

Abstract
Atherosclerosis is initiated by the accumulation of lipids in the arterial wall that trigger a complex and poorly understood network of inflammatory processes. At the same time, recent clinical findings reveal that targeting specific immune alterations in patients with cardiovascular disease (CVD) represents a promising approach to preventing recurrent cardiovascular events. In order to achieve these tailored therapies, it is critical to resolve the heterogenous environment of the atherosclerotic lesion and decipher the complex structural and functional changes which immune cells undergo throughout disease progression. Recently, single-cell approaches including single cell mass cytometry by time of flight (CyTOF), single cell RNA sequencing (scRNA-seq) and Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) have emerged as valuable tools in resolving cellular plasticity within atherosclerotic lesions. In this review, we will discuss the most important insights that have been gleaned from the application of these single-cell approaches to validated experimental models of atherosclerosis. Additionally, as clinical progress in treatment of the disease depends on the translation of discoveries to human tissues, we will also examine the challenges associated with the application of single-cell approaches to human vascular tissue and the discoveries made by the initial efforts in this direction. Finally, we will analyze the advantages and limitations of dissociative single-cell approaches and how novel in-situ technologies could advance the field by allowing for the investigation of individual cells while preserving the heterogenous architecture of the atherosclerotic lesion.
Graphical Abstract: Single-cell Analysis Reveals Novel Cellular Plasticity within the Atherosclerotic Lesion
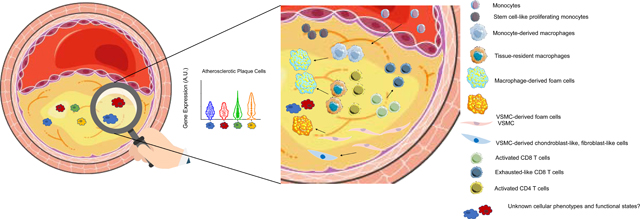
1. Introduction
The destabilization of vulnerable atherosclerotic plaques is the primary mechanism behind two of the most prevalent causes of cardiovascular death: heart attack and stroke [1]. Atherosclerosis is an inflammatory disease affecting focal regions of the arterial vasculature and is principally characterized by the accumulation of low-density lipoprotein (LDL) particles in the intima and migration of leukocytes across a dysfunctional endothelial monolayer [2]. The atherosclerotic lesion site becomes a complex microenvironment comprising heterogeneous immune cell populations and secreted products including cytokines and collagenous extracellular matrix [3]. Specialized immune cells play intricate roles in atherogenesis. Macrophages are a professional clean-up crew and ingest the accumulated LDL particles to form foam cells that gradually expire, leading to the development of a necrotic core [4–6]. Interestingly, these macrophages stem from multiple sources including vascular tissue resident populations, infiltrating blood-borne monocytes, and possibly from trans-differentiating vascular smooth muscle cells (VSMCs) [7–10]. Although most research has focused on this macrophage-driven process as the fundamental mechanism underlying atherogenesis, the tremendous diversity and abundance of other immune cells infiltrating the lesion site, including significant populations of T and B cells [11, 12], suggests that a broader network of cells govern the atherosclerotic niche. The impact of these cellular network on plaque growth and vulnerability and the molecular mechanisms through which they affect human disease are not fully understood.
Targeting inflammation to treat atherosclerosis is now a viable option that was underscored by results of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) and more recently confirmed by the Colchicine Cardiovascular Outcomes Trial (COLCOT) [13, 14]. The CANTOS study demonstrated that inhibiting interleukin-1β (IL1β) reduced cardiovascular (CV) events in high-risk post-myocardial infarction (MI) patients, and the more recent COLCOT study confirmed a key role for inflammation in driving cardiovascular events in high-risk patients. Collectively, these trials highlight the importance of inflammation in triggering cardiovascular events like heart attack and stroke. However, recent clinical trials also highlight the need for nuanced applications of anti-inflammatory medications in CVD. First, the negative results of the Cardiovascular Inflammation Reduction Trial (CIRT)[15], which broadly targeted inflammation using low dose of methotrexate, demonstrated that not all immunotherapies are atheroprotective. Second, results from the LoDoCo and LoDoCo2 trials show that low dose colchicine lowered the risk of composite adverse cardiovascular events among the study population but increased the risk of non-cardiovascular mortality [16, 17]. Finally, the Australian COPS Randomized Clinical Trial showed a higher mortality rate and no benefits on cardiovascular outcomes at 12 months in patients with acute coronary syndrome treated with colchicine [18].
Therefore, unravelling the complex immune environment within atherosclerotic lesions, and detailed phenotypic and functional differences between cellular subpopulations that may yield clinical relevance, is urgently needed. Indeed, recent studies have suggested complex roles not only for macrophages but also for CD4+, CD8+ T cells and VSMCs that may play pro- or anti- atherogenic roles within atherosclerotic plaques. Collectively, these findings suggest that understanding the functional diversity of subsets of distinct cell populations is key to deciphering their specific role in the disease [9, 19, 20].
To phenotypically and functionally characterize the immune cell diversity of the atherosclerotic niche, a need has emerged for high-dimensional analytical approaches capable of resolving the cellular composition of complex, heterogenous tissues. While traditional approaches such as immunohistochemistry [21] and flow cytometry [22–24] played integral roles in uncovering distinct immune subsets in atherosclerotic plaques, they are limited to the analysis of few markers and lack the resolution and throughput to fully dissect the heterogeneous cellular composition of tissues and detect phenotypic and functional differences between individual cells. Innovative single-cell approaches provide this resolution and represent a new and complementary way to study atherosclerosis. Technologies such as CyTOF [25], scRNA-seq [26], and CITE-Seq [27] allow for high-dimensional transcriptomics and proteomics with unprecedented sensitivity. These technologies are readily used to illuminate the diversity of cellular functions in blood and in specific tissue contexts and yield new insights into how cells communicate through cell-cell interactions [28, 29], particularly in other research fields like cancer [30–32]. Importantly, single-cell approaches are just beginning to surface in the context of atherosclerosis.
Although still at their infancy in the field of cardiovascular disease, single-cell analyses hold the potential to transform the way we characterize immune processes within experimental and human atherosclerotic lesions. These technologies promise to identify new cell targets and signaling pathways and discover molecular mechanisms of cell-cell communication that shape proatherogenic states. This knowledge has the potential to guide the development of molecularly targeted immunotherapies to treat inflammation in atherosclerosis and reduce the residual cardiovascular risk of optimally treated patients.
Single-cell technologies and analytical methodologies, which include preprocessing, visualization, clustering, differential gene expression analysis and other computational tools like trajectory and pseudotime analyses —which enable the study of dynamic changes in gene expression during cell differentiation— have been comprehensively described elsewhere [33–35], and an in-depth discussion of existing pipelines is beyond the scope of this review. Here, we summarize recent advances in single-cell analyses that are directly applicable to atherosclerosis. Furthermore, we describe findings from early efforts in this area and highlight opportunities for these approaches to uncover novel immunological processes underlying atherogenesis and plaque rupture.
2. Single-cell advances in proteomics and transcriptomics
The breakthrough of single-cell methods is now reinventing the way we define biological states of cells and provides several advantages for understanding both the proteome and transcriptome. The high parametrization and throughput of these technologies allows for the in-depth characterization of low-abundance cell populations and the unbiased identification of pathways or targets previously masked using bulk methods. Furthermore, because these single-cell technologies require relatively few cells to operate, they are ideal for working with limited sample sizes, such as patient biopsies, and support direct bench-to-patient personalized medicine. Here we will briefly discuss the most prevalent and modern techniques (Table 1).
