

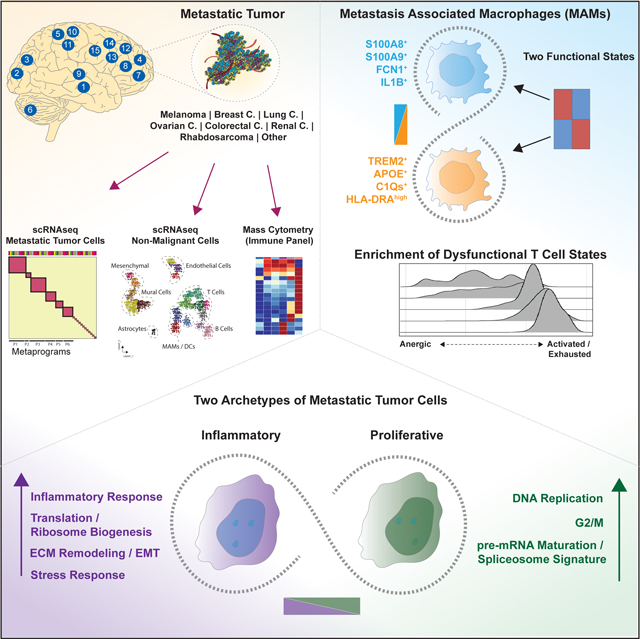
SUMMARY
Brain metastasis (BrM) is the most common form of brain cancer, characterized by neurologic disability and abysmal prognosis. Unfortunately, our understanding of the biology underlying human BrMs remains rudimentary. Here we present an integrative analysis of >100,000 malignant and non-malignant cells from 15 human parenchymal BrMs, generated by single-cell transcriptomics, mass cytometry, and complemented with mouse model- and in silico- approaches. We interrogated the composition of BrM niches, molecularly defined the blood-tumor interface, and reveal stromal immunosuppressive states, enriched with infiltrated T cells and macrophages. Specific single-cell interrogation of metastatic tumor cells provides a framework of 8 functional cell programs that coexist or anticorrelate. Collectively, these programs delineate two functional BrM archetypes, one proliferative and the other inflammatory, that are evidently shaped through tumor-immune interactions. Our resource provides a foundation to understand the molecular basis of BrM in patients with tumor cell-intrinsic and host environmental- traits.
Graphical Abstract
In Brief
Single cell analysis on human brain metastases from multiple cancer types, highlights the conserved cell types of immune and non-immune populations, and reveals two functional archetypes of metastatic cells that coexist in each tumor.
INTRODUCTION
Brain metastases (BrM) are the most common intracranial tumors in adults, approximately 20 to 40% of cancer patients will develop BrM (Kamp et al., 2018; Shojania et al., 2003). Currently, despite aggressive therapies, BrM is incurable with medians of 8.1 and 2.4 per cent overall survival, for 2- and 5-years respectively (Achrol et al., 2019; Hall et al., 2000). Further, BrM diagnosis is associated with extreme deterioration of quality of life.
Despite the urgency implicated by the high incidence, high morbidity, and poor prognosis of BrM, our current understanding about colonization of the human brain by metastatic tumor cells (MTCs) remains rudimentary. It has been proposed that MTCs may reside in a specialized ecosystem in patients that sustains their survival, termed brain metastatic niche (Lambert et al., 2017). As a function of this niche, MTCs may orchestrate functional programs that foster the colonization process. However, we lack detailed information about the specific composition of brain metastatic niches, their cellular diversities, and the functional programs adopted by MTCs in human tumors, remain speculative.
Recent studies applying high-dimensional, single cell analyses to primary tumors have had a profound impact on cancer biology (Rozenblatt-Rosen et al., 2020). Similar studies in human metastases are rare, largely due the clinical-, logistical- and technical- challenges inherent to collecting and profiling metastases at single cell level (Bova, 2017). Instead, studies thus far have either used experimental mouse models or bulk analyses of human tumors (Valiente et al., 2014; Zeng et al., 2019; Zhang et al., 2015).
Here, we extensively profiled MTCs and BrM-associated stromal cells from parenchymal human BrM tumors at single cell resolution using transcriptomics, mass cytometry, corresponding single cell analytical methods and complementary approaches. Our broad cancer type focus and deep analysis improves on existing knowledge of BrM biology.
RESULTS
Brain Metastases from Different Cancer Types Reveal Comparable Metastatic Niches
We obtained fresh parenchymal BrM specimens from fifteen patients with diagnosis of melanoma (n=3), breast cancer (n=3), lung cancer (n=3), ovarian cancer (n=2), colorectal cancer (CRC) (n=1), renal cell carcinoma (n=1), unknown primary carcinoma (n=1) and a case of adult rhabdomyosarcoma. All cases were located predominantly in brain cortex (Figure 1A) (clinical characteristics are listed in Table S1). Dissociated tumors were profiled by single cell RNA sequencing (scRNAseq) with the 10× drop-seq platform (10× Genomics). We achieved a median of 6,798 cells per tumor, with a mean of 79,609 read counts and a median of 2,865 genes per cell. For each sample we detected a median 23,525 total genes. In parallel, 11 of these 15 samples were analyzed by mass cytometry using an immune panel (Figure 1A). Single cell transcriptomes were analyzed using a custom computational pipeline based on the Seurat package (Butler et al., 2018) and visualized onto uniform manifold approximation and projection (UMAP) (Becht et al., 2018).
Figure 1. Cellular Census of Human Brain Metastases Using scRNAseq Analysis.
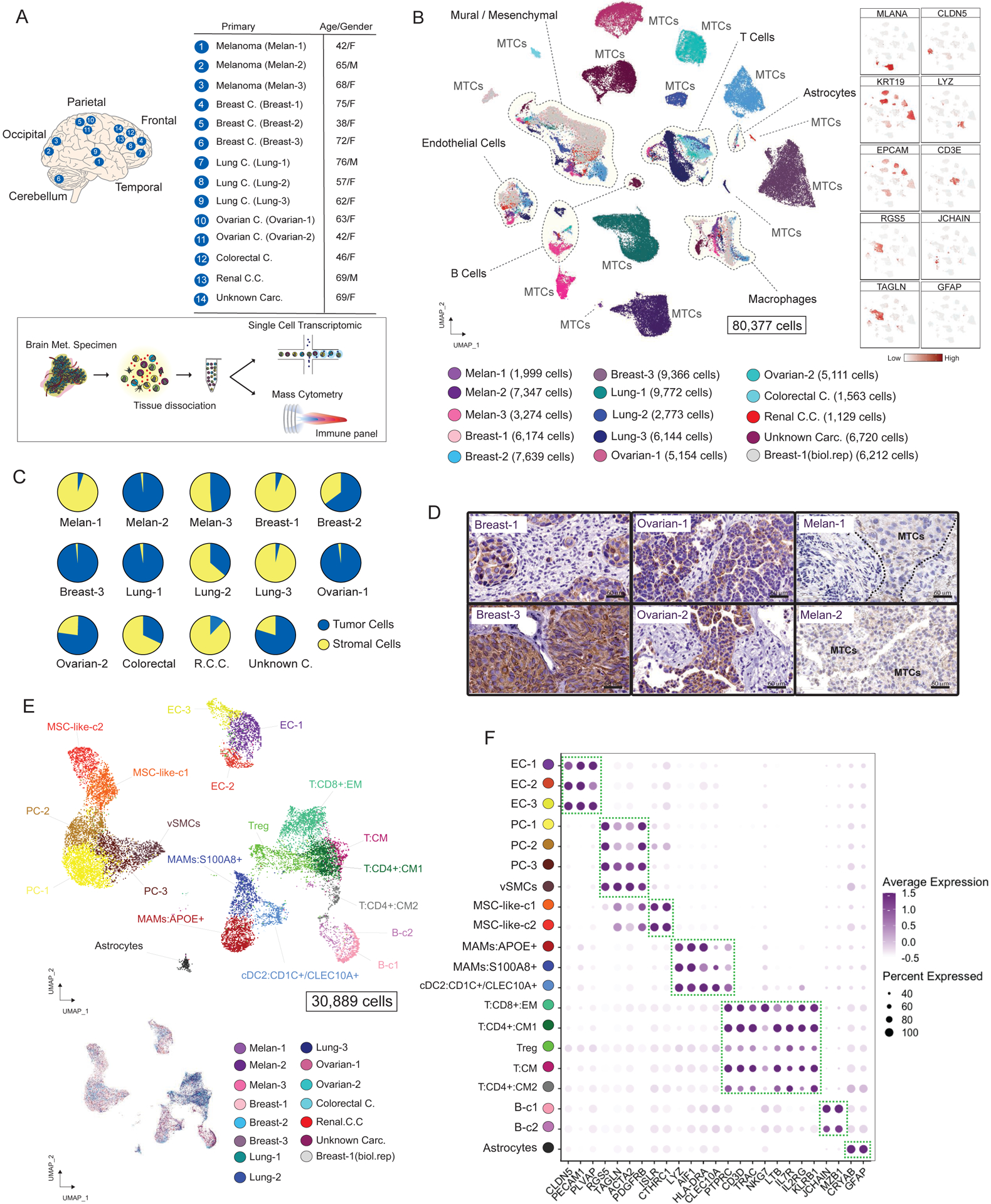
(A) Experimental approach. (B) Visualization of 80,377 malignant (MTCs) and non-malignant single cells. Cells are colored by sample and number of cells per sample are shown. Right panel displays feature plots for selected markers. (C) Pie charts illustrating the tumor purity of each BrM biopsy. (D) IHC staining showing the expression of KRT19 or Melan-A in selected carcinoma or melanoma BrMs (Scale bars, 60 μm). (E) Projection of 30,889 BrM-associated stromal cells including immune and non-immune fractions. (F) Dot plots of conserved and cell-type specific markers in BrM-associated stromal cells. See also Figure S1 and Table S2.
