

ABSTRACT
Antifolates targeting dihydrofolate reductase (DHFR) are antimalarial compounds that have long been used for malaria treatment and chemoprevention (inhibition of infection from mosquitoes to humans). Despite their extensive applications, a thorough understanding of antifolate activity against hepatic malaria parasites, especially resistant parasites, has yet to be achieved. Using a transgenic Plasmodium berghei harboring quadruple mutant dhfr from Plasmodium falciparum (Pb::Pfdhfr-4M), we demonstrated that quadruple mutations on Pfdhfr confer complete chemoprevention resistance to pyrimethamine, the previous generation of antifolate, but not to a new class of antifolate designed to overcome the resistance, such as P218. Detailed investigation to pinpoint stage-specific chemoprevention further demonstrated that it is unnecessary for the drug to be present throughout hepatic development. The drug is most potent against the developmental stages from early hepatic trophozoite to late hepatic trophozoite, but it is not effective at inhibiting sporozoite and early hepatic stage development from sporozoite to early trophozoite. Our data show that P218 also inhibited the late hepatic-stage development, from trophozoite to mature schizonts to a lesser extent. With a single dose of 15 mg/kg of body weight, P218 prevented infection from up to 25,000 pyrimethamine-resistant sporozoites, a number equal to thousands of infectious mosquito bites. Additionally, the hepatic stage of malaria parasite is much more susceptible to antifolates than the asexual blood stage. This study provides important insights into the activity of antifolates as a chemopreventive therapeutic which could lead to a more efficient and cost-effective treatment regime.
KEYWORDS: antifolates, malaria, drug resistance, dihydrofolate reductase, prophylaxis, sporozoite, hepatic stage
INTRODUCTION
Morbidity and mortality caused by malaria continue to be a heavy burden for humankind in many parts of the world. The World Malaria Report 2020 (1) revealed more than 200 million annual cases and 400,000 annual deaths from malaria worldwide in 2019 and 2020. Antimalarial treatments have long been used to reduce the burden of malaria infection in humans; however, emergence of parasites resistant to the antimalarials currently in clinical use requires the discovery and development of new antimalarial drugs. In addition, antimalarial drugs that can target only the asexual stage are insufficient to achieve the goal of malaria elimination, because the parasite undergoes a complex life cycle consisting of sexual and asexual stages in the human host and the mosquito vector. The ideal drug should be able to treat the symptoms of the disease, block the transmission from infected human to mosquito (transmission blocking), and prevent infection from infectious mosquito bites in humans (chemoprevention or prophylaxis).
The Plasmodium life cycle comprises multiple developmental stages in vertebrate and mosquito hosts, with each developmental stage having distinct physiology, metabolism, and function. Thus, antimalarial drugs might not be able to target more than one life stage if they do not target metabolism that is central to survival of the parasite. Malaria infection in humans occurs when Plasmodium-infected mosquitoes inject sporozoites into the human host during blood feeding. The injected sporozoites then infect hepatocytes in the liver, where the parasites undergo endomitotic replication to generate thousands of merozoites inside hepatic schizonts over a period of 1 week. Once the hepatic schizonts mature, the parasites rupture to release merozoites and invade red blood cells to start the erythrocytic stage, which is the stage that causes the clinical symptoms. In this stage, the parasites multiply and develop through cycles of ring, trophozoite, and schizont stages to generate billions of progenies inside the human host (2). A high replication rate is the common feature of developmental stages in different hosts, and an ideal antimalarial should target crucial factors involved in this process.
Enzymes in the folate pathway are well-defined clinically validated targets of antimalarial drugs that interfere with parasite DNA synthesis, especially the Plasmodium dihydrofolate reductase (DHFR). Antifolate compounds that target DHFR, such as pyrimethamine (PYR), have historically been used for treatment and prophylaxis against malaria. However, the treatment has been compromised due to the mutation on DHFR of the parasite which progressively develops in Plasmodium falciparum, with complete treatment failure of the drug in a quadruple mutant (N51I C59R S108N I164L) (3, 4). Recently, novel antifolate compounds have been rationally designed based on crystal structures to overcome antifolate resistance arising from DHFR mutations, with P218 as a lead compound (5).
Activity of antifolates as antimalarial drugs has been extensively studied only in the asexual blood stages due to the simplicity of in vitro drug testing assays. In contrast, antimalarial activity of antifolates against mosquito and liver stages has been investigated to a much lower extent, especially those with quadruple mutations of DHFR. This is due to the fact that Anopheles mosquito colonies are required for such experiments, and laboratory strains of quadruple mutant P. falciparum, such as the V1/S strain, cannot develop into gametocytes (6). In the rodent malaria models, pyrimethamine-resistant P. berghei and Plasmodium chabaudi strains were developed through drug pressure (7–9), but these mutations do not represent altered interactions between DHFR inhibitors and quadruple mutant P. falciparum DHFR (PfDHFR). With these limitations, there are still several knowledge gaps in the basic biology of antifolate chemopreventive activity, including (i) how much the quadruple mutations in DHFR contribute to chemoprevention resistance, (ii) whether recently developed antifolates can overcome the resistance mutations in the chemoprevention setting, (iii) whether it is necessary for the hepatic parasites to be exposed to the deployed antifolate throughout hepatic development, (iv) what specific hepatic developmental stages are susceptible to the antifolate, and (v) what the antifolate level of protection against sporozoite load is.
In this study, we used previously developed transgenic P. berghei with the wild-type dhfr (Pbdhfr) replaced by quadruple mutant Pfdhfr (10) to investigate chemoprevention activity of the previous generation as well as the new generation of antifolate compounds, represented by PYR and P218, respectively, against the quadruple mutant Pfdhfr parasite. This study also took an advantage of highly potent, short half-life properties of P218 to explore stage-specific activity of antifolate compounds against hepatic parasites. This study provides important new knowledge that can further help tailor drug administration schemes to fit malaria endemicity in targeted areas.
