

Abstract
We aimed to evaluate the value of ATN biomarker classification system (amyloid beta [A], pathologic tau [T], and neurodegeneration [N]) for predicting conversion from mild cognitive impairment (MCI) to dementia. In a sample of people with MCI (n = 415) we assessed predictive performance of ATN classification using empirical knowledge-based cut-offs for each component of ATN and compared it to two data-driven approaches, logistic regression and RUSBoost machine learning classifiers, which used continuous clinical or biomarker scores. In data-driven approaches, we identified ATN features that distinguish normals from individuals with dementia and used them to classify persons with MCI into dementia-like and normal groups. Both data-driven classification methods performed better than the empirical cut-offs for ATN biomarkers in predicting conversion to dementia. Classifiers that used clinical features performed as well as classifiers that used ATN biomarkers for prediction of progression to dementia. We discuss that data-driven modeling approaches can improve our ability to predict disease progression and might have implications in future clinical trials.
Keywords: Alzheimer’s disease, amyloid, biomarker profile, machine learning, mild cognitive impairment, neurodegeneration, predictive analytics, tau
1 |. NARRATIVE
1.1 |. Contextual background
Alzheimer’s disease (AD), an irreversible, progressive brain disorder, is the most common cause of dementia in the elderly.1 Despite the high burden of AD on patients and communities, disease-modifying treatments for AD are not available.2 High failure rates in AD clinical trials has been attributed to incomplete knowledge of the pathogenesis of AD (inappropriate or incomplete targets), biological heterogeneity in enrolled patients, poor reliability of outcome measurements, and design of clinical trials.3 A major reason for failure of trials is thought to be enrollment of participants at a disease stage that might be too advanced to benefit from the intervention being studied.3 Therefore, some trials have shifted toward secondary prevention, targeting participants at earlier clinical stages of disease, such as mild cognitive impairment (MCI). Other secondary prevention trials target stages that precede MCI.4,5 However, heterogeneity in participants with MCI, along with low rate of cognitive decline and low conversion rates to AD, makes it very difficult to detect differences between placebo and treatment groups, in clinical trials that focus on earlier clinical stages.6 In this article, we focus on predicting which MCI patients are more likely to decline based on their biomarker profiles.
The 2018 National Institute on Aging–Alzheimer’s Association (NIA-AA) Research Framework provides a biological staging model for AD known as the ATN system; it is based on neuropathologic changes or biomarkers.7 The classification uses three types of biomarkers: amyloid (A), tau (T), and neurodegeneration (N), which are presumed to be the central causal mechanisms for symptomatic AD. While using the framework helps with defining AD as a biological construct, it does not explicitly address the prediction of transitions among stages, an important need for clinical practice and clinical trials. Enrollment of participants in trials based on the criteria specified in the framework reduces the heterogeneity of the study population. However, to implement effective trials, the study population—in both placebo and treatment arms—has to be further enriched with participants that are expected to progress within the time period of the study. If the rate of decline is low in the placebo group in some individuals, it becomes more difficult to demonstrate that the active treatment slows decline.8
In MCI targeted secondary prevention trials, one approach for improving sensitivity to treatment effects is to enroll persons with MCI at high risk for progression, identified based on data-driven predictive models.8 While quantitative risk predictions for AD have been available for many years, these models have been incorporated into the design of AD clinical trials on a limited basis, primarily by targeting persons with a family history of AD, carriers of AD risk genes, or biomarkers of amyloid.4,5 In this study, we used data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) to investigate the performance of various modeling strategies for prediction of incident dementia among persons with MCI and to evaluate these models in the context of the ATN framework. We hypothesized that data-driven predictive models would provide improvements over classification criteria based on empirical cut-offs to define amyloid or tau positivity or the presence of neurodegeneration. Here, we used two progression-prediction approaches: (1)ATN-based classification using dichotomous biomarker levels and (2) data-driven predictive models (logistic regression [LR] and RUSBoost machine learning [ML]) using continuous biomarker features to classify participants into two classes. The data-driven models use baseline data to divide persons with MCI into two classes, one that resembles cognitively normal individuals (CN-like), and the other more similar to individuals with dementia (dementia-like). We assessed these classifications at baseline by assessing their ability to predict incident dementia. Subsequently, performance of all models was compared. Moreover, we describe the frequency of ATN profiles and their long-term clinical trajectory in CN-like and dementia-like classes, which were identified by data-driven models and clinical features.
1.2 |. Study conclusions and disease implications
Our results indicate that using a dichotomous biomarker levels approach, we can achieve either high sensitivity or high specificity in prediction of progression from MCI to dementia, but not both simultaneously. The overall performance of classifications derived from dichotomous use of biomarkers predicted incident dementia less well than the data-driven predictive models. Furthermore, we also showed that clinical features could be as informative as biomarkers for prediction of outcomes, a result discussed below. A higher rate of disease progression was observed in individuals with AD pathology compared to individuals with no pathological change (A–T–N–) or suspected non-AD pathology (SNAP, defined as A–T+N–, A–T–N+, and A–T+N+).
We showed that classification of persons with MCI using the dichotomous biomarker levels (the presence of any of the three cardinal features of AD pathologic change [classification-A: A+ or T+ or N+ vs. no AD pathology]) as differentiating factors, we predicted dementia onset with extremely high sensitivity but very low specificity. Using dichotomous biomarker levels to define the AD continuum based on amyloid positivity (classification-B: A+T±N± vs. others), the sensitivity of models decreased but specificity of the models improved. This method would identify virtually everyone with MCI who will progress (high sensitivity) but would also include many people who do not progress (modest specificity) over the timeframe of a typical clinical trial. Considering expected annual incidence of dementia, and due to low specificity and positive predictive value (PPV), using these classification approaches might not significantly improve patient selection for a secondary prevention trial. This finding corresponds to the observations that many cognitively normal older adults might have evidence for presence of one of the cardinal biomarkers of AD, but do not progress for extended periods of time.9
We also showed that dichotomous biomarker classification using the stricter criteria of presence of advanced AD pathology (classification-C: A+T+N+ vs. others), yields the highest specificities among models, but at the cost of a major reduction in sensitivity. This is compatible with prior studies indicating that highest rates of short-term progression are in individuals with the A+T+N+ biomarker profile.10 Therefore, using the latter classification as part of the enrollment criteria, one can improve the power of a trial. However, assuming that prevalence of A+T+N+ pathology in the general population is similar to that in our study (≈21%), many more participants would require biomarker assessments to identify sufficient individuals to enroll the study. And if we treat people with amnestic MCI (aMCI) who are A+T+N+ and treatment fails, failure might be attributed to treating too late.11
Data-driven approaches (LR and RUSBoost) had significantly higher performance than all dichotomous biomarker classification approaches. Performance of our models was on par with the majority of other studies that applied ML classifiers to clinical or biomarker data to detect MCI participants expected to progress to dementia.12,13 The observed differences among studies in prediction may be due to differences in feature selection, engineering methods, or choice of models. In line with many of the prior studies, our study shows that using continuous variables and a combination of different biomarkers can improve accuracy of predicting progression from MCI to dementia.13–16
Higher performance of data-driven classification over dichotomous biomarker classification is, in part, due to the fact that data-driven approaches do not rely solely on critical values to define positivity for a biomarker and use richer information that exists in full regional amyloid positron emission tomography (PET) standardized uptake value ratio (SUVR), regional metabolism, and full cerebrospinal fluid (CSF) biomarker profiles. A dichotomous approach lends itself to simple operational rules for enrollment but coarsens data and discards informative patterns of association among features.17 In our study, while performance of RUSBoost ML models was slightly superior to LR-based models, especially at later time points, the difference between these methods was not statistically significant. Prior studies comparing the performance of ML models versus “conventional” regression-based classifications, such as LR, have shown mixed results.12,18 The failure of ML models to consistently outperform regression-based methods may be attributable, at least in part, to the feature selection process. High dimensional data can introduce noise into multivariate models, and together with a small number of subjects (high feature/subject ratio), can result in over-fitting.19 In the current study, for LR-based models, we selected features purposefully to avoid multicollinearity and overfitting of data. We ran LR-based models without any feature selection and found that performance of LR-based models significantly decreases (results not shown). One potential advantage of ensemble ML models—such as RUSBoost—is that part of the feature-selection process is embedded in the models, which decreases the need for additional feature engineering steps.