Stromal cells from different tumors formed shared and well-separated clusters that allowed us to annotate cell types by cluster specific markers. In contrast, MTCs formed independent clusters without overlap between cases, revealing a high inter-patient transcriptional heterogeneity among MTCs (Figure 1B). We took advantage of carcinoma and melanoma markers, absent in brain tissue, to accurately identify MTCs in all analyzed cases and confirmed the annotations of malignant cells by evaluation of chromosomal aberrations estimating copy-number variations (CNV) in each tumor (STAR Methods). From carcinoma and melanoma BrMs, 80,377 single cell transcriptomes were analyzed, including 49,488 cells annotated as MTCs and 30,889 non-malignant BrM-associated cells. For downstream analyses, unless specified, we sought to reveal the commonalities across BrM tumors, with an emphasis on inflammatory immune and malignant cell states.
The cancer cell contribution of this BrM set ranged from 3.6% (Lung-3) to 98.1% (Melan-2), with a median of 56.6% across biopsies (Figure 1C), which was confirmed and further illustrated by single BrM sample clustering (Figure S1A) and immunostaining for KRT19 or MELAN-A to mark cancer cells (Figures 1D and S1B).
The unbiased clustering of non-malignant cells (STAR Methods) revealed a structure of the brain metastatic niche with UMAP clouds of vascular cells (endothelial and mural vascular cells), inflammatory immune cells, and mesenchymal progenitors (Figure 1E). Only very few astrocytes were detected in BrM lesions (Figure S1C). We observed comparable niche composition when clustering melanoma and carcinoma BrMs separately (Figures S1D and S1E).
We identified twenty distinct clusters that we assigned on the basis of marker gene expression as endothelial cells (EC-1, EC-2 and EC-3 clusters) with marker genes CLDN5 and PECAM1, mural vascular cells including pericytes (PC) and vascular smooth muscle cells (vSMCs) (PC-1, PC-2, PC-3 and vSMCs) with marker genes RGS5 and ACTA2, mesenchymal stromal cell-like cells (MSC-like-1 and MSC-like-2) with marker genes ISLR and CTHRC1 (Figures 1E and 1F). T cells (T:CD8+:EM, T:CD4+:CM1, T:CD4+:CM2, Tregs and T:CM) were identified with marker genes CD3D and IL7R, B cells (B-c1 and B-c2) with marker genes JCHAIN and MZB1, metastasis-associated macrophages (MAMs:APOE+ and MAMs:S100A8+) with marker genes AIF1 and LYZ, and dendritic cells (DCs) (cDC2:CD1C+/CLEC10A+) with marker genes CD1C and CLEC10A. We also identified one cluster of reactive astrocytes with marker genes GFAP and S100B (Figures 1E and 1F). Two ambiguous clusters characterized by mural cell markers and immune cell markers, detected in the Breast-1 and Unknown Carc. samples, were excluded from further analysis (Figure S1F). All markers and curated gene signatures used for annotations are listed in Table S2.
Our analysis of stromal composition indicates that human brain metastatic niches are comparable across multiple patients and cancer types (Figures 1E and S1A) but displays major differences at the immune composition level. Profiling of 6,325 cells from adult rhabdomyosarcoma BrM resected from brain cortex and unbiased clustering identified a stromal structure that mirrored the stroma in melanoma and carcinoma BrMs (Figure S1G). This well-annotated dataset of human parenchymal BrMs allowed us to next perform a deep characterization of the metastatic niche and malignant populations.
Molecular Definition of the Blood-Tumor-Interface in BrM
The blood-tumor-interface (BTI) is of relevance and problematic with chemotherapy in BrM patients (Arvanitis et al., 2020; Sprowls et al., 2019), but understanding of its cellular composition and properties remains incomplete. Here, we characterized the BrM-BTI through the capture of 12,521 endothelial and mural vascular cells (Figure 2A). RNAscope in situ hybridization confirmed the perivascular location of RGS5+ mural vascular cells (Figure S2A). Across tumors, the proportion of vascular cells ranged from 5% to 77%, with a median of 40% of the total stromal fraction (Figure 2B), we also confirmed by immunostaining for CD31 (PECAM1) (Figure S2B).
Figure 2. Uncovering the BrM-associated Blood-Tumor-Interface (BTI).
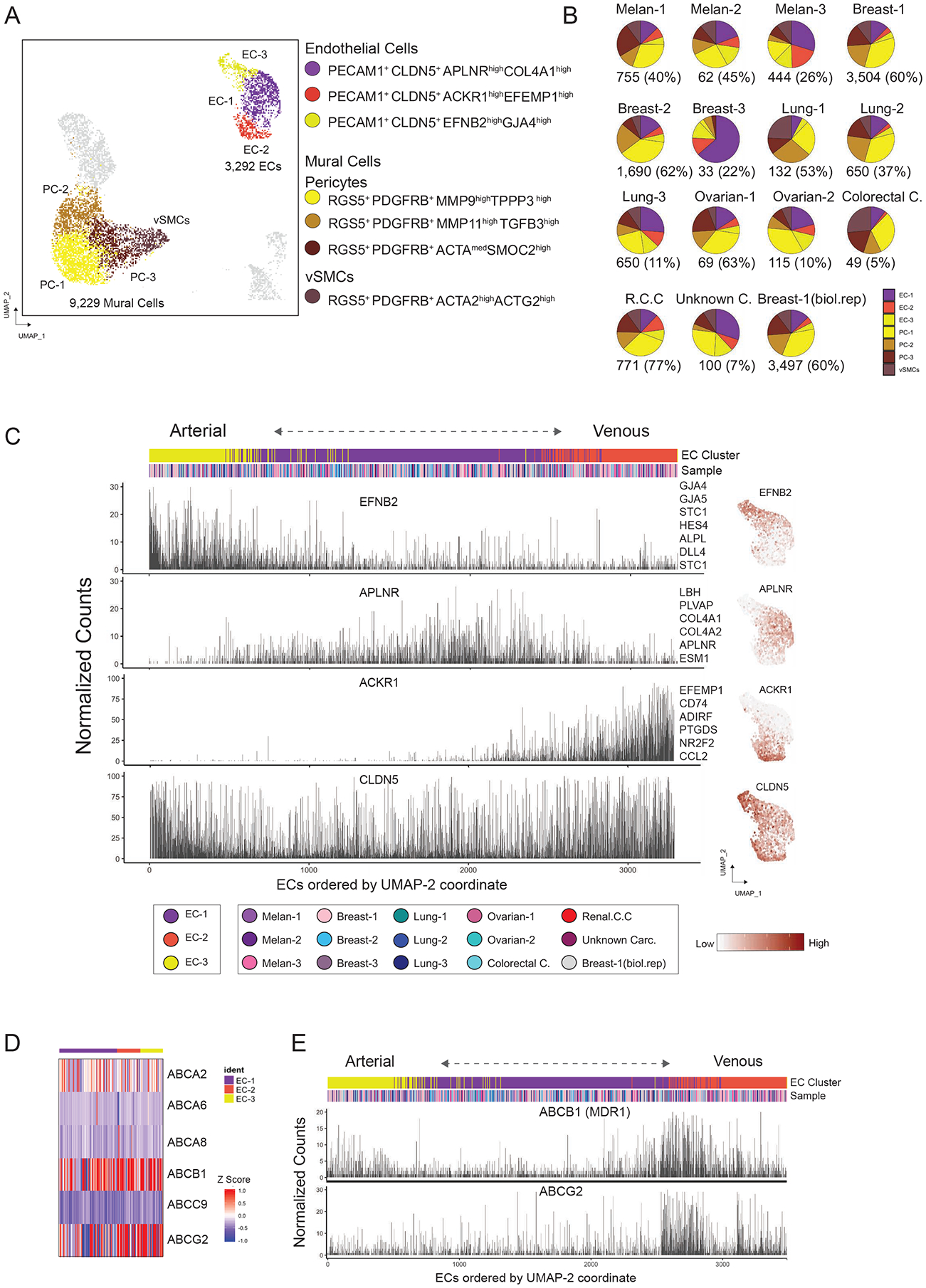
(A) Visualization of 3,292 endothelial cells and 9,229 mural vascular cells using UMAP embedding. (B) Pie charts illustrating the endothelial and mural vascular cell frequency per sample. Number of cells and percentage of the stromal fraction per sample are shown. (C) Zonal expression of arterial and venous transcripts across endothelial cells sorted by the UMAP-2 coordinate, with clusters and sample distribution shown above. Right panel illustrates feature plots for selected endothelial markers. (D) Expression of multi-specific ATP-binding cassette (ABC) transporters detected in BrM associated endothelial cells. (E) Expression of ABCB1 (MDR1) and ABCG2 across the arteriovenous axis sorted by the UMAP-2 coordinate. See also Figure S2.
Mural vascular cells were composed by three types of pericytes (PC) and a single cluster of vascular smooth muscle cells (vSMCs) (marked by ACTG2 and ACTA2 expression) (Figure 2A). Most marker genes are associated with tumor-associated processes, such as angiogenesis (e.g., MMP9) and extracellular matrix (ECM) remodeling (e.g., TINAGL1 and TGFB3) (Figure S2C) (Kessenbrock et al., 2010; Shen et al., 2019). In close relation to mural vascular cells, MSC-like cells upregulated the multifunctional secreted Wnt modulator CTHRC1 (Yamamoto et al., 2008) and the stem cell marker ISLR (Maeda et al., 2016) (Figure S2C), but expressed low levels of RGS5; RNAscope in situ hybridization for CTHRC1 reveals a perivascular location for these cells (Figures S2D and S2E). MSC-like cells were enriched for ECM related genes (e.g., POSTN, COL1A1 and PDGFRA).
