

Abstract

The trypanosome alternative oxidase (TAO), a mitochondrial enzyme involved in the respiration of the bloodstream form trypomastigotes of Trypanosoma brucei, is a validated drug target against African trypanosomes. Earlier series of TAO inhibitors having a 2,4-dihydroxy-6-methylbenzoic acid scaffold (“head”) and a triphenylphosphonium or quinolin-1-ium cation as a mitochondrion-targeting group (“tail”) were shown to be nanomolar inhibitors in enzymatic and cellular assays. We investigated here the effect of different mitochondrion-targeting cations and other scaffold modifications on the in vitro activity of this class of inhibitors. Low micromolar range activities were obtained, and the structure–activity relationship studies showed that modulation of the tail region with polar substituents is generally detrimental to the enzymatic and cellular activity of TAO inhibitors.
Keywords: Trypanosome alternative oxidase inhibitor, Trypanosoma brucei, benzamidine, imidazoline, glycolysis
African trypanosomes (Trypanosoma brucei sp.) are protozoan parasites that cause sleeping sickness (human African trypanosomiasis, HAT) in sub-Saharan Africa. Bloodstream form (BSF) trypomastigotes of T. brucei possess a unique energy metabolism as they only depend on glycolysis for energy supply.1,2 In the absence of a functional oxidative phosphorylation pathway, they use the trypanosome alternative oxidase (TAO) to reoxidize the NADPH that is formed during glycolysis.3 TAO is essential for the respiration of BSF trypomastigotes,3 is conserved among trypanosome subspecies,4 has no counterpart in mammalian cells, and has been validated as a drug target in trypanosomes.5
The localization of TAO at the interface of the inner mitochondrial membrane6,7 has inspired the development of potent 4-hydroxybenzoate- and 4-alkoxybenzaldehyde-based inhibitors that hold a lipophilic cation as the mitochondrion-targeting moiety.8−10 In particular, 2,4-dihydroxy-6-methylbenzoate derivatives were nanomolar range TAO inhibitors showing in vitro and in vivo trypanocidal activity in a mouse model of T. b. rhodesiense infection (Chart 1A).9 Mitochondrial localization of this class of inhibitors was confirmed by live-cell imaging with fluorescent analogues.11
Chart 1. (A) Example of Previously Reported Benzoate TAO Inhibitors with a Triphenylphosphonium Mitochondrion-Targeting Cation9 and (B) Structural Modifications Studied in This Work.

In the current study, new analogues of the benzoate lead compound were synthesized to extend the structure–activity relationship (SAR) of this class of TAO inhibitors. The first modification was the replacement of the ester bond by a more metabolically stable amide bond. Grady et al.12 showed that this structural modification produced inhibitors that were more soluble and more stable to serum hydrolases in vivo than the benzoate counterparts.5 Second, different cationic groups were tested in place of the bulky triphenylphosphonium (TPP+) and quinolin-1-ium cations that were used in the previous series, in which positive charge is highly delocalized.8,9
We observed previously, in model structures of TPP+-linked inhibitors binding to TAO, that the methylene linker (tail) engaged in hydrophobic interactions with the hydrophobic region of the enzyme cavity, whereas the large TPP cation extended outward into the solvent.9 In the present work, we tested the benzamidinium (1a–d), 2-phenylimidazolin-3-ium (2c), and 2-(phenylamino)imidazolin-3-ium (3c) cations as less bulky surrogates of TPP+ (Chart 1). Compounds containing these cationic groups, which are found in many trypanocidal drugs (e.g., pentamidine, diminazene) and investigational compounds, are known to strongly accumulate in the mitochondrial matrix of trypanosomes, against considerable concentration gradients.13−19 We hypothesized that smaller cations would insert themselves deeper into the enzyme cavity to promote favorable interactions of the 2,4-dihydroxy-6-methylbenzoic head with the enzyme active site. With the previous 4-hydroxybenzoate series, a methylene linker of less than C-14 between the TPP or quinolin-1-ium cation and the head region was detrimental to TAO inhibition.9,10 However, the imidazoline- and benzamidine-based cations used in this study are structurally different (i.e., shape, size, and electronic properties) to these cations and may present a different SAR. Hence, a methylene linker covering a wide range of lengths between the 2,4-dihydroxy-6-methylbenzoic scaffold and the cationic group were tested ((CH2)n, n = 3, 6, 12, 14). All of the compounds were assayed against recombinant TAO enzyme and wild-type (WT) and drug-resistant T. b. brucei strains.
Results and Discussion
The benzamidine derivatives with methylene linkers of 3, 6, 12, and 14 units (1a–d) were synthesized in five steps from the corresponding N-(n-bromoalkyl)phthalimide 4a–d (Scheme 1).
Scheme 1. Synthesis of Benzamidine Derivatives 1a–d.

