associated with microcephaly phenotypes (4). The underlying pathomechanism of microcephaly has to a large part been attributed to defective mitotic machinery affecting the proliferation and/or differentiation of neural precursor cells (31). Intriguingly, NUP85 was also reported to play a role in the proper cilia localization of NUP98, which regulates the diffusion of soluble molecules through the ciliary base (32). Mutations in other NUP genes (NUP37, NUP107 and NUP214) have been reported to cause microcephaly (5) and with our study NUP85 also joins the list of nucleoporins involved in brain development, which is in line with the expression of NUPs during early stages of brain development (33).
In conclusion, we extended the phenotypic spectrum of NUP85 to that of MCPH–SCKS disorders. Our data contributes to the existing evidence on the shared common molecular and cellular mechanisms among MCPH, SCKS, GMS, Microcephalic osteodysplastic primordial dwarfism (MOPD) spectrum disorders which includes a range of clinical phenotypes that may be present such as microcephaly with or without brain malformations, ID and growth restriction [Fig. 5; (34,35)]. SRNS is a feature also acknowledged in GMS (21,35). We discuss here that the disturbance in the cellular functions of NUP85 such as defective mitotic machinery and cytoskeletal morphology contribute to the common pathomechanisms underlying neurological disorders.
Figure 5 .
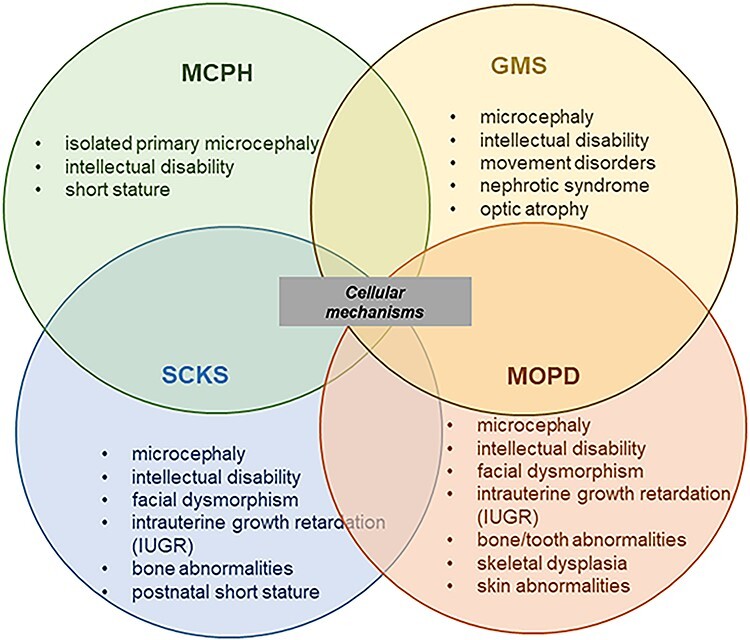
Representation of overlapping MCPH, SCKS, GMS and MOPD spectrum disorders. MCPH, SCKS, MGS and MOPD share microcephaly and ID as common phenotypes, and the disease-associated genes indicate shared cellular mechanisms underlying the disease spectrum phenotype.
Materials and methods
Patients
Written informed consent was obtained from the parents of the probands for the molecular genetic analysis, the publication of photos, and/or studies on primary fibroblasts and immortalized lymphocytes (LCLs). The human study was approved by the local ethics committees of the Charité (approval no. EA1/212/08; index family 1) and the Melbourne Health Human Research Ethics Committee as part of the Australian Genomics Health Alliance protocol (HREC/16/MH/251; index family 2).
