

Abstract

The GPVI platelet receptor was recently validated as a safe antiplatelet target for the treatment of thrombosis using several peptidic modulators. In contrast, few weakly potent small-molecule GPVI antagonists have been reported. Those that have been published often lack evidence for target engagement, and their biological efficacy cannot be compared because of the natural donor variability associated with the assays implemented. Herein, we present the first side-by-side assessment of the reported GPVI small-molecule modulators. We have characterized their functional activities on platelet activation and aggregation using flow cytometry as well as light transmission and electrical impedance aggregometry. We also utilized microscale thermophoresis (MST) and saturation transfer difference (STD) NMR to validate GPVI binding and have used this along with molecular modeling to suggest potential binding interactions. We conclude that of the compounds examined, losartan and compound 5 are currently the most viable GPVI modulators.
Keywords: Thrombosis, platelet inhibitor, GPVI, STD NMR, MST
Aberrant thrombus formation is the underlying cause of diseases such as deep vein thrombosis (DVT), stroke, and myocardial infarction (MI), with an MI occurring every 5 min in the UK, and every 39 s in the US.1,2 Despite the development of numerous antiplatelet and anticoagulant therapeutics, a safe and effective treatment for thrombosis remains elusive. Even the newer direct oral anticoagulants (DOACs), such as apixaban, rivaroxaban, and dabigatran, remain plagued by undesirable bleeding side effects and require the use of a reversal agent to prevent hemorraging.3−5
GPVI (glycoprotein VI) is a platelet receptor that adheres to exposed subendothelial collagen upon atherosclerotic plaque rupture, tethering platelets to the site of vascular injury. This subsequently leads to platelet activation and aggregation to form a stable vascular plug.6 There are numerous examples of epidemiological studies and preclinical experiments with genetic or peptide-induced GPVI deficient murine models validating GPVI as a safe and effective antiplatelet target.7−13 Moreover, two peptidic treatments: the soluble dimeric GPVI-Fc (fragment crystallizable) fusion protein Revacept, and the humanized antibody fragment against GPVI ACT017 (also known as Glenzocimab) have both successfully completed phase I clinical trials without reports of bleeding side effects.14−16 Notably, phase II clinical trials with Glenzocimab for patients with ischemic stroke (NCT03803007) are underway.17 Similarly, a phase II trial with Revacept for patients with coronary artery disease undergoing percutaneous coronary intervention was recently completed (NCT03312855).18 Importantly, preliminary results from a phase II clinical trial with Revacept for patients with symptomatic carotid artery stenosis, transient ischemic attacks, and stroke (NCT01645306) also showed no increase in bleeding complications.19 Despite this promising evidence validating GPVI as a safer antiplatelet target, relatively few direct GPVI small molecules have been reported (Table 1).20
Table 1. Summary of the Published Functional and Binding Data for Known Structurally Distinct GPVI Small-molecule Modulators21-26.
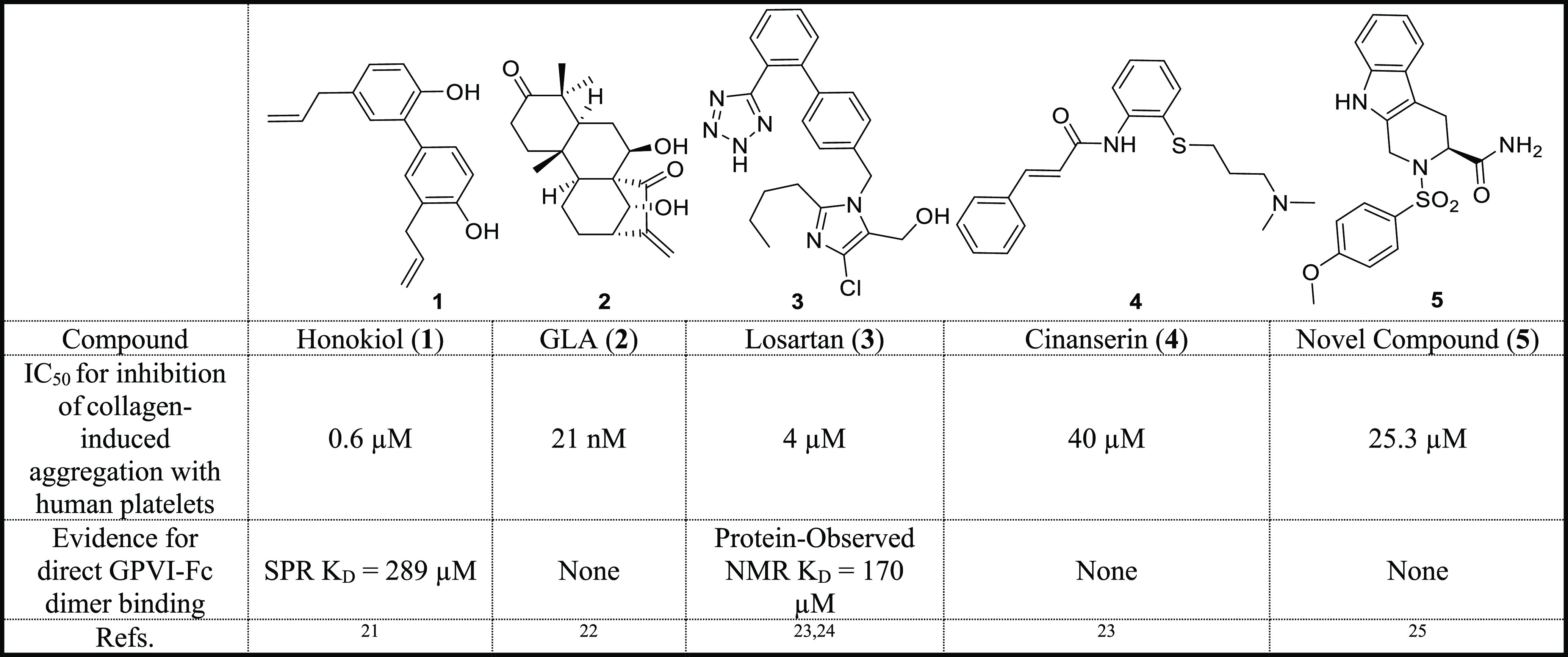
Honokiol and Glaucocalyxin A (GLA) are both natural products isolated from the bark of Magnolia officinalis and the perennial herb Rabdosia japonica, respectively.21,22 Losartan and cinanserin are repurposed medicines originally used for the treatment of hypertension and atypical pneumonia, respectively.23 Compound 5 is a novel inhibitor based on a tetrahydroisoindole scaffold.25 The activity of the known modulators in Table 1 has been separately quantified in the literature by the authors using analogous collagen-induced light transmission (LT) aggregometry assays. By directly comparing these values, most compounds are weakly potent against GPVI, with GLA being the most active. However, GLA’s activity was only measured at three concentrations, so the IC50 value was defined by extrapolation.22 Notably, the LT assay, and likewise any assay that uses human platelet samples, is subject to donor variation issues because the expression levels of GPVI on platelets varies greatly (typically 3000–4000 copies per human platelet).26 As such, it is not appropriate to directly compare the published literature values. Ideally, the compounds are tested under the same conditions with the same donor, although this can be challenging to achieve with the limited throughput nature of the functional assays employed. In addition, despite being described as GPVI modulators, often their binding to GPVI is insufficiently characterized. The compounds all reduce platelet aggregation induced by the GPVI specific agonists CRP-XL and/or convulxin suggesting that they are likely to be interacting at GPVI.20 However, only honokiol and losartan have evidence of directly interacting with the GPVI dimer (Table 1).21,24