The three endothelial clusters (EC-1, EC-2 and EC-3) encompass 3,292 cells. UMAP trajectories reflect a gradual progression along an arteriovenous axis (Figure S2F), in which cells with higher expression of arterial markers (e.g., EFNB2 and GJA5) or venous markers (e.g., ACKR1 and NR2F2) (Hirashima and Suda, 2006; Vanlandewijck et al., 2018) occupy more peripheral positions in the UMAP projection, consistent with a pattern of continuous phenotypic change (zonation), and confirmed using the molecular atlas of brain vasculature (Vanlandewijck et al., 2018) (Figures 2C and S2G). 60% of the total endothelial cells were assigned to a dominant EC-1 cluster EC-1 with tip cell markers APLNR and ESM1 (tip-like ECs) (Zhao et al., 2018) (Figures S2H and Table S2) and characterized by the enrichment of processes such as angiogenesis and collagen deposition. EC-2 (venous-like) expressed genes related to hypoxic, inflammatory and antigen-presentation related processes (Figures S2I and S2J), pointing to the role of the local intra-metastatic microenvironment in the phenotypic states of ECs.
Disappointing results in multiple clinical trials for BrM tumors, compared to extracranial metastases (Achrol et al., 2019), may stem from retention of blood-brain barrier (BBB) efflux properties by BrM-associated BTI. We explored gene expression that could relate to drug resistance and detected 6 multi-specific ATP-binding cassette (ABC) transporters expressed by BrM-ECs (Figure 2D), including ABCB1 (or multidrug resistance protein 1 (MDR1)) and ABCG2. Both transporters, crucial players in normal BBB, are expressed along the arteriovenous axis, with prominence in the zone where vein-like and tip-like ECs converge (Figure 2E).
scRNAseq of BrM-Infiltrated Immune Cells Reveals Heterogeneous T Cell Responses
Metastasis-associated innate- and adaptive- immune cells play major roles in determining the fate of MTCs in experimental mouse models of metastasis (Gonzalez et al., 2018a; Gonzalez et al., 2018b). Detailed information on cell types and states that integrate host immune responses in distant human metastases remains scarce (Friebel et al., 2020; Klemm et al., 2020). By annotating 14,499 single immune cell transcriptomes based on marker genes and curated gene signatures (Table S2), we identified five major T cell clusters, two macrophage clusters, one DCs cluster and two B cell clusters. All melanomas and carcinomas BrMs displayed mixed immune cell compositions and, thus, each immune cluster was comprised of cells from multiple tumors (Figures 3A and 3B). High frequencies of T cells and macrophages dominate the immune landscape of BrMs (Figure 3B), consistent with recent evidence (Friebel et al., 2020). Analysis of immune frequencies by cancer types, suggests that BrMs from different breast cancer patients display higher frequencies of macrophages. By contrast, lung cancers exhibited higher frequencies of T cells, while higher frequencies of B cells were observed in melanomas and ovarian cancer (Figures 3B and S3A). We postulated that BrM specific therapies could impact the observed immune clusters and immune frequencies but found no evidence for this (Table S1).
Figure 3. Immune Cell Types and Activation States Enriched in Human BrMs.

(A) UMAP plot of immune clusters. Bottom panel displays feature plots for selected markers. (B) Frequencies of immune clusters per sample, indicating the number of cells. (C) T cell clusters ordered by the diffusion component 1 score, visualized with density ridgeline plot. (D) Scatterplot showing the co-expression of the diffusion component 1 genes and a curated T cell activation signature (GO:0042110). T cell clusters are labeled colors. (E) Heatmap reporting the average expression of microenvironmental and metabolic gene signatures in each T cell cluster. (F) Boxplots displaying the normalized expression of known and new T cell anergic markers. (G) Heatmap of myeloid clusters, and representative markers. (H) 2D projection of the diffusion map analysis on macrophages clusters, which shows a continuum phenotypic change between two macrophages states. Bottom panel shows the normalized expression of selected markers. (I) RNA velocity analysis embedded in 2D diffusion map plot for samples Breast-1 and Melan-2. (J) Boxplots of transcriptional factors differentially expressed between macrophages clusters, with t-test statistics. (K) Heatmap showing the normalized expression level for selected markers for antigen presentation, inflammatory and migratory related processes in cDC2:CD1C+/CLEC10A+ cluster (879 cells). See also Figures S3, 4, S4 and Table S2.
The abundance and functional states of tumor infiltrated T cells are prognostic in several cancer types (Binnewies et al., 2018), and can impact cancer dissemination and metastatic outgrowth (Gonzalez et al., 2018a). Here, T cells showed the greatest degree of phenotypic heterogeneity among immune infiltrates in our BrM set. The 8,266 analyzed T cells were composed of CD8+ effector memory T cells (T:CD8+:EM), central memory T cells (T:CD4+:CM1, T:CD4+:CM2 and T:CM) and regulatory T cells (Tregs) (Figure S3B). Functionally, the T:CD8+:EM cluster was enriched in cytotoxic molecules (e.g., GZMA and IFNG) (Figure S3C). Markers for immunomodulation (e.g., LTB and IL32) and stress response (e.g., HSPA1A), defined similar but not identical T cell states (Figures S3C and S3D). For instance, the T:CD4+:CM cluster represented an activation state enriched of LTB, IL32 and IL7R, whilst the T:CM cluster that expressed low levels of CD4 and CD8A, displayed a similar state (LTB+ and IL32+) but differentiated by the enrichment of interferon related genes like ISG15 and MX1.
To better understand the functional transitions of these T cell states, we used diffusion maps, a non-linear dimensionality reduction technique that captures geometric structure and functional states in high dimensional data (Coifman et al., 2005; Haghverdi et al., 2015). This analysis revealed that the five T cell clusters are organized in a phenotypic variation coherently ordered by DC1 (diffusion component 1) (Figure 3C). Among the top 50 genes positive correlated with DC1 are molecules associated with lymphocyte activation (e.g., CORO1A and LCK) and chemotaxis and migration (e.g., CCR7 and CXCR4) (Figure S3E and Table S2). There is a strong correlation between expression of these top 50 DC1 genes and a curated T cell activation gene signature (GO:0042110) (Figure 3D), further supporting the notion that the activation state drives the observed diversity of T cell clusters.
To understand the context-dependent role of each T cell state, we evaluated expression of microenvironmental and metabolic gene signatures (Table S2). We observed variable expression for processes related to hypoxia, type-I and -II interferon responses, and inflammation (Figure 3E). The enrichment of interferon signature was most dominant in cluster T:CM. In contrast, enrichment in exhaustion signature associated with high T cell activation programs was most present in clusters T:CD8+:EM and T:CD4+:CM. Those T cell clusters with lower DC1 score (T:CD4+:CM2, Treg and T:CM) exhibited a strong anergic signature (Figure 3E). Interestingly, this phenotypic transition from activated to anergic T cell, coincides with a switch in metabolic signatures to lipid metabolism from glycolysis and the tricarboxylic acid (TCA) cycle (Figure 3E). Recent data supports the notion that impairment in TCA metabolism induces defective effector T cell function (Vardhana et al., 2020). Unexplored markers identified in the anergic T cell states included SERPINE1, PMP22 and NDRG1 (Figure 3F). In sum, our analyses revealed a spectrum of functional T cell states ranging from activated/exhausted to anergic, associated at single cell level with a concomitant metabolic and microenvironmental reprogramming.
Myeloid Composition of Brain Metastases
Myeloid cells, including MAMs (metastasis-associated macrophages) and DCs (dendritic cells) have been identified as important players in formation of the metastatic niche, either sustaining or restricting the metastatic outgrowth (Gonzalez et al., 2018a; Hagerling et al., 2019; Headley et al., 2016; Qian and Pollard, 2010). Here, we identified two major subsets of MAMs in our collection of BrMs; APOE+/C1QB+/TREM2+ MAMs (MAMs:APOE+) and IL1B+/FCN1+/S100A8+ MAMs (MAMs:S100A8+) (Figure 3G). MAMs:APOE+ express a core set of immunomodulatory molecules, including complement C1Q chains, SPP1 and HLA-related molecules, resembling TAMs (tumors associated macrophages) programs enriched in lung, liver and CRC tumors (Lavin et al., 2017; Zhang et al., 2020; Zhang et al., 2019). MAMs:S100A8+ express an inflammatory program characterized by high expression of S100A family genes, CXCL8, FCN1 and low expression of HLA-related genes (Figure 3G), resembling inflammatory FCN1+ myeloid-derived suppressor cells (MDSC) (Zhang et al., 2019). Thus, as previously reported in primary TAMs (Azizi et al., 2018), the identified BrM MAMs states are more complex than the classical M1/M2 polarization paradigm.
Functionally, a Gene Ontology (GO) analysis using the top 50 differentially expressed (DE) genes between MAMs subsets revealed a strong enrichment of antigen processing and presentation (P < 10–22, hypergeometric test) in MAMs:APOE+ versus an enrichment of cytokine-mediated signaling (P < 10–12, hypergeometric test) and response to interleukin 1 (P < 10–8, hypergeometric test) in MAMs:S100A8+ (Figure S3F). UMAP projection and diffusion map trajectories suggest that these two MAMs clusters represent differentiated cell states across a continuum of phenotypes (Figures 3A and 3H). For instance, expression of HLA-DQA1 gradually decreased across the DC1 dimension from MAMs:APOE+ to MAMs:S100A8+ (Figure 3H). RNA velocity analysis (La Manno et al., 2018) supported this notion and suggested a putative trajectory from MAMs:APOE+ to MAMs:S100A8+ (Figure 3I), thus MAMs:APOE+ and MAMs:S100A8+ subsets can scale across this continuum in a dynamic manner. Moreover, each MAMs program expressed distinctive transcription factors, for instance CREG1 and EGR2 were preferentially expressed in MAMs:APOE+, while ZFP36 and MAFF were preferentially expressed in MAMs:S100A8+ (Fig. 3J).
Finally, our myeloid compartment analysis identified one predominant DC cluster characterized by conventional type-2 DCs markers (CD1C and CLEC10A) (Figure 3G). Within this cluster, DCs with high expression of the migratory receptors (e.g., CXCR4 and GPR183) and high expression of the transcriptional regulators JUNB and NR4A2 downregulated most of the antigen processing and presentation molecules (e.g., CD74 and HLA-DPB1) and vice versa (Figure 3K). These DCs may exhibit mutually exclusive intra-metastatic migratory and antigen presenting properties.