Reagents and conditions. (i) PhtN–K+, DMF, rt, 20 h; (ii) PhtNH, Ph3P, DIAD, THF, 0 °C then rt, 20 h; (iii) K2CO3, CH3CN, 80 °C, 24 h; (iv) N2H4·H2O, EtOH, 80 °C, 12 h; (v) for 7a, 7c, and 7d, EDC·HCl, DMAP, CH3CN, 80 °C, 20 h; for 7b, PyBOP, DIPEA, DMF, rt, 18 h; (vi) NH2OH·HCl, tBuOK, DMSO, rt, 4 days; (vii) (1) Ac2O, AcOH, 15 min, (2) H2, 5% Pd–C, AcOH, rt, 12 h.
Compound 4a was commercially available, whereas 4b–d were synthesized, as shown in Scheme 1. A reaction of potassium phthalimide with an excess of 1,6-dibromohexane yielded 4b. Compounds 4c and 4d were obtained in good yields from phthalimide and 12-bromododecan-1-ol20 or 14-bromotetradecan-1-ol8 using the Mitsunobu protocol. Reaction of 4a–d with 4-cyanophenol and K2CO3 generated ethers 5a–d, which were converted to amines 6a–d using hydrazine monohydrate. The coupling of amines 6a–d with orsellinic acid was achieved with EDC hydrochloride and a catalytic amount of DMAP to give 7a, 7c, and 7d in low to moderate yields (15–40%). For the synthesis of 7a and 7b, another coupling agent (PyBOP) was tried, but no improvement of yield was observed (14 and 26%, respectively). Of note, the yield of this amide coupling seemed to decrease with the methylene chain length of the amine, reflecting more complex reaction crudes and, possibly, solubility issues (e.g., amine 6d was only soluble in hot acetonitrile).
Benzamidine synthesis was achieved in a two-step process involving the formation of benzamidoximes 8a–d followed by the catalytic hydrogenation of intermediate benzamidoximes in acetic acid/acetic anhydride to yield 1a–d.21 Imidazoline derivative 2c was synthesized in good yield (76%) by reaction of the cyano derivative 7c with ethylenediamine/P2S5 at 120 °C in a sealed tube (Scheme 2). The 2-aminoimidazoline analogue 3c was obtained in two steps from the amino precursor 12c using di-tert-butyl 2-thioxoimidazolidine-1,3-dicarboxylate (13) following a known protocol.13,22 Compound 12c was synthesized in four steps from 4c and 4-nitrophenol following the same route as described for the synthesis of 7a–d (Scheme 2).
Scheme 2. Synthesis of Imidazoline (2c) and 2-Aminoimidazoline (3c) Derivatives.

Reagents and conditions: (i) P2S5, 1,2-ethylenediamine, sealed tube, 120 °C, 2 h; (ii) K2CO3, CH3CN, 80 °C; (iii) N2H4·H2O, EtOH, 80 °C; (iv) EDC·HCl, DMAP, CH3CN, CH2Cl2, 80 °C; (v) H2, MeOH, Pd–C 5%; (vi) HgCl2, Et3N, DMF, 0 °C to rt; (vii) CH2Cl2, TFA, 0 °C.
We sought to understand the role of the amide bond in the binding to the TAO active site. To do so, we tried to prepare the “keto” analogue of 1d, with a carbonyl bond linking the methylene chain to the 5-methylresorcinol scaffold instead of an amide bond (Scheme 3a). 16-(4-Cyanophenoxy)hexadecanoic acid 18 was synthesized in three steps from 16-hydroxyhexadecanoic acid 15. Friedel–Crafts acylation of 5-methylresorcinol with 18 using AlCl3 gave a 23:20:57 mixture of three isomers 19/20/21 as detected by HPLC-MS. Compounds 20 and 21 were isolated (18 and 17% yield, respectively) and characterized by 1H and 13C NMR. However, we were unable to isolate the desired isomer 19 from the mixture due to very similar chromatographic behavior with 20 and 21. Attempts at the synthesis with a different Lewis acid (i.e., BF3–Et2O) led to the formation of more complex reaction mixtures. As an alternative route, the reaction of 2,4-dihydroxy-6-methylbenzaldehyde 22 with the Grignard reagent of 23 was tried several times using different conditions but without success (Scheme 3b). Hence, attempts to obtain sufficiently pure 19 were dropped. Nevertheless, the biological activity of intermediates 20 and 21, useful for SAR studies, is reported in Table 1.
Scheme 3. (a) Synthesis of Derivatives 19–21 and (b) Attempted Route toward Compound 19.