Genetic analyses
The probands and their parents were subjected to WES. The WES protocol was as follows for pedigree 1: Five microgram genomic DNA was enriched with the Agilent Human All Exon V3 kit (Agilent, Santa Clara, CA, USA) following the manufacturer’s protocol. The libraries were sequenced using Illumina HiSeq 2000 sequencer for 101 bp single-end reads. Coverage of coding regions was >92.6–93.4% with a minimal depth of 20-fold; with raw data processed as described previously (48). In brief, the raw sequences were aligned to human reference genome (hg19), and the alignment results were used to call variants of SNVs, indels and CNVs. Resulting variants were screened by known causal mutation databases (OMIM and HGMD), and polymorphism databases (1000 Genome, ESP6500 and dbSNP138) with matching variants not exceeding the prevalence cutoff of 0.5%, and the pathogenicity of variants was further evaluated (analysis pipeline: https://sourceforge.net/projects/merap/). Sanger sequencing of the NUP85 gene was performed to confirm the mutation in P1and establish the genotype in other family members. Primer sequences used for Sanger sequencing are: NUP85-F-TAGTTGAAATCCGGTGCCCT, NUP85-R- CTCACTCACCTGGGCATAGT. The healthy status of heterozygous parents argues against haploinsufficiency for this disease phenotype. For pedigree 2 (local reference PED113), trio WES was performed at the Genomics Platform at the Broad Institute (Boston, MA) from cerebellum (proband) and saliva (parents) DNA. In brief, coding regions were enriched using the TWIST Broad custom exome panel (37 Mb) and libraries were sequenced (2 × 150 bp) on an Illumina Novaseq 6000 instrument. Alignment of sequencing reads to b37 + decoy (hg19) using BWA resulted in an average coverage of 95× with 97.4% >20× (proband), 64.4× with 97% > 20× (mother) and 66.4× with 96.9% >20× (father). SNV and indel calling was performed using the GATK haplotypeCaller package 3.4, CNVs were called using an in-house (ACRF) pipeline. Variant filtering was performed to select for rare (≤1% gnomAD and in-house frequencies for recessive and ≤0.01% for dominant inheritance, and 0 in homozygosity) protein altering variants, which resulted in 12 variants (7 recessive and 5 dominant). The NUP85 variants were prioritized after removing variants with a low allelic balance (n = 5) and those located in genes that have yet been associated with a human disease (n = 5). Mutations in known microcephaly genes were excluded.
Fibroblasts culture and Epstein–Barr virus-transformed lymphocyte (LCLs)
LCLs were established from P1 (pedigree 1) and unrelated controls and cultured, as previously reported (36). Fibroblasts from P1-derived and unrelated controls were cultured and maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM GlutaMAX supplement pyruvate, Gibco, Paisley, Scotland) with 10% fetal bovine serum (FBS, Gibco, Paisley, Scotland), and 1% penicillin streptomycin (P/S, Gibco, Grand Island, USA) at 37°C.
Quantitative real-time PCR (qPCR)
RNA extraction and cDNA synthesis were performed as previously reported (36,37). We designed sets of primers using the Primer3 online software (www.primer3.ut.ee) to amplify NUP85 and the reference gene RPII (RNA polymerase II) cDNA. Primer sequences used for qPCR are: NUP85-F- ATCAAAGAGTGCAGCATCGC, NUP85-R- GATGAGCAAACAGTCCCGAG, RPII- F- GCACCACGTCCAATGACAT, RPII- R- TCATGAGTGGTCCTGTGTCC. qPCR experiments were performed using Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, Braunschweig, Germany) according to the manufacturer’s protocol. Quantification and statistical analysis were performed using GraphPad Prism 6 Software (Version 6.07; GraphPad Software Inc., La Jolla, CA, USA), as described previously (36).
Western blot
Protein extraction and Western blots were performed in triplicates with established methods reported previously; (36,38) The antibodies used in this study are anti-NUP85 (Proteintech, rabbit), anti-actin (Millipore, mouse).