CyTOF Analyses of Immune Cell Protein Markers in Brain Metastases
To extend our analysis with an orthogonal approach, we used high-dimensional mass cytometry (cytometry by time-of-flight, CyTOF) analysis to quantify 30+ protein parameters simultaneously in single cells (Table S3), for 11 of the 15 BrM biopsies with sufficient tissue. Distinct cell populations were projected in two dimensions with UMAP, representing a total of 20,900 cells from the 11 BrM biopsies (Figure 4A). Supervised analysis of abundance and activity state of the major immune cell subsets confirmed that T cells and macrophages are the dominant immune infiltrates in BrMs (Figure 4B). Proliferation of CD45+ cells, as evaluated by Ki67 expression, was modest in all samples, indicating a predominantly indolent immune microenvironment in BrMs (Figure 4B).
Figure 4. Mass Cytometry Analysis of BrM-associated Immune infiltrates.
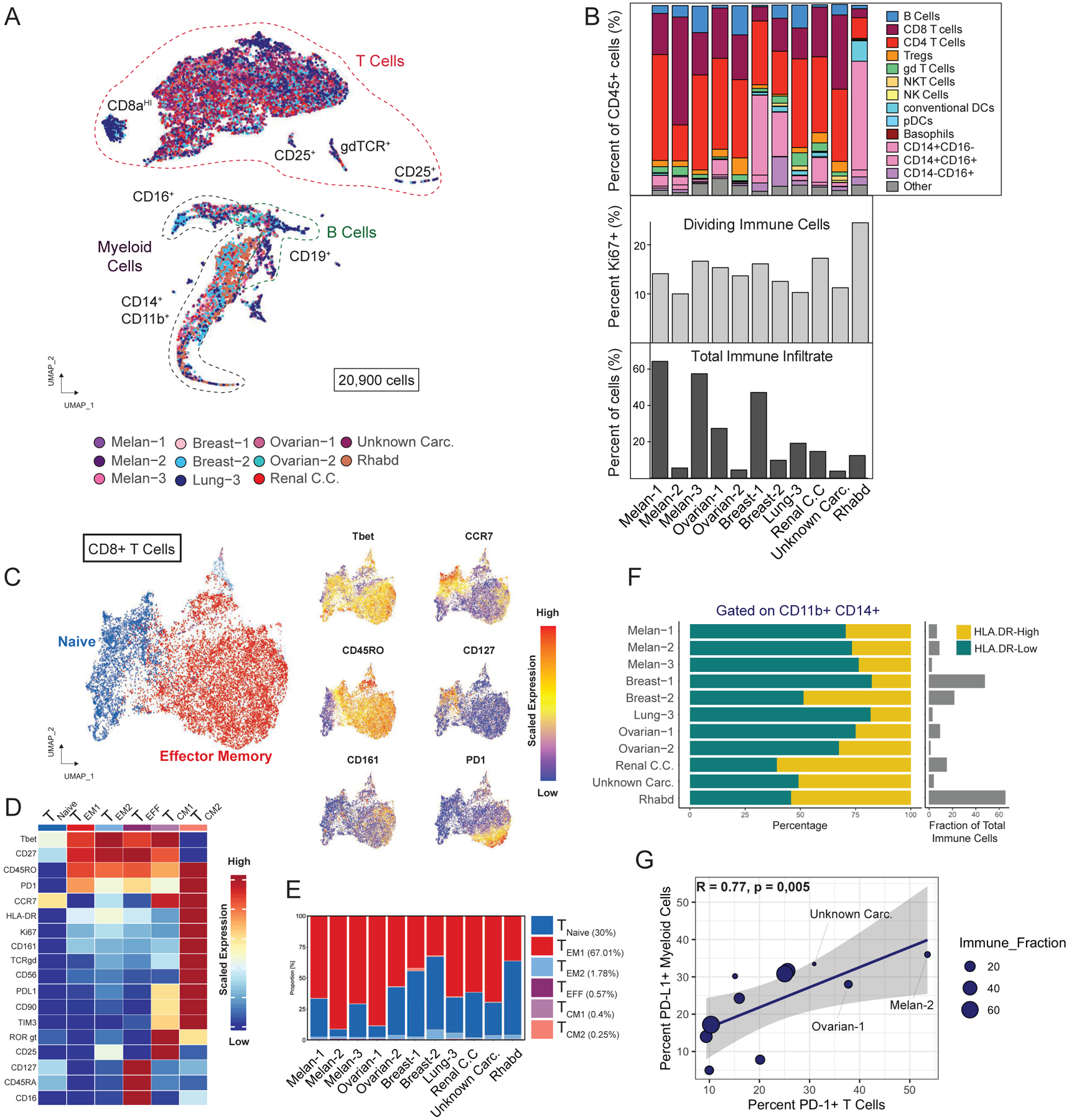
(A) UMAP projection of 20,900 immune cells showing the major immune cell populations. Cells are colored by sample. (B) Frequencies of major immune cell populations. (C) CD8+ T cell population clustered by FlowSOM and visualized by UMAP (see STAR Methods). (D) Functional and phenotypic median expression profiles for each CD8+ T cell clusters. (E) CD8+ T cell cluster proportions by patients. (F) Frequency of HLA-DR-high and HLA-DR-low cells within the CD11b+CD14+ population. (G) Scatterplot showing the correlated expression of PD-L1 by myeloid cells and PD-1 by T cells. See also Figure S4 and Table S3.
In depth analyses of T cells by UMAP and FlowSOM (Van Gassen et al., 2015) (STAR Methods), identified 6 clusters of CD8+ and CD4+ T cells on the basis of protein markers. Most CD8+ T cells (~69%) displayed a CD45ROhigh:CD45RAlow:CCR7neg:CD27high effector memory phenotype Figures 4C, 4D and 4E). CD4+ T cell clusters were phenotypically heterogenous, existing in a continuum across effector memory and central memory phenotypes (CD45ROhigh:CD45RAlow:CCR7pos:CD27low) (Figures S4A, S4B and S4C). Comparing both approaches, by and large, the T cell type annotations by single cell transcriptomics (Figures 3 and S3) are in line with T cell annotations observed by CyTOF. Similarly, gating on CD11b+/CD14+ cells (monocytes/macrophages) revealed two major subpopulations of cells, HLA-DRhigh and HLA-DRlow (Figure 4F), supporting the notion of the co-existence of two monocytes/macrophages states in each BrM tumor differentiated by antigen presenting properties.
Encouraging studies suggest that immune checkpoint inhibitors cross the blood-tumor-barrier and be effective to treat BrMs (Nieblas-Bedolla et al., 2021). We analyzed PD1 and its receptor (PD-L1), two of the most common targets for cancer immunotherapy (Sharma and Allison, 2015). PD1 expression was elevated in CD8+ and CD4+ T cell effector memory clusters (Figures 4D and S4B), associated with medium/low expression of activation marker HLA-DR and proliferation marker Ki67, and we also detected moderate expression of PD-L1 (Figures 4D and S4B). Increased PD-1 levels and PD-L1 co-expression likely represent increasingly exhausted T cell states (Wherry and Kurachi, 2015). Across all CD45+ T- and myeloid- cells, ~20% of T cells expressed PD1 and ~28% of myeloid cells expressed PD-L1 with significant correlation between PD-1+ T cells and PD-L1+ myeloid cells (Pearson’s r = 0.77, P = 0,005) across patients (Figure 4G), indicating a coordinated intra-metastatic pattern of immune checkpoint molecule expression.
Eight Functional Metaprograms in MTCs Define Brain Metastases
Successful metastasis depends on functional programs that allow metastatic tumor cells (MTCs) to survive, modify and ultimately dominate the parenchyma of distant organs while interacting with host-specific characteristics of the stroma (Lambert et al., 2017). Elucidating these programs has been challenging (Robinson et al., 2017).
We next performed a systematic analysis of MTC populations in BrMs to explore and characterize such potential programs. We first evaluated inter-patient heterogeneity by UMAP and transcriptional variance. As previously observed in Figure 1B, MTCs from all patients formed discrete individual clusters (Figure S5A), revealing a significant inter-patient heterogeneity (72% of transcriptional variance explained by the first 10 principal components) (Figure S5B). Moreover, 3D diffusion map analyses and unsupervised clustering of MTCs, pointed out a rich functional diversity within individual tumors. Examples are proliferation, ECM deposition, inflammation, or response to stress (Figures 5A, S5C and S5D).
Figure 5. Metastatic Cells Acquire Two Well-Defined Functional Archetypes.