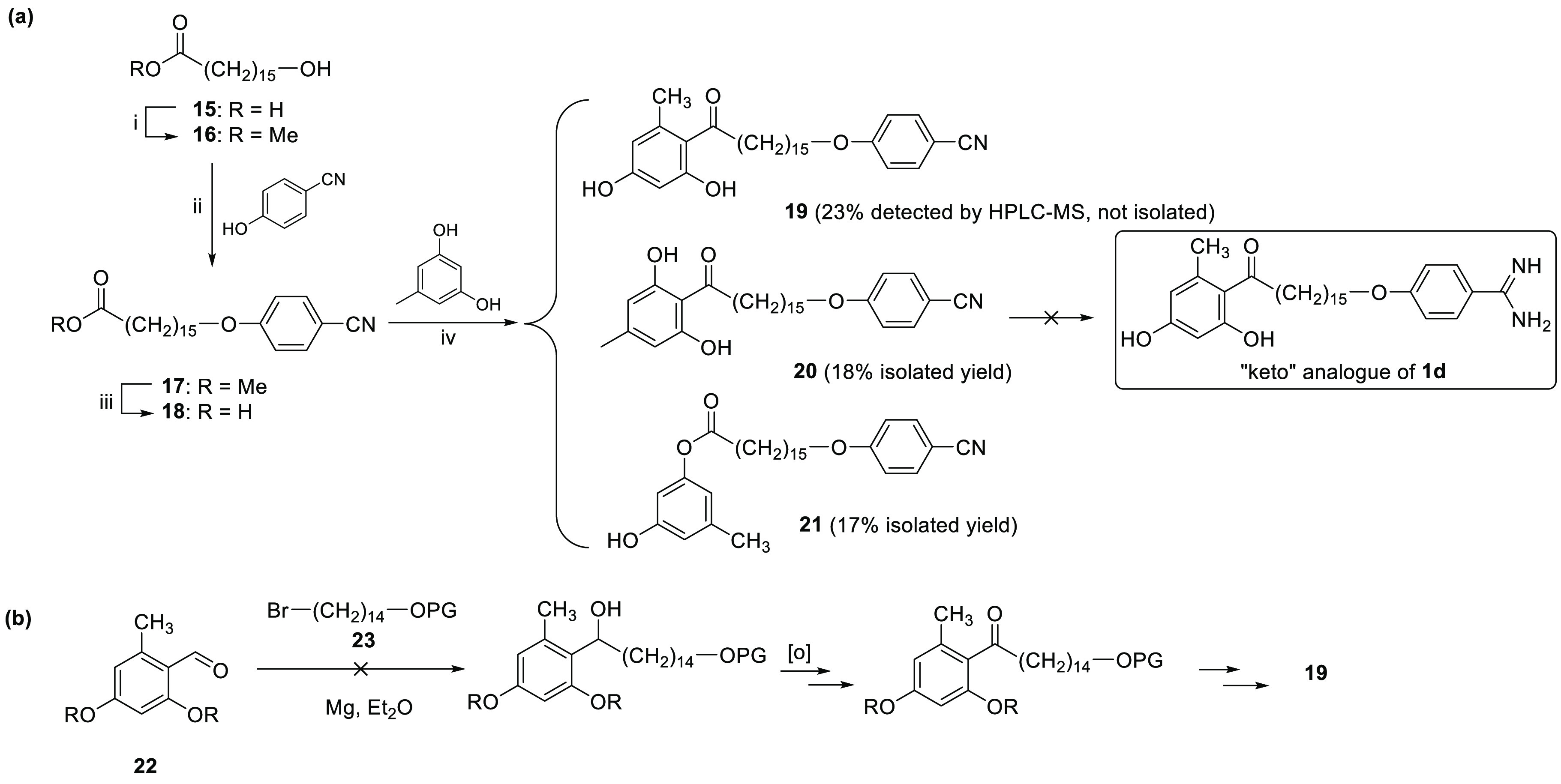
Reagents and conditions: (i) MeOH, TsOH–H2O, rt, 21 h, (96%); (ii) PPh3, DIAD, THF, 0 °C to rt, 4 days (58%); (iii) LiOH·H2O, THF/MeOH/H2O (2/1/1), rt (94%); (iv) AlCl3, 1,2-dichloroethane, 100 °C, 24 h.
Table 1. In Vitro Activity of Amidines (1a–d), Hydroxyamidines (8a–d), Imidazolines (2c, 3c, and 14c), and Synthetic Intermediates (7a–d, 11c, 12c, 20, and 21).