Relative to the sample cumulative incident dementia (or observed rate of dementia onset), RUSBoost models applied to biomarker feature set provided > 35% improvement in the prediction of dementia over 2 years (model’s PPV = 53.6 vs. observed rate of dementia onset = 18.0%) and > 45% over 4 years (model’s PPV = 68% vs. observed rate of dementia onset = 22.2%). Similarly, the RUSBoost model developed using clinical characteristics (clinical feature set) can provide > 32% improvement over 2 years (PPV = 50.5 vs. observed rate of dementia onset = 18.0%) and 45.1% improvement over 4 years (Mmodel’s PPV = 67.3% vs. observed rate of dementia onset = 22.2%) in prediction of progression to dementia.
In general, our findings indicate that using cheaper and more accessible clinical characteristics as predictors can provide high-performance models for the prediction of progression. These models perform similarly to those that use biological markers (i.e., ATN biomarkers) as model features. However, it should be noted that despite our attempt to minimize circularity in analysis (see Methods section), it is still expected that the best predictor of a specific entity (here, cognitive function) is going to be measures of that specific entity. High performance of models that used only clinical features indicate that collection of the whole panel of AD biomarkers is not necessary to identify MCI participants who are likely to progress. But this does not mean that clinical characteristics alone are sufficient to enroll participants in trials. As our survival analysis indicates, even after classification based on clinical features, individuals who are biomarker positive for AD pathology tend to have a higher rate of progression to dementia. In addition, enrolling patients with negative AD pathology (A– and T–) who are likely to have fast clinical decline in trials that target AD pathology, that is, therapies that target amyloid or tau, would decrease the power of trials if AD-targeted medications fail in these individuals. Characterization of the biological profile is essential to ensure the target pathology is present in the intervention group, while clinical benefit, that is, improvement in cognitive function or a slowing of decline, is essential for assessment of a trial’s success.
Based on the pathophysiological processes of AD, as proposed by Jack et al.,11 presence of amyloid PET usually precedes CSF tau positivity (at detectable threshold), which is typically followed by fluorodeoxyglucose (FDG) PET and magnetic resonance imaging (MRI) as the last biomarkers to become abnormal. In other words, the highest rates of short-term progression are expected to be among A+T+N+ and A+T+N– subgroups. Among individuals classified as CN-like, more than 30% are among A+T+N+ and A+T+N– subgroups. These individuals also have a higher rate of incident dementia based on the Cox proportional hazard models, though the rate of decline is not as high as those who have similar pathology and are classified as dementia-like. These findings are compatible with the proposed pathophysiological process of AD. However, some of the individuals who are at risk of disease progression, in the short term, are not among A+T+N+ or A+T+N– profiles, indicating the imperfect correlation between clinical outcomes and presence of AD pathology. This might be due to the role of other types of pathology that were not studied here.
In summary, this study indicates that data-driven predictive models are effective tools for prediction of disease progression among MCI individuals. Clinical features could be as informative as biomarkers for prediction purposes; however, biomarkers can add value to predictions and are essential for trials to enroll participants who actually have the disease pathology that is being targeted by the intervention.
1.3 |. Study limitations and recommendations for future studies
In the current study, we only focused on two data-driven models. Had we selected other classifiers or other feature-sets, results may have differed. In the ADNI sample, MCI and dementia diagnosis was not necessarily due to AD, and we did not exclude subjects based on presence or absence of AD pathology. We chose this approach (1) because ADNI used these clinical diagnoses for stratified enrollment of participants and (2) to avoid circularity problems that would arise if we used biological predictors to define outcomes. Lack of pathologically confirmed diagnosis makes it impossible for us to make conclusions regarding biological etiology of incident dementia. Furthermore, while ADNI is one of the largest studies collecting biomarker data from individuals in the AD spectrum, because there are eight distinct biomarker profiles, the smallest group (A+T–N+) included only four individuals. Therefore, assessment of longitudinal changes and predictive performance of models for individual ATN subgroups was not possible, and for survival analysis we focused on larger subgroups (A–T–N–, A− T±N±, and A+T±N±). As the next step, to further validate our findings, future studies should use data from large consortiums of AD (e.g., NIA-funded Alzheimer’s Disease Centers, iSTAGING cohorts, or the new AI4AD project).
Another limitation of our study is that we only used data from ADNI, a study with strict inclusion and exclusion criteria. This limitation on generalizability can be addressed by applying these methods to other study populations. One major challenge for assessment of reliability and generalizability of predictive models is limited access to data from clinical trials. While there are many large trials over the last decade, access to data has proven challenging. When older data become available, it has often ceased to be relevant due to shifting ideas about optimal biomarkers. To overcome this hurdle, we favor expanding public-private collaboration to promote timely data-sharing.20,21 We believe that application of predictive models in “real-world” practice has benefits that extend well beyond enrichment of prospective trials. We hypothesize that such models can be useful for at least two additional reasons: First, these models could be applied to data from concluded trials to identify patients who were expected to show decline. We hypothesize that this approach might identify subgroups who showed significant trends toward effectiveness of a drug and in extreme cases it might even revive some of the failed trials, especially if poor subject selection was the main reason for their failure. Second, once there are effective treatments for AD, these models could be used to identify individuals who would benefit from primary or secondary prevention.
Finally, we used an automated feature selection method to identify important features for inclusion in LR models. Automated feature selection engineering methods have proven to be essential for achieving high-performance predictive models22 and can lead to selection of features without prior knowledge of biological importance. This is the opposite of manual feature selection methods, in which features are all selected based on prior knowledge and based on biological importance. While this might be considered a limitation of automated feature engineering, it can be viewed as an opportunity for identifying new potential biological pathways and hypothesis generation, leading to new studies.
2 |. CONSOLIDATED RESULTS AND STUDY DESIGN
Approaches to predicting disease progression vary widely along at least three key dimensions: the operational definition of the outcome being predicted, the candidate features used for prediction, and the nature of the statistical model used to make the prediction. In this study, we used onset of dementia as the outcome of interest in persons with aMCI. While staging of AD based on clinical criteria and cognitive scores is not perfect,23 we use them to define the outcome as ADNI used these diagnoses for stratified enrollment of participants. Furthermore, similar diagnostic criteria are being used universally in the interventional trials of AD. We divided candidate predictors (features) into two sets, namely biomarker feature set, which comprised biomarkers including CSF, amyloid PET, and FDG-PET measures; and clinical feature set, which included demographics, apolipoprotein E (APOE) ε4 allele count, and cognitive measures. We used these feature -sets to predict clinical outcome using different statistical methods.