It has been suggested that caffeic acid phenethyl ester (CAPE) and S007-867 could also be GPVI modulators.27 However, CAPE was also found to inhibit aggretin-induced platelet aggregation, suggesting that this also interacts with integrin α2β1, another collagen adhesion receptor, so is unlikely to be specific for GPVI.28 Likewise S007-867 was determined to inhibit collagen-induced platelet aggregation, but this activity was markedly diminished with CRP-XL and abolished with convulxin (GPVI specific agonists), suggesting the compound likely targets another part of the collagen-mediated pathway.29 As such, CAPE and S007-867 were omitted from this study. The fragment hinokitkiol, likewise to honokiol has been reported to preferentially inhibit convulxin-induced aggregation compared to aggretin.21,30 These fragments are structurally similar, but honokiol has evidence for binding to GPVI by SPR and so was preferentially included in the study.
Herein, we have compared the known modulators in several orthogonal functional assays. We initially employed the commonly used LT human washed platelet functional assay. In addition, we assessed compound activity on platelet function in whole blood using multiplate electrical impedance aggregometry (MEA) and used flow cytometry (FC) to quantify changes to GPVI-mediated platelet activation. Unlike previous reports, we tested the various compounds in the same donor’s blood. Moreover, we investigated donor variability by repeating the experiment in triplicate with different donors. Importantly, we have used microscale thermophoresis (MST) and saturation-transfer difference (STD) NMR to verify which molecules bind to GPVI. These data were used to select binding pose predictions, which may help to guide the design of future analogues. We conclude by summarizing the functional and binding data collected from our controlled experiments, which could aid future drug discovery efforts.
Most of the known GPVI modulators were purchased with acceptable purity levels (>95% by HPLC) from commercial suppliers (Supporting Information (SI) Figure S1). Novel compound 5, however, was synthesized following the previously published method by Bhunia et al. with minor adapatations.25 In summary, the ester 6 (SI, Figure S2) was converted to the primary amide 7 and was then coupled with 4-methoxyphenylsulfonyl chloride to furnish 5 in an acceptable yield (Scheme 1).
Scheme 1. Synthesis of Novel Compound 5.

Reagents and conditions: (a) 7 M NH3 in MeOH, 60 °C; (b) 4-methoxyphenylsulfonyl chloride, dry acetone, TEA. Percent yields have been reported.
Unusually, the racemic mixture of compound 5 was previously found to be the most active platelet inhibitor.25 This was later rationalized by a cooperative or combination effect of the enantiomers interacting with GPVI at two distinct binding sites, which was suggested using molecular modeling studies.31 Herein, the S enantiomer was selectively chosen for the comparative study, as based on these molecular modeling predictions the S enantiomer interacted at the same site as losartan.24,31 Furthermore, compound 5 was previously determined to be the most potent enantiomer.
LT aggregometry was the commonly utilized technique (by the authors of the known modulators) to quantify potency against human platelet aggregation induced with collagen20 and involves stimulating a mixture of washed human platelets and inhibitor to aggregate with an agonist (collagen, CRP-XL, or convulxin). A light source is directed at this mixture, and as aggregation occurs light transmitted through the sample increases, with inhibitors showing decreased LT relative to DMSO controls. Unlike previous reports, herein the known modulators were assessed using the same donor’s blood (Figure 1), enabling comparisons between the compounds to be made. This procedure was repeated with two additional donors to assess donor variability.
Figure 1.