Immunocytology
Primary fibroblasts were plated on coverslips or crossbow-shaped micropatterns (CYTOO SA, Grenoble, France) and fixed with 4% PFA for 5 min, permeabilized with 0.5% (v/v) Triton X-100 in PBS for 5 min and then fixed again, or directly fixed for 10 min in 4% (w/v) PFA. LCLs were plated on poly-L-lysine (Sigma-Aldrich, Taufkirchen, Germany)-coated coverslips and fixed in 4% (w/v) PFA. Blocking was performed with blocking solution [2% (w/v) BSA/0.1% (v/v) Triton X-100 or 3% (w/v) BSA, 0.2% (w/v) gelatin, 0.25% (v/v) Triton X-100] in PBS for 30 min at room temperature (RT) followed by overnight incubation with primary antibodies and a 2 h incubation with the corresponding secondary antibodies. Antibodies used in this study are: Alpha-tubulin (Millipore, mouse), CDK5RAP2 (Sigma-Aldrich, rabbit), Mab414 (BioLegend, mouse), Phalloidin (Invitrogen). Nuclei were labeled with 4′,6-Diamidino-2-phenylindole (DAPI, 1:1000 (v/v), Sigma-Aldrich). Imaging was performed using a fluorescent Olympus BX51 microscope with Magnafire 2.1B software (2001; Olympus, Hamburg, Germany), a Zeiss Spinning Disc microscopy system CXU-S1 or a Zeiss LSM 710 (Zeiss, Oberkochen, Germany) confocal laser scanning microscope with Zeiss Plan-Apochromat 63x/1.4 oil objective. ZEN software (2012; Zeiss) was used with both Zeiss microscopes. Images were processed and quantified using Adobe Photoshop, ImageJ (Version 2.0.0-rc-69/1.52n) and GraphPad Prism 6 Software (Version 6.07; GraphPad Software Inc.).
Cell viability and proliferation assay
Fibroblasts were seeded at a density of 103 cells/well in 96-well plates. Cell viability (fluorimetric CellTiter-Blue Cell Viability Assay®, Promega, Madison, USA) and proliferation assay (colorimetric Cell Proliferation BrdU-ELISA, Roche Diagnostics, Mannheim, Germany) were performed as described previously (39). Measurements were performed using a SpectraMax iD3 plate reader (Molecular devices, San Jose, USA), and data were analyzed using GraphPad Prism 6 Software (Version 6.07; GraphPad Software Inc.).
Subcellular proteome fractionation
Cells were washed in ice-cold PBS and collected by centrifugation. Cell pellets were partially lysed in NP-40 lysis buffer [20 mm HEPES (pH 7.9), 0.5% NP-40, 0.25M Sucrose, 1.5 mm MgCl2 and 10 mm KCL] with protease inhibitor cocktail (Roche) and cytosolic (supernatant) and nuclear (pellet) fractions obtained by centrifugation. Supernatant was transferred to fresh tubes and pellets washed in ice-cold NP-40 lysis buffer. Nuclear fractions were further lysed in SDS before further use. Subcellular fractionation was accessed by shotgun proteomics and specific quantification of proteins with associated GO terms ‘Nucleus’ (GO:0005634) and ‘Cytoplasm’ (GO:0005737).
Mass spectrometry samples preparation and analysis
Proteins were ethanol precipitated and resuspended in a 2 M urea, 6 M thiourea and 0.1 M Tris pH 8 solution. Proteins were reduced with 10 mm DTT and alkylated with 55 mm iodoacetamide at 25°C. Next, proteins were incubated with lysyl endopeptidase (Lys-C, Wako-125-05061), at 25°C for 3 h. Three volumes of 50 mm ammonium bicarbonate solution were added, and proteins were further digested with trypsin (Promega, V5111) under constant agitation at 25°C for 16 h. Peptides were desalted with C18 Stage Tips (55) prior to liquid Chromatography with tandem mass spectrometry (LC–MS/MS) analysis. Prior to mass spectrometry analysis, peptides concentration was measured based on 280 nm UV light absorbance and total 1 μg of peptides loaded per run.
Reversed-phase liquid chromatography was performed using an EASY nLC II (Thermo Fisher Scientific, XX) and self-made C18 microcolumns (75 μm ID, packed with ReproSil-Pur C18-AQ 1.9 μm resin, Dr Maisch, Germany) connected on-line to the electrospray ion source (Proxeon, Denmark) of a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific). Peptide samples were eluted at a flow rate of 250 nl/min over 4 h with a 9–55.2% acetonitrile gradient in 0.1% (v/v) formic acid. The Q Exactive HF-X instrument was operated in data dependent mode with a full scan in the Orbitrap followed by up to 20 consecutive MS/MS scans. Settings for mass spectrometry analysis were as follows: one full scan (resolution, 60 000; m/z, 350–1800) followed by top 20 MS/MS scans using higher-energy collisional dissociation (resolution, 15 000; AGC target, 1e5; max. injection time, 22 ms; isolation width, 1.3 m/z; normalized collision energy, 26). Ions with an unassigned charge state, singly charged ions and ions with charge state higher than six were rejected. Former target ions selected for MS/MS were dynamically excluded for 30 s.