|
T. b. brucei |
cytotoxicity |
rTAOg % inhibition at 40 μM | rTAO | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) |
CC50 (μM) |
IC50 (μM) | ||||||||
| cmpd | n | WTa | B48b | RFc | AQP1-3 KOd | RFc | HEKe | SIf | ||
| 1a | 3 | >100 | >100 | nd | >200 | 48% | >40j | |||
| 1b | 6 | 15.5 ± 0.6 | 28.0 ± 0.5 | 1.8 | 17.4 ± 0.9 | 1.1 | >200 | >12.9 | 12.5% | >40 |
| 1c | 12 | 3.3 ± 0.2 | 3.6 ± 0.1 | 1.1 | nd | 43.5 ± 5.4 | 13.2 | 39.3% | >40 | |
| 1d | 14 | 18.6 ± 1.1 | 28.9 ± 5.2 | 1.6 | nd | >200 | 10.7 | 31.4% | >40 | |
| 2c | 12 | 1.7 ± 0.3 | nd | 2.9 ± 0.4 | 1.7 | 67.2 ± 0.8h | 39.1 | 10.0%i | >10 | |
| 3c | 12 | 2.7 ± 0.4 | nd | 3.92 ± 0.03 | 1.4 | >100h | >36.5 | 46.3%i | 22.5 ± 0.3 | |
| 7a | 3 | 37.2 ± 3.4 | 50.8 ± 2.7 | 1.4 | nd | >200 | >5.3 | 28.8% | >40 | |
| 7b | 6 | 30.4 ± 0.4 | 27.7 ± 2.0 | 0.9 | 30.5 ± 0.7 | 1.0 | >200 | 6.6 | 96.2% | 1.5 ± 0.1 |
| 7c | 12 | 15.6 ± 0.7 | 15.9 ± 0.7 | 1.0 | nd | 57.1 ± 0.1 | 3.7 | 27.4% | >40 | |
| 7d | 14 | 14.8 ± 0.5 | 15.7 ± 0.5 | 1.1 | nd | 56.8 ± 0.2 | 3.8 | 89.7% | 16.4 ± 0.7 | |
| 8a | 3 | >100 | >100 | nd | >200 | 14.1 | >40 | |||
| 8b | 6 | 19.5 ± 1.0 | 36.0 ± 1.8 | 1.9 | 21 ± 1 | 1.1 | 76.8 ± 7.2 | 3.9 | –14.9% | >40 |
| 8c | 12 | 8.4 ± 1.1 | 9.1 ± 0.5 | 1.1 | 108.9 ± 1.6 | 13 | 44.8 | >40 | ||
| 8d | 14 | 29.0 ± 2.2 | 27.1 ± 2.4 | 0.9 | nd | >200 | 6.9 | –3.4% | >40 | |
| 11c | 12 | 9.5 ± 0.5 | nd | 10.2 ± 0.7 | 1.1 | >100h | >10.5 | 53.8%i | 30.0 ± 1.5 | |
| 12c | 12 | 20.3 ± 1.3 | nd | 29.6 ± 2.2 | 1.5 | >100h | >4.9 | nd | nd | |
| 14c | 12 | 10.4 ± 0.7 | nd | 15.7 ± 0.3 | 1.5 | >100h | >9.6 | nd | nd | |
| 20 | 15 | 5.8 ± 0.7 | nd | 8.2 ± 0.9 | 1.4 | >100h | >17.1 | 13.4%i | >10 | |
| 21 | 15 | >100 | nd | >100 | >100h | 30.0%i | >10 | |||
| diminazene | 0.095 ± 0.011 | 0.107 ± 0.019 | 1.1 | |||||||
| pentamidine | 0.004 ± 0.001 | 0.208 ± 0.021 | 49.5 | 0.046 ± 0.003 | 10.8 | |||||
| phenylarsine oxide | 0.9 ± 0.1 | |||||||||
| ascofuranone | 100% | |||||||||
Bloodstream form trypomastigotes of T. b. brucei strain 427 (n = 3).
T. b. brucei strain resistant to pentamidine (n = 3).
Resistance factor relative to WT.
T. brucei cell line from which all aquaporins were knocked out (n = 3).
Cytotoxicity on human embryonic kidney cells (n = 3).
Selectivity index (SI) = CC50/EC50 (WT).
Purified recombinant trypanosome alternative oxidase (ΔMTS-TAO)9 from T. b. brucei (n = 3); compound concentration = 40 μM.
n = 2.
Compound tested at 10 μM concentration.
No reliable IC50 could be obtained for inhibitors with less than 40% single-point inhibition as a sigmoidal curve could not be generated.
Biology
The trypanocidal activity of compounds 1a–d, 2c, 3c, 7a–d, and 8a–d and synthetic intermediates 11c, 12c, 14c, 20, and 21 against wild-type (s427) and drug-resistant strains of T. b. brucei (i.e., B48, AQP1-3 KO) was determined in vitro using a resazurin-based assay.8,9 In general, a methylene linker of 12 carbons gave the lowest EC50 values against T. brucei (compare 1a–d/ 7a–d/ 8a–d). Target compounds 1c, 2c, and 3c were the most effective compounds of the series with EC50 values of <4 μM against T. brucei (Table 1). Among them, the 2-phenylimidazolin-3-ium derivative 2c was marginally more active with EC50 = 1.72 μM. This finding was in agreement with previous reports on TAO inhibitors showing that a decrease in efficacy against T. b. brucei growth inhibition was observed as chain length decreased.8,9,23 Hence, compounds with short linkers (C-3) were poorly active (7a) or inactive (1a, 8a) against T. brucei. Substituents in the para position of the phenoxy group affected the trypanocidal activity in the following order: 2-phenylimidazolin-3-ium (2c) > 2-(phenylamino)imidazolin-3-ium (3c) ≈ benzamidinium (1c) > N-hydroxyamidine (8c) ≈ 4-NO2 (11c).