RESULTS
Quadruple mutation on P. falciparum dhfr gene renders PYR ineffective for chemoprevention.
We utilized a transgenic P. berghei parasite harboring P. falciparum dhfr-4M (Pb::Pfdhfr-4M parasite) developed by our group (10) to determine chemoprevention activity of PYR against the quadruple mutant parasite. We have confirmed that the transgenic Pb::Pfdhfr-4M parasite still maintains its ability to develop into mosquito and liver stages (Fig. 1).
FIG 1.
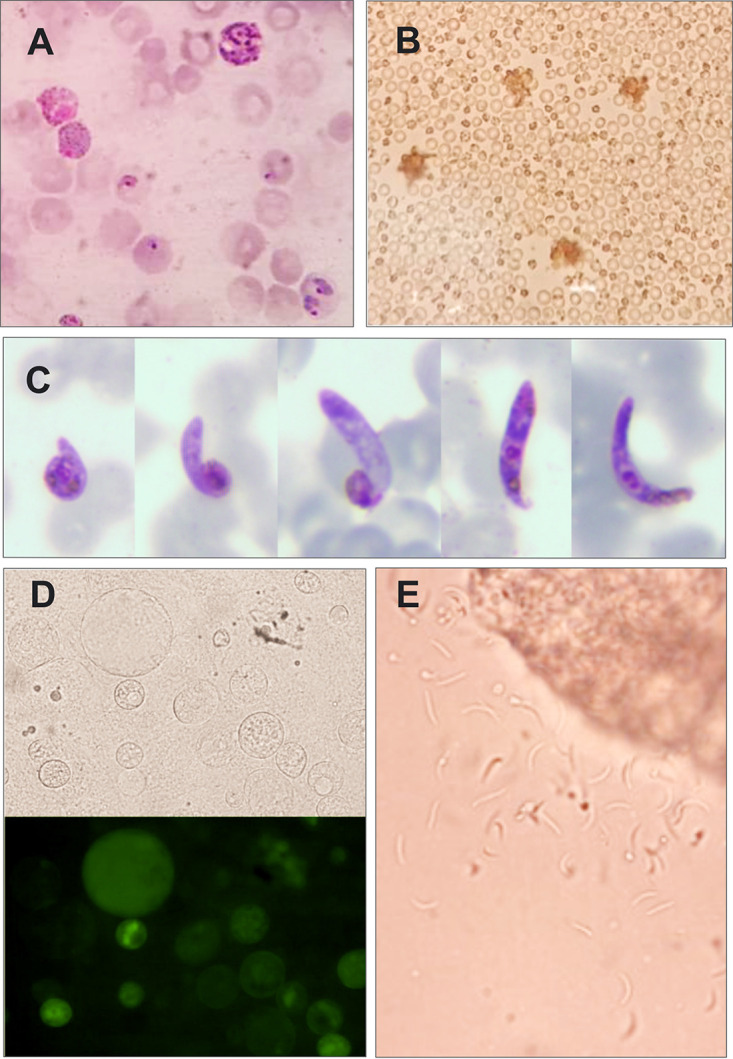
The transgenic parasite maintains an ability to develop to mosquito stages. (A) Mature male and female gametocytes; (B) male gametocyte exflagellation under bright-field microscopy; (C) Giemsa-stained ookinete; (D) midgut oocysts under bright-field and fluorescence microscopy; (E) sporozoites released from squashed salivary gland under bright-field microscopy.
First, two repeated doses of 30 mg/kg PYR were administered 1 day before and 1 day after intravenous injection of 2,000 wild-type or transgenic Pb::Pfdhfr-4M sporozoites (Fig. 2A). Using this administration scheme, the sporozoites were exposed to the drug as soon as they entered the circulatory system and also after hepatocyte invasion. All five mice in the control (carrier) group became infected by either parasite following the challenge (Fig. 2B). While 30 mg/kg PYR could inhibit wild-type P. berghei, the drug failed to prevent infection by transgenic Pb::Pfdhfr-4M, as all five mice became infected after challenge (Fig. 2B). After sporozoite injection, P. berghei growth pattern showed a peak of asexual-stage parasitemia which dropped and rose again later in the course of infection (Fig. 2B) (11). Time to the first peak of parasitemia can be delayed and parasitemia level at the first peak can be decreased upon liver stage parasite suppression. The time to peak parasitemia of all the Pb::Pfdhfr-4M-injected mice in the carrier and PYR treatment groups were 8 days, and the levels of parasitemia at the first peak were not statistically different between these groups (Fig. 2C). These results demonstrated that the quadruple mutations in Pfdhfr cause complete chemoprevention resistance to PYR, the first generation of antifolate.
FIG 2.

Quadruple mutation of the Plasmodium falciparum dhfr gene confers complete resistance to previous-generation antifolate. (A) Schematic diagram of in vivo chemoprevention assay to test activity of antifolates against wild-type and quadruple mutant Pfdhfr transgenic parasites. Five mice were used for each treatment and control group. Mice in the treatment group were orally treated with two doses of 30 mg/kg PYR or P218 1 day before and 1 day after sporozoite injection. Mice in the control group were given a carrier for drug administration using the same scheme as for the treatment group (0.5% [wt/vol] hydroxypropylmethylcellulose, 0.8% [vol/vol] Tween 80 in sterile water). (B) Asexual-stage parasitemia of the wild-type P. berghei (ANKA) and the transgenic Pb::Pfdhfr-4M 4 to 14 days after sporozoite challenge (intravenous injection with 2,000 sporozoites). (C) Box and scatterplot of the parasitemia level at the first peak of infection of the mice in the carrier and 30-mg/kg PYR groups. Each dot represents parasitemia of each individual mouse. Statistical analysis was conducted using Wilcoxon rank sum test.