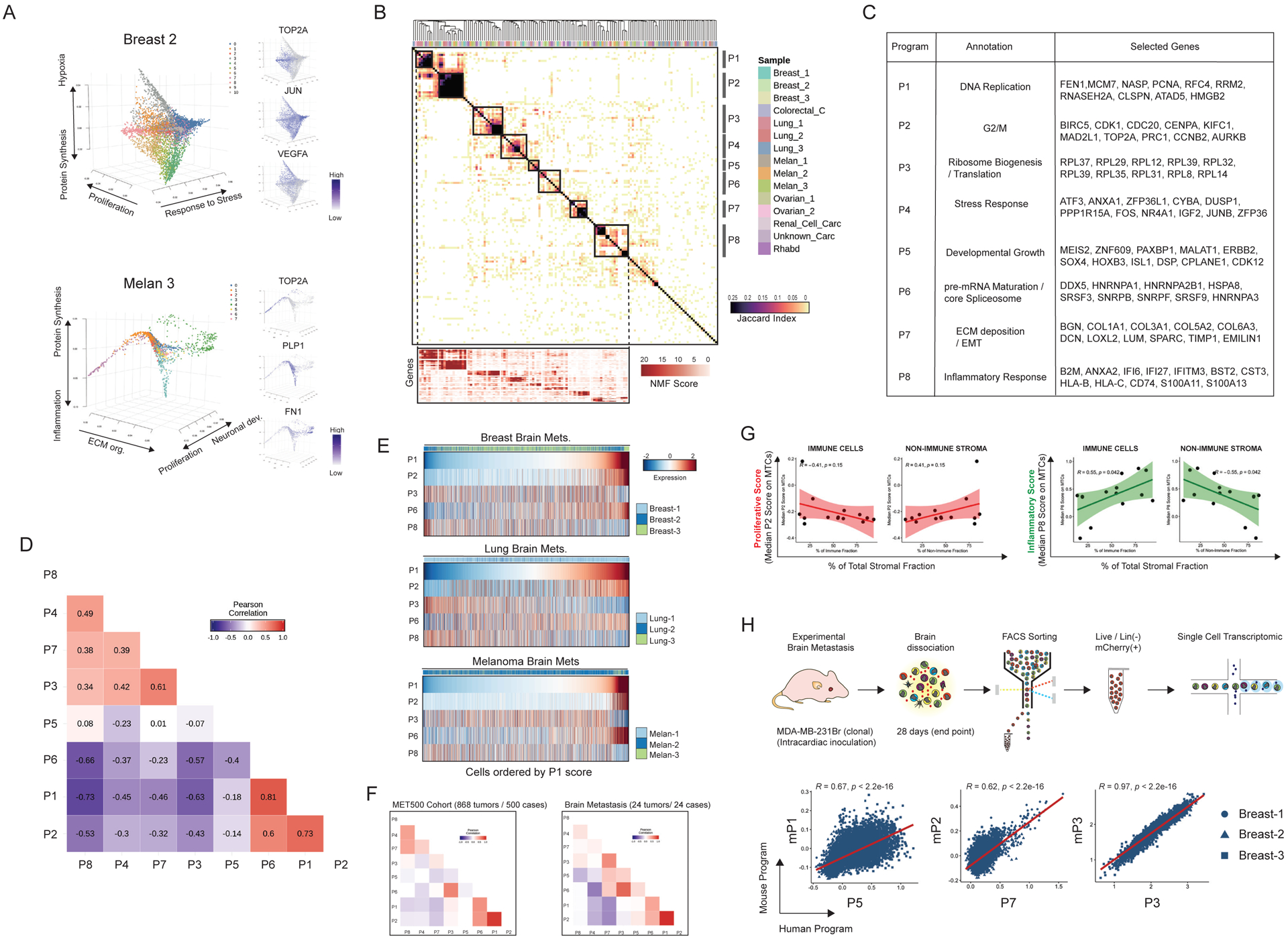
(A) Intra-tumoral diversity of metastatic cells in two selected samples evaluated by diffusion map analysis and clustering. Right panel shows the normalized expression of selected markers. (B) Hierarchical clustering of pairwise similarities between NMF programs identified across metastatic cells from all the analyzed samples. Bottom panel displays the NMF scores of signature genes (rows) for each metaprogram (columns). (C) Annotation and selected top genes for each metaprogram. (D) Heatmap displays the Pearson correlation coefficients calculated between the single-cell gene signature scores of NMF metaprograms. (E) Heatmap showing the expression of selected metaprograms. Cells were plotted by cancer type (breast, lung, and melanoma) and ordered by the P1 score. (F) Correlation coefficients calculated by scoring the 868 samples from MET500 cohort for the NMF metaprograms. Samples were plotted including all (n: 868) metastatic samples (left) and just BrM (n: 24) (right). (G) Scatter plot comparing the median score value for inflammatory (P8) or proliferative (P2) metaprograms on MTCs, and the composition of the stromal fraction in each sample. (H) Interrogation of the functional equivalence of human vs mouse BrM programs. Top panel shows the schematic illustration of the murine brain metastasis and scRNAseq analysis approach. Scatterplots display the co-expression of human NMF metaprograms scores (x axis) and mouse programs scores (y axis) in single cells from the 3 breast cancer BrM samples. See also Figure S5, Figure S6 and Table S4.
Next, to define coherent gene sets (programs) within single cell populations and to explore the existence of common programs that define MTCs among different patients, we applied non-negative matrix factorization (NMFs) (Puram et al., 2017). Hierarchical clustering based on shared genes of 150 transcriptional programs (10 programs of each patient) identified 8 metaprograms (P1–P8) recurrent in multiple or all BrMs (Figure 5B). The observed metaprograms were functionally annotated using their top-50 genes ranked on the basis of NMF scores (metagenes) (Table S4). Metaprograms P1 and P2 were classified as cell cycle programs; the P1 program was enriched for genes of the DNA replication (e.g., MCM7, FEN1 and RFC4), whilst P2 was characterized by G2/M phase genes (e.g., BIRC5, CDK1 and AURKB) that were highly similar across samples, defining conserved proliferation programs in BrMs (Figure 5C). Metaprogram P3 was enriched by structural ribosomal proteins (RPs) of the 60S ribosome (e.g., RPL37, RPL29 and RPL12) and identified in 14 out of 15 samples. P4 was present in 12 out of 15 samples and characterized by stress response genes (e.g., ATF3, DUSP1 and CYBA). P5 represents markers of developmental processes, for example transcription factors such as MEIS2, PAXBP1, SOX4 and HOXB3 and was identified in 5 carcinoma BrMs. Metaprogram P6 encompasses pre-mRNA maturation and core spliceosome genes (e.g., DDX5, HNRNPA1 and SRSF3). P7 represents ECM components such as COL1A1, COL3A1, MMP2 and SPARC, and was enriched for epithelial-to-mesenchymal transition (EMT) markers (e.g., VIM, MYL9 and FN1) (Figure 5C and Table S4). The final metaprogram (P8) was present in all 15 analyzed samples and defined by inflammatory and interferon related genes (e.g., IFI6, HLA-A and S100A11). Of note, we also examined the genetic heterogeneity of MTCs in all samples, by estimation of large-scale copy number variations (CNVs) (Tirosh et al., 2016b). Consistent with recent evidence (Reiter et al., 2020), we observed that BrM lesions are predominantly composed by genetically homogeneous populations of MTCs (Figures S5E and S5F). In sum, the selective analysis of MTC populations revealed eight recurrent metaprograms (P1–P8) associated with highly specialized, functional processes.
Two Archetypes of MTCs Support Brain Metastases
Deeper analysis of the 8 metaprograms (STAR Methods) revealed that these are not utilized in random fashion, instead, they delineate two well-defined functional archetypes (Figure 5D); For instance, P3 (ribosome biogenesis / translation program) positively correlated with P7 (ECM deposition and EMT program) (Pearson’s r = 0.61, p < 2.2e–16). Interestingly, P3 and P7 followed a strong pattern of co-exclusion with P1 (p < 2.2e–16; p < 2.2e–16) and P2 (p < 2.2e–16; p < 2.2e–16) cell cycle metaprograms (Figure 5D). Furthermore, there was a striking positive correlation between P1 and P2 (proliferation) and P6 (pre-mRNA splicing program) (p < 2.2e–16; p < 2.2e–16) and concomitant mutual exclusivity with P4 (stress) (p < 2.2e–16) and P8 (inflammatory) (p < 2.2e–16), either in combined analyses or across individual BrMs (Figures 5D and 5E). Our analyses of the eight metaprograms put forth the model that there are two underlying single-cell archetypes that co-exist in BrMs, one high in proliferation and pre-mRNA splicing (P1, P2 and P6), the other high in stress, inflammation, translation, and ECM deposition (P3, P4, P7 and P8). In addition, plotting single cell data by P1 score showed a continuum of intermediate states between these two archetypes (Figure 5E). These results imply that those MTCs that are not proliferating, may get reprogramed into the other archetype (enriched in P3, P4, P7 and P8). Importantly, scoring bulk transcriptomes from 868 multi-organ metastatic tumors or 24 BrMs available in the MET500 cohort dataset (Robinson et al., 2017) through our 8 metaprograms, showed similar patterns of metaprogram co-expression and anti-correlation (Figure 5F).
Next, we interrogated the two MTCs archetypes in terms of immune and stromal phenotypes described in this study. Expression of the inflammatory metaprogram (P8) by MTCs was positively associated with the percentage of immune cells (r=0.55, p = 0.042), and negatively associated with non-immune stroma (r= −0.55, p = 0.042) (Figure 5G). Among immune cells, the T cell clusters with higher activation and low anergic signatures T:CD8+:EM, T:CD4+:CM1 (See Figure 3E), and B-c1, showed positive associations with P8 expression by MTCs. In contrast, MAMs:APOE+, Treg, and the highly anergic cluster T:CM2 (See Figure 3E), exhibit negative associations with the expression of P8 by MTCs (Figure S6A and S6B). These observations suggest a correlation between the composition of the stromal niche and the generation of the two described MTCs archetypes.
Finally, we interrogated the specificity of the P1–P8 metaprograms in publicly available single cell datasets of primary breast cancer, lung cancer, melanoma, and glioblastoma (Kim et al., 2020; Neftel et al., 2019; Tirosh et al., 2016a; Wu et al., 2021), by analyzing the expression of the top 30 genes of each program. We observed that programs such as P1/P2 (Cell Cycle), P3 (Protein Synthesis) and P6 (pre-mRNA Maturation / Core Spliceosome) were clearly identified in primary tumors (Figure S6C). In contrast, a fragmented expression of programs P8, P5 and P4 was observed. The most striking difference was the scarce expression of metaprogram P7 (ECM deposition / EMT) in primary tumors. More importantly, the co-expression patterns of programs differ between primary tumors and MTCs. For instance, the co-expression of P6 and P1/P2 metaprograms observed on MTCs appears absent in primary tumors. We made similar observations related to P8 and P3 with cell cycle programs P1 and P2 (Figure S6C). These comparisons suggest that the expression of the MTCs metaprograms, and their coordinated pattern of expression are a specific characteristic of human metastatic cells.