Apparently, the effect of changing the amide connecting group in 7d with a keto bond (20) was favorable for anti-T. brucei activity, as shown by the 2.5-fold lower EC50 of 20 (5.8 μM) versus 7d (14.8 μM). However, because 7d and 20 are slightly different isomers (2,4-dihydroxy-6-methyl and 2,6-dihydroxy-4-methyl, respectively), an isomer-dependent effect cannot be ruled out. Wild-type and drug-resistant strains showed virtually the same susceptibility toward these compounds (within 2-fold difference), indicating that the compounds, unlike some other benzamidines such as pentamidine,24 are not dependent on aquaporins, or on the aminopurine transporter TbAT1, for uptake by T. brucei. Cytotoxicity against HEK cells was low, resulting in selectivity indexes of >10 for 1b–d to >36.5 for 2c and 3c.
The compounds were screened at a single concentration (either 10 or 40 μM) as inhibitors of the ubiquinol oxidase activity of purified ΔMTS-TAO. The IC50 values of the compounds displaying the best percentage of inhibition were also determined (Table 1).9 In general, the benzamide derivatives reported here were poor TAO inhibitors, with IC50 values in the micromolar range compared to the nanomolar range inhibitors reported previously.8,9 The best inhibitors were the uncharged 4-cyanophenoxy analogues 7b (IC50 = 1.52 μM, C-6 methylene linker) > 7d (IC50 = 16.4 μM, C-14 methylene linker). More polar substituents in the para position of the phenoxy group such as 2-(phenylamino)imidazolin-3-ium (3c) or 4-NO2 (11c) gave less potent inhibitors (IC50 = 22.5 and 30 μM, respectively). These SAR results regarding TAO activity were consistent with previous work showing that the introduction of polar substituents in the tail region of TAO inhibitors is not well-tolerated, leading to a strong decrease in inhibitory potency.23 This effect seems to be counterbalanced when lipophilic cations such as TPP+ or quinolin-1-ium are used, but in that case, the linker length in the tail region must be long enough (≥C-14) to allow the bulky TPP cation to remain outside the enzyme active site, giving rise to low nanomolar TAO inhibitors.5,9 For the benzamidine-based TAO inhibitors 1a–d, a linkage of 14 methylene units did not improve TAO inhibition versus the C-12 linker, as opposed to the previous series having a quinolinium or TPP cations.9 In that case, the aromatic moieties of TPP and quinolinium cations interact with the surface of the enzyme, and the linker length must give the flexibility to the aromatic ring to orient itself optimally. Apparently, such interactions may not happen for the benzamidine compounds reported here.
Unfortunately, our efforts to isolate pure keto analogue 19, which would have informed about the effect of the amide bond on TAO inhibitory activity, were unsuccessful. However, the lack of TAO inhibition by the structurally close analogue 20 seems to indicate that the keto connection is not substantially superior to the amide linkage.
A positive correlation between clogP and the cellular activity against T. brucei was observed for the cationic derivatives 1a–d, 2c, 3c, and 8a–d (Figure 1a) and the noncationic derivatives 7a–d, 11c, 12c, 14c, and 20 (Figure 1b), although this was disconnected from inhibition of purified rTAO. A similar trend was observed by West and co-workers in a series of noncationic TAO inhibitors structurally related to ascofuranone. However, in this case, clogP also correlated with TAO inhibition.23 The positive effect of compound lipophilicity on the efficacy against T. brucei of the derivatives possibly reflects an increase in the permeability of the compounds through the cell and/or mitochondrial membranes, in agreement with previous studies on mitochondrion-targeted antiparasitic compounds.8−10 As reported previously, the accumulation of cationic compounds (e.g., 1a–d, 2c, 3c, and 8a–d) in the T. brucei mitochondrion is expected to affect the mitochondrial membrane potential Ψm by disruption of mitochondrial functions involved in maintaining the ion gradients.11,25 Hence, the absence of correlation between rTAO inhibition and T. brucei growth is probably the result of several factors including activity against multiple targets.
Figure 1.

Correlations of cLogP versus T. b. brucei pIC50 growth inhibition for (a) cationic and (b) noncationic derivatives.
To conclude, this study showed that the replacement of TPP+ or quinolin-1-ium groups with imidazoline- and benzamidine-based mitochondrion-targeting cations was detrimental to the enzymatic and cellular activity of TAO inhibitors compared with previous series having the same 2,4-dihydroxy-6-methylbenzoic acid head. The comparatively weak micromolar range activity against TAO of these compounds illustrates the difficulty of modulating the tail region of TAO inhibitors with polar substituents without losing efficacy. Nevertheless, the 2-(phenylamino)imidazolin-3-ium group (3c) provided an inhibitor that was active against TAO and T. brucei in the low micromolar range, with adequate selectivity versus mammalian HEK cells.