Novel antifolate potently inhibits liver stage of the quadruple mutant parasite.
Using P218 as a model for a new class of antifolates, subsequent experiments established that this lead compound can inhibit liver-stage infection of both the wild-type and quadruple mutant parasites. Oral administration of 30 mg/kg P218 on day −1 and day 1 relative to sporozoite challenge prevented asexual-stage infection in both the wild-type and the transgenic PYR-resistant parasites (Fig. 2B).
After chemoprevention activity of P218 against the wild-type and quadruple mutant parasite was confirmed at 30 mg/kg, we then further investigated how much quadruple mutations alter parasite sensitivity to P218 using various doses of the drug. Our preliminary testing showed that 12-mg/kg, 5-mg/kg, and 2-mg/kg doses successfully inhibited the wild-type parasite (data not shown); therefore, the 4-fold serial dilution from 2 mg/kg was tested for the wild type. Based on the previous information on P218 dose-response curve against the asexual-stage parasite (10), the half-maximal effective dose (ED50) against the transgenic quadruple mutant Pb::Pfdhfr-4M was approximately 8 times higher than that against the wild-type parasite. Therefore, the highest dose, 16 mg/kg, was used for the transgenic parasite.
In the wild-type parasite, P218 could completely prevent parasite development into blood stage when orally treated with at least 0.5 mg/kg P218 (Fig. 3B). Although not statistically significant, the median time to peak parasitemia was extended and parasitemia at the first peak was lower in the groups treated with P218 at the dose lower than 0.5 mg/kg than for the carrier group (Fig. 3C and D). Even though complete blocking of infection was not achieved, the lower doses of P218 could improve survival rate at 14 days postinfection (dpi). At 0.125 mg/kg, none of the mice died at 14 dpi, and 2 out of 4 mice became infected (Fig. 3B). These results demonstrate that although P218 at a dose lower than 0.5 mg/kg was not able to completely prevent parasite development into blood stage, the drug could still reduce the burden of malaria.
FIG 3.

Rationally designed antifolate, P218, is highly potent for chemoprevention against both wild-type and PYR-resistant parasites. (A) Schematic diagram of in vivo dose-response analysis for chemoprevention against wild-type and quadruple Pfdhfr mutant transgenic parasites. For biological replicates, 4 or 5 mice were used in each group. Each mouse was inoculated with 2,000 wild-type and transgenic sporozoites. Mice infected by the wild-type P. berghei ANKA were orally treated with 2 doses of 0.0078, 0.031. 0.125, 0.5, or 2 mg/kg P218, while those infected with PYR-resistant parasites were orally treated with 2 doses of 0.0625, 0.25. 1, 4, or 16 mg/kg P218. The control groups were treated with a carrier used for oral drug delivery (0.5% [wt/vol] hydroxypropylmethylcellulose, 0.8% [vol/vol] Tween 80 in sterile water). (B and E) Asexual-stage parasitemia of the wild-type P. berghei (B) and the transgenic Pb::Pfdhfr-4M (E) 5 to 14 days after sporozoite challenge and treatment with various doses of P218. (C and F) Box and scatterplot representing the time to the first peak of parasitemia for the wild-type P. berghei (C) and the transgenic Pb::Pfdhfr-4M (F). (D and G) Box and scatterplot of the parasitemia level at the first peak of infection for the mice infected with the wild-type P. berghei (D) and the transgenic Pb::Pfdhfr-4M (G). Each dot represents parasitemia of each individual mouse. Statistical analyses were conducted using Kruskal-Wallis analysis of variance (ANOVA) by rank test followed by multiple-pairwise comparisons with Dunn’s post hoc test.
P218 is potent for chemoprevention against PYR-resistant parasites in vivo. At an oral dose of 1 mg/kg, P218 could completely prevent transgenic PYR-resistant P. berghei development into blood stage in all mice (Fig. 3E). The doses of 0.25 and 0.0625 mg/kg reduced the mortality at 14 dpi (Fig. 3E), demonstrating that these lower doses could reduce burden from PYR-resistant malaria, although complete blocking was not achieved. The median time to the first peak parasitemia of the groups treated with 0.25 and 0.0625 mg/kg was significantly extended (Fig. 3F), while the parasitemia at first peak was marginally lower than in the carrier group (Fig. 3G). This MIC of P218 against PYR-resistant parasite is only 2-fold higher than those of the PYR-sensitive parasite, suggesting that the rationally designed antifolate can overcome resistance from quadruple mutation in Pfdhfr and should be effective for chemoprevention against the resistant parasites prevalent in the field. However, the real-world efficacy and administration scheme in the natural infections of resistant parasite have yet to be validated.
Development from hepatic ring to trophozoite is the stage the most susceptible to antifolate.
Characteristics of P218, including high potency, rapid absorption (time to maximum concentration of drug in serum [Tmax] in mice = 0.25 h), and short half-life (oral half-life in mice = 4 h) (5), allow us to dissect the specific hepatic developmental stage that is the most susceptible to antifolate. When administered at minimum inhibitory dose, the compound should enter the circulation almost immediately and be quickly cleared from the body, thus being exposed only to that specific developmental stage. Previous studies showed that P. berghei develops from sporozoite to ring stage during the first day, ring to late trophozoite stage during the second day, and late trophozoite to schizont during the third day (Fig. 4A) (12). In the following experiment, a single dose of P218 was administered at the MIC (1 mg/kg) on various days from a day before sporozoite injection to 3 days after infection, as depicted in Fig. 4A.