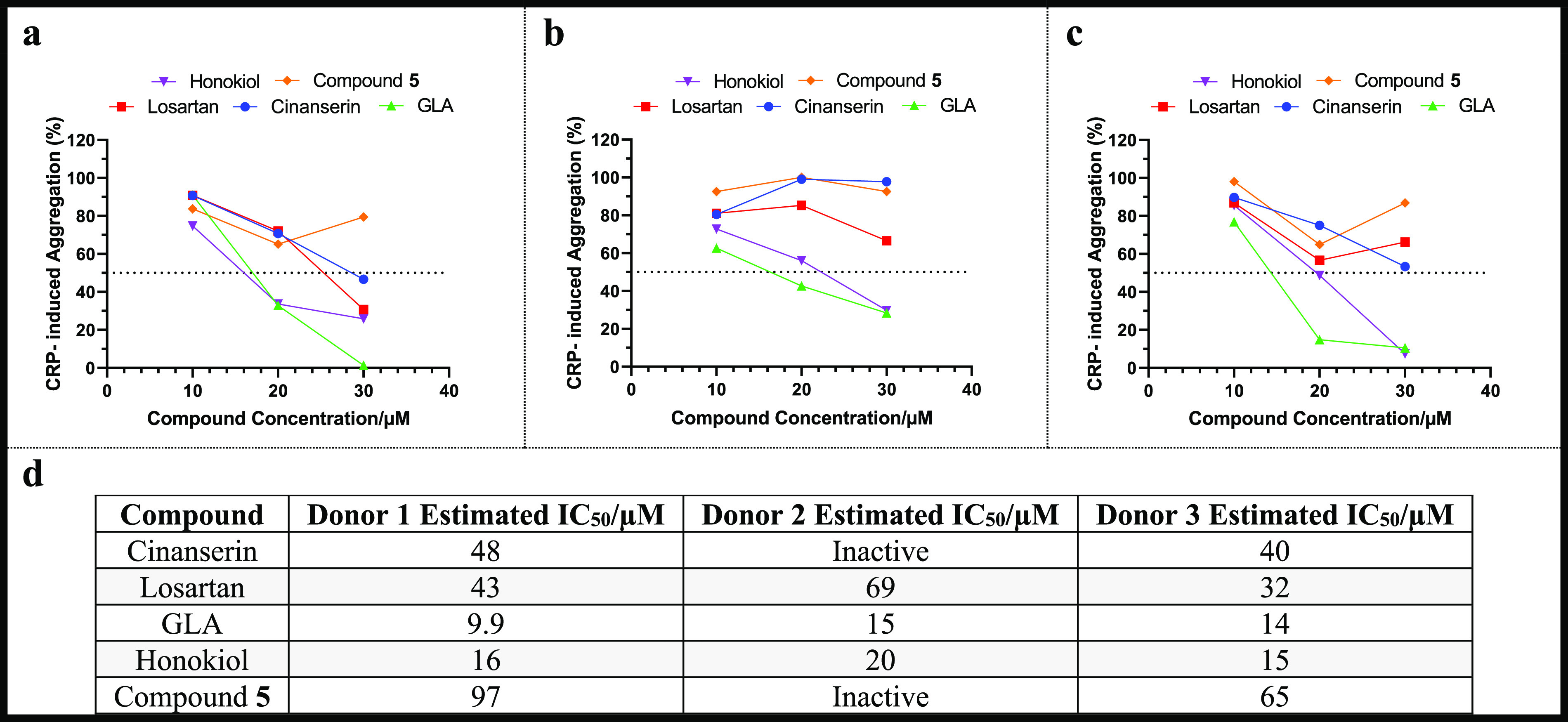
Light transmission aggregometry results for the known GPVI modulators with CRP-XL (2 μg/mL). Three donor repeats were recorded: (a) donor 1, (b) donor 2, and (c) donor 3 with n = 1 for each data point. Percentage aggregation was calculated relative to DMSO negative controls (= 100% aggregation) diluted in the same manner as the modulators (n = 1). (d) Estimation of IC50 values derived from fitting the data with a nonlinear regression curve that assumes a hill slope of −1.
By comparing the estimated IC50 values across the donors (Figure 1), GLA was the most potent inhibitor, followed by honokiol and then losartan. Contrastingly, cinanserin and compound 5 reliably showed weaker inhibitory activity against platelet aggregation when tested at a top concentration of 30 μM.
Upon atherosclerotic plaque rupture, GPVI tethers to subendothelial collagen, initiating the release of calcium from intracellular stores.6 The calcium concentration is further enhanced by extracellular influxes via store-operated calcium channels, following platelet activation by various agonists (thromboxane A2 (TXA2), ADP and thrombin).32 This leads to the degranulation of α granules and surface expression of P-selectin, which can be assessed by FC by utilization of an anti-CD62p antibody with a fluorescent conjugate (CD62p-PE).33,34 By stimulating activation with the GPVI specific agonist CRP-XL, the alteration in P-selectin expression can be quantified by changes in the PE fluorescence levels, therefore FC was utilized to assess alterations to GPVI-mediated platelet activation with the known modulators (Figure 2).
Figure 2.

Influence of the five known GPVI modulators (100 μM) on GPVI-specific platelet activation determined by assessing changes to surface expression of P-selectin. Compounds were incubated with whole human blood at 37 °C for 5 min before incubating with CRP-XL (3 μg/mL) for 20 min. Three donor repeats were recorded with n = 3 for each data point.
Herein, GLA was determined to be the most potent inhibitor followed by cinanserin (Figure 2). However, compound 5, honokiol and losartan showed negligible inhibition of GPVI-mediated platelet activation when tested at 100 μM.
Although LT aggregometry is currently the most popular primary functional assay used to quantify GPVI modulators in the literature, it is a low throughput, high volume method. Preparing washed human platelets for the assay is time-consuming, limiting the number of experiments that can be performed before the platelets are no longer viable. The wider adoption of FC as the primary functional assay within the field may therefore expediate the development of future modulators because its throughput is higher. This is accounted for by the reduction in sample preparation time, as whole blood can be used in place of washed platelets. Moreover, prepared samples are fixed in paraformaldehyde prior to analysis, eliminating the time constraint of platelet viability. Finally, the LT aggregometry assay only yields a single data point, whereas in the FC assay 10 000 individual platelet events are recorded, so this better assesses changes to the overall platelet population.
To further assess the functional effects of the known modulators on GPVI, their ability to disrupt platelet aggregation in a whole blood sample was evaluated using MEA (Figure 3).
Figure 3.