All raw files were analyzed with MaxQuant software [v1.6.0.1; (40)] with default parameters. Match between runs was allowed. Search parameters included two missed cleavage sites, cysteine carbamidomethyl fixed modification and variable modifications including methionine oxidation and protein N-terminal acetylation. Database search was performed with Andromeda (41) against UniProt/Swiss-Prot human database (downloaded on January 2019) with common contaminants provided by MaxQuant. FDR was set to 1% at peptide spectrum match (PSM) and protein levels. Minimum peptide count required for protein quantification was set to two. Mass spectrometry proteomics data have been deposited to ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD018838.
Structural analysis of NUP85
Structural interpretation was performed and figures were generated using PyMOL (Schrödinger LLC). A model of the complete yeast NPC has been recently described and is available for visualization (PDB-Dev numbers 10, 11 and 12, (https://pdb-dev.wwpdb.org/) 8). Structural alignment was achieved using standard settings and structures from the PDB: 3f3f [S. cerevisiae; (42)], 5a9q [H. sapiens; (26)] 4ycz [M. thermophila, (25)]. Moreover, in order to gain a more detailed insight into the fold, a partial model using Phyre2 was obtained using the full-length sequence (43).
Scanning electron microscopy
To expose primary fibroblast nuclei for high resolution imaging we used our previously published protocol (17,44). In brief, control and P1-derived fibroblasts were grown to 70% confluence, detached by trypsinization, resuspended in a hypotonic buffer in the absence of detergents and passed through a 21-gauge needle. The specimens were then gently centrifuged onto the surface of 5 × 5 mm2 silicon chips (#16008, Ted Pella, Redding, CA), fixed and further processed for electron microscopy including controlled dehydration, critical-point drying on a K850 apparatus (Quorum Technologies, Redding, CA). Specimens were then coated with a ~ 1 nm thick layer of iridium using a Q150T turbo-pumped sputter coater (Quorum Technologies) and imaged on a Merlin field emission scanning electron microscope (Zeiss, Redding, CA) equipped with a secondary electron in-lens detector.
Supplementary Material
Acknowledgments
The authors thank the index families for their participation in this study. We acknowledge K. Blaesius, B. Hartmann, J. Fassbender, Cynthia Gutierrez de Velazco for their contribution of clinical data, technical help and discussions. We also thank the members of the Genomic Autopsy Network, Genetics and Molecular Pathology Department, SA Pathology for their support and the staff of the Broad Institute’s Genomic Platform and the Broad Center for Mendelian Genomics’ team for WES of index family 2. We thank providing sequencing by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG).
Conflict of Interest statement. All authors declare no conflict of interest in the preparation or publication of the data in this manuscript.
Contributor Information
Ethiraj Ravindran, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany; Charité - Universitätsmedizin Berlin, Department of Pediatric Neurology, Berlin, 13353, Germany; Charité - Universitätsmedizin Berlin, Center for Chronically Sick Children (Sozialpädiatrisches Zentrum, SPZ), Berlin 13353, Germany.
Ramona Jühlen, Institute of Molecular Biology and Medicine, Université Libre de Bruxelles, Charleroi, 6041, Belgium.
Carlos H Vieira-Vieira, Proteome Dynamics Lab, Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association, Berlin, 13125, Germany; Humboldt-Universität zu Berlin, Faculty of Life Sciences, Berlin, 10099, Germany.
Thuong Ha, Genetics and Molecular Pathology Research Laboratory, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, Australia; ACRF Cancer Genomics Facility, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, SA, Australia.
Yuval Salzberg, Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel.