Table 1:
Dissociative single-cell approaches
| Method | Type of data | Parameters | Flow rate | References |
|---|---|---|---|---|
| Flow Cytometry | Proteomic | 4 to 18 | ≤10,000 cells / sec | [36] |
| Cytek Aurora | Proteomic | 40 parameters | 35,000 events / sec | [37] |
| CyTOF | Proteomic | 40+ parameters | ~250–500 cells/sec | [25] |
| SCoPE-MS | Proteomic | 1000+ | [40] | |
| Method | Type of data | Cell isolation | Minimum cell requirement | |
|
| ||||
| SMART-seq2 | Transcriptomic | FACS | None | [46] |
| MARS-seq | Transcriptomic | FACS | None | [46] |
| 10X Chromium | Transcriptomic | 10X Controller | [46, 49] | |
| Method | Type of data | Parameters | ||
|
| ||||
| CITE-seq | Proteomic + Transcriptomic | Dependent on commercial availability of antibodies | [27] | |
| REAP-seq | Proteomic + Transcriptomic | Dependent on commercial availability of antibodies | [54] | |
2.1. scProteomics
Flow cytometry and CyTOF are among the most popular single-cell approaches and allow researchers to gather proteomic data from individual cells. Flow cytometry has seen significant advances in recent years that have increased the number of parameters that can be simultaneously analyzed up to 40 [36, 37]. Additionally, the simultaneous development of antibodies tagged with transition metal isotopes allows CyTOF to detect more than 40 parameters at single-cell resolution while dramatically reducing spectral spillover between channels [25, 38]. The web-based platform Cytobank [39] incorporates several computational methods used to analyze CyTOF data including traditional gating, viSNE, FlowSOM, SPADE, and CITRUS, providing a user-friendly platform for bench scientists.
Despite robust advances in antibody-based approaches to single-cell proteomics, these methods are nonetheless limited by the commercial availability of antibodies, epitope accessibility, and permeability of target cells [40, 41]. Alternatively, Single Cell Proteomics by Mass Spectrometry (SCoPE-MS) is a new method to detect proteomic heterogeneity of single-cells and has the capacity to quantify thousands of proteins per cell. SCoPE-MS achieves single-cell proteomics by introducing a “carrier” channel comprising protein from roughly 200 cells, which allows for peptide sequence identification and subsequent single-cell proteomic analyses of individual cells in 8 remaining channels [40]. Though this technology is in early stages of development and is currently limited by high costs per cell, early applications have produced promising results and resolved proteomic heterogeneity among differentiating mouse embryonic stem cells [40]. Furthermore, the developers, Budnik et al., report that the underlying technologies can be extended to detect most proteins within mammalian cells [40]. Recently, improvements to this platform have been proposed with novel SCoPE2 technology, which aims to reduce the operating costs and increase the quantities of cells and proteins analyzed [42]. An early application of this approach demonstrated that the macrophage proteome spans a continuous gradient between cellular phenotypes [42].
2.2. scTranscriptomics
In addition to advances in proteomics, single-cell approaches are also increasing the sensitivity and granularity with which researchers can investigate transcriptomic physiological changes and dysregulations associated with disease. In traditional bulk RNA sequencing (RNA-seq), tissue is homogenized, and the extracted RNA represents an average of millions of cellular transcriptomes. Therefore, it is difficult to identify transcriptional differences between individual cells, and transcripts of low abundance cells may be undetected through this approach [43]. In the last decade however, the development of single-cell RNA-seq technologies has offered a solution to this issue allowing sequencing of individual cells. These transcriptional data are not only able to reveal functional differences within populations of heterogeneous cells based on canonical markers but also among cells bearing homogenous proteomes [44].
Currently, numerous scRNA-seq techniques exist, many of which proceed through a common methodology in which individual cells are isolated and lysed. This releases cellular RNA which is converted to cDNA by reverse transcription. Finally, sequencing libraries are created by amplifying this cDNA [45]. Despite these similarities, there are important distinctions between different scRNA-seq approaches. SMART-seq2 and MARS-seq incorporate flow cytometry (FACS) to isolate cells prior to transcriptomic analysis [46]. This upstream sorting allows researchers to precisely select cells based on viability, size, shape, canonical surface markers and desired number; however, this process is also mechanically strenuous on cells and has been shown to significantly impact their metabolic states and impart transcriptomic stress response signatures [47, 48]. These issues of upstream sorting are being resolved with the introduction of novel sorters based on either microchip- or image-based droplets technologies that allow single-cell isolation with reduced stress and high cell viability. Alternatively, the Fluidigm C1 HT microfluidics system is capable of automatically placing up to 800 cells into microwells where they are lysed and a template for sequencing analysis is made. More recently, Drop-seq and the 10x Chromium technology have improved sequencing capabilities to many thousands of cells, each one inserted into individual droplets [49]. Depending on the number of cells to be analyzed, throughput requirements, cost, and the possibility for long-term storage of cells, selecting the optimal scRNA-seq protocol is critical. Although a comprehensive review of these methods is beyond the scope of this review, other publications exist to aid in pairing an scRNA-seq protocol to experimental needs and provide useful framework for benchmarking the most suitable technique to best answer different research questions [26, 46, 50].
In addition to the variety of scRNA-seq protocols in use today, computational pipelines to analyze the data remain in their early stages and gold-standard approaches for standardized analyses of patient samples from distinct datasets will need to be implemented to advance new clinically relevant discoveries [26]. Presently, data analysis techniques exist including unsupervised clustering algorithms which group cells based on the similarity of their gene expression profiles before assigning cell identities to distinct clusters [51]. In addition to clustering algorithms, differential expression analyses may be used to identify genes with different levels of expression across experimental conditions. Finally, trajectory inference methods approach cellularity heterogeneity as a fluid and continuous process, incorporating dynamic models of gene expression to illustrate transitions of cellular identities [51, 52]. In the future, standardization of these approaches and the development of solutions to current impediments such as batch effects will simplify the use of scRNA-seq and facilitate the sequencing of larger numbers of cells [53]. For an in-depth review of best practices in scRNA-seq data analysis, we refer readers to extremely comprehensive publications by Hwang et al. [26] and Luecken and Theis [51].
The recent advances in single-cell proteomics and transcriptomics have also yielded novel methods that combine the two. CITE-Seq is a single-cell phenotyping method in which oligonucleotide-labeled antibodies, also known as antibody-derived tags (ADTs), bind to proteins of interest. Upon cell lysis in droplets, ADTs and cellular mRNA are converted to cDNA and sequenced, providing both proteomic and transcriptomic measurements within a single-cell readout [27]. Because of this ADT system, CITE-seq is capable of detecting a virtually unlimited number of cell surface markers and is constrained only by the commercial availability of antibodies. Of note, RNA Expression and Protein Sequencing Assay (REAP-Seq) proceeds through a similar methodology [54]. Both of these technologies represent mergers of single-cell proteomics and transcriptomics and thereby resolve distinct cell types with greater clarity than either method could individually. While both CITE-seq and REAP-seq are limited to the analysis of extracellular protein, novel technologies including INs-seq (intracellular staining and sequencing) demonstrate the feasibility of merging single-cell transcriptomics and intracellular proteomics [55].
Collectively, these technologies provide researchers with the tools to detect cell-to-cell variations in tissues that are otherwise masked in bulk analyses (Figure 1A). As described by Altschuler and Wu[56], populations of “seemingly identical” cells based on canonical surface marker expression may actually contain a heterogenous diversity of phenotypic and transcriptional differences. Furthermore, these variations within populations can give rise to subpopulations of cells with distinct and important functions. As the field of atherosclerosis turns its attention to the diverse spectrum of leukocytes infiltrating plaque tissue, single-cell approaches present critical tools to uncover the roles of distinct immune cell types in atherogenesis and plaque destabilization.