Experimental Section
T. brucei Susceptibility Assays
BSF trypanosomes of monomorphic strains Lister 427 (WT), multi-drug-resistant clone B48, and the AQP1-3 KO, which lacks all aquaglyceroporins,26 were grown in complete HMI-9 with 10% fetal bovine serum, exactly as described, and tested using a standard resazurin-based assay with 23 doubling dilutions for each compound starting at 100 μM.27 Human embryonic kidney (HEK) cells were cultured and assayed with a resazurin-based assay exactly as described previously.9 EC50 values were calculated by nonlinear regression with an equation for a sigmoid curve with variable slope (Prism 8.0, GraphPad).
Inhibition of rTAO
The test compounds were assayed as inhibitors of the ubiquinol oxidase activity of purified ΔMTS-TAO by recording the absorbance change of ubiquin-1-ol at 278 nm exactly as previously described.9 Briefly, determination of ΔMTS-TAO activity was performed on a V-630 Jasco UV–vis spectrophotometer (Jasco Corporation, Tokyo, Japan) by measuring the change in absorbance of the substrate ubiquinol (ε278 = 15,000 M–1 cm–1) at 278 nm over a period of 2 min in a 1 cm cuvette. The recombinant enzyme was preincubated for 2 min in a 50 mM Tris-HCl (pH 7.4) buffer containing the detergent octaethylene glycol monododecylether (0.05% (w/v)) in a total reaction volume of 1 mL at 25 °C. Reactions were initiated by the addition of ubiquinol to the cuvette. The inhibition reaction assay was performed by preincubating a fixed amount of rTAO with varying amounts of the inhibitor for 2 min in the same buffer before adding the substrate. Ascofuranone was used as positive control whereas DMSO was used as negative control. Control experiments were also carried out throughout the experiment to verify that there was no autoxidation of ubiquinol in the medium. Residual activities were plotted against the corresponding inhibitor concentration to generate the IC50 value using GraphPad Prism.
Acknowledgments
D.C. was a recipient of a JAE-intro fellowship financed by CSIC, JAE program.
Glossary
Abbreviations
- BSF trypanosome
bloodstream form trypanosome
- DIAD
diisopropylazodicarboxylate
- DMAP
4-dimethylaminopyridine
- HAT
human African trypanosomiasis
- MTS
mitochondrion-targeting sequence
- NADPH
nicotinamide adenine dinucleotide phosphate
- PyBOP
benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- RF
resistance factor
- TAO
trypanosome alternative oxidase
- TPP
triphenylphosphonium
- WT
wild-type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00717.
Synthesis and NMR spectra of all the target compounds 1a–d, 2c, 3c, 20, and 21 (PDF)
Author Present Address
⊥ Centro de Astrobiología (CAB), Instituto Nacional de Técnica Aeroespacial (INTA), Torrejón de Ardoz, Madrid, Spain
This work was funded by Ministerio de Ciencia, Innovación/Agencia Estatal de Investigación (MCIN/AEI/10.13039/501100011033), through the project RTI2018-093940-B-I00 (co-funded by European Regional Development Fund, ERDF, “A way to build Europe”). This research was also supported by a fellowship grant from the Japan Society for the promotion of Science (JSPS Grant No. 17F17420) to G.U.E. M.M.A. and H.A.A.E. were funded by studentships from the Libyan government. M.A.U. was funded by the Petroleum Technology Development Fund of Nigeria.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters virtual special issue “Medicinal Chemistry in Portugal and Spain: A Strong Iberian Alliance”.