FIG 4.

Development from early trophozoite to early schizonts is the hepatic stage most susceptible to antifolate. (A) Schematic diagram of an in vivo experiment to determine the hepatic stage most susceptible to antifolate. Mice were orally treated with a single dose of 1 mg/kg P218 on different days from day −1 to day 3 relative to sporozoite challenge. The oral administration on day 0 was conducted 3 h before sporozoite challenge. All mice were intravenously infected with 2,000 transgenic Pb::Pfdhfr-4M sporozoites, and 4 or 5 mice were used for each group. (B) Asexual-stage parasitemia of the transgenic Pb::Pfdhfr-4M 5 to 14 days after sporozoite challenge and treatment with a single dose of 1 mg/kg P218 on day −1 to day 3 relative to sporozoite challenge. (C) Schematic diagram of an in vivo experiment to determine the effect of an increased dose (15 mg/kg) on early- and late-hepatic-stage parasites. (D) Asexual stage parasitemia of the transgenic Pb::Pfdhfr-4M 5 to 14 days after sporozoite challenge and treatment with a single dose of 15 mg/kg P218 on day 0 (3 h before) or day 3 relative to sporozoite challenge.
A single oral dose of 1 mg/kg P218 was able to completely clear the P. berghei liver-stage infection, with no recrudescence of parasites (monitored with thin blood smear until day 28 post-sporozoite injection) when administered on day 1 (Fig. 4B), demonstrating that a single dose of P218 is sufficient for chemoprevention when administered with this scheme. The treatment with single dose of P218 on day 1 resulted in a complete prevention of parasite infection, similar to the treatment with two doses in Fig. 3.
Administration of the compound 1 day before (day −1) or 3 h before (day 0) had no impact on liver-stage infection in terms of both infection prevalence and intensity (Fig. 4B). Although all mice became infected when P218 was administered 2 or 3 days after sporozoite challenge, lower parasitemia and slower parasite development into erythrocytic stage were observed (Fig. 4B). These results suggested that the hepatic developmental stage 1 day after sporozoite challenge, namely, from ring to late trophozoite, is the developmental stage most susceptible to P218.
The maximum dose of P218 used during clinical development in humans was 1,000 mg, which could be translated into 13 to 14 mg/kg (13, 14). Therefore, we next investigated if a single oral administration of 15 mg/kg P218 could improve the chemoprevention when the compound was treated on the nonoptimal days (same day and 3 days after sporozoite challenge) as demonstrated in Fig. 4C. Although the increased dose of P218 administered 3 days after sporozoite challenge could not improve chemoprevention compared with the treatment at 1 mg/kg, the increased dose prevented infection of all the mice when administered on the day of sporozoite challenge (Fig. 4D). These results demonstrate that the increased dose of P218 could prolong the chemoprevention activity of P218 administered earlier than the optimum time and emphasize that chemoprevention activity of antifolates is the most active against the intrahepatic ring to trophozoite developmental stage.
P218 could prevent infection by a large number of PYR-resistant sporozoites.
Information on how many sporozoites the chemopreventive drug can protect against will be beneficial to tailor drug administration schemes in regions where malaria is endemic and determine the protection in relation to the number of potential infectious bites in the areas of endemicity. In the subsequent experiment, mice were infected with 5,000, 10,000, and 25,000 transgenic PYR-resistant P. berghei sporozoites to determine if P218 is potent enough in chemoprevention against high numbers of PYR-resistant sporozoites (Fig. 5A). With the single oral dose of 15 mg/kg on day 1, P218 completely prevented infection in all mice infected with up to 10,000 transgenic sporozoites (Fig. 5B). In the group injected with 25,000 transgenic sporozoites, no blood-stage parasites were detected in four out of five mice, suggesting a borderline limit of chemoprevention at this number (Fig. 5B).
FIG 5.

P218 prevents infection by a large number of PYR-resistant sporozoites. (A) Schematic diagram of an in vivo experiment to determine the number of PYR-resistant sporozoites for which 15 mg/kg P218 can completely prevent infection. Mice were intravenously infected with 5,000, 10,000, and 25,000 sporozoites and then orally treated with 15 mg/kg P218 1 day after the challenge. The control group was infected with 2,000 sporozoites, and then a carrier used for oral delivery was administered 1 day after the challenge. (B) Asexual-stage parasitemia of the transgenic Pb::Pfdhfr-4M 5 to 14 days after a challenge with 5,000, 10,000, or 25,000 sporozoites followed by an oral dose of 15 mg/kg on the next day.
DISCUSSION
The resistance to antifolates, such as pyrimethamine, has long been a problem, and it started soon after the drug was introduced many decades ago. A recent study demonstrated that the resistant parasites persist in the areas where malaria is endemic even after the long-term withdrawal of antifolate use (15). Although the hepatic development and blood-stage development follow the same pattern from rings to trophozoites and schizonts, the hepatic schizogeny contains many more rounds of DNA replication and results in a much larger number of merozoites (16). It is possible that this might lead to a difference in susceptibility to antifolate between the hepatic and blood stages, especially in the antifolate-resistant parasites.
This study sought to characterize antifolates as chemopreventive antimalarial drugs, especially against the quadruple mutant Pfdhfr parasite. Using transgenic P. berghei harboring quadruple mutant Pfdhfr in place of the wild-type Pbdhfr as an in vivo antifolate-resistant model, we demonstrated that the quadruple mutation on Pfdhfr not only leads to PYR treatment failure (10) but also confers complete resistance to PYR chemoprevention. Antifolates of the previous generation are then no longer effective for both applications in most areas where malaria is endemic, thus prompting the development of alternative treatments. The benefit of having a well-known target is that it allows characterization of the resistance mechanism and rational design of new classes of antifolates that can overcome the resistance (5). Although the new class of antifolates, such as P218, can inhibit asexual-stage and male gametocyte exflagellation of quadruple mutant parasites (5, 6, 10), how potent these new compounds are for the chemoprevention activity against quadruple mutant parasites has never been experimentally demonstrated due to the lack of a reliable liver-stage model of the resistant parasite.