Figure 1:
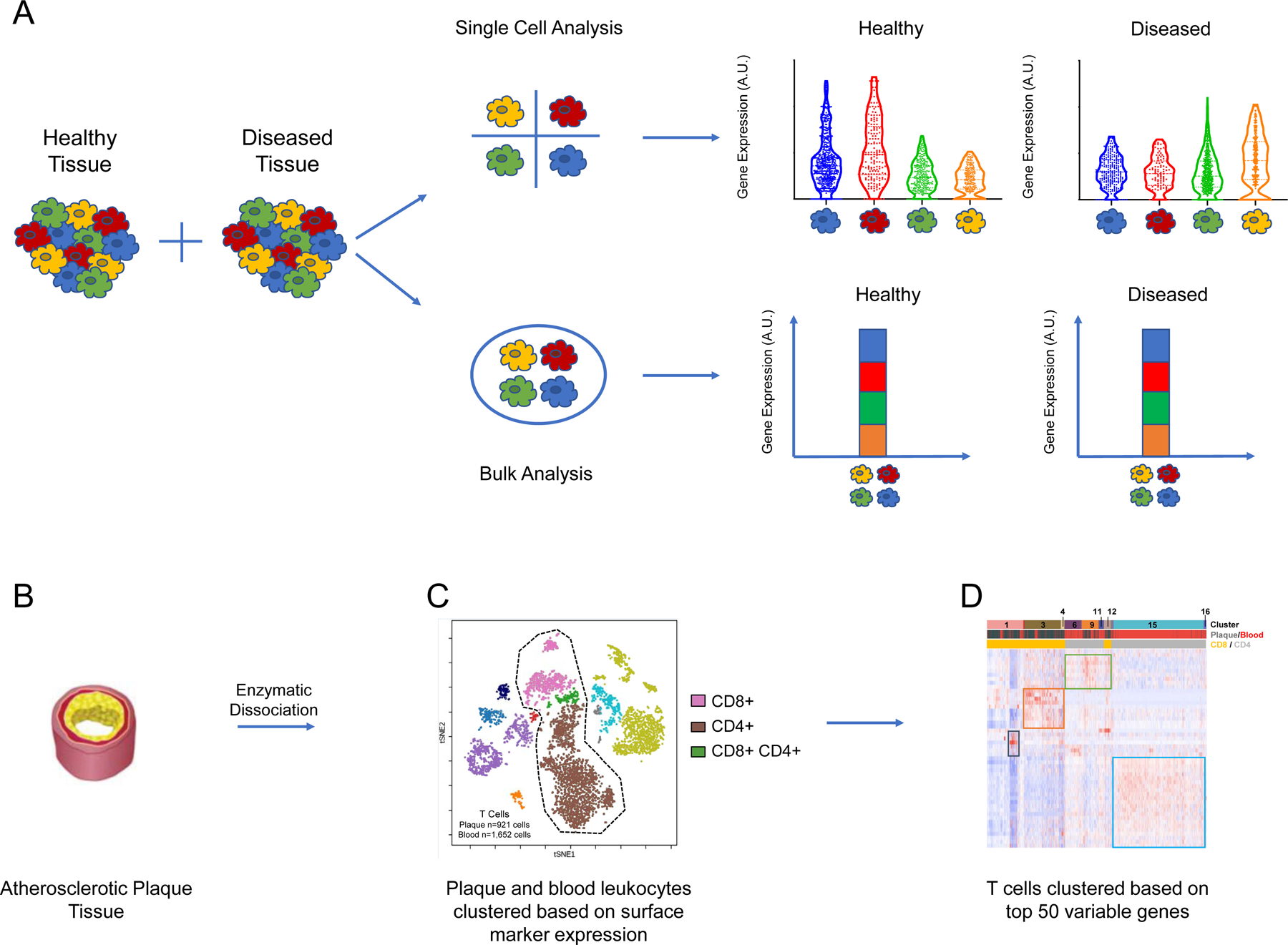
Single-cell approaches unveil disease-relevant heterogeneity among cellular populations
(A) Single cell approaches reveal heterogeneity between individual cells that is obscured by bulk approaches. In this example experiment, distinct transcriptomic profiles are revealed between subpopulations of cells in healthy versus diseased tissues. (C) tSNE plot displaying cells derived from plaque and blood of patients with atherosclerosis (n = 5,362 cells). Cells are clustered using a 21-marker antibody-derived tag (ADT) panel. This proteomic analysis reveals major populations of cells including CD4+, CD8+, and double positive T cells. (C) Heatmap of T cells from plaque and blood (n = 2,573 cells) hierarchically clustered based on top 50 variable genes. The first banner denotes the 16 distinct clusters of T cells that were uncovered. The second banner indicates the cells’ origins from plaque or blood, and the bottom banner indicates the cluster’s identity as CD8+ or CD4+ T cells. The heat map was made using the Fernandez et al. 2019 dataset.
3. Uncovering mechanisms of atherosclerosis using single-cell methods
Building on the evolving understanding of the staggering diversity of cell types which comprise healthy vascular tissue [57], recent studies have employed CyTOF and single-cell RNA-seq to characterize the landscape of immune cells in diseased tissue from ApoE−/− and Ldlr−/− mice. Collectively, the results from these studies generate a novel in-depth profile of the immune landscape of experimental atherosclerosis (Figure 2A and Table 2). Employing a 35-marker CyTOF panel to investigate CD45+ cell populations from ApoE−/− aortas, Cole et al. [58] identified 13 broad populations of leukocytes and found that mice fed on a high-fat diet exhibited increases in monocyte, pDC, and CD11c+ macrophages, compared with ApoE−/− mice fed a standard chow diet. The proportions of CD206+CD169+ subsets of macrophages were significantly reduced, as were type 2 conventional dendritic cells (cDC2) in the same mice fed a high-fat diet. This study is complemented by the findings of Cochain et al. [59], who employed single-cell RNA-seq to identify three distinct macrophage populations in aortic tissue from ApoE−/− and Ldlr−/− mice. One cluster of resident-like macrophages was shared between diseased and healthy mice, while two additional clusters of inflammatory and TREM2hi macrophages appeared exclusively in atherosclerotic tissue and presented a transcriptional profile similar to that of intimal foamy macrophages described by Kim et al. [60]. Winkels et al. [61] employed scRNA-seq followed by an unsupervised clustering algorithm to reveal a greater diversity of leukocytes in diseased aortic tissue from ApoE−/− and Ldlr−/− mice compared to wild type mice. They subsequently employed CyTOF to validate the phenotypic diversity of these leukocyte subsets, based on the expression of canonical surface protein markers, and found high phenotypic overlap between the scRNA-seq and CyTOF clusters. Results from this study demonstrate the compatibility of combined scRNA-seq and CyTOF approaches in resolving diversity of cell types in tissue.