Boris Fichtman, Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel.
Lena Luise-Becker, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany; Charité - Universitätsmedizin Berlin, Department of Pediatric Neurology, Berlin, 13353, Germany; Charité - Universitätsmedizin Berlin, Center for Chronically Sick Children (Sozialpädiatrisches Zentrum, SPZ), Berlin 13353, Germany.
Nuno Martins, Institute of Molecular Biology and Medicine, Université Libre de Bruxelles, Charleroi, 6041, Belgium.
Sylvie Picker-Minh, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany; Charité - Universitätsmedizin Berlin, Department of Pediatric Neurology, Berlin, 13353, Germany; Charité - Universitätsmedizin Berlin, Center for Chronically Sick Children (Sozialpädiatrisches Zentrum, SPZ), Berlin 13353, Germany.
Paraskevi Bessa, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany.
Peer Arts, Genetics and Molecular Pathology Research Laboratory, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, Australia.
Matilda R Jackson, Genetics and Molecular Pathology Research Laboratory, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, Australia; Australian Genomic Health Alliance, Melbourne, VIC, 3052, Australia.
Ajay Taranath, Department of Medical imaging, South Australia Medical Imaging, Women’s and Children’s Hospital, North Adelaide, 5006, SA, Australia.
Benjamin Kamien, Genetic Services of Western Australia, Perth, 6008, Australia.
Christopher Barnett, Australian Genomic Health Alliance, Melbourne, VIC, 3052, Australia; Paediatric and Reproductive Genetics Unit, South Australian Clinical Genetics Service, Women’s and Children’s Hospital, North Adelaide, 5006, SA, Australia; School of Medicine, University of Adelaide, Adelaide, 5000, SA, Australia.
Na Li, Laboratory of Medical Systems Biology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, 510623, China.
Victor Tarabykin, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany; Research Institute of Medical Genetics, Tomsk National Research Medical Center of the Russian Academy of Sciences, Tomsk, 634009, Russia.
Gisela Stoltenburg-Didinger, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany.
Amnon Harel, Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel.
Matthias Selbach, Proteome Dynamics Lab, Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association, Berlin, 13125, Germany.
Achim Dickmanns, Department of Molecular Structural Biology, Institute for Microbiology and Genetics (GZMB), Georg-August-University Göttingen, Göttingen, 37073, Germany.
Birthe Fahrenkrog, Institute of Molecular Biology and Medicine, Université Libre de Bruxelles, Charleroi, 6041, Belgium.
Hao Hu, Laboratory of Medical Systems Biology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, 510623, China; Third Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052 , China.
Hamish Scott, Genetics and Molecular Pathology Research Laboratory, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, Australia; ACRF Cancer Genomics Facility, Centre for Cancer Biology, An alliance between SA Pathology and the University of South Australia, Adelaide, 5000, SA, Australia; Australian Genomic Health Alliance, Melbourne, VIC, 3052, Australia; School of Medicine, University of Adelaide, Adelaide, 5000, SA, Australia; UniSA Clinical and Health Sciences, University of South Australia, Adelaide, 5000, Australia; School of Biological Sciences, University of Adelaide, Adelaide, 5000, SA, Australia.
Angela M Kaindl, Charité - Universitätsmedizin Berlin, Institute of Cell Biology and Neurobiology, Berlin, 10117, Germany; Charité - Universitätsmedizin Berlin, Department of Pediatric Neurology, Berlin, 13353, Germany; Charité - Universitätsmedizin Berlin, Center for Chronically Sick Children (Sozialpädiatrisches Zentrum, SPZ), Berlin 13353, Germany.