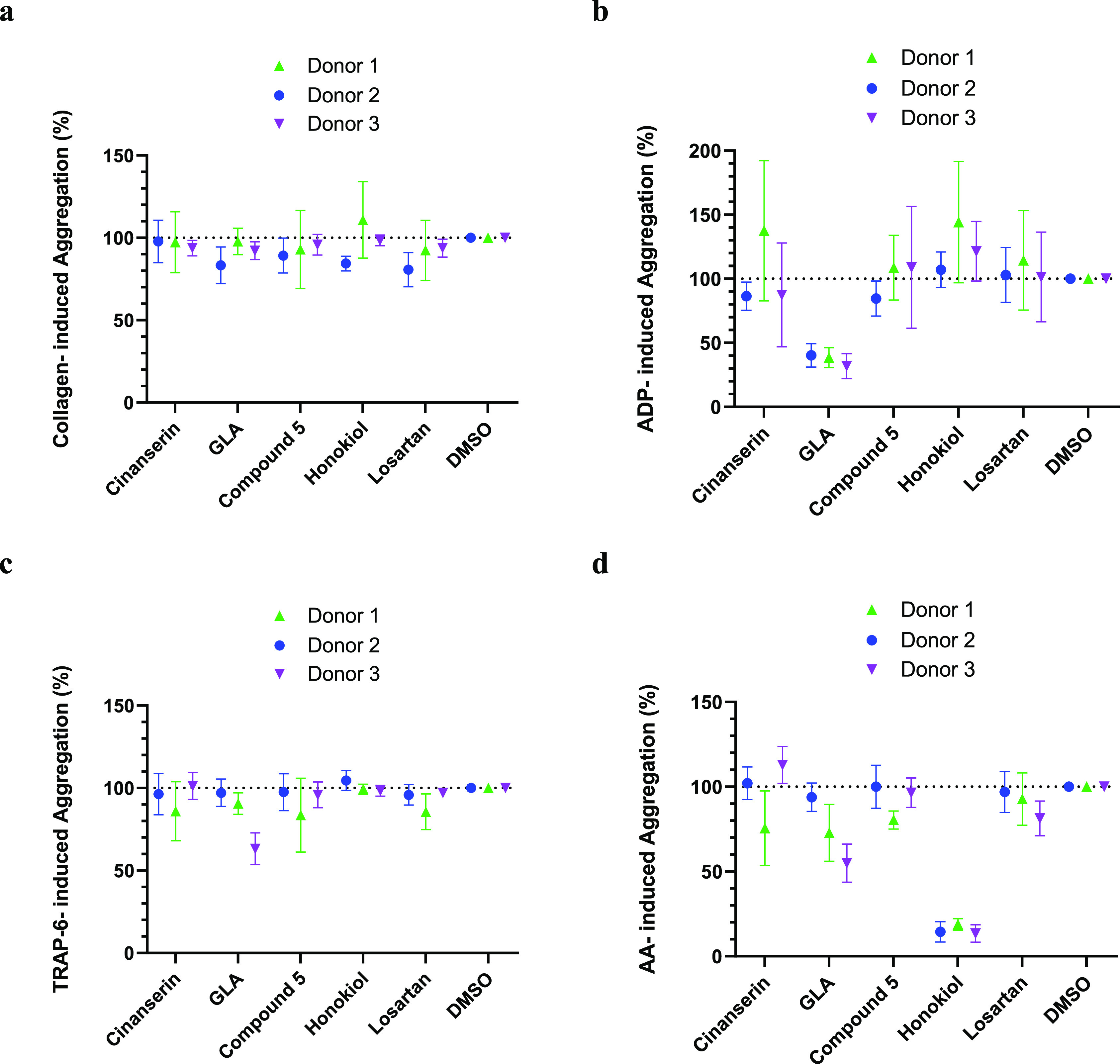
Multiplate electrical impedance aggregometry results with the known GPVI modulators (100 μM) with various agonists: (a) collagen (100 μg/mL), (b) ADP (6.5 μM), (c) TRAP6 (32 uM), and (d) arachidonic acid (AA) (0.5 mM). Percentage aggregation was calculated relative to DMSO negative controls (= 100% aggregation) with n ≥ 3. Three donor repeats were recorded with n ≥ 3 for each data point.
In brief, a sample of human whole blood was incubated with the compound of interest before the addition of collagen. The mixture is in contact with two wire prongs, if aggregation occurs the resistance (measured as area under curve (AUC) from a plot of resistance over time) between the wires increases, with inhibitors showing reduced resistance. As collagen is a GPVI agonist, alterations to collagen-induced aggregation can be used to assess changes to GPVI activity. However, collagen is also a direct and indirect ligand for numerous other platelet receptors including α2β1 integrin,36,37 hence the use of orthogonal functional and binding assays in conjunction with MEA was essential to verify that the compounds are likely to be interacting at the desired receptor. All the known modulators showed minimal inhibitory activity against collagen-induced aggregation in whole blood (Figure 3A). The lack of activity in the FC and MEA assays is likely accounted for by the increase in complexity of the testing system, leading to a reduction in effective drug concentration from the influence of plasma protein binding, which was not alleviated despite increasing the compound concentration (100 μM) relative to the LT aggregometry assay (≤30 μM).
Owing to decreased sample preparation time and higher throughput capability compared to LT aggregometry, a platelet receptor selectivity study was also performed using MEA. This was achieved using alternative agonists to collagen. If the modulator was able to reduce aggregation stimulated by either TRAP-6, arachidonic acid (AA) or ADP, this suggested that the compound may also modulate the PAR1, TP, P2Y1, or P2Y12 receptors, respectively, and hence are not GPVI-selective. If the compound was membrane permeable, a reduction in AA-induced aggregation may also suggest that the compound inhibits COX1 or another intermediate enzyme in this activation pathway. Most modulators were found to be ineffective at inhibiting TRAP-6- and AA-induced aggregation (Figure 3C and D), with GLA potentially showing some weak inhibitory activity against both. However, honokiol significantly reduced AA-induced aggregation compared to collagen-induced aggregation when tested at 100 μM. Similarly, GLA was determined a more potent inhibitor of ADP-induced aggregation compared to collagen-induced (Figure 3B).
The potent inhibitory activity of GLA observed in both the LT and FC assays, and honokiol in the LT assay could hence be accounted for by off-target activity at the P2Y1/P2Y12 and TP receptors (and/or COX1 and the intermediate enzymes involved in this pathway), respectively. Despite using a GPVI specific agonist (CRP-XL) in these assays to assess GPVI activity, the platelet activation pathways are interconnected, working harmoniously to assist thrombus formation.38−40 As such, even when using CRP-XL, the other pathways beyond GPVI are activated (albeit likely to a lesser extent), and so may also be inhibited by the modulators, influencing assay readouts models. Notably, honokiol is a CLEC-2 inhibitor41 because AA is a secondary mediator of CLEC-2 the inhibition of AA-induced aggregation may be linked to off-target activity at CLEC-2, but requires further investigation.42 However, this effect was not observed for losartan, which was also reported to be a CLEC-2 modulator.41
While direct binding data does not confer information pertaining to efficacy of platelet inhibition in biological terms, it is critical to assess the binding properties of these small-molecule modulators to the target to verify specificity. Therefore, this comparative assessment study presents both functional and binding data for a comprehensive comparison. Here we assessed if the known modulators bound to the GPVI-Fc dimer by the use of MST (Figure 4).35
Figure 4.