Figure 2:
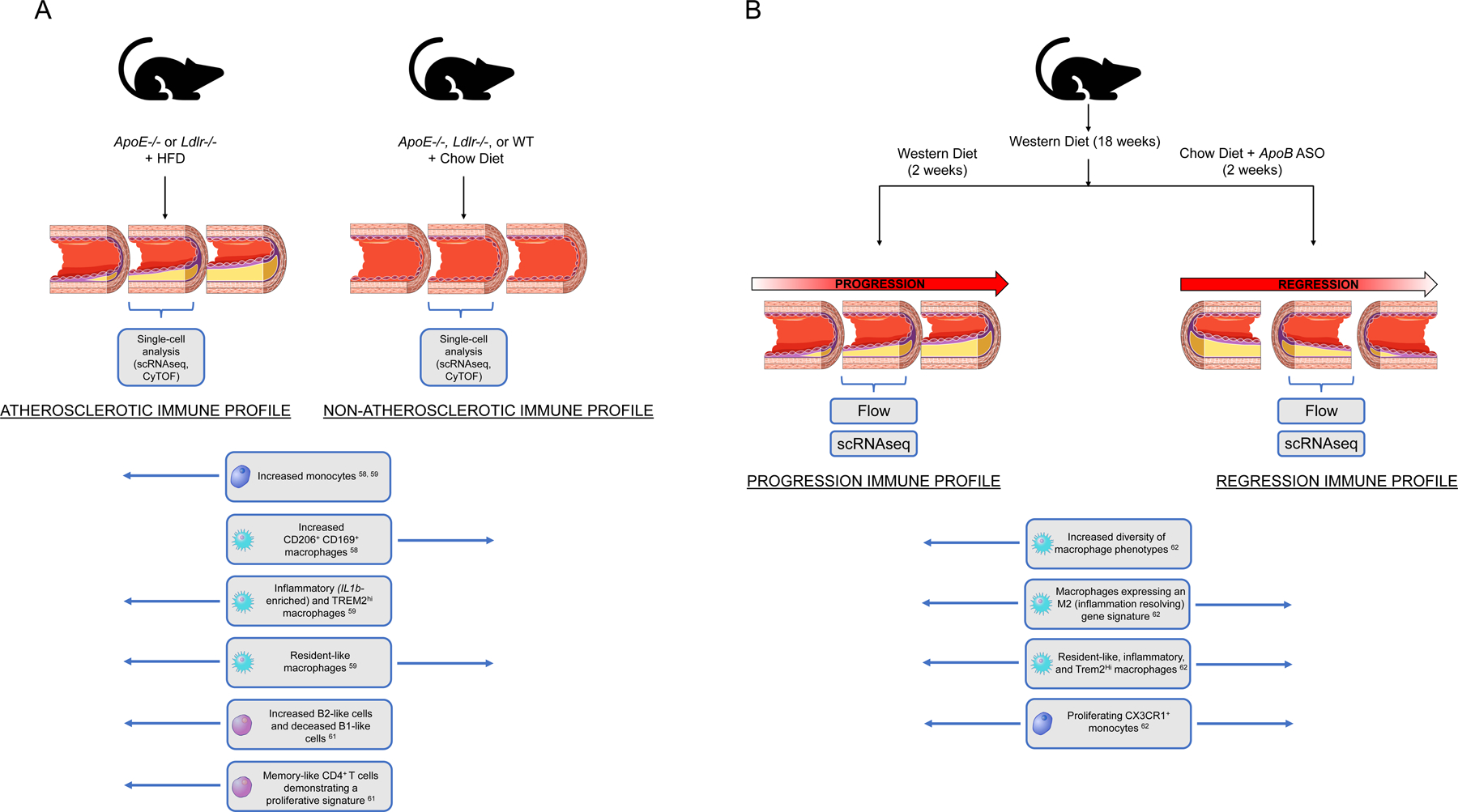
Single-cell investigations reveal distinct immune profiles between (A) non-atherosclerotic and atherosclerotic murine aortas and (B) models of progressing and regressing atherosclerotic lesions, as reported by Lin et al. [62]
Table 2:
Applications of SC approaches to preclinical models of atherosclerosis
| Tissue | Mouse model | Cell type focus | SC platform | Key findings | References |
|---|---|---|---|---|---|
| Aorta (arch, thoracic, abdominal) | ApoE −/− | Macrophages | CyTOF | - High-fat diet triggered increases in monocyte, pDC, and CD11c+ macrophages. - CD206+CD169+ subsets of macrophages were significantly reduced, as were type 2 conventional dendritic cells (cDC2). |
[58] |
| Aorta | Ldlr−/− & ApoE−/− | Macrophages | 10X Genomics | - Inflammatory IL1b-enriched macrophages and TREM2Hi macrophages were specific to the atherosclerotic aorta and presented lipid-metabolism-related functions. | [59] |
| Whole aorta (arch, thoracic, abdominal) | Ldlr−/− & Apoe−/− | Macrophages | 10x Genomics + bulk RNA seq and Lipid Staining–Based Flow Cytometric | - Foamy macrophages were positively correlated with severity of atherosclerosis, expressing few inflammatory genes, but many lipid-processing gene. | [60] |
| Whole aorta | Ldlr−/− & Apoe−/− | 10X Genomics + CyTOF | - Greater leukocyte diversity in disease vs. healthy aortas - CyTOF revealed rare γ/δ T-cells, (~1% of leukocytes) - 3 subsets of B-cells were detected. |
[61] | |
| Aortic arch | Models of progression / regression using BL6 mice injected with AAVmPCSK9 and fed WD for 18 weeks before branching into chow vs WD. | Macrophages | FACS sorting followed by 10X Genomics | - Increased macrophage phenotypes in progressing plaques compared to regressing. - Both stages revealed greater macrophage diversity than is explained by the classic M1/M2 definition. - Identified inflammatory and TREM2-high macrophages in both stages. |
[62] |
| Aortic root and ascending aorta | ApoE −/− | VSMCs | 10x Genomics + CITE-Seq | - SMC phenotypic modulation occurs along a continuous trajectory from a contractile SMC towards a fibroblast-like cell. - TCF21 influences the atheroprotective transition of VSMCs to fibroblast-like cells. - Deletion of TCF21 reduced the number of VSMC-derived fibromyocytes and resulted in a thinner, fibrous cap. |
[89] |
| Medial layer of mouse aorta and whole aorta. | Myh11-CreERt2, Rosa26-Confetti (Confetti), Rosa26-EYFP (EYFP), ApoE−/−, Sca1-GFP | VSMCs | Fluidigm C1, Smart-seq2, 10X Chromium | - Identified distinct transcriptional profiles between VSCMs in the disease-prone aortic arch and those of the relatively protected descending aorta. - Propose that Sca1 expression during plaque development could mark an intermediate VSMCs state and indicates phenotypic switching. |
[104] |
VSMCs: Vascular smooth muscle cells
In addition to unveiling the diversity of cell types in diseased versus health vasculature, a recent study by Lin et al. [62] employed single-cell methods to characterize the dynamic changes of plaque immune cells in two distinct phases of disease: progression and regression of atherosclerotic lesions (Figure 2B). Employing validated models of each phase, [63] Lin et al. combined scRNA-seq with genetic fate mapping to resolve the origin and diversity of macrophage transcriptional states in the murine aortic arch tissue. Providing enhanced clarity to previous studies, which emphasize the importance of local macrophage proliferation to overall macrophage accumulation in atherosclerotic lesions [64, 65], Lin et al. revealed an increased number of distinct macrophage phenotypes in progressing plaques than in regressing ones. Overall, the authors showed that both progressing and regressing plaques exhibit a more complex spectrum of macrophage activation states than is explained by the traditional M1/M2 definition. Their analysis also revealed the presence of proliferating monocytes with a stem cell–like signature in atherosclerotic tissue. Considered within the context of the prevailing model of atherosclerosis, in which monocytes differentiate immediately upon entering the tissue, these results suggest that cellular plasticity may be more complex than previously thought and propose a possible role of self-renewing monocytes in atherosclerotic progression.
Results from Lin et al. [62] also complement findings from other groups by confirming the presence of distinct immune cell populations and characterizing their role in disease progression or regression. Specifically, Lin et al. identified populations of inflammatory and TREM2high macrophages analogous to those observed in diseased tissue by Cochain et al. [59]. Because Lin et al. detected these cells in both progressing and regressing plaques, they concluded that these populations may represent general inflammatory features of atherosclerosis, rather than specific drivers of pathogenesis or healing. Additionally, the appearance of macrophages in the progression model which expressed M2 features, such as an IL-4 signature, suggests that the previously proposed assignment of M1 macrophages to plaque progression and M2 macrophages to plaque regression does not capture the full dynamic complexity of macrophage plasticity within the lesion.
While discrete applications of scRNA-seq to atherosclerosis in murine models have individually pried into the diversity of leukocytes within the lesion, there is a clear need for analytical efforts to combine the collective resources compiled across these studies. This was recently accomplished by Zernecke et al. [66] in a meta-analysis of 9 scRNA-seq and 2 CyTOF studies. Employing Harmony [67] to analyze a total of 15,288 cells from various laboratories, the authors assigned proinflammatory roles to interferon-inducible cell (IFINC) and inflammatory macrophages, while concluding that Trem2+ macrophages do not express a proinflammatory signature. Together with the findings from Lin et al. [62] that Trem2hi macrophages are present in both progressing and regressing plaques, these data suggest that these macrophages may have more complex roles in atherosclerosis than previously established. Zernecke et al. also reported that half of all foam cells present in the atherosclerotic mouse aorta are derived from smooth muscle cells. These data support previous observations that a large portion of plaque foam cells derive from smooth muscle cells [10, 68] but are in contrast with a recent scRNA-seq analysis of murine and human atherosclerotic plaques revealing that smooth muscle cells differentiate into unique fibroblast-like cells rather than macrophages and foam cells. Overall, these findings highlight that cell plasticity and adaptation to the atherosclerotic microenvironment remain poorly understood, and that single-cell studies can contribute to resolve this complex biology.