Funding
German Research Foundation (SFB665, SFB1315, FOR3004 to A.M.K.); the Sonnenfeld Stiftung (A.M.K.); the Berlin Institute of Health (BIH, CRG1 to A.M.K.); the Charité (A.M.K., E.R., L.L.B., S.P.M., G.S.D.); the Israel Science Foundation (985/15 to A.H.); National Health and Medical Research Council (NHMRC) grant (APP1123341 to H.S. and C.B.); the Australian Genomic Health Alliance NHMRC Targeted Call for Research into Preparing Australia for the Genomics Revolution in Healthcare (GNT1113531 to H.S. and C.B.); Beat Cancer Principal Research Fellowship (H.S.) and Medical Research Future Fund (GHFM76777 to H.S. and C.B.); the Major Medical Collaboration and Innovation Program of Guangzhou Science Technology and Innovation Commission (201604020020); the National Natural Science Foundation of China (81671067, 81974163 and 81701451); Russian Science Foundation (21-65-00017); National Human Genome Research Institute; the National Eye Institute; and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm.
Author contributions
AMK was responsible for project conception. AMK, BK, AT, CB and MRJ recruited subjects, gathered patient history as well as clinical information and contributed clinical samples. HH, NL, TH, PA and HS generated and analyzed WES. CV obtained and analyzed proteomic data supervised by MS. ER, RJ, YS, BF, LLB, NM, SPM, PB, GDS, VT, AH, AD, BF and AMK performed functional experiments and/or interpreted data. NM developed Macros for ImageJ/Fiji to analyze NPC density. ER, RJ and CV performed statistical analysis. AMK and ER drafted the manuscript that was revised and accepted by all coauthors.
A. Kaindl, H. Scott, B. Fahrenkrog, A. Dickmanns, A. Harol, C. H. Vierra-Vierra have full access to all of the data in the study and take responsibility for the integrity of their data and accuracy of the data analysis.
References
- 1. Verloes, A., Drunat, S., Gressens, P. and Passemard, S. (2013) Primary Autosomal Recessive Microcephalies and Seckel Syndrome Spectrum Disorders – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. In Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., (eds), GeneReviews®. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- 2. Kaindl, A.M., Passemard, S., Kumar, P., Kraemer, N., Issa, L., Zwirner, A., Gerard, B., Verloes, A., Mani, S. and Gressens, P. (2010) Many roads lead to primary autosomal recessive microcephaly. Prog. Neurobiol., 90, 363–383. [DOI] [PubMed] [Google Scholar]
- 3. Kaindl, A.M., Passemard, S. and Gressens, P. (2009) Autosomal recessive primary microcephalies (MCPH). Eur. J. Paediatr. Neurol., 13, 458. [DOI] [PubMed] [Google Scholar]
- 4. Zaqout, S., Morris-Rosendahl, D. and Kaindl, A.M. (2017) Autosomal recessive primary microcephaly (MCPH): an update. Neuropediatrics, 48, 135–142. [DOI] [PubMed] [Google Scholar]
- 5. Braun, D.A., Lovric, S., Schapiro, D., Schneider, R., Marquez, J., Asif, M., Hussain, M.S., Daga, A., Widmeier, E., Rao, J. et al. (2018) Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Invest., 128, 4313–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck, M. and Hurt, E. (2017) The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol., 18, 73–89. [DOI] [PubMed] [Google Scholar]
- 7. Isabelle Loı¨odice, A.A., Gwe’nae¨l, R., van Overbeek, M., Ellenberg, J., Sibarita, J.-B. and Vale’rie, D. (2004) The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol. Biol. Cell, 15, 3333–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zuccolo, M., Alves, A., Galy, V., Bolhy, S., Formstecher, E., Racine, V., Sibarita, J.B., Fukagawa, T., Shiekhattar, R., Yen, T. et al. (2007) The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J., 26, 1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raices, M. and D'Angelo, M.A. (2017) Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol., 46, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szymborska, A., de Marco, A., Daigle, N., Cordes, V.C., Briggs, J.A. and Ellenberg, J. (2013) Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science, 341, 655–658. [DOI] [PubMed] [Google Scholar]