Changes in thermophoresis (quantified by changes to FNorm (0/00)) associated with the known modulators interacting with the GPVI-Fc dimer: (a) cinanserin, (b) losartan, (c) honokiol, (d) compound 5, and (e) GLA, with 20% MST and 40% LED power (n = 1). Experiment was performed in triplicate, each with freshly labeled FITC–GPVI dimer.
Thermophoresis refers to the directed movement of molecules in a temperature gradient and is commonly stimulated by directing a laser beam at a fluorescently labeled protein. The resulting movement of the protein either toward or away from the laser beam is referred to as negative and positive thermophoresis, respectively, and is monitored via the fluorescence of the protein. In this study, if a ligand binds to FITC-labeled GPVI, the ability of the resulting complex to move in response to the laser beam is altered, thus the fluorescence detected should be substantially different from the DMSO negative controls. Honokiol (Figure 4C) and compound 5 (Figure 4D) both showed a dose-dependent change in thermophoresis at higher compound concentrations, suggesting that they may bind to GPVI. Conversely, the interaction of GPVI with cinanserin, GLA, and losartan did not lead to a change in thermophoresis. The binding of losartan to GPVI has been previously verified by protein-observed NMR experiments, so attempts were made to optimize the MST results with losartan, but these were unsuccessful (SI, Figure S7).24
STD NMR was employed to validate the weak binders from the MST assay and to determine which regions of the ligands interact with GPVI (Figure 5). In the absence of a protein–ligand crystal structure, designing future analogues can be challenging. Understanding the ligand binding epitope for the known modulators would therefore be invaluable to this process.
Figure 5.

1H STD-NMR results for the known modulators (100 μM) with GPVI–Fc dimer (1 μM): (a) losartan, (b) honokiol, (c) compound 5, (d) cinanserin, and (e) GLA. The top spectrum is a water suppressed 1H NMR, and below is the corresponding STD NMR difference spectrum. Solvent and other minor impurities have been indicated with an asterisk.
The 1H STD-NMR applies specific NMR pulses that saturate protons across the protein but not proteins associated with the ligand of interest. This protein saturated signal can be transferred to a bound ligand by spin diffusion, reducing the signal intensity of the interacting protons in the corresponding proton NMR spectrum. Binding is observed in a difference spectrum produced by subtraction between two acquired STD NMR 1H NMR spectra, differing only in whether protein saturation is on or off. To validate that the STD was a result of the protein–ligand interaction, the STD experiment was repeated in the absence of the GPVI-Fc dimer (SI, Figure S3).
In agreement with previous protein-observed NMR data, losartan’s Ha protons (Figure 5A), located on the 1,2-substituted phenyl ring, were found to interact with GPVI.24 In addition, we found that the Hb and Hc protons, located on the 1,4-substituted phenyl ring, and the terminal methyl group also interacted with GPVI. In keeping with published SPR data, and the MST data reported herein (Figure 4C), we then demonstrated that honokiol can to interact with GPVI (Figure 5B), with nearly all proton signals being retained in the STD NMR spectrum.21 Finally, in line with our MST results (Figure 4D), our STD-NMR result provided further evidence to support that compound 5 can bind to GPVI (Figure 5C). Interestingly, all aromatic protons of the aforementioned compounds were involved in the binding process, perhaps illustrating the importance of hydrophobic interactions to small-molecule GPVI binding. Cinanserin had few sharp peaks in the STD NMR, perhaps suggesting a weaker interaction with GPVI, which could therefore account for the negative result observed in the MST assay reported herein (Figure 4A). In agreement with the MST results herein, GLA was also determined to be a nonbinder in the STD NMR experiment (Figure 5E).
Currently there are four published GPVI crystal structures (PDBs 2GI7, 5OU8, 5OU9, and 7NMU), all of which lack a cocrystallized small molecule. Hence, the location of the small-molecule ligand binding site remains elusive, making modeling studies challenging.20 However, suggestions for the small-molecule binding site have been made using convincing protein-observed NMR experiments with losartan.24 We used this information to guide the generation of a predictive binding model (SI, Figures S4–S6) to suggest how the other known GPVI modulators are interacting with the protein (Figure 6). GLA was not modeled into GPVI, as the MEA, MST, and STD NMR data presented herein (Figures 3–5) suggested that it is nonspecific for GPVI.
Figure 6.

Predicted binding poses for: (a) losartan, (b) honokiol, (c) compound 5, (d) cinanserin-pose 1, and (e) cinanserin-pose 2. Generated using induced fit docking with with SP, protonated ligands, and with water in the crystal structure (PDB 2GI7). Polar contacts have been highlighted with yellow dashes.
Poses were selected that agreed with the STD NMR results presented herein (Figure 5). For example, it was deduced that the biphenyl group and terminal methyl of the butyl chain interacted with GPVI (Figure 5A), hence a pose was selected that appeared to facilitate these interactions (Figure 6). This selection criteria accounts for the difference between our predicted pose for compound 5 and the previously published one.25
All modeled compounds appeared to form hydrophobic interactions with the Leu39 and the backbone amides of Gln48 and Lys41. Notably, this agreed with the previously reported protein-observed NMR results for losartan.24 Similarly, all modulators could hydrogen bond with Arg46 (Figure 6A–D). In addition, losartan’s tetrazole can interact ionically with Arg46. Losartan, compound 5, and cinanserin (Figure 6A,C, and E) all form an edge-face π interaction with Tyr66, however, honokiol did not (Figure 6B). This perhaps accounts for the reduction in predicted potency (Glide GScore) for honokiol compared to the other modulators. Compound 5 had the best Glide GScore (Figure 6C), which can perhaps be explained by the number of additional hydrogen bonds that it can form with GPVI compared to the other modulators (amide carbonyl to Ser44, and amide NH2 to the backbone amide of Ser61). Ultimately, these molecular modeling theories may guide the design of future compounds, but these theories first require crystallographic confirmation.
Herein we have assessed the known GPVI modulators in a series of controlled binding and functional assays including: STD NMR, MST, MEA, FC, and LT aggregometry (Table 2).
Table 2. Summary of the Comparative Study.