Our first method was confined to the biomarker feature set and was developed based on dichotomous classification of biomarkers to create profiles defined in the 2018 NIA-AA research framework. We note that the ATN framework is not developed for the purpose of prediction of clinical outcomes, but many studies use cut-offs on global measures of amyloid, tau, or neurodegeneration to classify individuals based on their ATN profile to predict clinical progression or enroll participants in the study.24–27 Using dichotomous biomarker levels, we classified aMCI individuals to four different groups (see section 3.1.6). This approach yielded classifications with either high sensitivities and low specificities or low sensitivities and high specificities.
The second method was using data-driven models and continuous features to predict disease progression. We used two different data-driven approaches: (1) multivariate LR and (2) the RUSBoost classification tree, which is a ML model. Using baseline data from CN and dementia participants, LR or RUSBoost models were trained to differentiate CN participants from individuals diagnosed with dementia. Subsequently, the trained model was applied to aMCI participants to classify them as “CN-like” (participants who are more similar to CNs and more likely to remain cognitively stable) or “dementia-like” (participants who are more similar to people with dementia and more likely to progress to dementia), based on probability of belonging to one group or the other (cut-off = 0.5). Performance of models and the accuracy of the predicted outcomes for the aMCI population—namely, likely to remain stable versus likely to progress to dementia—was evaluated using available longitudinal data. Compared to the first method (ATN-based classification), both LR- and RUSBoost models had better performance in prediction of disease progression: Sensitivity of LR-based models decreased from 73.6% at 6 months to 48.0% at 4 years, while specificity increased from 77.6% at 6 months to 90.6% at 4 years of follow-up. For the RUSBoost model the sensitivity declined from 73.6% at 6 months to 61.5% at 4 years, and the specificity increased from 75.1% to 91.7% at 4 years of follow-up. There was no significant difference between performance of LR models and RUSBoost models at any timeframe.
Subsequently, we applied LR and RUSBoost models to the clinical feature set to predict disease progression in aMCI individuals. Performance of these models was comparable to models using the biomarker feature set. Sensitivity for LR models decreased from 81.8% at 6 months to 63.4% at 4 years, and specificity increased from 72.4% at 6 months to 90.1 at 4 years of follow-up. RUSBoost models showed declining sensitivity from 90.9% at 6 months to 63.4% at 4 years of follow-up and increasing specificity from 73.7% to 91.2%.
Cox proportional hazard models showed that in the CN-like subgroup identified by data-driven methods and the clinical feature set, individuals with AD pathology had higher risk of conversion to dementia than the group with normal biomarkers (reference group).
3 |. DETAILED METHODS AND RESULTS
3.1 |. Methods
3.1.1 |. ADNI study design
The data used for this analysis were downloaded from the ADNI database in January 2020. ADNI is an ongoing effort, which was launched in 2003 as a public–private partnership with the primary goal of testing whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and mild dementia. For up-to-date information on ADNI, see adni.Ioni.usc.edu. The individuals included in the current study were initially recruited as part of ADNI-GO, and ADNI-2. ADNI data collection was approved by the institutional review boards of all participating institutions. Informed written consent was obtained from all participants at each site.
3.1.2 |. Participants
A total of 787 participants, including 250 CN, 415 aMCI, and 122 with dementia diagnosis at baseline were eligible for this study. Inclusion criteria in this study were having amyloid PET (A), CSF p-tau examination (T), and FDG PET(N) at baseline and at least one wave of follow-up. Figure 1 provides a flowchart of study participants.
FIGURE 1.

Flowchart of study participants. A, amyloid; ADNI, Alzheimer Disease Neuroimaging Initiative; CN, cognitively normal; CSF, cerebrospinal fluid; FDG, 18F-fluorodeoxyglucose; MCI, mild cognitive impairment; N, neurodegeneration; PET, positron emission tomography; T, tau
CN participants had Mini-Mental State Examination (MMSE) scores of 24 or higher and a Clinical Dementia Rating (CDR) score of 0. All MCI participants were diagnosed as having aMCI; this diagnostic classification required MMSE scores between 24 and 30 (inclusive), a memory complaint, objective memory loss measured by education-adjusted scores on the Wechsler Memory Scale Logical Memory II, a CDR of 0.5, absence of significant impairment in other cognitive domains, essentially preserved activities of daily living, and absence of dementia. All CN participants selected for this study remained cognitively normal within the first 6 months of follow-up during the first year of follow-up. The subjects with dementia had to satisfy the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria for clinically defined probable AD, and have MMSE scores between 20 and 26 (inclusive), and CDR of 0.5 or 1.
3.1.3 |. Cognitive and functional scores
Neurocognitive tests included Trail Making Test part B (TMT-B), the Functional Activities Questionnaire (FAQ), the MMSE, Alzheimer’s Disease Assessment Scale Cognitive subscale (ADAS-cog), the Rey’s Auditory Vocabulary List Test (RAVLT), Montreal Cognitive Assessment (MoCA), Logical Memory II, and the CDR.
3.1.4 |. CSF measurements
CSF amyloid beta (Aβ)42, tau, and phosphorylated tau (p-tau) were measured at the ADNI Biomarker Core Laboratory (University of Pennsylvania) using the multiplex xMAP Luminex platform (Luminex Corporation) with Innogenetics (INNO-BIA AlzBio3; for research use only reagents) immunoassay kit-based reagents.27 All CSF biomarker assays were performed in duplicate and averaged.
3.1.5 |. Neuroimaging
Amyloid PET imaging was collected with 18-F florbetapir as the radiotracer; images were averaged, spatially aligned, interpolated to a common voxel size (1.5 mm3), and smoothed to a common resolution of 8-mm full width at half maximum.26 FDG-PET data were acquired and reconstructed according to a standardized protocol (http://adni.loni.usc.edu/). Spatial normalization of each individual’s PET image to the standard MNI template was conducted using SPM5.25
Amyloid abnormal (A+) and normal (A–) groups were determined by applying a cut-off value of 1.11 for the global florbetapir SUVR.27 The global 18F-florbetapir SUVR was calculated by averaging the 18F-florbetapir retention ratio from four large cortical gray matter regions (frontal, anterior cingulate, precuneus, and parietal cortex) using the cerebellum as a reference region.
Whether tau pathology was abnormal (T+) or normal (T–) w determined by a cut-off value of 23 pg/mL for CSF p-tau level.27
Based on the NIA-AA ATN system, we can evaluate neurodegeneration status using either FDG-PET or structural MRIs, but it is not clear which of these methods leads to superior results. Here, for simplicity, we defined neurodegeneration status (N+ or N–) using the average of angular, temporal, and posterior cingulate FDG PET values with a cut-off value at 1.21.25 Results presented in the body of the article are based on this method. In a secondary analysis, abnormal N was defined as hippocampal volume adjusted for total intracranial volume (HVa) of less than 6,723 mm328 (supporting information).
3.1.6 |. Statistical analysis
Two sets of features were used for data analysis:
Biomarker feature set (imaging and CSF biomarkers) included variables from all modalities that contribute to creation of ATN biomarker profile: (1) all CSF biomarkers (Aβ1–42,tau, p-tau181p), (2) regional amyloid PET measures (florbetapir SUVR of 75 brain regions), (3) regional FDG PET (FDG measures from five different brain regions). All variables were included in models as continuous variables unless stated otherwise. The full list of variables is provided in the supporting information.