Interestingly, non-vascular applications of single-cell methods are yielding important insights for atherosclerosis, notably the effects of obesity and hypercholesterolemia on other tissues of the body. Recent studies have investigated the immune mechanisms governing inflammation of visceral adipose tissue (VAT), a process which is associated with type 2 diabetes in obese subjects. Findings reveal that a dietary switch from high-fat diet to caloric restriction impacts the relative frequencies of distinct macrophage populations in VAT [69]. Furthermore, the population, which is dominant throughout caloric restriction, demonstrates upregulation of phagocytotic genes and may contribute to lipid clearance and the resolution of inflammation. A complementary study implicates Netrin-1 in the governance of adipose tissue macrophages and showed that the myeloid-specific deletion of the gene enhances lipid handling and reduces adipose inflammation [70]. Additionally, Trem2 signaling, which is implicated in atherosclerosis as discussed previously, has been shown to serve as a mechanism by which macrophages respond to the loss of metabolic homeostasis in multiple tissues [71].
The application of single-cell approaches to atherosclerosis in mice is an opportunity to further explore and build on the known vascular biology of well validated experimental models with unprecedented resolution. As these studies demonstrate, expanded antibody panels and the ability to detect cell types using a combination of canonical markers and transcriptional profiles allow researchers to resolve new cellular phenotypes and functional states over spectrums and trajectories of differentiation that are undetectable using traditional low-resolution approaches. Furthermore, genetic and dietary manipulation of preclinical models reveal the functional specialization of cellular subpopulations and their contributions to disease pathology. Despite these advantages, mouse models represent incomplete recreations of human atherosclerosis and pathophysiology. Principally, mouse models of atherosclerosis do not exhibit spontaneous plaque rupture [72–74], and the atherosclerotic lesions that they develop bare intrinsic differences to the human immune microenvironment [72, 73, 75]. The argument has been made that the extreme levels of hypercholesterolemia exhibited by mouse models of atherosclerosis render them more analogous to rare human diseases such as homozygous familial hypercholesterolemia than to the more common and multifactorial clinical atherosclerosis [76, 77]. Other reasons span from the lack of genetic diversity of most experimental models to species-specific differences in non-conserved enhancers and long-non coding RNAs [78]. Recent studies have also highlighted how the lack of microbial experience of laboratory mice may limit adequate modelling of immunological events relevant to humans [79].
Building on the insights gleaned from these models, the next step toward developing novel immunotherapies necessitates complementary human studies in humans to facilitate the successful transition of future treatments to patients.
4. Translating single-cell approaches to human disease
Presently, the use of single-cell approaches to dissect human atherosclerotic disease lags behind other areas of biomedical research, including the groundbreaking and clinically relevant insights into cancer. For example, Lavin et al. [28] combined CyTOF and single-cell transcriptomics to resolve the immune composition of early lung adenocarcinoma lesions. By investigating cells from the tumor site, non-involved lung tissue, and blood, this study identified tumor-specific alterations within the T cell, NK cell and myeloid compartments that likely degrade anti-tumor T cell function and present promising targets for immunotherapies to bolster immune responses to early lung adenocarcinoma. Furthermore, the heightened cellular resolution provided by single-cell technologies has allowed researchers to construct models of the tumor immune microenvironment which can predict immunotherapeutic responses in patients [80].
As researchers strive to harness single-cell methods in atherosclerosis to advance new treatments for atherosclerotic cardiovascular disease, analyses of blood samples from patients with coronary artery disease (CAD) have yielded novel insights into the composition of disease-associated leukocytes in circulation. Hamers et al. [81] employed CyTOF followed by the FlowSOM clustering algorithm to reveal heterogeneous populations of nonclassical monocytes whose relative frequencies are altered in the setting of severe CAD. These analyses uncovered new monocyte adaptations to disease state comprising 8 distinct clusters of circulating monocytes and suggest interesting clinical implications as a particular subset of Slan+CXCR6+ nonclassical monocytes was expanded and correlated with disease severity.
Although single-cell methods are highly suited to analyses of circulating leukocytes, application of these techniques to human atherosclerotic vascular tissue has been challenging. Researchers seeking to investigate the mechanisms of atherogenesis and plaque rupture in humans have faced several difficulties including the limited availability of fresh diseased samples and the significant challenges in tissue dissociation of such complex tissue. Because prompt access to fresh vascular tissue from patients is rare, previous histopathological studies of plaque morphology have focused on atherosclerotic coronary and carotid specimens from deceased patients or surgical samples [82]. These studies revealed that atherosclerotic plaque represents a heterogenous environment, containing cellular and acellular components including immune and non-immune cells, a necrotic core, and fibrotic tissue [82, 83]. Although these studies vastly expanded our understanding of end-stage disease in humans, significant knowledge gaps remain in the heterogeneous cellular composition and morphology of healthy, control tissue and tissue in the developing stages of disease.
Single-cell-driven investigations of human atherosclerotic tissue are in their infancy, and just beginning to emerge (Figure 3). Fernandez et al. [84] employed a combination of single-cell approaches including scRNA-seq, CyTOF, and CITE-seq to identify immune dysregulations associated with advanced atherosclerosis and clinical CV events. By performing paired analyses of plaque and blood from the same patients, this study uncovered an immune microenvironment at the lesion site containing specialized cellular subtypes and behaviors not found in the circulation. Results showed that plaques of symptomatic patients who had suffered a recent stroke were enriched in a specific population of CD4+ effector memory T cells. Furthermore, by employing CITE-seq to merge proteomic and transcriptomic analyses of T cells in the blood and plaque, this study revealed that even subsets of cells that appear homogenous based on marker analysis may in fact demonstrate heterogenous transcriptional profiles likely reflecting a complex spectrum of functional differentiation at the plaque site (Figure 1B–D). Interestingly, T cells in both symptomatic (stroke) and asymptomatic (no stroke) patients exhibited transcriptional signatures associated with activation, but the co-expression of markers of exhaustion and differentiation were found predominantly in T cells of symptomatic patients. The presence of activated T cells in the plaque was confirmed in a subsequent study of human atherosclerosis by Depuydt et al. [85]. In this study, the authors applied scRNA-seq and scATAC-seq, a method to assess chromatin accessibility in individual cells, to plaque-derived immune and non-immune cells. In T cells, the appearance of open chromatin at cytokine gene loci, including IFNG, suggests possible cell-cell interactions and classical activation of proinflammatory macrophages by neighboring T cells in the lesion. Interestingly, these dynamic processes within the T cell compartment also appear to cross boundaries within the field of vascular disease and have been observed in diseased tissues of ascending aortic aneurysms [86].
Figure 3:
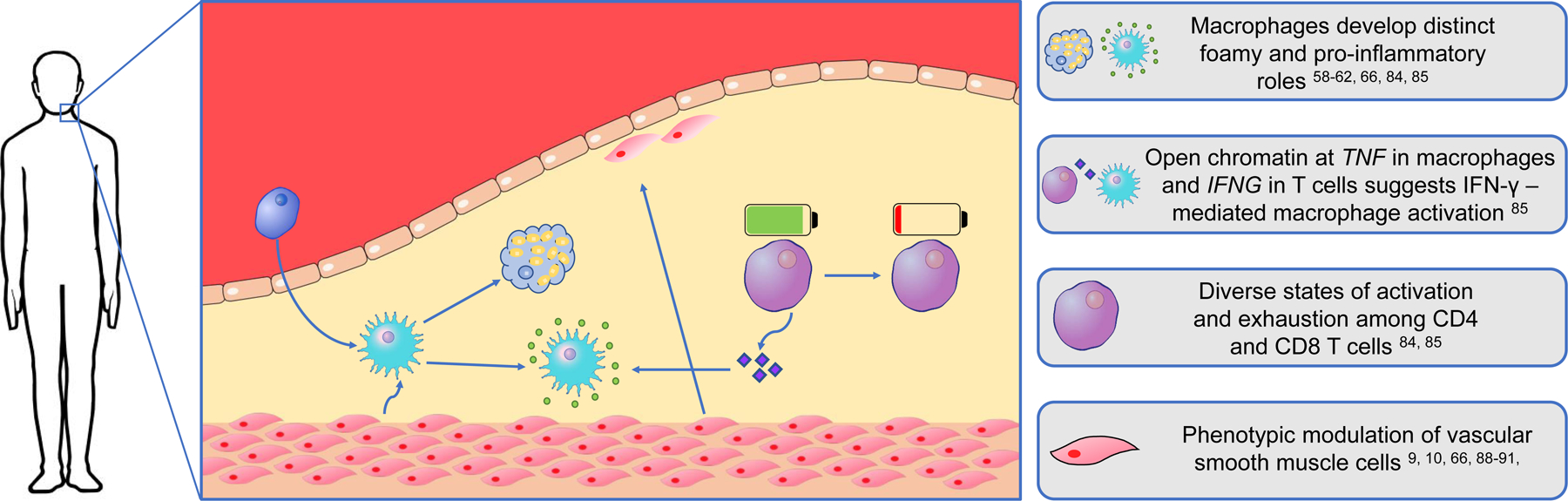
Cellular processes in the human atherosclerotic lesion uncovered by single-cell approaches
In addition to dysregulations of the adaptive immune system, early applications of single-cell approaches to human atherosclerotic tissue have also revealed disease-associated attributes of the innate immune system. Fernandez et al. [84] identified macrophage subsets in symptomatic patients with stroke that displayed transcriptional signatures associated with both pro-inflammatory and reparative functions, which may contribute to plaque healing post-rupture. Additionally, Depuydt et al. [85] identified foam cell-like macrophages expressing TREM2 and a profibrotic phenotype suggestive of a role in plaque stabilization. Collectively, these studies provided cellular atlases of human atherosclerosis and suggest new targets to be further mechanistically investigated that could contribute to the development of precise immunotherapies and advance current treatment options.