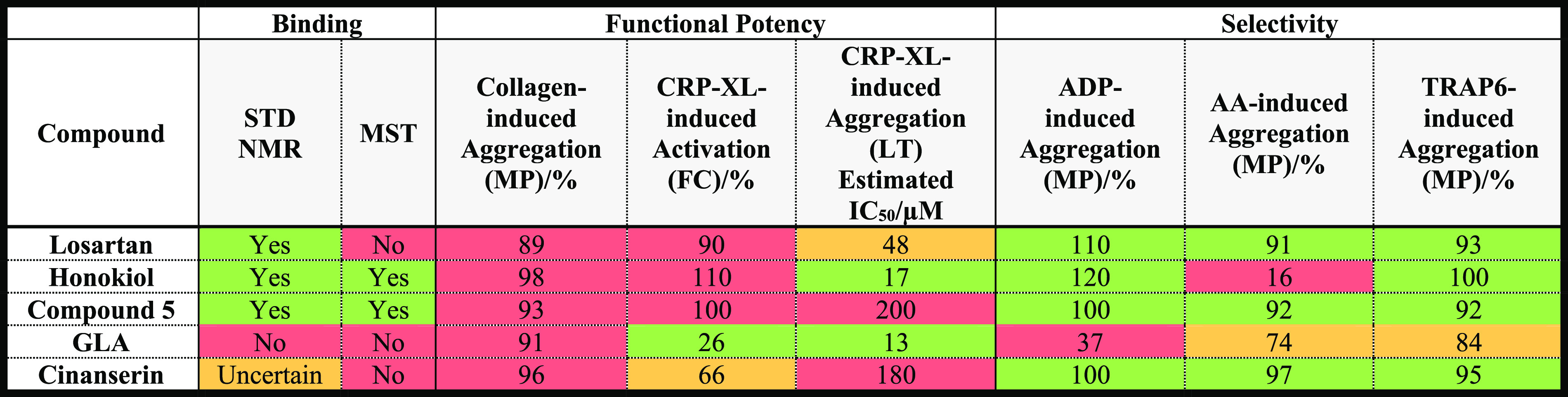
Results have been colored on a red to green scale to indicate negative and positive results, respectively, to allow for easy comparisons between the compounds. For the binding assays, a binary quantification has been provided to indicate which compounds interact with the GPVI–Fc dimer. The functional assay results for the three independent donors were averaged to give the potency and selectivity results presented. In the MP and FC functional assays, all compounds were tested at a final concentration of 100 μM.
Despite GLA being a potent inhibitor of GPVI-mediated aggregation and activation, it showed no evidence to bind to GPVI. In fact, it was a more potent inhibitor of ADP-induced aggregation, suggesting that it may preferentially modulate the P2Y1 and P2Y12 receptors, or dense granule secretion. Similarly, despite binding to GPVI, honokiol displayed a greater preference for inhibiting AA-induced aggregation compared to collagen-induced aggregation in the comparative MEA assays (Figure 3), suggesting that honokiol more selectively inhibits the TP platelet receptor, COX1, or the intermediate enzymes involved in this pathway. Although cinanserin was a reasonably potent inhibitor of GPVI-mediated activation, relatively few protons were determined to be part of the binding epitope by STD NMR, making its engagement with GPVI questionable.
Conversely, losartan and compound 5 were both determined to be clear binders by STD NMR, although MST did not confirm this for losartan. Both compounds appeared to be relatively selective inhibitors of CRP-XL-induced aggregation, and we conclude that of the compounds investigated, losartan and compound 5 are the most genuine small-molecule GPVI antagonists. Notably, their inhibitory activity in the light transmission aggregometry assay (Figure 1) appears modest due to the low testing concentration (30 μM) used to compare the modulators. When tested at 100 μM (above the estimated IC50 values for both compounds), they were both potent inhibitors of platelet aggregation (SI, Figure S8). On the basis of the molecular modeling presented herein, interactions from the ligand to Arg46, Gln48, Lys41, and Leu39 appear important for effective modulation of GPVI, although this hypothesis requires crystallographic confirmation. Future work should focus on optimizing the potency and selectivity of losartan and compound 5 for GPVI. In addition, it would be interesting to investigate whether these compounds disrupt the GPVI–fibrin(ogen) interaction, as this is another important interaction supporting platelet activation and thrombus consolidation.20
Acknowledgments
We thank the BHF (British Heart Foundation) for funding (grant no. FS/17/66/33480), and the many willing blood donors, without whom this work would not be possible. The Cytoflex was funded by BBSRC BB/R000352/1 and the microscale thermophoresis instrument by 105615/Z/14/Z. We also thank Dr. James N. Ayres for his help proofreading the manuscript.
Glossary
Abbreviations
- AA
arachidonic acid
- AUC
area under curve
- CAPE
caffeic acid phenethyl ester
- DOACs
direct oral anticoagulants
- DVT
deep vein thrombosis
- FC
flow cytometry
- glaucocalyxin A
GLA
- GPVI
glycoprotein six
- HRMS
high resolution mass spectrometry
- LT
light transmission
- MEA
multiplate electrical impedance aggregometry
- MFI
mean fluorescence intensity
- MI
myocardial infarction
- MST
microscale thermophoresis
- PBS
phosphate buffered saline
- STD
saturation-transfer difference
- TBG
Tyrode’s buffer supplemented with glucose
- TXA2
thromboxane A2;
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00414.
Experimental procedures for: chemical synthesis, functional assays, and binding assays; HPLC chromatograms; STD NMR control spectra; details on the development of a GPVI molecular model; losartan MST optimization experiments; LT aggregometry results with a higher concentration of losartan and compound 5(PDF)
Author Contributions
H.F. designed and performed the experiments, analyzed the data, and also wrote the manuscript in consultation with R.F., H.P., and C.W. R.F., H.P., and C.W. were involved in planning and supervised the work. R.F., H.P., C.W., M.J.H., I.M., R.A., K.N., and L.R.V. supported the experimental designs and aided in interpreting the results. C.W. prepared the washed human platelets. J.S.G. and R.X. prepared the GPVI-Fc dimer.
The authors declare no competing financial interest.