Our current study demonstrates a high potency of P218 against the PYR-resistant parasite both in terms of drug concentration required to completely inhibit the liver-stage parasite and in terms of the number of sporozoites it can inhibit. In our model, the quadruple mutation of the Pfdhfr gene resulted in an approximately 2-fold increase in P218 minimum inhibitory dose required for complete chemoprevention, from 0.5 mg/kg for the sensitive parasite to 1 mg/kg for the quadruple mutant, suggesting that the rationally designed compound can overcome resistance in chemoprevention. It should be noted that our experiments used the wild-type parasite carrying P. berghei dhfr, not the wild-type P. falciparum dhfr. However, previous studies showed that P218 has comparable blood-stage inhibitory concentrations against the wild-type P. berghei (0.9 nM [17]) and wild-type P. falciparum (1 nM [Medicines for Malaria Venture, personal communication]). Therefore, we speculate that the chemoprevention minimum inhibitory dose obtained from the wild-type P. berghei can be representative of a parasite harboring wild-type Pfdhfr.
Detailed understanding of the chemoprevention activity of antifolate allows optimization of the drug administration scheme for the most cost-effective and efficient malaria control in areas where malaria is endemic. P218 administration at 3 h before sporozoite injection exposed two parasite stages to this potent antifolate compound: sporozoites, which were immediately exposed to this potent antifolate compound in the circulation, and early hepatic parasites, during the development from sporozoite to hepatic ring stage. Despite these exposures during the first day, the parasite could complete its intrahepatic development and the outcome of infection was not different from that in the untreated group, suggesting that antifolate compounds are not active against sporozoites and the very early intrahepatic stages. During approximately the first 20 h after the invasion of the hepatocyte, P. berghei develops into hepatic ring and early trophozoite, and the parasites remain haploid with a single nucleus in G1 phase (18). Since antifolate compounds target the pathway involved in DNA synthesis, these compounds cannot inhibit these nonreplicative early hepatic stages.
Around 20 h after sporozoite invasion into hepatocytes, the parasites develop into trophozoites as the cell cycle stage goes from G1 to M1 phase. During trophozoite development, each single parasite continue DNA synthesis and nuclear division to generate 20,000 to 30,000 new parasites in approximately 30 h (18). Consequently, DNA synthesis and nucleotide metabolism crucial for this developmental stage. P218 treatment at 1 day (24 h) after P. berghei sporozoite injection completely inhibited parasite development into blood stage, confirming that the development from trophozoite to schizonts (schizogeny) stage is extremely susceptible to antifolate due to their highly replicative nature. Interestingly, although P218 was cleared from the circulation in the following days due to its short half-life, no blood-stage parasite was detected in any of the treated mice. This suggests that if the parasite cannot accumulate enough DNA, they cannot stay dormant and quickly perish. This might be due to the parasite undergoing several checkpoints prior to schizogeny, including S and G2 checkpoints (check for successful DNA replication or DNA damage) as well as M checkpoint (check for chromosome attachment to the spindle) (16).
Once the hepatic schizogeny is completed or almost completed, the antifolate no longer affects the parasite’s ability to further progress to blood stages as demonstrated by the fact that P218 could no longer inhibit the matured hepatic parasite even at an increased dose (15 mg/kg). Due to a much higher number of parasites to be inhibited after progression to blood stage, multiple doses of P218 are needed for malaria treatment, as demonstrated in previous studies (10, 14).
It is interesting that the folate pathway is also involved in other biological processes, such as methionine synthesis, but the compounds could not inhibit specific developmental stages with high protein synthesis, such as the development from ring to trophozoite. Any other antifolate inhibitory activity against other biological processes during hepatic development is likely to be negligible to be considered as an additional mode of action of the compounds. Our study is consistent with previous findings (19) that antifolates mainly target the parasites’ DNA synthesis during blood-stage development.
This study provides insights into the nature of chemoprevention of P218 that complement data from a P218 clinical trial on sporozoite challenge (14) that may be beneficial in improving drug regimes in clinical use. First, the clinical trial by Chughlay et al. (14) stated that it was unclear how much chemoprevention activity of P218 was due to activity against liver-stage versus activity against blood-stage parasites. Our stage-specific experiment clearly demonstrated that the activity against the hepatic parasite is the main contributor to chemoprevention. Second, our study demonstrated that P218 is highly active against the quadruple mutant parasite, thus providing data to support its potential use in the real world, where resistant parasites are predominant. Third, this study confirms that antifolates have superior potency against the liver stage compared to the blood stage. We previously demonstrated that 10 mg/kg P218 was required to completely clear asexual-stage parasites using the standard 4-day suppressive test (10), while only a single dose of 1 mg/kg P218 is required for complete chemoprevention. It is possible that the higher sensitivity is due to a combination of more rounds of endomitosis during hepatic development and the lower number of parasites to be inhibited. Fourth, the P218 plasma concentration required for chemoprevention in the real-world setting might be overestimated due to the number of sporozoites used for challenge in most preclinical and clinical studies. Our study demonstrated a correlation between dose required for chemoprevention and the number of sporozoites during the infection: 1 mg/kg P218 could prevent infection from 2,000 sporozoites and 15 mg/kg P218 could prevent infection from 25,000 sporozoites. During transmission from infected mosquitoes to humans, the majority of mosquitoes ejected, on average, 10 sporozoites per bite (20). Thus, it is likely that even lower doses are required for complete chemoprevention.