As single-cell approaches probe with new depth into the complex immune environment of atherosclerotic tissue, they are also providing new transcriptional insights into the plasticity of other cell types in the vasculature. Interestingly, recent applications of scRNA-seq have uncovered new mechanisms governing the differentiation of vascular smooth muscle cells (VSMCs) at the atherosclerotic site. It has previously been observed that VSMCs respond to atherosclerotic risk factors and lesion development and subsequently differentiate into distinct lineages that may be either atherogenic or atheroprotective. For example, Shankman et al. [9] demonstrated that Krüppel-like factor 4 (KLF4), which has previously been shown to drive VSMC transdifferentiation and promote aortic aneurysms [87], also promotes the transition of VSMCs toward a macrophage-like phenotype which is believed to increase atherosclerotic plaque size and risk of rupture. Conversely, VSMCs are also known to form fibroblast-like cells which secret matrix proteins and cytokines, strengthening the fibrous cap and reducing the risk of plaque rupture [19, 88]. Complementing these discoveries, Wirka et al. [89] identified the transcription factor TCF21 as an important factor governing the atheroprotective transition of VSMCs to fibroblast-like cells, termed “fibromyocytes.” Employing scRNA-seq, they observed that the deletion of TCF21 in ApoE−/− mice significantly reduced the number of VSMC-derived fibromyocytes and resulted in a thinner, fibrous cap. Furthermore, to determine whether the atheroprotective function of TCF21 is present in humans, the authors employed a similar technique to identify analogous populations of fibromyocytes in human tissues. To confirm these findings, they incorporated large GWAS datasets and found that individuals with decreased TCF21 expression resulting from genetic variants (SNPs) were at increased risk of CVD events. Recently, this trans-differentiation of VSMCs was further confirmed in human coronary arteries by Ma et al. [90], who applied pseudotemporal ordering to an existing scRNA dataset to reveal distinct VSMC-derived chondroblast-like and fibroblast-like lineages. Importantly, the clinical implications of this phenomenon are underscored by a recent study by Pan et al. [91] which demonstrated that blocking the differentiation of VSMCs to an intermediate phenotype (SEM) reduces atherosclerotic burden and enhances lesion stability.
The application of single-cell approaches to human tissue represents the next step in discovering new mechanisms of human disease and in developing precise immunotherapies and other strategies to achieve plaque regression or stabilization in atherosclerosis. Based on these initial scRNA-seq studies, it is evident that distinct but phenotypically similar cell types may play opposing roles in the disease that may be dictated by both local environmental cues and systemic cross-tissue influences due to underlying cardiometabolic co-morbidities in patients. Furthermore, plasticity and inter-conversion between functional phenotypes reinforces the need to resolve the atherosclerotic cellular environment with exceptional granularity in different subtypes of patients and age groups. In order to bring about the next era of clinical atherosclerotic treatment, larger datasets will help to reveal functionally relevant cell types and cellular interactions whose contributions to disease can be further studied and validated in experimental models.
5. On the horizon: non-dissociative methods and in-situ single-cell analyses
Single-cell approaches are rapidly opening new fields of research and empowering researchers to investigate cellular functions with exceptional resolution. Despite the potential of these massive contributions to current research, significant limitations remain. Principally, the enzymatic dissociation techniques required to convert whole tissues to single-cell suspensions for high throughput single-cell analyses present potential drawbacks including impacts on the viability of recovered cells, possible disproportionate recovery of different cell types, and inadvertent effects on gene expression [92]. Fortunately, solutions to these challenges appear to be on the horizon. Protocols are being developed to minimize the effects of dissociation on gene expression [93]. Furthermore, as nuclei can often be isolated from tissue more easily than whole cells, single-nucleus RNA-sequencing (snRNA-seq) can be used discriminate between related cell types with minimal transcriptomic alterations and sensitivity comparable to scRNA-seq [94]. Application of these approaches to atherosclerosis may be of particular benefit, because cells in plaque tissue are embedded within a variety of calcific and fibrous microenvironments.
Novel technologies also expand the current capacity to study cells while preserving their spatial contextualization in tissue (Table 3). Plaques represent extremely heterogenous environments, and researchers have previously investigated the effect of cellular localization on atherogenesis through gross anatomical separation of plaque regions. Single-cell analysis of isolated adventitial tissue during early-stage atherosclerosis revealed novel communications by resident macrophages leading to increased myeloid infiltration [95]. Additionally, another recent study separated atherosclerotic lesions into core and proximal adjacent regions and revealed processes through which peripheral cells undergo cellular transdifferentiation in response to inflammatory signaling to become matrix-secreting core cells [96]. These studies highlight that spatial considerations are critical to understanding atherosclerotic pathophysiology. Unfortunately, conventional dissociation approaches are poorly suited to deeper investigations of the effect of localization on the role of distinct leukocyte populations. Once again however, technologies that are currently being implemented in other fields suggest solutions to this challenge.
Table 3:
In-situ single-cell approaches
| Method | Type of data | Simultaneous parameters | Max parameters per specimen | Resolution | References |
|---|---|---|---|---|---|
| IMC | Proteomic | 40 | 40 | ~1000 nm | [105] |
| MIBI | Proteomic | 40 | 40 | ~260 nm | [105] |
| CODEX | Proteomic | 1–5 | ~60 (via serial staining) | Subcellular | [98] |
| Method | Type of data | Genes per cell | Resolution | ||
|
| |||||
| seqFISH+ | Transcriptomic | >10000 | [106] | ||
| Slide-seq | Transcriptomic | 100s | ~10um | [101] | |
Multiplexed ion beam imaging (MIBI) provides researchers with the unique opportunity to study cells in situ. Similar to CyTOF, MIBI involves the staining of samples with antibodies bound to pure elemental metal reporters. Secondary ion mass spectrometry is then employed to analyze samples, collecting proteomic data from individual cells as well as information detailing cellular morphology and localization [97]. Additionally, conventional fluorescence microscopes can now be converted into high dimensional imaging devices thanks to the recent development of CODEX. With this method, a tissue is stained with antibodies labeled with oligonucleotide duplexes [98]. Through iterative cycles, cells are presented with a solution containing non-fluorescent “index” nucleotides and fluorescent nucleotides which are repeatedly imaged and removed [98]. Importantly, analogous innovations are also occurring in the field of spatial transcriptomics. Sequential Fluorescence in Situ Hybridization (seqFISH) allows researchers to identify mRNA transcripts within their spatial contexts by labeling transcripts directly in tissue sections using fluorescent probes [99]. Another approach, Slide-seq, adapted the original Drop-seq technology to capture mRNA on a lattice of barcoded beads [99]. This technology achieved detection of RNAs with a spatial resolution of 10 μm, and recent modifications to the protocol have yielded improved RNA capture efficiency in the updated version, Slide-seqV2 [100]. Although these technologies are currently limited in terms of their spatial resolution and the number of transcripts that can be simultaneously analyzed, early applications of them in murine brain tissue have demonstrated the capacity of these approaches to obtain spatially resolved gene expression data [101, 102]. Undoubtedly, these technologies will break new ground in atherosclerosis, where spatial proteomic and transcriptomic methods promise to expand our understanding of plaque morphology and resolve the spatial relationships of cells across these complex and heterogenous tissues.