Testing Our Metaprograms of Metastasis with Two Experimental BrM Models
As functional exploration of our BrM metaprograms, we investigated if experimental mouse models of BrM can recapitulate these programs. Here we used the breast cancer cell (human MDA-MB-231Br) as case study, a model widely used to study BrM formation (Bos et al., 2009; Chen et al., 2016; Gril et al., 2018; Priego et al., 2018; Valiente et al., 2014; Zeng et al., 2019; Zhang et al., 2015). 3,194 high quality single cell transcriptomes from sorted metastatic MDA-MB-231Br cells were profiled (STAR Methods). We uncovered 6 mouse transcriptional programs (NMF programs (mP1-mP6)) in this experimental MDA-MB-231Br model (Figures S7A, S7B and Table S4). mP4 and mP6 reflected proliferation processes, mP2 a hybrid program was defined by developmental processes and collagen biosynthesis, mP1 represented a developmental program, mP3 reflects a translation and ribosome biogenesis process, and mP5 reflects an angiogenic program (Figure S7B). The mP1, mP2 and mP3 programs partially recapitulated the functional counterpart of P5, P7 and P3 metaprograms in breast cancer BrM patients (Figure 5H). On the other hand, the MDA-MB-231Br experimental model lacked inflammatory and stress response programs, or a discrete ECM / EMT remodeling program as observed in patients.
Secondly, we explored the 4T1Br breast cancer model that is immunocompetent (STAR Methods). Single cell profiling of 1,472 sorted metastatic cells and NMF pipeline analysis revealed 6 transcriptional programs (mP1_4T1Br to mP6_4T1Br) (Figures S7C, S7D, S7E and Table S4). As in the MDA-MB-231Br model, we identified cell cycle programs, developmental growth program, translation, angiogenic programs, and new program associated with oxidative phosphorylation (Figure S7E and Table S4). We did not observe a discrete inflammatory, stress or ECM remodeling / EMT programs. Staining for metastatic cells and immune cells on mouse brain sections at endpoint (Figure S7F) suggests that the absence of inflammatory and stress programs stems from lack of anti-tumoral immune response in metastatic lesions.
An Immune Evasive Program in MTCs Supports Tumor Cell Proliferation
The anticorrelations in the P1-, P2-, and P6- metaprograms with those in P4, P3, P7 and P8, prompted us to understand the mechanistic underpinnings of the anticorrelated archetypes. Combined principal component analysis (PCA) revealed two predominant functional trajectories; cells with high PC1 score (proliferative processes) and cells with low PC2 score (inflammatory processes) and a continuum of states in between (Figure 6A). Immunostaining for Ki67 protein confirmed the existence of heterogeneously distributed, proliferating MTCs and with variable levels of proliferating cells across samples (Figures 6B). We derived a conserved core signature (excluding cell cycle genes, see STAR Methods) that specifies the transition (or reactivation) from quiescence to active proliferation. This core signature contains 190 genes in cycling MTCs among patients, 89 downregulated and 101 upregulated genes (Table S5). Downregulated genes in actively cycling MTCs reflected processes related to inflammatory stress such as major histocompatibility complex (MHC) class I molecules (e.g., HLA-A and B2M), inflammation (e.g., S100A6 and IFI6), macro-autophagy genes (e.g., NUPR1 and SQSTM1) and the exosome marker CD63 (Figure 6C). Among the genes specifically upregulated in cycling cells were members of the core spliceosome and MYC targets (Table S5). Of note, two known imprinted genes were identified among the top hits, MEST (Mesoderm-Specific Transcript) and PEG10 (Paternally Expressed Gene 10), both previously implicated in poor prognosis in multiple cancer types (Ishii et al., 2017; Li et al., 2016; Pedersen et al., 1999; Shapovalova et al., 2019; Vidal et al., 2014) (Figure 6C). We confirmed these patterns by immunostaining for PEG10 and S100A6 in cycling (Ki67+) MTCs (Figure 6D). The core signature has 12 transcriptional regulators that distinguish these two cell states. For instance, the transcriptional repressor HES1 and its repressor HES6 (Bae et al., 2000) followed a pattern of mutual exclusion. Interestingly, the DNA methyl transferase DNMT1 that was found to be upregulated in cycling MTCs (Figure 6C and Table S5); further, the enrichment of DNMT1 has been recently causally associated to immune evasion by cancer cells (Luo et al., 2018; Peng et al., 2015).
Figure 6. Reconstruction of the Transition from Quiescence to Proliferation in Metastatic Cells.

(A) Principal component analysis (PCA) of all metastatic cells colored by patient. (B) Immunohistochemical staining of the proliferative marker Ki67 in two selected samples. Two representative images per sample are shown. Scale bars, 100 μm. (C) Heatmap showing the conserved core signature that describes the transition from cell cycle arrest to proliferative reactivation in metastatic cells. Cells are ordered from left to right by cycling score (CS). (D) Validation at protein level of the patterns observed in scRNAseq. Images of dual immunofluorescent staining of Ki67 and PEG10 or S100A6 in multiple BrM samples. DAPI denotes nuclear staining. For each field, representative cells that denote the differential expression of S100A6 and PEG10 in cycling and non-cycling cells-are indicated by arrowheads. Scale bars, 50 μm. See also Figure 7 and Table S5.
We interpreted the core signature as a reflection of the immune effector response imposed on MTCs and their ability to adapt and escape of such control. In support of this idea, when we inferred the magnitude of leukocyte infiltration from bulk RNAseq in the MET500 cohort by Mimmscore estimation (Robinson et al., 2017), we found that leukocyte infiltration was positively correlated with genes downregulated in cycling MTCs, while genes upregulated in cycling MTCs were negative correlated with immune infiltration (Figure 7A). In addition, the estimation of immune cell types in the MET500 cohort (Newman et al., 2019) revealed that metastatic tumors expressing higher level of genes downregulated in cycling MTCs were enriched of monocytes/macrophages and T cells (Figure 7B). We therefore assessed the level of tumor proliferation, immune infiltrates, and selected markers of the core signature by immunostaining in an independent cohort of 13 patients diagnosed with melanoma- (n:5), lung cancer- (n:5) and breast cancer BrMs (n:3). We confirmed the negative correlation between proliferating metastatic cells and immune infiltrates (Spearman’s rank correlation coefficient, r= −0.69, P = 0,009) (Figures 7C and 7D). Furthermore, in this cohort, biopsies with higher proliferation score and lower grade of immune infiltrates, upregulated PEG10 (up in cycling MTCs) and downregulated CD63 (down in cycling MTCs) at protein levels. The inverse correlation was observed in biopsies with low proliferation and high immune infiltrates (Figure 7E). In sum, we identified a conserved core gene signature, that we believe illustrates escape from control imposed by effector immune response and allows the proliferation of MTCs in patients.
Figure 7. Immune Evasive State of Proliferating Metastatic Cells.

(A) Scatterplots showing the correlation of the expression of genes downregulated and up-regulated in cycling metastatic cells (y axis) with the magnitude of immune infiltration (MimmScore) across 868 metastatic tumors (MET500 cohort) (x axis). (B) Relative proportions of immune infiltrates in MET500 samples with high immune composition evaluated by CIBERSORT with the estimation confidence (empirical p-val <0.05). The color represents the Spearman correlation coefficients between estimated cell type fractions and the expression of a signature called “genes down in cycling MTCs”. (C-D) Immunohistochemical staining and quantification of the proliferative marker Ki67 and the immune marker CD45 in an external cohort of 13 BrM samples (Lung = 5, Melanoma = 5, Breast = 3). Scale bars, 100 μm. Bottom panel shows the correlation between the percentage of proliferation (x axis) and immune infiltration (y axis). (E) Immunohistochemical staining of the proliferative marker Ki67, immune marker CD45, markers down-regulated (CD63) and up-regulated (PEG10) in cycling metastatic tumor cells. Two BrM cases patients are shown. Scale bars, 100 μm. See also Figure 6 and Table S5.
DISCUSSION
Here, we generated and analyzed a comprehensive catalog of the functional cell types and cellular states present in human parenchymal BrMs as a resource for the research community.
In the first part we focused on immunoprofiles of BrM. We identified two macrophages states, MAMs:APOE+ and MAMs:S100A8+, in BrM. Recent evidence from melanoma and carcinoma patients suggests that BrM-associated macrophages may be derived from both, resident microglia cells (~ 10–50%) and infiltrated monocytes (~ 25–75%) (Friebel et al., 2020). The MAMs:APOE+ population expresses SPP1 and SPP1+ TAMs have been described as macrophages in CRC linked to therapy resistance and angiogenesis (Zhang et al., 2020). Relevant to the MAMs:S100A8+ we describe here, enrichment of S100A8+/S100A9+ TAMs is associated with immunosuppression and shorter survival in patients with head and neck cancer (Kwak et al., 2020) and poor response to checkpoint inhibitors in patients with metastatic melanoma (Wagner et al., 2019). T cells are a dominant immune cell type in BrM and we uncovered correlations between functional T cell states and metabolic and microenvironmental signatures. Recent evidence points to the role of hypoxic and acidic tumor microenvironments in sustaining immunosuppressive T cell states (Watson et al., 2021) and promoting dysfunctional effector T cells (Scharping et al., 2016). Continuous antigen stimulation coordinated with hypoxia results in impaired mitochondrial function and promotes terminal T cell exhaustion in melanoma (Scharping et al., 2021).
In the second part, we revealed two well-defined cellular archetypes by the selective characterization of MTCs that resulted in a framework of the P1–P8 metaprograms. Previous pan-cancer transcriptomic analyses identified two major categories of metastases in patients: highly proliferative or highly inflammatory (Robinson et al., 2017). Our observations support and expand this notion by identifying that proliferative and inflammatory processes coexist as opposite major cell states, and suggest that immune scape and proliferation act as concerted events in MTCs. Our work here, and that of others, supports the theory that distant metastases are formed by genetically homogeneous populations (Priestley et al., 2019; Reiter et al., 2020). Our study with single-cell resolution analysis of MTCs, stromal, and immune cell types puts forth testable hypotheses to understand the interplay between tumor cell-intrinsic traits and host environment traits in human metastasis.