Lastly, the stage-specific experiment agrees with the clinical trial that the longer exposure of P218 throughout hepatic development is not necessary to maintain chemoprevention. In the real-world setting, it is impossible to predict when each person will be bitten by infectious mosquitoes. When translated into clinical use, the candidate drug might be administered at longer intervals that have potential to target the development from hepatic ring to early schizonts. In addition, since P218 is highly effective against the parasite and does not require high plasma concentration for chemoprevention, it will be of greater benefit to develop a slow-release formulation that can sustain moderate levels of the candidate drug over time.
One interesting additional application of new antifolates, especially P218, is to use these drugs to assist immunization by attenuated sporozoites, which has proved to provide a long-lasting immunity against malaria, thus offering a potential vaccine candidate. The attenuation of sporozoites has been achieved by irradiation or genetic modification, but previous reports also demonstrated a potential to use chemopreventive drugs such as primaquine or pyrimethamine to attenuate the hepatic parasite (21). Since the P218 is safe and highly potent in suppressing a large number of liver-stage parasites, it could potentially be used in such an application. Our results demonstrated that P218 did not kill sporozoites but rather inhibited development of hepatic stages. Consequently, parasites are still able to invade hepatocytes and are attenuated during intrahepatic development. These attenuated parasites in infected cells are likely to be processed through the antigen presentation by class I major histocompatibility complex (MHC) and triggers cytotoxic T-cell responses, an important part of antimalarial immunity (22). It will be interesting to compare immune responses between irradiated sporozoites as well as other sporozoite-targeted vaccines to the infection by active sporozoites that could enter the hepatic stage and subsequently be killed by P218 treatment.
MATERIALS AND METHODS
Ethics statement.
This study was strictly carried out in accordance with the Guide for the Care and Use of Laboratory Animals (23) and Thailand’s National Research Council. The animal protocol was approved by the BIOTEC Institutional Animal Care and Use Committee (permit numbers BT-Animal 27/2562 and 08/2564).
Animal and parasite strains.
Healthy 8-week-old female ICR mice were used for all the experiments in this study. The mice were obtained from the National Laboratory Animal Center, Mahidol University, Thailand. The mice were acclimatized for at least 3 days before experiments and were maintained at 22 ± 2°C with a 12/12-h light/dark cycle. Water and food were provided ad libitum.
The wild-type Pbdhfr P. berghei ANKA strain 676m1cl1 (MRA-868) was obtained through BEI Resources, NIAID, NIH, contributed by Chris J. Janse and Andrew P. Waters. P. berghei with a quadruple dhfr mutant from P. falciparum replacing the wild-type dhfr was generated in a previous study (10).
Laboratory strains of Anopheles dirus were originally collected in Mae Sod District, Tak Province, Thailand. A colony of A. dirus was maintained at the Department of Parasitology, Chiang Mai University, and BIOTEC’s insectary at 27°C with 70% humidity and a 12-h day/night, 30-min dusk/dawn lighting cycle. The larvae were fed a diet of powdered fish food (Tetrabit, Germany). Adults were maintained on a 10% sucrose solution supplemented with 5% multivitamin syrup (Seven Seas, UK) ad libitum (24).
P. berghei propagation.
Asexual stages of the P. berghei ANKA 676m1cl1 expressing green fluorescent protein (GFP)-luciferase (wild-type P. berghei dhfr [MRA-868]) and transgenic P. berghei ANKA Pb::Pfdhfr-4M harboring quadruple mutant Pfdhfr were propagated by passaging in ICR mice by intraperitoneal (i.p.) injection of 150 μL frozen glycerol parasite stock. These parasites were passaged in mice for no longer than 8 generations, without passaging in mosquito to ensure that the parasites maintained mosquito infectivity. To collect frozen parasite stocks, parasitemia in mice was monitored by thin blood smears daily until reaching 10 to 20%. Parasitized blood was then collected by cardiac puncture, mixed with 0.1 volume of 0.25 mM heparin and an equal volume of freezing medium (sterile 30% glycerol in phosphate-buffered saline [PBS]), and then stored in liquid N2.
P. berghei sporozoite culture.
To start P. berghei sporozoite culture, 150 μL frozen P. berghei stock was thawed at room temperature and then i.p. injected into a mouse. On the same day, a second mouse was i.p. injected with 0.2 mL 6-mg/mL phenylhydrazine (Sigma, USA; catalog no. P26252) to stimulate reticulocyte production. The parasitemia and gametocytemia were monitored until reaching 10 to 15% (from days 3 to 5 postinfection). Infected blood was then collected using heparin as an anticoagulant and then diluted to a concentration of 5 × 107 infected red blood cells/mL in 1× PBS buffer. The phenylhydrazine-treated mouse was then infected with 5 × 106 infected red blood cells. Exflagellation of the parasite in the second mouse was determined by mixing 2 μL blood collected from the tail vein in 37 μL exflagellation medium (RPMI 1640 supplemented with 25 mM HEPES [Sigma; catalog no. H4034], 2 mM glutamine [Sigma; catalog no. G7513], 100 mM xanthurenic acid [Sigma; catalog no. D120804]) and then incubating the mixture for 10 min at room temperature. Exflagellation foci were then counted under a light-contrast microscope. Mice with 10 to 15% parasitemia and >20 exflagellation sites per field of view (×100 magnification) were used for mosquito feeding.