The application of single-cell approaches to vascular biology and atherosclerosis also contributes to several large-scale projects aiming to definitively profile the cellular composition of the human body. In particular, the Human Cell Atlas [103] and the NIH Human BioMolecular Atlas Program (HuBMAP) are employing single-cell methods to construct comprehensive reference maps of healthy and diseased tissues. With focuses on clinical care, these projects aim to establish references against which clinical specimens may be compared and through which new diagnostic approaches can emerge.
6. Conclusion
Single-cell approaches are broadening our understanding of the complexity and diversity of cell types that exist within all tissues. The morphological complexity of atherosclerotic plaques and the vast diversity of cells involved in the atherogenesis and plaque destabilization represent ripe opportunities for single-cell-driven discovery leading to advances in clinical care. By deciphering the roles of distinct cell types, single-cell methods have the potential of moving the field one step closer to the development of novel immunotherapies to treat advanced atherosclerosis and reduce risk of clinical cardiovascular events.
Highlights.
Atherosclerosis is initiated by the accumulation of lipids in the arterial wall that trigger a complex network of inflammatory processes.
Recent clinical trials revealed that targeting inflammation in patients with cardiovascular disease is a promising approach to preventing recurrent clinical events.
Single-cell technologies can resolve the heterogenous environment of the atherosclerotic lesion and the functional changes of several immune cells throughout disease progression and regression.
Acknowledgements
Dr. Giannarelli acknowledges research support from the NIH-NHLBI (R01 HL153712-01), NIH-NCATS (UH3TR002067), AHA (20SFRN35210252) and CZI (NFL-2020-218415). Dr. Fernandez was supported by the NIH T32HL007824.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Heistad DD, Unstable Coronary-Artery Plaques. New England Journal of Medicine, 2003. 349(24): p. 2285–2287. [DOI] [PubMed] [Google Scholar]
- 2.Lusis AJ, Atherosclerosis. Nature, 2000. 407(6801): p. 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yurdagul A Jr., et al. , The arterial microenvironment: the where and why of atherosclerosis. The Biochemical journal, 2016. 473(10): p. 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore KJ, Sheedy FJ, and Fisher EA, Macrophages in atherosclerosis: a dynamic balance. Nature reviews. Immunology, 2013. 13(10): p. 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koelwyn GJ, et al. , Regulation of macrophage immunometabolism in atherosclerosis. Nature immunology, 2018. 19(6): p. 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore KJ and Tabas I, Macrophages in the pathogenesis of atherosclerosis. Cell, 2011. 145(3): p. 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu X-H, et al. , Foam cells in atherosclerosis. Clinica Chimica Acta, 2013. 424: p. 245–252. [DOI] [PubMed] [Google Scholar]
- 8.Flynn MC, et al. , Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Frontiers in Pharmacology, 2019. 10(666). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shankman LS, et al. , KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine, 2015. 21(6): p. 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, et al. , Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol, 2019. 39(5): p. 876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grivel J-C, et al. , Activation of T Lymphocytes in Atherosclerotic Plaques. Arteriosclerosis, Thrombosis, and Vascular Biology, 2011. 31(12): p. 2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonasson L, et al. , Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis: An Official Journal of the American Heart Association, Inc, 1986. 6(2): p. 131–138. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, et al. , Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med, 2017. 377(12): p. 1119–1131. [DOI] [PubMed] [Google Scholar]
- 14.Tardif JC, et al. , Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med, 2019. 381(26): p. 2497–2505. [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM, et al. , Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med, 2019. 380(8): p. 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nidorf SM, et al. , Colchicine in Patients with Chronic Coronary Disease. N Engl J Med, 2020. 383(19): p. 1838–1847. [DOI] [PubMed] [Google Scholar]
- 17.Nidorf Stefan M, et al. , Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. Journal of the American College of Cardiology, 2013. 61(4): p. 404–410. [DOI] [PubMed] [Google Scholar]
- 18.Tong DC, et al. , Colchicine in Patients With Acute Coronary Syndrome: The Australian COPS Randomized Clinical Trial. Circulation, 2020. 142(20): p. 1890–1900. [DOI] [PubMed] [Google Scholar]
- 19.Bennett MR, Sinha S, and Owens GK, Vascular Smooth Muscle Cells in Atherosclerosis. Circulation research, 2016. 118(4): p. 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cochain C and Zernecke A, Protective and pathogenic roles of CD8+ T cells in atherosclerosis. Basic Research in Cardiology, 2016. 111(6): p. 71. [DOI] [PubMed] [Google Scholar]
- 21.Finn AV, et al. , Concept of Vulnerable/Unstable Plaque. Arteriosclerosis, Thrombosis, and Vascular Biology, 2010. 30(7): p. 1282–1292. [DOI] [PubMed] [Google Scholar]
- 22.Swirski FK, et al. , Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of clinical investigation, 2007. 117(1): p. 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koltsova EK, et al. , Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. The Journal of clinical investigation, 2012. 122(9): p. 3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galkina E, et al. , Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. The Journal of experimental medicine, 2006. 203(5): p. 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spitzer MH and Nolan GP, Mass Cytometry: Single Cells, Many Features. Cell, 2016. 165(4): p. 780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang B, Lee JH, and Bang D, Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine, 2018. 50(8): p. 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoeckius M, et al. , Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 2017. 14: p. 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavin Y, et al. , Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell, 2017. 169(4): p. 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar MP, et al. , Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Reports, 2018. 25(6): p. 1458–1468.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sierant MC and Choi J, Single-Cell Ssequencing in Cancer: Recent Applications to Immunogenomics and Multi-omics Tools. Genomics & informatics, 2018. 16(4): p. e17–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puram SV, et al. , Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell, 2017. 171(7): p. 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tirosh I, et al. , Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (New York, N.Y.), 2016. 352(6282): p. 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zappia L, Phipson B, and Oshlack A, Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput Biol, 2018. 14(6): p. e1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strzelecka PM, Ranzoni AM, and Cvejic A, Dissecting human disease with single-cell omics: application in model systems and in the clinic. Disease models & mechanisms, 2018. 11(11): p. dmm036525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulkarni A, et al. , Beyond bulk: a review of single cell transcriptomics methodologies and applications. Current Opinion in Biotechnology, 2019. 58: p. 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendall SC, et al. , A deep profiler’s guide to cytometry. Trends in Immunology, 2012. 33(7): p. 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park LM, Lannigan J, and Jaimes MC, OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytometry Part A, 2020. 97(10): p. 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gadalla R, et al. , Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Frontiers in Oncology, 2019. 9(415). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotecha N, Krutzik PO, and Irish JM, Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom, 2010. Chapter 10: p. Unit10 17. [DOI] [PMC free article] [PubMed]
- 40.Budnik B, et al. , SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology, 2018. 19(1): p. 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcon E, et al. , Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nature Methods, 2015. 12: p. 725. [DOI] [PubMed] [Google Scholar]
- 42.Specht H, et al. , Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biology, 2021. 22(1): p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaitin DA, et al. , Each cell counts: Hematopoiesis and immunity research in the era of single cell genomics. Seminars in Immunology, 2015. 27(1): p. 67–71. [DOI] [PubMed] [Google Scholar]
- 44.Szabo PA, et al. , Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nature Communications, 2019. 10(1): p. 4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsen TK and Baryawno N, Introduction to Single-Cell RNA Sequencing. Current Protocols in Molecular Biology, 2018. 122(1): p. e57. [DOI] [PubMed] [Google Scholar]
- 46.See P, et al. , A Single-Cell Sequencing Guide for Immunologists. Frontiers in Immunology, 2018. 9(2425). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binek A, et al. , Flow Cytometry Has a Significant Impact on the Cellular Metabolome. J Proteome Res, 2019. 18(1): p. 169–181. [DOI] [PubMed] [Google Scholar]