Supplementary Material
References
- Heart Statistics—heart and Circulatory Diseases in the UK; British Heart Foundation, 2021; https://www.bhf.org.uk/what-we-do/our-research/heart-statistics (accessed 2021-02-09.
- Heart and Stroke Statistics; American Heart Association, 2021; https://www.heart.org/en/about-us/heart-and-stroke-association-statistics (accessed 2021-05-21).
- Lobraico-Fernandez J.; Baksh S.; Nemec E. Elderly bleeding risk of direct oral anticoagulants in nonvalvular atrial fibrillation: a systematic review and meta-analysis of cohort studies. Drugs R&D 2019, 19, 235–245. 10.1007/s40268-019-0275-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lip G. Y. H.; Keshishian A.; Kamble S.; Pan X.; Mardekian J.; Horblyuk R.; Hamilton M. Real-world comparison of major bleeding risk among non-valvular atrial fibrillation patients initiated on apixaban, dabigatran, rivaroxaban, or warfarin. A propensity score matched analysis. Thromb. Haemostasis 2016, 116, 975–986. 10.1160/TH16-05-0403. [DOI] [PubMed] [Google Scholar]
- Vinogradova Y.; Coupland C.; Hill T.; Hippisley-Cox J. Risks and benefits of direct oral anticoagulants versus warfarin in a real world setting: cohort study in primary care. BMJ. 2018, 362, k2505. 10.1136/bmj.k2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroi M.; Jung S. M. Platelet glycoprotein VI: its structure and function. Thromb. Res. 2004, 114, 221–233. 10.1016/j.thromres.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Nieswandt B.; Schulte V.; Bergmeier W.; Mokhtari-Nejad R.; Rackebrandt K.; Cazenave J. P.; Ohlmann P.; Gachet C.; Zirngibl H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 2001, 193, 459–469. 10.1084/jem.193.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachel C.; Mathes D.; Arias-Loza A. P.; Heitzmann W.; Nordbeck P.; Deppermann C.; Lorenz V.; Hofmann U.; Nieswandt B.; Frantz S. Inhibition of platelet GPVI protects against myocardial ischemia-reperfusion injury. Arterioscler., Thromb., Vasc. Biol. 2016, 36, 629–635. 10.1161/ATVBAHA.115.305873. [DOI] [PubMed] [Google Scholar]
- Lockyer S.; Okuyama K.; Begum S.; Le S.; Sun B.; Watanabe T.; Matsumoto Y.; Yoshitake M.; Kambayashi J.; Tandon N. N. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb. Res. 2006, 118, 371–380. 10.1016/j.thromres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Kato K.; Kanaji T.; Russell S.; Kunicki T. J.; Furihata K.; Kanaji S.; Marchese P.; Reininger A.; Ruggeri Z. M.; Ware J. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood 2003, 102, 1701–1707. 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- Goebel S.; Li Z.; Vogelmann J.; Holthoff H. P.; Degen H.; Hermann D. M.; Gawaz M.; Ungerer M.; Münch G. The GPVI - Fc fusion protein revacept improves cerebral infarct volume and functional outcome in stroke. PLoS One 2013, 8, e66960. 10.1371/journal.pone.0066960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschnitz C.; Pozgajova M.; Pham M.; Bendszus M.; Nieswandt B.; Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007, 115, 2323–2330. 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- Arthur J. F.; Dunkley S.; Andrews R. K. Platelet glycoprotein VI-related clinical defects. Br. J. Haematol. 2007, 139, 363–372. 10.1111/j.1365-2141.2007.06799.x. [DOI] [PubMed] [Google Scholar]
- Lebozec K.; Jandrot-Perrus M.; Avenard G.; Favre-Bulle O.; Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. MAbs 2017, 9, 945–958. 10.1080/19420862.2017.1336592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voors-Pette C.; Lebozec K.; Dogterom P.; Jullien L.; Billiald P.; Ferlan P.; Renaud L.; Favre-Bulle O.; Avenard G.; Machacek M.; Plétan Y.; Jandrot-Perrus M. Safety and tolerability, pharmacokinetics, and pharmacodynamics of ACT017, an antiplatelet GPVI (glycoprotein VI) Fab. Arterioscler., Thromb., Vasc. Biol. 2019, 39, 956–964. 10.1161/ATVBAHA.118.312314. [DOI] [PubMed] [Google Scholar]
- Ungerer M.; Rosport K.; Bültmann A.; Piechatzek R.; Uhland K.; Schlieper P.; Gawaz M.; Münch G. Novel antiplatelet drug revacept (dimeric glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 2011, 123, 1891–1899. 10.1161/CIRCULATIONAHA.110.980623. [DOI] [PubMed] [Google Scholar]
- Acute Ischemic Stroke Interventional Study. ClinicalTrials.gov; NIH U.S. National Library of Medicine, 2021; https://clinicaltrials.gov/ct2/show/NCT03803007 (accessed 2020-04-17).
- Intracoronary Stenting and Antithrombotic Regimen: Lesion Platelet Adhesion As Selective Target of Endovenous Revacept ClinicalTrials.gov; NIH U.S. National Library of Medicine, 2020; https://clinicaltrials.gov/ct2/show/NCT03312855 (accessed 2019-06-14).
- Revacept in Symptomatic Carotid Stenosis (Revacept/CS/02). ClinicalTrials.gov; NIH U.S. National Library of Medicine. 2021; https://www.clinicaltrials.gov/ct2/show/NCT01645306 (accessed 2021-09-04).