- 11. Belgareh, N., Rabut, G., Bai, S.W., van Overbeek, M., Beaudouin, J., Daigle, N., Zatsepina, O.V., Pasteau, F., Labas, V., Fromont-Racine, M. et al. (2001) An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J. Cell Biol., 154, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orjalo, A.V., Arnaoutov, A., Shen, Z., Boyarchuk, Y., Zeitlin, S.G., Fontoura, B., Briggs, S., Dasso, M. and Forbes, D.J. (2006) The Nup107-160 nucleoporin complex is required for correct bipolar spindle assembly. Mol. Biol. Cell, 17, 3806–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakano, H., Wang, W., Hashizume, C., Funasaka, T., Sato, H. and Wong, R.W. (2011) Unexpected role of nucleoporins in coordination of cell cycle progression. Cell Cycle, 10, 425–433. [DOI] [PubMed] [Google Scholar]
- 14. Lupu, F., Alves, A., Anderson, K., Doye, V. and Lacy, E. (2008) Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev. Cell, 14, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nofrini, V., Di Giacomo, D. and Mecucci, C. (2016) Nucleoporin genes in human diseases. Eur. J. Hum. Genet., 24, 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shamseldin, H.E., Makhseed, N., Ibrahim, N., Al-Sheddi, T., Alobeid, E., Abdulwahab, F. and Alkuraya, F.S. (2019) NUP214 deficiency causes severe encephalopathy and microcephaly in humans. Hum. Genet., 138, 221–229. [DOI] [PubMed] [Google Scholar]
- 17. Fichtman, B., Harel, T., Biran, N., Zagairy, F., Applegate, C.D., Salzberg, Y., Gilboa, T., Salah, S., Shaag, A., Simanovsky, N. et al. (2019) Pathogenic variants in NUP214 cause “plugged” nuclear pore channels and acute febrile encephalopathy. Am. J. Hum. Genet., 105, 48–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basel-Vanagaite, L., Muncher, L., Straussberg, R., Pasmanik-Chor, M., Yahav, M., Rainshtein, L., Walsh, C.A., Magal, N., Taub, E., Drasinover, V. et al. (2006) Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann. Neurol., 60, 214–222. [DOI] [PubMed] [Google Scholar]
- 19. Zanni, G., De Magistris, P., Nardella, M., Bellacchio, E., Barresi, S., Sferra, A., Ciolfi, A., Motta, M., Lue, H., Moreno-Andres, D. et al. (2019) Biallelic variants in the nuclear pore complex protein NUP93 are associated with non-progressive congenital ataxia. Cerebellum, 18, 422–432. [DOI] [PubMed] [Google Scholar]
- 20. Sandestig, A., Engstrom, K., Pepler, A., Danielsson, I., Odelberg-Johnsson, P., Biskup, S., Holz, A. and Stefanova, M. (2020) NUP188 Biallelic loss of function may underlie a new syndrome: nucleoporin 188 insufficiency syndrome? Mol. Syndromol., 10, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujita, A., Tsukaguchi, H., Koshimizu, E., Nakazato, H., Itoh, K., Kuraoka, S., Komohara, Y., Shiina, M., Nakamura, S., Kitajima, M. et al. (2018) Homozygous splicing mutation in NUP133 causes Galloway-Mowat syndrome. Ann. Neurol., 84, 814–828. [DOI] [PubMed] [Google Scholar]
- 22. Tullio-Pelet, A., Salomon, R., Hadj-Rabia, S., Mugnier, C., de Laet, M.H., Chaouachi, B., Bakiri, F., Brottier, P., Cattolico, L., Penet, C. et al. (2000) Mutant WD-repeat protein in triple-A syndrome. Nat. Genet., 26, 332–335. [DOI] [PubMed] [Google Scholar]
- 23. Maroun, L.L. and Graem, N. (2005) Autopsy standards of body parameters and fresh organ weights in nonmacerated and macerated human fetuses. Pediatr. Dev. Pathol., 8, 204–217. [DOI] [PubMed] [Google Scholar]
- 24. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelley, K., Knockenhauer, K.E., Kabachinski, G. and Schwartz, T.U. (2015) Atomic structure of the Y complex of the nuclear pore. Nat. Struct. Mol. Biol., 22, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Appen, A., Kosinski, J., Sparks, L., Ori, A., DiGuilio, A.L., Vollmer, B., Mackmull, M.T., Banterle, N., Parca, L., Kastritis, P. et al. (2015) In situ structural analysis of the human nuclear pore complex. Nature, 526, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thery, M., Pepin, A., Dressaire, E., Chen, Y. and Bornens, M. (2006) Cell distribution of stress fibres in response to the geometry of the adhesive environment. Cell Motil. Cytoskeleton, 63, 341–355. [DOI] [PubMed] [Google Scholar]