- 48.Llufrio EM, et al. , Sorting cells alters their redox state and cellular metabolome. Redox biology, 2018. 16: p. 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen QH, et al. , Experimental Considerations for Single-Cell RNA Sequencing Approaches. Frontiers in Cell and Developmental Biology, 2018. 6(108). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziegenhain C, et al. , Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell, 2017. 65(4): p. 631–643.e4. [DOI] [PubMed] [Google Scholar]
- 51.Luecken MD and Theis FJ, Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology, 2019. 15(6): p. e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanay A and Regev A, Scaling single-cell genomics from phenomenology to mechanism. Nature, 2017. 541: p. 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Svensson V, Vento-Tormo R, and Teichmann SA, Exponential scaling of single-cell RNA-seq in the past decade. Nature Protocols, 2018. 13: p. 599. [DOI] [PubMed] [Google Scholar]
- 54.Peterson VM, et al. , Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnology, 2017. 35(10): p. 936–939. [DOI] [PubMed] [Google Scholar]
- 55.Katzenelenbogen Y, et al. , Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell, 2020. 182(4): p. 872–885.e19. [DOI] [PubMed] [Google Scholar]
- 56.Altschuler SJ and Wu LF, Cellular Heterogeneity: Do Differences Make a Difference? Cell, 2010. 141(4): p. 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalluri Aditya S, et al. , Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation, 2019. 140(2): p. 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cole JE, et al. , Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovascular Research, 2018. 114(10): p. 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cochain C, et al. , Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circulation Research, 2018. 122(12): p. 1661–1674. [DOI] [PubMed] [Google Scholar]
- 60.Kim K, et al. , Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ Res, 2018. 123(10): p. 1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winkels H, et al. , Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circulation Research, 2018. 122(12): p. 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin J-D, et al. , Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight, 2019. 4(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peled M, et al. , A wild-type mouse-based model for the regression of inflammation in atherosclerosis. PloS one, 2017. 12(3): p. e0173975–e0173975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Randolph GJ, Proliferating macrophages prevail in atherosclerosis. Nature Medicine, 2013. 19: p. 1094. [DOI] [PubMed] [Google Scholar]
- 65.Robbins CS, et al. , Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nature medicine, 2013. 19(9): p. 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zernecke A, et al. , Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ Res, 2020. 127(3): p. 402–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korsunsky I, et al. , Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods, 2019. 16(12): p. 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allahverdian S, et al. , Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation, 2014. 129(15): p. 1551–9. [DOI] [PubMed] [Google Scholar]
- 69.Weinstock A, et al. , Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism, 2019. 1: p. e190008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma M, et al. , Netrin-1 Alters Adipose Tissue Macrophage Fate and Function in Obesity. Immunometabolism, 2019. 1(2): p. e190010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaitin DA, et al. , Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell, 2019. 178(3): p. 686–698.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jawien J, The role of an experimental model of atherosclerosis: apoE-knockout mice in developing new drugs against atherogenesis. Curr Pharm Biotechnol, 2012. 13(13): p. 2435–9. [PubMed] [Google Scholar]
- 73.von Scheidt M, et al. , Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metabolism, 2017. 25(2): p. 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Heiden K, et al. , Animal models for plaque rupture: a biomechanical assessment. Thromb Haemost, 2016. 115(3): p. 501–8. [DOI] [PubMed] [Google Scholar]
- 75.Pasterkamp G, et al. , Human Validation of Genes Associated With a Murine Atherosclerotic Phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology, 2016. 36(6): p. 1240–1246. [DOI] [PubMed] [Google Scholar]
- 76.Oppi S, Lüscher TF, and Stein S, Mouse Models for Atherosclerosis Research—Which Is My Line? Frontiers in Cardiovascular Medicine, 2019. 6(46). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bentzon JF and Falk E, Atherosclerotic lesions in mouse and man: is it the same disease? Current Opinion in Lipidology, 2010. 21(5). [DOI] [PubMed] [Google Scholar]
- 78.von Scheidt M, et al. , Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab, 2017. 25(2): p. 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abolins S, et al. , The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun, 2017. 8: p. 14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Binnewies M, et al. , Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature medicine, 2018. 24(5): p. 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hamers AAJ, et al. , Human Monocyte Heterogeneity as Revealed by High-Dimensional Mass Cytometry. Arterioscler Thromb Vasc Biol, 2019. 39(1): p. 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Virmani R, et al. , Pathology of the Vulnerable Plaque. Journal of the American College of Cardiology, 2006. 47(8, Supplement): p. C13–C18. [DOI] [PubMed] [Google Scholar]
- 83.Virmani R, et al. , Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol, 2002. 15(6): p. 439–46. [DOI] [PubMed] [Google Scholar]
- 84.Fernandez DM, et al. , Single-cell immune landscape of human atherosclerotic plaques. Nature Medicine, 2019. 25(10): p. 1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Depuydt MAC, et al. , Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circulation research, 2020. 127(11): p. 1437–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, et al. , Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation, 2020. 142(14): p. 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen PY, et al. , Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell, 2020. 26(4): p. 542–557.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan H and Reilly MP, A protective smooth muscle cell transition in atherosclerosis. Nature Medicine, 2019. 25(8): p. 1194–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wirka RC, et al. , Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nature Medicine, 2019. 25(8): p. 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma WF, et al. , Single-cell RNA-seq analysis of human coronary arteries using an enhanced workflow reveals SMC transitions and candidate drug targets. bioRxiv, 2020: p. 2020.10.27.357715.
- 91.Pan H, et al. , Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation, 2020. 142(21): p. 2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van den Brink SC, et al. , Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods, 2017. 14(10): p. 935–936. [DOI] [PubMed] [Google Scholar]
- 93.Lambrechts D, et al. , Phenotype molding of stromal cells in the lung tumor microenvironment. Nature Medicine, 2018. 24(8): p. 1277–1289. [DOI] [PubMed] [Google Scholar]
- 94.Bakken TE, et al. , Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLOS ONE, 2018. 13(12): p. e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gu W, et al. , Adventitial Cell Atlas of wt (Wild Type) and ApoE (Apolipoprotein E)-Deficient Mice Defined by Single-Cell RNA Sequencing. Arteriosclerosis, thrombosis, and vascular biology, 2019. 39(6): p. 1055–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alsaigh T, et al. , Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. bioRxiv, 2020: p. 2020.03.03.968123. [DOI] [PMC free article] [PubMed]
- 97.Angelo M, et al. , Multiplexed ion beam imaging of human breast tumors. Nature medicine, 2014. 20(4): p. 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goltsev Y, et al. , Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell, 2018. 174(4): p. 968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burgess DJ, Spatial transcriptomics coming of age. Nature Reviews Genetics, 2019. 20(6): p. 317–317. [DOI] [PubMed] [Google Scholar]
- 100.Stickels RR, et al. , Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nature Biotechnology, 2020. [DOI] [PMC free article] [PubMed]
- 101.Rodriques SG, et al. , Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science, 2019. 363(6434): p. 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eng CL, et al. , Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature, 2019. 568(7751): p. 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adlung L and Amit I, From the Human Cell Atlas to dynamic immune maps in human disease. Nature Reviews Immunology, 2018. 18(10): p. 597–598. [DOI] [PubMed] [Google Scholar]
- 104.Dobnikar L, et al. , Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nature Communications, 2018. 9(1): p. 4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baharlou H, et al. , Mass Cytometry Imaging for the Study of Human Diseases—Applications and Data Analysis Strategies. Frontiers in Immunology, 2019. 10(2657). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eng C-HL, et al. , Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature, 2019. 568(7751): p. 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]