Supplementary Material
References
- Grant P. T.; Sargent J. R. L-alpha-Glycerophosphate dehydrogenase, a component of an oxidase system in Trypanosoma rhodesiense. Biochem. J. 1961, 81, 206–14. 10.1042/bj0810206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P. T.; Sargent J. R. Properties of L-alpha-glycerophosphate oxidase and its role in the respiration of Trypanosoma rhodesiense. Biochem. J. 1960, 76, 229–37. 10.1042/bj0760229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson A. B. Jr.; Bienen E. J.; Pollakis G.; Grady R. W. Respiration of bloodstream forms of the parasite Trypanosoma brucei brucei is dependent on a plant-like alternative oxidase. J. Biol. Chem. 1989, 264, 17770–17776. 10.1016/S0021-9258(19)84639-2. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Fujioka S.; Fukumoto S.; Inoue N.; Sakamoto K.; Hirata H.; Kido Y.; Yabu Y.; Suzuki T.; Watanabe Y.-i.; Saimoto H.; Akiyama H.; Kita K. Trypanosome alternative oxidase, a potential therapeutic target for sleeping sickness, is conserved among Trypanosoma brucei subspecies. Parasitol. Int. 2010, 59, 560–564. 10.1016/j.parint.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Ebiloma G. U.; Balogun E. O.; Cueto-Díaz E. J.; de Koning H. P.; Dardonville C. Alternative oxidase inhibitors: Mitochondrion-targeting as a strategy for new drugs against pathogenic parasites and fungi. Med. Res. Rev. 2019, 39, 1553–1602. 10.1002/med.21560. [DOI] [PubMed] [Google Scholar]
- Menzies S. K.; Tulloch L. B.; Florence G. J.; Smith T. K. The trypanosome alternative oxidase: a potential drug target?. Parasitology 2018, 145, 175–183. 10.1017/S0031182016002109. [DOI] [PubMed] [Google Scholar]
- Shiba T.; Kido Y.; Sakamoto K.; Inaoka D. K.; Tsuge C.; Tatsumi R.; Takahashi G.; Balogun E. O.; Nara T.; Aoki T.; Honma T.; Tanaka A.; Inoue M.; Matsuoka S.; Saimoto H.; Moore A. L.; Harada S.; Kita K. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Nat. Acad. Sci. 2013, 110, 4580–4585. 10.1073/pnas.1218386110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meco-Navas A.; Ebiloma G. U.; Martín-Domínguez A.; Martínez-Benayas I.; Cueto-Díaz E. J.; Alhejely A. S.; Balogun E. O.; Saito M.; Matsui M.; Arai N.; Shiba T.; Harada S.; de Koning H. P.; Dardonville C. SAR of 4-Alkoxybenzoic Acid Inhibitors of the Trypanosome Alternative Oxidase. ACS Med. Chem. Lett. 2018, 9, 923–928. 10.1021/acsmedchemlett.8b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebiloma G. U.; Díaz Ayuga T.; Balogun E. O.; Abad Gil L.; Donachie A.; Kaiser M.; Herraiz T.; Inaoka D. K.; Shiba T.; Harada S.; Kita K.; de Koning H. P.; Dardonville C. Inhibition of trypanosome alternative oxidase without its N-terminal mitochondrial targeting signal (ΔMTS-TAO) by cationic and non-cationic 4-hydroxybenzoate and 4-alkoxybenzaldehyde derivatives active against T. brucei and T. congolense. Eur. J. Med. Chem. 2018, 150, 385–402. 10.1016/j.ejmech.2018.02.075. [DOI] [PubMed] [Google Scholar]
- Fueyo González F. J.; Ebiloma G. U.; Izquierdo García C.; Bruggeman V.; Sánchez Villamañán J. M.; Donachie A.; Balogun E. O.; Inaoka D. K.; Shiba T.; Harada S.; Kita K.; de Koning H. P.; Dardonville C. Conjugates of 2,4-dihydroxybenzoate and salicylhydroxamate and lipocations display potent anti-parasite effects by efficiently targeting the Trypanosoma brucei and Trypanosoma congolense mitochondrion. J. Med. Chem. 2017, 60, 1509–1522. 10.1021/acs.jmedchem.6b01740. [DOI] [PubMed] [Google Scholar]
- Cueto-Díaz E. J.; Ebiloma G. U.; Alfayez I. A.; Ungogo M. A.; Lemgruber L.; González-García M. C.; Giron M. D.; Salto R.; Fueyo-González F. J.; Shiba T.; González-Vera J. A.; Ruedas Rama M. J.; Orte A.; de Koning H. P.; Dardonville C. Synthesis, biological, and photophysical studies of molecular rotor-based fluorescent inhibitors of the Trypanosome Alternative Oxidase. Eur. J. Med. Chem. 2021, 220, 113470. 10.1016/j.ejmech.2021.113470. [DOI] [PubMed] [Google Scholar]
- Grady R. W.; Bienen E. J.; Dieck H. A.; Saric M.; Clarkson A. B. Jr. N-n-alkyl-3,4-dihydroxybenzamides as inhibitors of the trypanosome alternative oxidase: activity in vitro and in vivo. Antimicrob. Agents Chemother. 1993, 37, 1082–1085. 10.1128/AAC.37.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardonville C.; Nué Martínez J. J. Bis(2-aminoimidazolines) and Bisguanidines: Synthetic Approaches, Antiparasitic Activity and DNA Binding Properties. Curr. Med. Chem. 2017, 24, 3606–3632. 10.2174/0929867324666170623091522. [DOI] [PubMed] [Google Scholar]