To prepare mosquitoes for blood feeding, female A. dirus mosquitoes were separated into a waxed paper cup and then starved overnight. To infect mosquitoes, an infected mouse was anesthetized with 0.6 mL 2% 2,2,2-tribromoethanol (Sigma; catalog no. T48402). When the mouse was nonresponsive after footpad pinching, it was placed on top of the mosquito cup, and the mosquitoes were allowed to feed for 30 to 45 min at 19 ± 1°C. Blood-fed mosquitoes were then aspirated into a new cup and then provided with 10% sucrose supplemented with 0.05% para-aminobenzoic acid (PABA; Sigma; catalog no. A9878) and maintained at 19 ± 1°C. On days 8 to 10, GFP fluorescent oocysts in the bloodfed mosquitoes were observed under a fluorescence microscope. Salivary glands containing sporozoites were then dissected between 20 and 23 days after infectious bloodmeal, similar to previously published protocols (25, 26). Briefly, infected mosquitoes were surface sterilized with 70% ethanol and then washed twice with 1× PBS. Salivary glands were then dissected in a drop of 1× PBS and then collectively transferred to 100 μL ice-cold Schneider’s insect cell culture medium without NaHCO3 (Sigma; catalog no. S9895). Sporozoites were then released from salivary glands by gentle grinding with a sterile plastic pestle, followed by pipetting 100 times. The sporozoites were then counted with a hemacytometer before dilution to the desired number in an incomplete RPMI 1640 medium for intravenous injection.
In vivo antimalarial chemoprevention assay.
Chemoprevention activity of antifolates was conducted by measuring the ability of the drugs to prevent asexual-stage development in mice. Briefly, antifolates such as PYR and P218 were prepared in a drug carrier (0.5% [wt/vol] hydroxypropylmethylcellulose [HPMC], 0.8% [vol/vol] Tween 80 in sterile water) and then orally administered following a regime specific to each experiment, and 3 to 5 mice were used as biological replicates for each experiment. Sporozoites were inoculated into mice via the tail vein with the number as specified in each experiment. Drug carrier (0.5% HPMC and 0.4% Tween 80 in water) was used as an untreated control of each experiment. To determine asexual-stage development, a Giemsa-stained thin blood smear of each mouse was monitored between 4 to 14 days after sporozoite injection. Time to first peak of asexual-stage parasitemia after sporozoite injection and parasitemia at the first peak were used to compare chemopreventive activities between the treatment and carrier groups. Statistical analyses were conducted using Kruskal-Wallis analysis of variance (ANOVA) by rank test followed by Dunn’s post hoc multiple-pairwise-comparisons test with the rstatix package, version 0.7.0.999 in R.
ACKNOWLEDGMENTS
This work was financially supported by a research chair grant (P-1850116) from the National Science and Technology Development Agency (NSTDA), Thailand, to S.K., and a BIOTEC research unit director initiative grant to N.J. (P-1851424).
P218 compound was kindly provided by Medicine for Malaria Venture (MMV). P. berghei ANKA 676m1cl1 expressing GFP-luciferase (MRA-868), contributed by Chris J. Janse and Andrew P. Waters, was obtained through BEI Resources, NIAID, NIH. We also thank Warangkhana Songsungthong for assistance with the initial setup of the insectary and P. berghei culture. We are grateful to Patchara Sriwichai from the Department of Medical Entomology, Faculty of Tropical Medicine, Mahidol University, who kindly provided A. dirus adults used to establish the colonies used in this study to A. Saeung. We also thank José L. Ramirez from the USDA for editorial assistance with the manuscript.
We declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Contributor Information
Sumalee Kamchonwongpaisan, Email: sumaleek@biotec.or.th.
Natapong Jupatanakul, Email: natapong.jup@biotec.or.th.
REFERENCES
- 1.World Health Organization. 2020. World malaria report 2020. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Cowman AF, Healer J, Marapana D, Marsh K. 2016. Malaria: biology and disease. Cell 167:610–624. 10.1016/j.cell.2016.07.055. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed A, Das MK, Dev V, Saifi MA, Wajihullah, Sharma YD. 2006. Quadruple mutations in dihydrofolate reductase of Plasmodium falciparum isolates from Car Nicobar Island, India. Antimicrob Agents Chemother 50:1546–1549. 10.1128/AAC.50.4.1546-1549.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sirawaraporn W. 1998. Dihydrofolate reductase and antifolate resistance in malaria. Drug Resist Updat 1:397–406. 10.1016/S1368-7646(98)80015-0. [DOI] [PubMed] [Google Scholar]
- 5.Yuthavong Y, Tarnchompoo B, Vilaivan T, Chitnumsub P, Kamchonwongpaisan S, Charman SA, McLennan DN, White KL, Vivas L, Bongard E, Thongphanchang C, Taweechai S, Vanichtanankul J, Rattanajak R, Arwon U, Fantauzzi P, Yuvaniyama J, Charman WN, Matthews D. 2012. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci USA 109:16823–16828. 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Posayapisit N, Pengon J, Prommana P, Shoram M, Yuthavong Y, Uthaipibull C, Kamchonwongpaisan S, Jupatanakul N. 2021. Transgenic pyrimethamine-resistant Plasmodium falciparum reveals transmission-blocking potency of P218, a novel antifolate candidate drug. Int J Parasitol 51:635–642. 10.1016/j.ijpara.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Hayton K, Ranford-Cartwright LC, Walliker D. 2002. Sulfadoxine-pyrimethamine resistance in the rodent malaria parasite Plasmodium chabaudi. Antimicrob Agents Chemother 46:2482–2489. 10.1128/AAC.46.8.2482-2489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nuralitha S, Siregar JE, Syafruddin D, Roelands J, Verhoef J, Hoepelman AIM, Marzuki S. 2016. Within-host selection of drug resistance in a mouse model of repeated incomplete malaria treatment: comparison between atovaquone and pyrimethamine. Antimicrob Agents Chemother 60:258–263. 10.1128/AAC.00538-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Dijk MR, McConkey GA, Vinkenoog R, Waters AP, Janse CJ. 1994. Mechanisms of pyrimethamine resistance in two different strains of Plasmodium berghei. Mol Biochem Parasitol 68:167–171. 10.1016/0166-6851(94)00163-4. [DOI] [PubMed] [Google Scholar]
- 10.Koonyosying P, Jupatanakul N, Vanichtanankul J, Saeyang T, Pethrak C, Pengon J, Tipsuwan W, Yuthavong Y, Kamchonwongpaisan S, Uthaipibull C. 2020. Transgenic pyrimethamine resistant Plasmodium berghei as a model for in vivo anti-DHFR drug testing. bioRxiv 2020.09.30.281055.