Clinical feature set (demographics, clinical characteristics and APOE status) included (1) demographics: age, sex, education; (2) number of APOE ε4 alleles; and (3) cognitive measures: ADAS-Cog11, RAVLT (immediate recall, 30-min delayed recall, and recognition), TMT-B, FAQ total score, MoCA. To decrease circularity with clinical diagnostic criteria, MMSE, logical memory, and CDR scores were not included in this feature set.
The goal of predictive modeling was to partition persons with aMCI into two groups: one that did not progress to dementia, and another that would progress to dementia within the next 4 years of follow-up. Two different methods were used to predict progression:
LR-based classifiers were used as a traditional multivariate statistical method for predicting progression.
RUSBoost: While many ML models have proven to be effective tools for predictions of outcomes in AD, in a previous study we showed that the ensemble ML models perform better than other ML models in predicting clinical outcomes.16 RUSBoost is a hybrid sampling/boosting algorithm for learning from skewed training data. In our case, there were 250 CN and 122 AD participants.
The traditional approach to develop clinical risk prediction models involves the use of regression models—specifically, logistic regression to predict disease presence (diagnosis) or disease outcomes (prognosis).29 Definitions of what constitutes “traditional” multivariate statistical modeling for prediction and its differences with ML have been discussed at length in the literature, yet the distinction is not clear-cut.30 Studies comparing performance of ML models versus traditional multivariate models such as LRs have shown mixed results.29,30
Feature selection is a dimensionality reduction technique that selects only a subset of measured features (predictors) that provide the best predictive power in modeling the data. It is particularly useful when dealing with very high-dimensional data or when modeling with all features is undesirable. Purposeful feature selection is particularly important for LR models as multicollinearity and high dimensionality of features has proven to substantially decrease performance of these models.31
For feature selection for LR models, as the first step (and to avoid multicollinearity), pairs of variables for which the correlation coefficient was > 0.8 were identified and all but one of such variables were deleted. Next, we followed the method proposed by Bursac et al.31 to select the final features included in LR models. The final list of features selected after these steps is provided in the supporting information.
For feature selection for the RUSBoost model, considering that this tree-based ensemble uses multiple weak learning and boosting methods to identify the best features, we did not need to use any additional feature engineering and all available features were included as predictors in this model.
To predict disease progression from aMCI to dementia, MCI participants were classified using one of the following methods:
- ATN-based classification using dichotomous biomarker levels. Classification was based on dichotomizing biomarker measures using the cut-offs mentioned above. As will be depicted in Table 2, the sample size of some the eight ATN subgroups was very small, and therefore, evaluating predictive value of individual ATN subclasses was not possible. Therefore, we used the following criteria and the larger ATN groupings for classification:
- Dichotomous ATN classification A: any pathology (A+ or T+ or N+) vs. no AD pathology (A–T–N–);
- Dichotomous ATN classification B: AD continuum (A+/T±/N±) vs. others;
- Dichotomous ATN classification C: advanced AD pathology (A+T+N+) vs. others; and
- Dichotomous ATN classification D: suspect non-AD pathology (SNAP; A–T+N± or A–T±N-) vs. others.
Data-driven logistic regression. Using baseline data from CN and dementia participants, a LR model was trained to differentiate participants with dementia from healthy controls. A 10-fold cross-validation procedure was used in all models for testing validity of the LR models. Cross-validation is an established statistical method for validating a predictive model, which involves training several parallel models, each based on a subset of the training data. Then, the model performance is evaluated based on the average accuracy in predicting the labels of the omitted portion of the training data.32 Cross-validation can detect if models are overfitted by determining how well the model generalizes to other subsets of datasets by partitioning the data. Subsequently, the trained model was applied to aMCI participants to classify them as “CN-like” (participants who are more [> 50%) similar to CNs and expected to remain cognitively stable) or “dementia-like” (participants who are more [> 50%) similar to people with dementia).
Data-driven RUSBoost. Using data from CN and dementia participants, RUSBoost models were applied to data from CN and dementia groups for training. Similar to LR models, a 10-fold cross-validation procedure was used in all models for testing validity of these models. Trained models were applied to aMCI participants to classify them as “CN-like” or “dementia-like.”
TABLE 2.
Characteristics of 415 aMCI participants by ATN biomarker classification
| Classification | Normal blomarker | Suspect non-AD pathology (SNAP) | AD continuum | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Variable | A−T−N− | A−T+N− | A−T−N+ | A−T+N+ | A+T−N− | A+T+N− | A+T−N+ | A+T+N+ |
| N* | 79 (19.0) | 71 (17.1) | 26 (6.2) | 13 (3.1) | 15 (3.6) | 117 (28.1) | 4 (1.0) | 90 (21.6) |
| Age, years | 68.21 (7.34) | 69.66 (8.20) | 73.20 (7.70) | 76.07 (7.22) | 71.26 (5.00) | 72.64 (7.29) | 77.80 (6.60) | 73.00 (6.42) |
| Male* | 41 (51.9) | 36 (50.7) | 17 (65.4) | 9 (69.2) | 7 (46.7) | 65 (55.6) | 3 (75.0) | 52 (57.8) |
| APOE ε4 carriers* | 19 (24.1) | 18 (24.6) | 6 (23.1) | 4 (30.8) | 6 (40.0) | 76 (65.0) | 3 (75.0) | 67 (74.6) |
| Education, years | 16.73 (2.36) | 16.45 (2.46) | 15.85 (2.75) | 15.00 (2.44) | 15.47 (2.90) | 15.90 (2.70) | 17.00 (3.83) | 16.31 (2.82) |
| MMSE score | 28.84 (1.30) | 28.61 (1.39) | 28.50 (1.86) | 26.69 (1.65) | 28.67 (1.11) | 27.88 (1.82) | 28.75 (0.96) | 27.19 (1.87) |
| ADAS-Cog score | 6.86 (3.35) | 7.87 (3.34) | 8.73 (3.50) | 10.31 (1.01) | 8.13 (4.73) | 9.24 (3.99) | 11.75 (2.63) | 12.49 (5.06) |
| Amyloid PET, SUVR | 1.01 (0.05) | 1.02 (0.05) | 1.01 (0.07) | 1.01 (0.06) | 1.25 (0.19) | 1.37 (0.17) | 1.13 (0.03) | 1.42 (0.14) |
| CSF p-tau | 17.67 (3.89) | 35.3 (11.2) | 15.85 (3.65) | 38.2 (12.83) | 18.64 (3.13) | 49.86 (21.5) | 13.92 (3.3) | 57.58 (21.5) |
| FDG | 1.35 (0.09) | 1.33 (0.09) | 1.15 (0.07) | 1.14 (0.04) | 1.32 (0.07) | 1.32 (0.09) | 1.14 (0.05) | 1.11 (0.08) |
Note: All values are mean (standard deviation) unless otherwise stated.
Values represent: number (%).
Progression rate to dementia or expected prevalence.
Abbreviations: A−/+, amyloid status using amyloid PET; ADAS-Cog, Alzheimer Disease Assessment Scale–Cognitive subscale 11 score; aMCI, amnestic mild cognitive impairment; CSF, cerebrospinal fluid; FDG, 18F-fluorodeoxyglucose; MMSE, Mini-Mental State Examination; N−/+, neurodegeneration or neuronal injury normal using FDG-PET; PET, positron emission tomography; SUVR, standardized uptake value ratio; T−/+, tau status using CSF p-tau.