- Foster H.; Wilson C.; Philippou H.; Foster R. Progress towards a glycoprotein VI modulator for the treatment of thrombosis. J. Med. Chem. 2020, 63, 12213–12242. 10.1021/acs.jmedchem.0c00262. [DOI] [PubMed] [Google Scholar]
- Lee T.-Y.; Chang C.-C.; Lu W.-J.; Yen T.-L.; Lin K.-H.; Geraldine P.; Li J.-Y.; Sheu J.-R. Honokiol as a specific collagen receptor glycoprotein VI antagonist on human platelets: functional ex vivo and in vivo studies. Sci. Rep. 2017, 7, 40002. 10.1038/srep40002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Tang X.; Yi W.; Li Q.; Ren L.; Liu X.; Chu C.; Ozaki Y.; Zhang J.; Zhu L. Glaucocalyxin A inhibits platelet activation and thrombus formation preferentially via GPVI signaling pathway. PLoS One 2013, 8, e85120. 10.1371/journal.pone.0085120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L.; Vasudevan S. R.; Jones C. I.; Gibbins J. M.; Churchill G. C.; Campbell R. D.; Coxon C. H. Discovery of novel GPVI receptor antagonists by structure-based repurposing. PLoS One 2014, 9, e101209. 10.1371/journal.pone.0101209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Ueda H.; Yoshizawa Y.; Akazawa D.; Tanimura R.; Shimada I.; Takahashi H. Structural basis for platelet antiaggregation by angiotensin II type 1 receptor antagonist losartan (DuP-753) via glycoprotein VI. J. Med. Chem. 2010, 53, 2087–2093. 10.1021/jm901534d. [DOI] [PubMed] [Google Scholar]
- Bhunia S. S.; Misra A.; Khan I. A.; Gaur S.; Jain M.; Singh S.; Saxena A.; Hohlfield T.; Dikshit M.; Saxena A. K. Novel glycoprotein VI antagonists as antithrombotics: synthesis, biological evaluation, and molecular modeling studies on 2,3- disubstituted tetrahydropyrido(3,4-b)indoles. J. Med. Chem. 2017, 60, 322–337. 10.1021/acs.jmedchem.6b01360. [DOI] [PubMed] [Google Scholar]
- Best D.; Senis Y. A.; Jarvis G. E.; Eagleton H. J.; Roberts D. J.; Saito T.; Jung S. M.; Moroi M.; Harrison P.; Green F. R.; Watson S. P. GPVI levels in platelets: relationship to platelet function at high shear. Blood 2003, 102, 2811–2818. 10.1182/blood-2003-01-0231. [DOI] [PubMed] [Google Scholar]
- Damaskinaki F. N.; Moran L. A.; Garcia A.; Kellam B.; Watson S. P. Overcoming challenges in developing small molecule inhibitors for GPVI and CLEC-2. Platelets 2021, 32, 744–752. 10.1080/09537104.2020.1863939. [DOI] [PubMed] [Google Scholar]
- Hsiao G.; Lee J. J.; Lin K. H.; Shen C. H.; Fong T. H.; Chou D. S.; Sheu J. R. Characterization of a novel and potent collagen antagonist, caffeic acid phenethyl ester, in human platelets: in vitro and in vivo studies. Cardiovasc. Res. 2007, 75, 782–792. 10.1016/j.cardiores.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Anil Kumar K. S.; Misra A.; Siddiqi T. I.; Srivastava S.; Jain M.; Bhatta R. S.; Barthwal M.; Dikshit M.; Dikshit D. K. Synthesis and identification of chiral aminomethylpiperidine carboxamides as inhibitor of collagen induced platelet activation. Eur. J. Med. Chem. 2014, 81, 456–472. 10.1016/j.ejmech.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Lu W.-J.; Wu M.-P.; Lin K.-H.; Lin Y.-C.; Chou H.-C.; Sheu J.-R. Hinokitiol is a novel glycoprotein VI antagonist on human platelets. Platelets 2014, 25, 595–602. 10.3109/09537104.2013.863856. [DOI] [PubMed] [Google Scholar]
- Bhunia S. S.; Saxena A. K. Molecular modelling studies in explaining the higher GPVI antagonistic activity of the racemic 2-(4-methoxyphenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide than its enantiomers. SAR QSAR Environ. Res. 2017, 28, 783–799. 10.1080/1062936X.2017.1396247. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D.; Braun A.; Nieswandt B. Calcium signaling in platelets. J. Thromb. Haemostasis 2009, 7, 1057–1066. 10.1111/j.1538-7836.2009.03455.x. [DOI] [PubMed] [Google Scholar]
- Estevez B.; Du X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. 10.1152/physiol.00020.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S. H.; Sim E. H.; Goh R. Y.; Park J. I.; Han J. Y. Platelet activation: the mechanisms and potential biomarkers. BioMed Res. Int. 2016, 2016, 9060143. 10.1155/2016/9060143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek-Willemsen M.; André T.; Wanner R.; Roth H. M.; Duhr S.; Baaske P.; Breitsprecher D. Microscale thermophoresis: interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. 10.1016/j.molstruc.2014.03.009. [DOI] [Google Scholar]
- Slater A.; Di Y.; Clark J. C.; Jooss N. J.; Martin E. M.; Alenazy F. O.; Thomas M. R.; Ariëns R. A.; Herr A. B.; Poulter N. S.; Emsley J.; Watson S. P. Structural characterization of a novel GPVI-nanobody complex reveals a biologically active domain-swapped GPVI dimer. Blood 2021, 137, 3443–3453. 10.1182/blood.2020009440. [DOI] [PubMed] [Google Scholar]
- Nieswandt B.; Watson S. P. Platelet-collagen interaction: is GPVI the central receptor?. Blood 2003, 102, 449–461. 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- Stalker T. J.; Newman D. K.; Ma P.; Wannemacher K. M.; Brass L. F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 210, 59–85. 10.1007/978-3-642-29423-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onselaer M. B.; Hardy A. T.; Wilson C.; Sanchez X.; Babar A. K.; Miller J. L. C.; Watson C. N.; Watson S. K.; Bonna A.; Philippou H.; Herr A. B.; Mezzano D.; Ariëns R. A. S.; Watson S. P. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. 2017, 1, 1495–1504. 10.1182/bloodadvances.2017007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Yao J.; Abildgaard F.; Dyson H. J.; Oldfield E.; Markley J. L.; Sykes B. D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Onselaer M. B.; Nagy M.; Pallini C.; Pike J. A.; Perrella G.; Quintanilla L. G.; Eble J. A.; Poulter N. S.; Heemskerk J. W. M.; Watson S. P. Comparison of the GPVI inhibitors losartan and honokiol. Platelets 2020, 31, 187–197. 10.1080/09537104.2019.1585526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badolia R.; Inamdar V.; Manne B. K.; Dangelmaier C.; Eble J. A.; Kunapuli S. P. Gq pathway regulates proximal C-type lectin-like receptor-2 (CLEC-2) signaling in platelets. J. Biol. Chem. 2017, 292, 14516–14531. 10.1074/jbc.M117.791012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.