- Ríos Martínez C. H.; Nué Martínez J. J.; Ebiloma G. U.; de Koning H. P.; Alkorta I.; Dardonville C. Lowering the pKa of a bisimidazoline lead with halogen atoms results in improved activity and selectivity against Trypanosoma brucei in vitro. Eur. J. Med. Chem. 2015, 101, 806–17. 10.1016/j.ejmech.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Rodríguez F.; Rozas I.; Kaiser M.; Brun R.; Nguyen B.; Wilson W. D.; García R. N.; Dardonville C. New bis(2-aminoimidazoline) and bisguanidine DNA minor groove binders with potent in vivo antitrypanosomal and antiplasmodial activity. J. Med. Chem. 2008, 51, 909–923. 10.1021/jm7013088. [DOI] [PubMed] [Google Scholar]
- Wang X.; Dong Y.; Cal M.; Kaiser M.; Wittlin S.; Vennerstrom J. L. Antiprotozoal Selectivity of Diimidazoline N-Phenylbenzamides. ACS Infect. Dis. 2015, 1, 135–139. 10.1021/id500034v. [DOI] [PubMed] [Google Scholar]
- Lanteri C. A.; Tidwell R. R.; Meshnick S. R. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 2008, 52, 875–882. 10.1128/AAC.00642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim H. M. S.; Al-Salabi M. I.; El Sabbagh N.; Quashie N. B.; Alkhaldi A. A. M.; Escale R.; Smith T. K.; Vial H. J.; de Koning H. P. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J. Antimicrob. Chemother. 2011, 66, 111–125. 10.1093/jac/dkq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan C. R.; Acosta-Reyes F. J.; Lagartera L.; Ebiloma G.; Lemgruber L.; Nué Martinez J. J.; Saperas N.; Dardonville C.; de Koning H.; Campos J. L. Functional and structural analysis of AT-specific minor groove binders that disrupt DNA-protein interactions and cause disintegration of the Trypanosoma brucei kinetoplast. Nucleic Acids Res. 2017, 45, 8378–8391. 10.1093/nar/gkx521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J. M.; Heuft M. A.; Rabbat P. Solvent Effects on the Monobromination of α,ω-Diols: A Convenient Preparation of ω-Bromoalkanols. J. Org. Chem. 2000, 65, 5837–5838. 10.1021/jo000291u. [DOI] [PubMed] [Google Scholar]
- Judkins B. D.; Allen D. G.; Cook T. A.; Evans B.; Sardharwala T. E. A Versatile Synthesis of Amidines from Nitriles Via Amidoximes. Synth. Commun. 1996, 26, 4351–4367. 10.1080/00397919608003838. [DOI] [Google Scholar]
- Dardonville C.; Goya P.; Rozas I.; Alsasua A.; Martin M. I.; Borrego M. J. New aromatic iminoimidazolidine derivatives as alpha1-adrenoceptor antagonists: a novel synthetic approach and pharmacological activity. Bioorg. Med. Chem. 2000, 8, 1567–1577. 10.1016/S0968-0896(00)00089-4. [DOI] [PubMed] [Google Scholar]
- West R. A.; Cunningham T.; Pennicott L. E.; Rao S. P. S.; Ward S. E. Toward More Drug Like Inhibitors of Trypanosome Alternative Oxidase. ACS Infect. Dis. 2018, 4, 592–604. 10.1021/acsinfecdis.7b00218. [DOI] [PubMed] [Google Scholar]
- Alghamdi A. H.; Munday J. C.; Campagnaro G. D.; Gurvic D.; Svensson F.; Okpara C. E.; Kumar A.; Quintana J.; Martin Abril M. E.; Milic P.; Watson L.; Paape D.; Settimo L.; Dimitriou A.; Wielinska J.; Smart G.; Anderson L. F.; Woodley C. M.; Kelly S. P. Y.; Ibrahim H. M.; Hulpia F.; Al-Salabi M. I.; Eze A. A.; Sprenger T.; Teka I. A.; Gudin S.; Weyand S.; Field M.; Dardonville C.; Tidwell R. R.; Carrington M.; O’Neill P.; Boykin D. W.; Zachariae U.; De Koning H. P. Positively selected modifications in the pore of TbAQP2 allow pentamidine to enter Trypanosoma bruce. elife 2020, 9, e56416 10.7554/eLife.56416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhaldi A. A. M.; Martinek J.; Panicucci B.; Dardonville C.; Zíková A.; de Koning H. P. Trypanocidal action of bisphosphonium salts through a mitochondrial target in bloodstream form Trypanosoma brucei. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 23–34. 10.1016/j.ijpddr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeacock L.; Baker N.; Wiedemar N.; Mäser P.; Horn D. Aquaglyceroporin-null trypanosomes display glycerol transport defects and respiratory-inhibitor sensitivity. PLOS Pathogens 2017, 13, e1006307 10.1371/journal.ppat.1006307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenko B.; Van Der Burg A. M.; Wanner M. J.; Kaiser M.; Brun R.; Gould M.; De Koning H. P.; Koomen G. J. 2,N6-disubstituted adenosine analogs with antitrypanosomal and antimalarial activities. Antimicrob. Agents Chemother. 2007, 51, 3796–3802. 10.1128/AAC.00425-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.