Performance of models and the accuracy of the predicted outcomes for the aMCI population—namely, likely to remain stable versus likely to progress to dementia—was evaluated using available longitudinal data. Sensitivity, specificity, PPV, and negative predictive value (NPV) for each model were calculated. Considering change in available longitudinal data (due to drop outs, death, etc.), the performance of models is reported separately for each follow-up at 6, 12, 24, 36, and 48 months.
To judge classification performance, we used the McNemar test33 to compare the pairwise performance of classifications at 95% confidence level (α = 0.05): z = where Sab refers to the samples correctly classified in classification a, but incorrectly classified in classification b, and Sba indicates the samples that are misclassified in classification a, but correctly classified in classification b.
For biomarker profiling and longitudinal outcome assessment, characteristics and the biomarker profile of each class identified by the ML model (CN-like vs. dementia-like) are reported. In addition, to assess the risk of progression from aMCI to dementia, we constructed unadjusted Cox proportional hazard models.
All statistical analyses were conducted using MATLAB© (version 2020a) or SPSS statistical software (version 25, IBM statistics).
3.2 |. Results
3.2.1 |. Study characteristics
The demographic, clinical, and imaging characteristics of all participants by diagnostic group are shown in Table 1. Characteristics of aMCI participants by ATN biomarker classification are summarized in Table 2. The mean (standard deviation [SD]) age of the participants was 72.4 years; 52.7% were men; 98.7% had 12 years or more of education; and 47% had an APOE ε4 allele. Among 415 individuals with aMCI, 388 participants were assessed at 6 months, 392 participants at 1 year, 339 participants at 2 years, 291 at 3 years, and 234 at 4 years. The cumulative proportions of individuals who progressed from aMCI to AD was 5.7% at 6 months, 9.9% at 1 year, 18.0% at 2 years, 21.3% at 3 years, and 22.2% at 4 years of follow-up. Of persons with aMCI, 79 (19.0%) had normal biomarkers, 110 (26.5%) had SNAP, and 226 (54.5%) had AD pathological changes. Characteristics of aMCI participants were also calculated using HVa as the N measure (Table S1 in supporting information). Biomarker characteristics of aMCI participants based on follow-up diagnosis (stable aMCI vs. converters to dementia) are summarized in Table S2 in supporting information.
TABLE 1.
Characteristics of study population by clinical diagnosis
| Variables | CN | aMCI | Dementia |
|---|---|---|---|
| N | 250 | 415 | 122 |
| Age, years | 72.8 (6.1) | 71.5 (7.4) | 74.7 (8.3) |
| Male* | 114 (45.6) | 230 (55.4) | 71 (58.2) |
| APOE ε4 carriers* | 72 (28.8) | 199 (48.0) | 80 (65.6) |
| Education, years | 16.6 (2.5) | 16.2 (2.6) | 15.7 (2.7) |
| MMSE score | 29.1 (1.2) | 28.0 (1.7) | 23.1 (2.1) |
| ADAS-Cog score | 5.7 (3.0) | 9.2 (4.4) | 20.7 (7.0) |
Note: All values are mean (standard deviation) unless otherwise stated.
Values represent: number (%).
Abbreviations: ADAS-Cog, Alzheimer Disease Assessment Scale–Cognitive subscale 11 score; aMCI, amnestic mild cognitive impairment; APOE, apolipoprotein E; CN, cognitively normal; MMSE, Mini-Mental State Examination.
3.2.2 |. Prediction performance of different methods
Dichotomous ATN classification A (any pathology [A+ or T+ or N+] versus no AD pathology [A–T–N–]): This approach classifies participants into a group with no AD biomarker and another group with any of the amyloid, tau, or neurodegeneration biomarkers. This classification showed very high sensitivities (range: 96.8%–100%) but specificities were very low (range: 19.1%–26.9%) across all timeframes (Table 3).
TABLE 3.
Performance of predictive models in prediction of progression from aMCI to dementia at different follow-up time points. Models developed using biomarker feature set
| Model | Follow-up, years | Sensitivity, % | Specificity, % | PPV, % | NPV, % | AUC | Observed rate, %a |
|---|---|---|---|---|---|---|---|
| ATN classification-Ab | 0.5 | 100 | 19.1 | 6.9 | 100 | – | 5.7 |
| 1 | 97.44 | 20.4 | 11.9 | 98.6 | – | 9.9 | |
| 2 | 98.36 | 22.6 | 21.8 | 98.4 | – | 18.0 | |
| 3 | 96.77 | 24.8 | 25.8 | 96.6 | – | 21.3 | |
| 4 | 98.08 | 26.9 | 27.7 | 98.0 | – | 22.2 | |
| ATN classification-Bb | 0.5 | 86.3 | 46.7 | 8.8 | 98.2 | – | 5.7 |
| 1 | 84.6 | 48.7 | 15.4 | 96.6 | – | 9.9 | |
| 2 | 88.5 | 54.6 | 30.0 | 95.6 | – | 18.0 | |
| 3 | 87.1 | 57.6 | 35.7 | 94.2 | – | 21.3 | |
| 4 | 80.7 | 61.0 | 37.1 | 91.7 | – | 22.2 | |
| ATN classification-Cb | 0.5 | 45.4 | 83.3 | 14.0 | 96.2 | – | 5.7 |
| 1 | 51.2 | 85.8 | 28.5 | 94.1 | – | 9.9 | |
| 2 | 54.1 | 91.3 | 57.8 | 90.0 | – | 18.0 | |
| 3 | 43.5 | 93.4 | 64.2 | 85.9 | – | 21.3 | |
| 4 | 48.0 | 95.6 | 75.7 | 86.5 | – | 22.2 | |
| ATN classification-Db | 0.5 | 13.6 | 72.4 | 2.8 | 93.3 | – | 5.7 |
| 1 | 12.8 | 71.6 | 4.7 | 88.1 | – | 9.9 | |
| 2 | 9.8 | 67.9 | 6.3 | 77.4 | – | 18.0 | |
| 3 | 9.6 | 67.2 | 7.4 | 73.3 | – | 21.3 | |
| 4 | 17.3 | 65.9 | 12.6 | 73.6 | – | 22.2 | |
| Logistic regression | 0.5 | 73.6 | 77.6 | 14.5 | 97.2 | 0.71 | 5.7 |
| 1 | 69.2 | 81.3 | 29.0 | 95.9 | 0.75 | 9.9 | |
| 2 | 67.2 | 88.4 | 56.1 | 92.4 | 0.78 | 18.0 | |
| 3 | 50.0 | 90.3 | 58.4 | 86.9 | 0.70 | 21.3 | |
| 4 | 48.0 | 90.6 | 59.5 | 85.9 | 0.69 | 22.2 | |
| RUSBoost Tree | 0.5 | 73.6 | 75.1 | 13.3 | 97.1 | 0.69 | 5.7 |
| 1 | 69.2 | 78.1 | 25.9 | 95.8 | 0.74 | 9.9 | |
| 2 | 72.1 | 86.3 | 53.6 | 93.3 | 0.79 | 18.0 | |
| 3 | 64.5 | 90.3 | 64.5 | 90.3 | 0.77 | 21.3 | |
| 4 | 61.5 | 91.7 | 68.0 | 89.3 | 0.77 | 22.2 |
Observed rate of dementia onset (or cumulative incident dementia) based on longitudinal data at each timeframe.