LIMITATIONS OF THE STUDY
Our study suggests that our findings may not be driven by recent therapies. However, we have not definitely determined the impact of previous therapies. It is common for cancer patients to develop distant metastases long after treatment and removal of primary tumors. Likewise, we have not determined the prognostic impact of our findings for BrM patients. Future efforts assembling single cell datasets with clinical information from larger cohorts will be required to infer clinical outcomes, and to study the impact of the therapies aimed to treat primary cancer, on the programs observed in metastases.
Although our single-cell transcriptomic dataset exceeds the scale of previous efforts to systematically characterize BrM tumors, or any other distant metastases for that matter, our study does not have a large enough sample size to demonstrate unique features for cancer type, cancer subtypes, or BrM specificity due a lack of comparisons with matched primary tumors or extracranial metastases. Much remains to be gained by further increasing the sample size. Metastatic biopsies remain rare and small specimens, the poor patient prognosis makes it difficult to collect them, and those that are obtained often are not profuse enough to offer multiple layers of information (Bova, 2017). Future efforts in collecting and biobanking fresh resected specimens will facilitate cancer specific studies at single cell resolution, especially in metastatic biopsies from less studied metastatic cancers such as: ovarian, renal, colorectal or sarcomas.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Hugo Gonzalez Velozo (Hugo.GonzalezVelozo@ucsf.edu).
Material Availability
This study did not generate new reagents.
Data and Code Availability
Raw mapped counts from patients, raw mapped counts and FASTQ data from experimental mouse models, and normalized expression data with cell type annotations can be found in the National Center for Biotechnology information Gene Expression Omibus (GEO) (accession number GSE186344).
We did not generate original codes in this study. Codes used are available upon request without restrictions.
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human Specimens
Human specimens from fifteen patients who were pathologically diagnosed with brain metastases, were enrolled in this study after approvals by the Ethics Committee of University of California San Francisco, IRB Number 10–01318. Resources were provided by the UCSF Brain Tumor SPORE Biorepository NIH/NCI 5P50CA097257–18 (J.J.P.). The sample R.C.C. (renal cell carcinoma) brain metastasis was collected and sent from UCSD by Dr. Clark Chen. All patients in this study provided written informed consent for sample collection and data analyses. Their ages ranged from 42 to 76, with a median age of 63 years. Eleven of the cases were women and four men. With the exception of patients Breast-1 and Breast-3 that received gamma radiation 24hrs before surgery, none of the patients was treated with chemotherapy or radiation aimed to treat brain metastasis prior to surgery. The time to brain metastasis progression since initial cancer diagnosis ranged from 0 months (7 cases) to 171 months (Ovarian-1). Anatomically, fourteen cases were located in brain cortex and one (Breast-3) in the cerebellum. The available clinical characteristics of these patients are summarized in Table S1. Fresh tumor sizes ranged from 10–200mg.
Formalin-fixed and paraffin-embedded of 13 cases of brain metastases from melanoma and cancer patients diagnosed at Skåne University Hospital, Sweden, were used for immunohistochemical staining. The ethical permit was obtained from the regional ethical committee at Lund University (Dnr 2019-04998).
Experimental brain metastasis
Female athymic nude or BALB/c mice (4–6 weeks old) were obtained from Charles River (Wilmington, MA, USA) and maintained under specific pathogen-free conditions. The University of California, San Francisco Institutional Animal Care and Use Committee (IACUC) reviewed and approved all animal experiments. MDA-MB-231Br cells were kindly donated by Dr. Patricia S. Steeg (National Cancer Institute, Maryland). We expanded a single clonal MDA-MB-231BR cells expressing luciferase and the mCherry marker (Addgene, Plasmid #29783), cells were harvested, washed and re-suspended in sterile phosphate buffered saline (PBS). 0,25×106/100ml cells were injected into the left ventricle of the heart of each mouse under anesthesia. To stablish a syngeneic brain metastasis model, 0,1×106/100ml cells of parental breast cancer 4T1 cells expressing luciferase and mCherry (Addgene, Plasmid #29783), were intracardiac inoculated in female BALB/c mice, after reaching endpoint, 4T1Br cells were isolated from brain lesions by mCherry expression, grown in culture (4T1Br P1), and again inoculated in mice. This procedure was repeated 3 times. To monitor the growth of brain metastases, mice were intraperitoneally injected with D-luciferin (100 mg/kg), and then imaged and analyzed using the IVIS Spectrum imaging system (Caliper Life Sciences) twice a week after the injection. Following these protocols, mice reached human endpoint (20% body weight, seizures or paralysis) 4–5 weeks for MDA-MB-231BR and 12 days for 4T1Br models. At endpoint, mice were euthanized and perfused with 40 ml of saline buffer via left ventricle to eliminate blood and circulating tumor cells. To mimic the treatment used with human BrM biopsies, fresh brains were dissected and maintained in cold RPMI-1610 medium (Invitrogen), cut into approximately 0.5 mm3 pieces and stored by freezing in 90% FBS and 10% dimethylsulfoxide (DMSO) in liquid nitrogen, the period since euthanasia and cryopreservation ranged from 10 to 15 minutes.
METHOD DETAILS
Sample collection and single cell RNAseq library preparations
Fresh tumor samples were maintained in cold RPMI-1610 medium (Invitrogen) after surgery, cut into approximately 0.5 mm3 pieces and stored by freezing in 90% FBS and 10% dimethylsulfoxide (DMSO) in liquid nitrogen, the period of time since surgical resection to cryopreservation ranged from 30 to 60 minutes. Briefly, the day of single cell RNAseq processing, tissues were washed in cold RPMI to eliminate the remanent DMSO, mechanically chopped with scalpels, placed in culture medium (RPMI with 2% FBS, 5 μg ml−1 insulin (UCSF Cell Culture Facility), 50 ng ml−1 gentamycin (UCSF Cell Culture Facility) containing 0.2 mg ml−1 collagenase-1 (Sigma) and supplemented with 2 U μl−1 DNase for 45 mins. After washing with cold PBS, samples were dissociated into single cells by a gentle treatment with trypsin at 0.0025% (dilution in PBS of original stock) supplemented with 2 U μl−1 DNase for 2 min. After dissociation, residual erythrocytes were lysed with red blood cell lysis buffer for 1 min at room temperature. All samples were filtered through a 70 μm cell strainer. Prior to scRNAseq processing, the dead cells were removed using dead cell removal kit (Miltenyi). Samples were washed twice and resuspended in PBS with 0.05% BSA at a concentration of 1,000 cells per ul. Single-cell libraries were generated via the Chromium Controller and the Single Cell Reagent Kit 3’ v2 (10× Genomics, Pleasanton, CA) according to the manufacturer’s instructions. For each patient, 25,000 cells or the total number of cells from smaller biopsies, were loaded for encapsulation aiming to capture ~10,000 cells. After sequencing, Cell Ranger count (Version 3.0.2) was used to map the FASTQ files to the human genome GRCh38. We achieved a median of 6,798 cells per tumor, with a mean of 79,609 read counts and a median of 2,865 genes per cell. Per sample we detected a median 23,525 total genes. All sequencing metrics are deposited with the corresponding scRNAseq dataset in Gene Expression Omibus (GEO).
Single cell sorting of metastatic cells from experimental models and library preparation
On the day of scRNAseq processing, mouse brain tissues were washed in cold RPMI to eliminate the remanent DMSO, mechanically chopped with scalpels, placed in culture medium (RPMI with 2% FBS, 5 μg ml−1 insulin (UCSF Cell Culture Facility), 50 ng ml−1 gentamycin (UCSF Cell Culture Facility) containing 0.2 mg ml−1 collagenase-1 (Sigma) and supplemented with 2 U μl−1 DNase for 45 mins. After washing with cold PBS, samples were dissociated into single cells by a gentle treatment with trypsin at 0.0025% (dilution in PBS of original stock) supplemented with 2 U μl−1 DNase for 2 min. Afterward, the homogenate was filtered through a 70 mm cell strainer and washed in cold PBS for 5 mins at 300 g. Myelin was removed by gradient centrifugation with 20% / 80% Percoll (Sigma-Aldrich) in PBS (1,200 × g for 30 minutes at 4C; without brakes during deceleration). The middle transparent layer (containing mononuclear cells including tumor cells) was collected and washed in cold PBS for 5 mins at 300g and processed for fluorescence-activated cell sorting (FACS) staining. Briefly, for mouse lineage (Lin) antigens, mCD45 (FITC, eBioscience), mTer119 (FITC, eBioscience), mCD31 (FITC, eBioscience) were used. To detect human cells, hCD298 (PE, Biolegend) was used. The antibody staining for FACS was performed in DMEM/2% FBS. After 30 min on ice, stained cells were washed of excess unbound antibodies and resuspended in DMEM/2% FBS. Viable (Ghost Dye negative) MDA-MB-231Br (hCD298+, mCherry+, mCD45-, mTer119-, mCD31-) cells were sorted by FACS into RPMI with 2% FBS. Similarly, Viable (Ghost Dye negative) 4T1Br cells (mCherry+, mCD45-, mTer119-, mCD31-) were sorted by FACS into RPMI with 2% FBS. After sorting, cells were washed twice and resuspended in PBS with 0.05% BSA at a concentration of 1,000 cells per ul. Single-cell libraries were generated via the Chromium Controller and the Single Cell Reagent Kit 3’ v2 (10x Genomics, Pleasanton, CA) according to the manufacturer’s instructions. For each replicate, we aimed to capture ~5,000 cells. After sequencing, Cell Ranger count (Version 6.1.1) was used to map the FASTQ files to the human GRCh38 or mouse mm10 genomes. All sequencing metrics, mapped counts and raw FASTQ data from experimental models are deposited in Gene Expression Omnibus.
Mass cytometry staining
Cell preparation and viability staining:
Single-cell suspensions (see above) were washed with PBS + 5mM EDTA and re-suspended 1:1 with PBS + 5mM EDTA and 100 mM cisplatin (Enzo Life Sciences) for 60s before quenching 1:1 with PB + 5mM EDTA + 0.5% BSA to determine viability as previously described (cite PMID 22577098). Cells were centrifuged at 500g for 5 min at 4 °C and re-suspended in PBS/EDTA/BSA at a density between 1 × 106 and 10 × 106 cells per ml. Suspensions were fixed for 10 min at room temperature using 1.6% paraformaldehyde (PFA) and washed twice with cell-staining media (CSM, PBS with 0.5% BSA and 0.02% NaN3).
