
Figure 7.
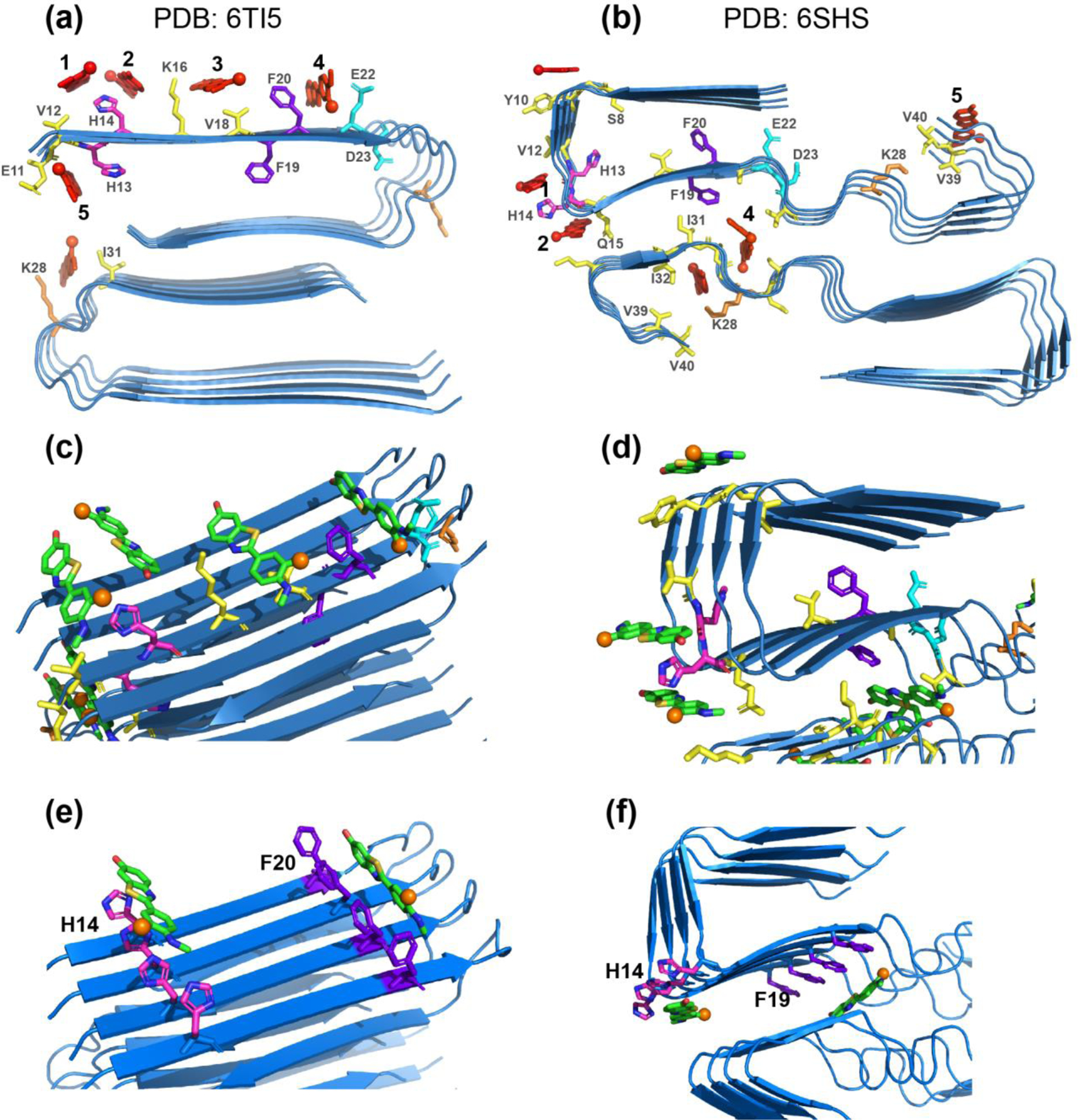
Experimentally constrained molecular docking of flutemetamol to two-fold symmetric Aβ40 structural models. The common docked sites are numbered as 1–5. H13 and H14 are shown in magenta, F19 and F20 in purple, E22 and D23 in cyan, K28 in orange, and all other residues in yellow. Flutemetamol is shown in red. (a) Two-fold symmetric in vitro Aβ40 structure from solid-state NMR 63 (PDB: 6TI5). (b) Two-fold symmetric meningeal AD brain Aβ40 structure 2 (PDB: 6SHS). (c-d) Additional views of flutemetamol binding sites in the solid-state NMR structure (c) and the cryo-EM structure (d). A single active residue was used in each simulation. Flutemetamol (green sticks) straddles several β-strands in most docked poses. The fluorine atom is shown in orange. (e-f) Simulations using one active residue per peptide and hence four active residues in each simulation. (e) Docking results at H14 and F20 for the two-fold symmetric solid-state NMR structure. (f) Docking results at H14 and F19 for the two-fold symmetric AD brain Aβ40 structure. The ligand binding pose is similar to the pose obtained when a single active residue was used in each simulation.