ATN classification-A, classifies participants based on presence of any AD pathology (any AD pathology vs. normal AD pathology); ATN classification-B, classifies participants based on presence of any pathology (A+T±N± vs. others); ATN classification-C, classifies participants based on presence of advanced AD pathology (A+T+N+ vs. others); and ATN classification-D, classifies participants based on presence of any non-AD pathology (A−T+N± or A−T±N− vs. others).
Abbreviations: AD, Alzheimer’s disease; aMCI, amnestic mild cognitive impairment; ATN, amyloid, tau, neurodegeneration; AUC, area under the curve; NPV, negative predictive value; PPV, positive predictive value.
Dichotomous ATN classification B (AD continuum [A+T±N±] versus others): This approach classifies persons with MCI and amyloid positivity into one group (AD pathologic change) and all others into a second pathologic group (amyloid negative). This approach had high sensitivities and low specificities for progression prediction of dementia (Table 3). Sensitivities at 6 months, 1 year, 2 years, 3 years, and 4 years of follow-up were 86.3%, 84.6%, 88.5%, 87.1%, and 80.7%, respectively. Specificity gradually increased with longer follow-up periods and was 46.7% at 6 months, 48.7% at 1 year, 54.6% at 2 years, 57.6% at 3 years, and 61.0% at 4 years. This method favored sensitivity over specificity for progression.
Dichotomous ATN classification C (advanced AD pathology [A+T+N+] versus others): In this approach, positivity for amyloid, tau, and neurodegeneration was required to classify the high-risk group. Those missing a single biomarker were considered low risk. This method yielded low sensitivity and very high specificity for progression-prediction across all timeframes. Sensitivities at 6 months, 1 year, 2 years, 3 years, and 4 years of follow-up were 45.4%, 51.2%, 54.1%, 43.5%, and 48.0%, respectively. Specificities were of 83.3% at 6 months, 85.8% at 1 year, 91.3% at 2 years, 93.4% at 3 years, and 95.6% at 4 years of follow-up.
Dichotomous ATN classification D (SNAP [A–T+N± or A–T±N–] versus others): This approach assesses progression prediction for individuals with non-AD pathology to dementia. This approach yielded very low sensitivity (range: 9.6–17.3) and modest specificities (range: 65.9–72.4) across different timeframes.
All dichotomous ATN classifications were redeveloped using a hippocampal volume cut-off to define neurodegeneration (Table S1).
Data-driven models using biomarker feature set: Performance of data-driven classifiers in differentiating cognitively normal from AD participants using biomarker feature set is summarized in Table S3 in supporting information. Data-driven methods showed better performance in detecting individuals who progressed to dementia (Table 3). Sensitivity of LR-based models decreased from 73.6% at 6 months to 48.0% at 4 years, and specificity increased from 77.6% at 6 months to 90.6% at 4 years. For the RUSBoost model the sensitivity declined from 73.6% at 6 months to 61.5% at 4 years, and the specificity increased from 75.1% to 91.7% at 4 years.
The McNemar test showed that both data-driven methods outperformed ATN-based models in prediction of disease progression at all timeframes (P < 0.001 for all). Performance of data-driven methods did not significantly differ from each other for prediction of disease progression at any timeframe (P > 0.05 at all timeframes). Data-driven models were also created using MRI brain region volumes (derived from FreeSurfer instead of FDG PET measures, producing comparable performance [Table S4 in supporting information]).
Data-driven models using clinical feature set: The LR model had sensitivity of 81.8% at 6 months, 76.9% at 1 year, 72.1% at 2 years, 66.1% at 3 years, and 63.4% at 4 years of follow-up, and specificity of 72.4% at 6 months, 75.0% at 1 year, 80.9% at 2 years, 86.0% at 3 years, and 90.1% at 4 years of follow-up (Table 4). RUSBoost using the clinical feature set also showed declining sensitivity from 90.9% at 6 months to 63.4% at 4 years of follow-up and increasing specificity from 73.7% to 91.2% (Table 4). The McNemar test did not show any significant difference between the two data-driven models at any of the five considered conversion intervals (P > 0.05 at all timeframes).
TABLE 4.
Performance of predictive models in prediction of progression from aMCI to dementia at different follow-up time points
| Model | Follow-up time, years | Sensitivity, % | Specificity, % | PPV, % | NPV, % | Accuracy, % | AUC | Observed rate, %a |
|---|---|---|---|---|---|---|---|---|
| Logistic regression | 0.5 | 81.8 | 72.4 | 15.1 | 98.5 | 72.9 | 0.77 | 5.7 |
| 1 | 76.9 | 75.0 | 25.4 | 96.7 | 75.2 | 0.76 | 9.9 | |
| 2 | 72.1 | 80.9 | 45.3 | 92.9 | 79.3 | 0.77 | 18.0 | |
| 3 | 66.1 | 86.0 | 56.1 | 90.3 | 81.7 | 0.76 | 21.3 | |
| 4 | 63.4 | 90.1 | 64.7 | 89.6 | 84.1 | 0.77 | 22.2 | |
| RUSBoost Tree | 0.5 | 90.9 | 73.7 | 17.2 | 99.2 | 74.7 | 0.82 | 5.7 |
| 1 | 76.9 | 76.4 | 26.5 | 96.7 | 76.5 | 0.77 | 9.9 | |
| 2 | 73.7 | 84.1 | 50.5 | 93.6 | 82.3 | 0.79 | 18.0 | |
| 3 | 72.5 | 89.0 | 64.2 | 92.3 | 85.5 | 0.81 | 21.3 | |
| 4 | 63.4 | 91.2 | 67.3 | 89.7 | 85.0 | 0.77 | 22.2 |
Note: Models developed using clinical feature set.
Observed rate of dementia onset (or cumulative incident dementia) based on longitudinal data at each timeframe.
Abbreviations: AD, Alzheimer’s disease; aMCI, amnestic mild cognitive impairment; ATN, amyloid, tau, neurodegeneration; AUC, area under the curve; NPV, negative predictive value; PPV, positive predictive value.
Prevalence of stable versus converter MCIs within CN-like and dementia-like classes for LR and RUSBoost models is summarized in Table S5 in supporting information.
3.2.3 |. Biomarker profile in each aMCI class (CN-like versus dementia-like) CN-like versus dementia-like)
Table 5 summarizes characteristics of aMCI participants classified as CN-like and dementia-like using the data-driven approaches and clinical feature-set predictors (demographics, APOE ε4, cognitive scores). Using both approaches, the proportion of abnormal amyloid, tau, and neurodegeneration was significantly higher in the dementia-like group compared to the CN-like group. Figure 2 summarizes the ATN biomarker profile of CN-like and dementia-like groups as classified by RUSBoost models. Among the dementia-like group, 74.6% of participants had A+T+N± pathology, compared to 40% in the CN-like group. The proportion of A+T+N+ was 12.1% in the CN-like group and 45.8% in the dementia-like group. SNAP accounted for 15.2% of the dementia-like group and 31.4% of the CN-like group. The proportion of normal biomarker profile (A–T–N–) was 24.9% in the CN-like group and only 4.2% in the dementia-like group.
TABLE 5.