- 11.Conteh S, Anderson C, Lambert L, Orr-Gonzalez S, Herrod J, Robbins YL, Carter D, Bin Shamamba Karhemere S, Pyana P, Büscher P, Duffy PE. 2017. Grammomys surdaster, the natural host for Plasmodium berghei parasites, as a model to study whole-organism vaccines against malaria. Am J Trop Med Hyg 96:835–841. 10.4269/ajtmh.16-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng S, Schwartz RE, March S, Galstian A, Gural N, Shan J, Prabhu M, Mota MM, Bhatia SN. 2015. Human iPSC-derived hepatocyte-like cells support Plasmodium liver-stage infection in vitro. Stem Cell Rep 4:348–359. 10.1016/j.stemcr.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chughlay MF, Rossignol E, Donini C, Gaaloul ME, Lorch U, Coates S, Langdon G, Hammond T, Möhrle J, Chalon S. 2020. First-in-human clinical trial to assess the safety, tolerability and pharmacokinetics of P218, a novel candidate for malaria chemoprotection. Br J Clin Pharmacol 86:1113–1124. 10.1111/bcp.14219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chughlay MF, El Gaaloul M, Donini C, Campo B, Berghmans P-J, Lucardie A, Marx MW, Cherkaoui-Rbati MH, Langdon G, Angulo-Barturen I, Viera S, Rosanas-Urgell A, Van Geertruyden J-P, Chalon S. 2021. Chemoprotective antimalarial activity of P218 against Plasmodium falciparum: a randomized, placebo-controlled volunteer infection study. Am J Trop Med Hyg 104:1348–1358. 10.4269/ajtmh.20-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugaram R, Suwannasin K, Kunasol C, Mathema VB, Day NPJ, Sudathip P, Prempree P, Dondorp AM, Imwong M. 2020. Molecular characterization of Plasmodium falciparum antifolate resistance markers in Thailand between 2008 and 2016. Malar J 19:107. 10.1186/s12936-020-03176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthews H, Duffy CW, Merrick CJ. 2018. Checks and balances? DNA replication and the cell cycle in Plasmodium. Parasit Vectors 11:216. 10.1186/s13071-018-2800-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swann J, Corey V, Scherer CA, Kato N, Comer E, Maetani M, Antonova-Koch Y, Reimer C, Gagaring K, Ibanez M, Plouffe D, Zeeman A-M, Kocken CHM, McNamara CW, Schreiber SL, Campo B, Winzeler EA, Meister S. 2016. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect Dis 2:281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graewe S, Stanway RR, Rennenberg A, Heussler VT. 2012. Chronicle of a death foretold: Plasmodium liver stage parasites decide on the fate of the host cell. FEMS Microbiol Rev 36:111–130. 10.1111/j.1574-6976.2011.00297.x. [DOI] [PubMed] [Google Scholar]
- 19.Dieckmann A, Jung A. 1986. Stage-specific sensitivity of Plasmodium falciparum to antifolates. Z Parasitenkd 72:591–594. 10.1007/BF00925479. [DOI] [PubMed] [Google Scholar]
- 20.Graumans W, Jacobs E, Bousema T, Sinnis P. 2020. When is a Plasmodium-infected mosquito an infectious mosquito? Trends Parasitol 36:705–716. 10.1016/j.pt.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesen J, Borrmann S, Matuschewski K. 2011. Induction of antimalaria immunity by pyrimethamine prophylaxis during exposure to sporozoites is curtailed by parasite resistance. Antimicrob Agents Chemother 55:2760–2767. 10.1128/AAC.01717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurup SP, Butler NS, Harty JT. 2019. T cell-mediated immunity to malaria. Nat Rev Immunol 19:457–471. 10.1038/s41577-019-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 24.Choochote W, Saeung A. 2013. Systematic techniques for the recognition of Anopheles species complexes, p 57-79. In Manguin S (ed), Anopheles mosquitoes—new insights into malaria vectors. InTech, Rijeka, Croatia. [Google Scholar]
- 25.Roth A, Adapa SR, Zhang M, Liao X, Saxena V, Goffe R, Li S, Ubalee R, Saggu GS, Pala ZR, Garg S, Davidson S, Jiang RHY, Adams JH. 2018. Unraveling the Plasmodium vivax sporozoite transcriptional journey from mosquito vector to human host. Sci Rep 8:12183. 10.1038/s41598-018-30713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth A, Maher SP, Conway AJ, Ubalee R, Chaumeau V, Andolina C, Kaba SA, Vantaux A, Bakowski MA, Thomson-Luque R, Adapa SR, Singh N, Barnes SJ, Cooper CA, Rouillier M, McNamara CW, Mikolajczak SA, Sather N, Witkowski B, Campo B, Kappe SHI, Lanar DE, Nosten F, Davidson S, Jiang RHY, Kyle DE, Adams JH. 2018. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat Commun 9:1837. 10.1038/s41467-018-04221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]