- 28. Sakuma, S. and D'Angelo, M.A. (2017) The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin. Cell Dev. Biol., 68, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandez-Martinez, J., Kim, S.J., Shi, Y., Upla, P., Pellarin, R., Gagnon, M., Chemmama, I.E., Wang, J., Nudelman, I., Zhang, W. et al. (2016) Structure and function of the nuclear pore complex cytoplasmic mRNA export platform. Cell, 167, e1225, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waters, A.M., Asfahani, R., Carroll, P., Bicknell, L., Lescai, F., Bright, A., Chanudet, E., Brooks, A., Christou-Savina, S., Osman, G. et al. (2015) The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J. Med. Genet., 52, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jayaraman, D., Bae, B.I. and Walsh, C.A. (2018) The genetics of primary microcephaly. Annu. Rev. Genomics Hum. Genet., 19, 177–200. [DOI] [PubMed] [Google Scholar]
- 32. Endicott, S.J. and Brueckner, M. (2018) NUP98 sets the size-exclusion diffusion limit through the Ciliary Base. Curr. Biol., 28, 1643, e1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reza, N., Khokha, M.K. and Del Viso, F. (2016) Nucleoporin gene expression in Xenopus tropicalis embryonic development. Int. J. Dev. Biol., 60, 181–188. [DOI] [PubMed] [Google Scholar]
- 34. Megraw, T.L., Sharkey, J.T. and Nowakowski, R.S. (2011) Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol., 21, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colin, E., Huynh Cong, E., Mollet, G., Guichet, A., Gribouval, O., Arrondel, C., Boyer, O., Daniel, L., Gubler, M.C., Ekinci, Z. et al. (2014) Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am. J. Hum. Genet., 95, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ravindran, E., Hu, H., Yuzwa, S.A., Hernandez-Miranda, L.R., Kraemer, N., Ninnemann, O., Musante, L., Boltshauser, E., Schindler, D., Hubner, A. et al. (2017) Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet., 13, e1006746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neitzel, H. (1986) A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet., 73, 320–326. [DOI] [PubMed] [Google Scholar]
- 38. Issa, L., Kraemer, N., Rickert, C.H., Sifringer, M., Ninnemann, O., Stoltenburg-Didinger, G. and Kaindl, A.M. (2013) CDK5RAP2 expression during murine and human brain development correlates with pathology in primary autosomal recessive microcephaly. Cereb. Cortex, 23, 2245–2260. [DOI] [PubMed] [Google Scholar]
- 39. von Bernuth, H., Ravindran, E., Du, H., Frohler, S., Strehl, K., Kramer, N., Issa-Jahns, L., Amulic, B., Ninnemann, O., Xiao, M.S. et al. (2014) Combined immunodeficiency develops with age in immunodeficiency-centromeric instability-facial anomalies syndrome 2 (ICF2). Orphanet J. Rare Dis., 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cox, J. and Mann, M. (2008) Max Quant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 41. Cox, J., Neuhauser, N., Michalski, A., Scheltema, R.A., Olsen, J.V. and Mann, M. (2011) Andromeda: a peptide search engine integrated into the Max Quant environment. J. Proteome Res., 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- 42. Debler, E.W., Ma, Y., Seo, H.S., Hsia, K.C., Noriega, T.R., Blobel, G. and Hoelz, A. (2008) A fence-like coat for the nuclear pore membrane. Mol. Cell, 32, 815–826. [DOI] [PubMed] [Google Scholar]
- 43. Kelley, L.A., Mezulis, S., Yates, C.M., Wass, M.N. and Sternberg, M.J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc., 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fichtman, B., Shaulov, L. and Harel, A. (2014) Imaging metazoan nuclear pore complexes by field emission scanning electron microscopy. Methods Cell Biol., 122, 41–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.