Characteristics of aMCI participants based on logistic regression (LR) models and RUSBoost machine learning classification using clinical feature-set (demographics, APOE ε4, and cognitive scores)
| Model | RUSBoost |
LR |
|||||
|---|---|---|---|---|---|---|---|
| Classification | CN-like | Dementia-like | P-valueb | CN-like | Dementia-like | P-valueb | |
| N, % | 297 (71.5) | 118 (28.5) | – | 292 (70.4) | 123 (29.6) | – | |
| Clinical feature set | Age, years | 70.80 (7.4) | 73.30 (7.09) | 0.002 | 70.4 (7.32) | 74.05 (7.04) | <0.001 |
| Malea | 156 (52.5%) | 74 (62.7%) | 0.060 | 152 (52.1%) | 78 (63.4%) | 0.034 | |
| APOE ε4 carriersa | 128 (43.1%) | 71 (60.2%) | <0.001 | 116 (39.7%) | 83 (67.4%) | <0.001 | |
| Education, years | 16.29 (2.54) | 15.96 (2.85) | 0.242 | 16.14 (2.56) | 16.34 (2.79) | 0.471 | |
| MMSE score | 28.44 (1.55) | 27.14 (1.78) | <0.001 | 28.48 (1.50) | 27.12 (1.83) | <0.001 | |
| ADAS-Cog score | 7.36 (2.90) | 13.97 (4.1) | <0.001 | 7.49 (2.99) | 13.41 (4.57) | <0.001 | |
| Biomarker feature set | Amyloid PET, SUVR | 1.16 (0.20) | 1.33 (0.24) | <0.001 | 1.15 (0.20) | 1.35 (0.22) | <0.001 |
| CSF p-tau | 34.84 (21.0) | 49.29 (23.2) | <0.001 | 35.07 (21.50) | 48.16 (22.53) | <0.001 | |
| FDG | 1.30 (0.12) | 1.18 (0.12) | <0.001 | 1.31 (0.14) | 1.17 (0.16) | <0.001 | |
Note. All values are mean (standard deviation) unless otherwise stated.
Values represent: number (%).
Using t-test for continuous variables and chi-square test for categorical variables.
Abbreviations: ADAS-Cog, Alzheimer Disease Assessment Scale–Cognitive subscale 11 score; aMCI, amnestic mild cognitive impairment; APOE, apolipoprotein E; CN, cognitively normal; CSF, cerebrospinal fluid; FDG, 18F-fluorodeoxyglucose; LR, logistic regression; MMSE, Mini-Mental State Examination; PET, positron emission tomography; SUVR, standardized uptake value ratio.
FIGURE 2.

ATN biomarker profile of aMCI participants in different classification groups. Classification model is developed by applying RUSBoost ML models (top) or logistic regression (LR) models (bottom) to demographics, APOE ε4 status, and neuropsychological tests. Numbers represent the percentage in a particular color. A, amyloid; aMCI, amnestic mild cognitive impairment; APOE, apolipoprotein E; CN, cognitively normal; FDG, 18F-fluorodeoxyglucose; ML, machine learning; N, neurodegeneration; SUVR, standardized uptake value ratio; T, tau
3.2.4 |. Longitudinal clinical outcomes in aMCI classes based on ATN profiles
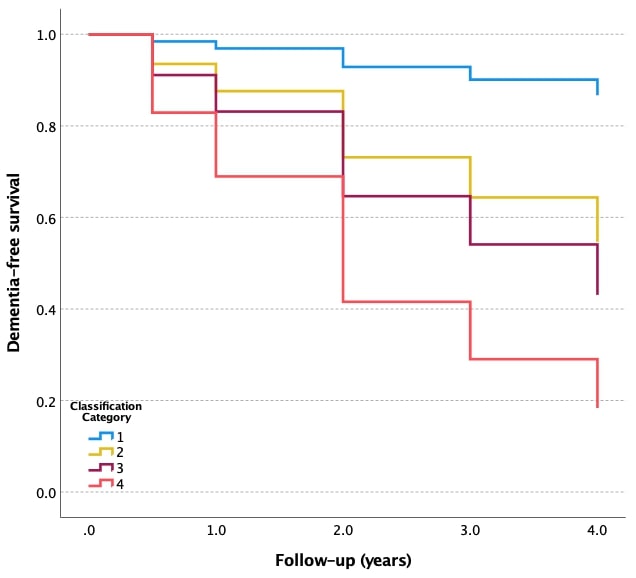
Cox proportional hazards models were conducted to estimate incident dementia, for three different ATN profiles in each class of aMCI (CN-like vs. dementia-like), as identified by data-driven models and the clinical feature set. In the CN-like class, the subgroup with AD pathology had a significantly higher risk of conversion to dementia (LR model: hazard ratio [HR] = 17.4, 95% confidence interval [CI] = 2.36–128.1, P = 0.005; RUSBoost model: HR = 16.5, 95% CI = 2.24–121.8, P = 0.006) compared to the group with normal biomarkers. There was no significant difference between ATN profiles in the dementia-like class as defined by either data-driven model. Figure 3 exhibits the Kaplan-Meier survival curves.
FIGURE 3.

Kaplan-Meier curves showing cumulative probability of disease progression from aMCI to dementia. Classification model is developed by applying logistic regression (A) and RUSBoost models (B) to demographics, APOE ε4, and neuropsychological tests. AD, Alzheimer’s disease; aMCI, amnestic mild cognitive impairment; APOE, apolipoprotein E; CN, cognitively normal
Using the biomarker feature set, a total of 36 participants were classified into dementia-like only by RUSBoost model, 22 participants were classified into dementia-like only by LR model,75 individuals were classified into dementia-like by both models, and 282 were classified into CN-like by both models. To evaluate the differences between these models and how those impacted the long-term progression predictions, we ran additional Cox regression models stratifying the sample into four groups of CN-like, RUSBoost dementia-like only, LR dementia-like, and both-model dementia-like. Results indicate that compared to the group identified as CN-like by both models, individuals who were identified as dementia-like by one or both models are more likely to progress to dementia (Figure S1 in supporting information). There was no significant difference between progression rate among those who were classified as dementia-like by RUSBoost versus those classifies as dementia-like only by LR model.
Supplementary Material
{kind=link}
RESEARCH IN CONTEXT.
Systematic review: Clinical trials frequently use specific clinical criteria and different biomarkers for enrichment of the study population. These criteria are usually based on cut-offs or index scores derived from previous studies. However, it has been suggested that data-driven predictive models can further improve the enrichment process.
Interpretation: These studies indicate that predictive models such as logistic regression and machine learning classifiers are effective tools for prediction of disease progression.
Future directions: This study provides an approach to improve prediction of disease progression using a variety of clinical features and biomarkers. Future research should be conducted using data from clinical trials to explore validity and generalizability of our models in the setting of trials.
ACKNOWLEDGMENTS
Data collection and sharing for the ADNI project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LL.; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Authors of this study were supported by grants from the National Institute of Health (NIA K23 AG063993 [A.E]; P01-AG003949 [R.B.]); P41 EB015922, U01 AG024904, P01 AG026572, P01 AG055367, R56 AG058854, RF1 AG051710 (P.M.T); U01 AG024904 [P.M.T and C.D.]); the Alzheimer’s Association (2019-AACSF-641329; A.E.); Cure Alzheimer Fund (A.E. & R.B.L.), the Leonard and Sylvia Marx Foundation (R.B.L.). Sponsors played no role.
Funding information
the National Institute of Health, Grant/Award Numbers: NIA K23AG063993 [A.E],P01AG003949[R.B.], P41EB015922, U01 AG024904, P01AG026572, P01AG055367, R56AG058854, RF1AG051710(P.M.T); the Alzheimer’s Association, Grant/Award Number:2019-AACSF-641329; Cure Alzheimer’s Fund (A.E.&R.B.L.); Leonard and Sylvia Marx Foundation (R.B.L.)
CONFLICTS OF INTEREST
Ali Ezzati has served on the advisory board of Eisai. Ahmed Abdulkadir has received research support from Swiss National Science Foundation and served as an external expert for the European Research Commission. Clifford R. Jack Jr. has research support from NIH unrelated to this manuscript and serves on advisory boards for Biogen, Eisai, and Roche. Paul M. Thompson is supported in part by a research grant from Biogen, Inc. (Boston, USA) for research unrelated to this manuscript. He receives consulting fees from Kairos Venture Capital, Inc. Danielle J. Harvey receives research support as a biostatistician from the following sources unrelated to this manuscript: NIH: P30 AG010129, U54 NS079202, R01 AG048252, R01 AG029672, R01 AG051618, R01 HD093654, UH2/UH3 NS100608, R01 AG062240, R01 AG062689, R01 AG064688, 2U54 HD079125; DoD: W81XWH-13-1-0259, W81XWH-12-2-0012, W81XWH-14-1-0462; California Department of Public Health: 1910611-0. Monica Truelove-Hill has nothing to disclose. Lasya P. Sreepada has nothing to disclose. Christos Davatzikos receives research support from the following sources unrelated to this manuscript: NIH: R01NS042645, R01MH112070, R01NS042645, U24CA189523; and medical legal consulting work unrelated to this paper. Richard B. Lipton receives research support from the following sources unrelated to this manuscript: NIH: 2PO1 AG003949 (mPI), 5U10 NS077308 (PI), R21 AG056920 (Investigator), 1RF1 AG057531 (Site PI), RF1 AG054548 (Investigator), 1RO1 AG048642 (Investigator), R56 AG057548 (Investigator), U01062370 (Investigator), RO1 AG060933 (Investigator), RO1 AG062622 (Investigator), 1UG3FD006795 (mPI), 1U24NS113847 (Investigator), K23 NS09610 (Mentor), K23AG049466 (Mentor), K23 NS107643 (Mentor). He also receives support from the Migraine Research Foundation and the National Headache Foundation. He serves on the editorial board of Neurology, is senior advisor to Headache, and associate editor to Cephalalgia. He has reviewed for the NIA and NINDS; holds stock options in eNeura Therapeutics and Biohaven Holdings; serves as consultant, advisory board member, or has received honoraria from: Abbvie (Allergan), American Academy of Neurology, American Headache Society, Amgen, Avanir, Biohaven, Biovision, Boston Scientific, Dr. Reddy’s (Promius), Electrocore, Eli Lilly, eNeura Therapeutics, Equinox, GlaxoSmithKline, Grifols, Lundbeck (Alder), Merck, Pernix, Pfizer, Supernus, Teva, Trigemina, Vector, Vedanta. He receives royalties from Wolff’s Headache 7th and 8th editions, Oxford Press University, 2009, Wiley, and Informa. He receives consulting fees from Impel NeuroPharma and Novartis and has stock or options in Control M.
Footnotes
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found here.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Thies W, Bleiler L. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–245. [DOI] [PubMed] [Google Scholar]
- 2.Gauthier S, Albert M, Fox N, et al. Why has therapy development for dementia failed in the last two decades?. Alzheimers Dement. 2016;12:60–64. [DOI] [PubMed] [Google Scholar]
- 3.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bateman RJ, Benzinger TL, Berry S, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimers Dement. 2017;13:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Donohue MC, Raman R, et al. Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol. 2020;77:735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275:251–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ezzati A, Lipton RB. Machine learning predictive models can improve efficacy of clinical trials for Alzheimer’s disease 1, 2. Alzheimers Dement. 2020:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ottoy J, Niemantsverdriet E, Verhaeghe J, et al. Association of short-term cognitive decline and MCI-to-AD dementia conversion with CSF, MRI, amyloid-and 18F-FDG-PET imaging. NeuroImage. 2019;22:101771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR, Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falahati F, Westman E, Simmons A. Multivariate data analysis and machine learning in Alzheimer’s disease with a focus on structural magnetic resonance imaging. J Alzheimers Dis. 2014;41:685–708. [DOI] [PubMed] [Google Scholar]
- 13.Ansart M, Epelbaum S, Bassignana G, et al. Predicting the progression of mild cognitive impairment using machine learning: a systematic, quantitative and critical review. Med Image Anal. 2020:101848. [DOI] [PubMed] [Google Scholar]
- 14.Shah ND, Steyerberg EW, Kent DM. Big data and predictive analytics: recalibrating expectations. JAMA. 2018;320:27–28. [DOI] [PubMed] [Google Scholar]
- 15.Franzmeier N, Koutsouleris N, Benzinger T, et al. Predicting sporadic Alzheimer’s disease progression via inherited Alzheimer’s disease-informed machine-learning. Alzheimers Dement. 2020;16:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezzati A, Zammit AR, Harvey DJ, et al. Optimizing machine learning methods to improve predictive models of Alzheimer’s disease. J Alzheimers Dis. 2019;71:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ. 2006;332:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jie M, Collins GS, Steyerberg EW, Verbakel JY, van Calster B. A systematic review shows no performance benefit of machine learning over logistic regression for clinical prediction models. J Clin Epidemiol. 2019. [DOI] [PubMed] [Google Scholar]
- 19.McEvoy LK, Holland D, Hagler DJ Jr, Fennema-Notestine C, Brewer JB, Dale AM. Mild cognitive impairment: baseline and longitudinal structural MR imaging measures improve predictive prognosis. Radiology. 2011;259:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson KL, Collins FS. Sharing and reporting the results of clinical trials. JAMA. 2015;313:355–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo B Sharing clinical trial data: maximizing benefits, minimizing risk. JAMA. 2015;313:793–794. [DOI] [PubMed] [Google Scholar]
- 22.Zheng A, Casari A. Feature Engineering for Machine Learning: Principles and Techniques for Data Scientists. O’Reilly Media, Inc; 2018. [Google Scholar]
- 23.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickerson BC, Wolk D. Biomarker-based prediction of progression in MCI: comparison of AD-signature and hippocampal volume with spinal fluid amyloid-β and tau. Front Aging Neurosci. 2013;5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landau S, Harvey D, Madison C, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landau SM, Lu M, Joshi AD, et al. Comparingpositron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid.Ann Neurol. 2013;74:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014;71:1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosner DW, Lemeshow S. Applied Logistic Regression. New York: Jhon Wiley & Son; 1989. [Google Scholar]
- 30.Breiman L Statistical modeling: the two cultures (with comments and a rejoinder by the author). Stat Sci. 2001;16:199–231. [Google Scholar]
- 31.Bursac Z, Gauss CH, Williams DK. Hosmer DW. Purposeful selection of variables in logistic regression. Source Code Biol Med. 2008;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hastie T, Tibshirani R, Friedman J. Model Assessment and Selection. The Elements of Statistical Learning. Springer; 2009:219–259. [Google Scholar]
- 33.Dietterich TG. Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput. 1998;10:1895–1923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.