Mass-tag cellular barcoding:
Mass-tag cellular barcoding was performed as previously described (Zunder et al., 2015). Briefly, cells from each sample were barcoded with distinct combinations of stable Pd isotopes in 0.02% saponin in PBS. Cells were washed once with cell-staining media (PBS with 0.5% BSA and 0.02% NaN3) and once with 1× PBS and pooled into a single fluorescence-activated cell sorting (FACS) tube (BD Biosciences). After data collection, each condition was deconvoluted using a single-cell debarcoding algorithm (Zunder et al., 2015).
RNAscope
RNA in situ hybridizations were performed according to the manufacturer’s instructions, using the RNAscope Multiplex Fluorescent kit (Advanced Cell Diagnostics) for paraffin fixed tissue. Following probes with suitable combinations were used (indicated with gene target name for mouse and respective channel, all Advanced Cell Diagnostics): hs-CTHRC1, hs-RGS5 and hs-CLDN5. All sections were mounted with Prolong Diamond Antifade Mountant (Thermo Fisher Scientific).
Histology, Immunohistochemistry, and Immunofluorescence
Human BrM tissue were fixed overnight in 4% paraformaldehyde (PFA) and processed for paraffin embedding tissue sections of 5um were used. Following deparaffinization and rehydration, antigen retrieval was performed by submerging the slides in Antigen Unmasking Solution, Tris-based (Vector Laboratories).
For immunohistochemistry (IHC), endogenous peroxidase activities were inactivated in 3% H2O2 for 5 min at room temperature. Tissue sections were blocked with 1% BSA, 2% goat serum in PBS- 0.2% Triton X-100 for 1 h at room temperature and incubated with primary antibodies for 1 h in blocking buffer. After washes with PBS, biotinylated secondary antibodies were applied for 1 h, followed by treatment with avidin/biotinylated enzyme complex and substrate/chromogen incubation (Vector laboratories). Slides were counterstained with hematoxylin. For immunofluorescence, tissue sections were stained with primary antibodies overnight. After washes with PBS, fluorescently labelled secondary antibodies were used. Nuclei were counterstained with DAPI. Mouse immunofluorescence for mCherry and CD45 were performed in frozen brain sections (20 μm) as described before (Gonzalez et al., 2013). Images were obtained using Keyence BZ-X800 microscope at 20× resolution. Primary antibodies used and dilution ratios are: KRT19 (1:500), MELAN-A (1:200), PECAM1 (1:500), PEG10 (1:200), CD63 (1:250), S100A6 (1:200), anti-hCD45 (1:300), anti-mCD45 (1:200), anti-mCherry (1:100) and anti-Ki67 (1:50).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Pre-processing with Seurat Package
The Seurat pipeline was applied to each sample and for combined analyses (Butler et al., 2018). Genes that were expressed in less than 5 cells and cells that expressing less than 500, or more than 9,000 genes (outliers), or with a percentage of mitochondrial genes higher than 10% were excluded from further analyses. To accurately separate tumor cells and stromal cells in each sample, we merged datasets from different biopsies, this strategy outperforms single sample clustering and efficiently separates stromal cells in samples with higher tumor purities and low stromal fractions (e.g., Melan-2, Breast-3, Lung-1 and Ovarian-1) and also improve the identification of rare cell types (e.g., astrocytes). Following identification with known marker genes, we extracted all stromal cells for each patient.
To reveal the shared sources of biological variation (conserved sub-structures) between BrM-associated stromal cells, we used the integration tool for scRNAseq data sets provided by Seurat package. The top 2,000 highly variable genes of each sample were detected by variance stabilizing transformation method in FindVariableFeatures. Anchors across the 14 datasets were then identified and the datasets were integrated using the default parameters. Before clustering, the number of counts and percentage of mitochondrial genes were regressed out using a negative binomial model (function vars.to.regress). We did not regress cell cycle genes because we did not observe cell cycle genes driving clusters in the stroma. Principal component analysis (PCA) was performed with PCA function and dataset dimensionality kept for downstream analysis was determined heurustically with Elbowplot and ScoreJackStraw functions. A UMAP dimensional reduction was performed on the scaled matrix using the first 18 PCA components to obtain a two-dimensional representation. For clustering, we used the function FindClusters that implements SNN (shared nearest neighbor) modularity optimization-based clustering algorithm on the first 18 PCA components with resolution 0.5 – 1.5. A resolution of 1.3 was chosen for the analysis, in our hands the most informative resolution but the result was generally robust to hyperparameter choices.
For clustering of individual samples, a UMAP dimensional reduction was performed on the scaled matrix using the first 10 PCA components and a FindClusters function was set at resolution of 0.5. For clustering of all combined MTCs, normalized counts of annotated MTCs of all samples were merged, centered, and scaled, dataset dimensionality for downstream analysis was determined heurustically with Elbowplot and ScoreJackStraw functions, finally the UMAP dimensional reduction was performed using the first 20 PCA components.
Identification of Cluster-Specific Genes in Stromal cells and Marker-Based Classification
To identify marker genes, the FindAllMarkers function was used with likelihood-ratio test for single cell gene expression. For each cluster, only genes that were expressed in more than 25% of cells with at least 0.25-fold difference were considered (Top 30 genes per cluster can be found in Table S2). To characterize clusters, we used marker genes and curated gene signatures (Table S2).
CNV analysis
CNV was estimated using R package CONICSmat (Muller et al., 2018). Count matrix was normalized by taking log2(CPM/10+1) and genes expressed in very few cells (< 5) were excluded from the analysis. A normalization factor for calculated for each cell by taking the mean expression of the cell. The average expression in each cell was centered using the previously calculated normalization factor. Then, the z-score of the centered gene expression across all cells was calculated. To quantitatively determine the CNV of cells, we fitted a Gaussian mixture model based on the mean z-scores of 44 chromosome arms (sex chromosomes were not included). Each cell was assigned a posterior probability of belonging to one of the copy number group of each chromosome arm.
To infer subclones in each tumor, we first identified candidate chromosome arms that showed a clear bimodal expression pattern in the cancer cell populations. Chromosome arms were chosen if more than 1000 cells or 20% of the cancer cells belonged to the minor copy number group (posterior probability > 0.95). Then we assigned a genotype of each candidate chromosome arm for each cancer cell using the posterior probabilities. Cancer cells were grouped into clones based on genotypes and clones with more than 1000 cells or 20% of the cancer cells were identified.
For visualization purpose, a sliding window approach was used as previously described (Patel et al., 2014). Briefly, a window of 121 genes within each chromosome was applied to calculate the mean expression of genes sorted by genomic coordinates. Genes with average expression larger than 0.4 were included in the analysis. To limit the influence of any particular gene on the moving average, we limited the relative expression values to [−1, 1] by replacing all values above/below this threshold (x>1/x<(−1)) to 1 /−1. The plotChromosomeHeatmap function was used in heatmap visualization.
Definition of single-cell gene signature scores
Given a set of genes (Gj), for each cell i, a score SCj(i) reflecting the level of expression of Gj in cell i was calculated similar to previously described (Tirosh et al., 2016a) (Seurat AddModuleScore function) (Butler et al., 2018). Briefly, a control gene set is defined by first binning all analyzed genes into 24 bins of aggregate expression levels and then, for each gene in Gj, randomly selecting 100 genes from the same expression bin. The expression score is defined as the average expression levels of Gj subtracted by the aggregated expression of control gene set: SCj(i)=average[ER(Gj,i)] – average[ER(Gjcont,i)].
Diffusion component analysis
We used diffusion map (Coifman et al., 2005), a nonlinear dimensionality reduction approach, to identify the major components of variation across tumor cells. The count matrix of each tumor was first normalized using global scaling log normalization followed by PCA on the highly variable genes selected by mean variability plot method in FindVariableFeatures. We then computed the diffusion components in each tumor using the first 10 principal components as input of the DiffusionMap function of R package destiny 2.14.0 (Angerer et al., 2016). Uninformative components representing isolated clusters with very few cells were not included in the visualization. To identify genes associated with each diffusion component, we calculated the Pearson correlation coefficient between diffusion components and the scaled expression of each gene and retained the top 50 positively/negatively correlated genes. Biological processes and pathways were annotated based on Metascape annotation (Zhou et al., 2019) of the top correlated genes and manual inspection.
Identification of recurrent expression programs in human brain metastases with Non-Negative Matrix Factorization (NMF)
For tumor cells in each sample, Non-Negative Matrix Factorization (Matlab nnmf function, with number of factors set to 10) was applied to the scaled and centered expression data (Seurat ScaleData function), after converting all negative values to zero. For each of the resulting 10 factors, 50 genes with the highest NMF scores were defined as a signature. In order to identify recurrent expression programs across human brain metastasis, we aggregated 150 signatures from 15 samples and did hierarchical clustering using 1 minus Jaccard index as the distance metric. This revealed the 8 meta-programs described in this study. For each meta-program, we calculated the expression score using genes in the meta-program and ranked all the genes by their correlation with the expression score. Each meta-program was then redefined using the top 30 correlated genes. To infer the co-occurrence of programs, Pearson correlation coefficients were calculated between the single-cell gene signature scores of NMF programs.
Principal component analysis
We performed principal components analysis (PCA) on the normalized count data (Seurat NormalizeData function) of all cancer cells. To identify common features across samples and minimize the impact of intertumoral variability, we scaled and centered the data within each sample as previously described (Venteicher et al., 2017). PCA was then performed on the top 2000 highly variable genes selected by Seurat’s variance stabilizing transformation method in FindVariableFeatures. Biological processes and pathways associated with each PC were annotated based on Metascape annotation (Zhou et al., 2019) of genes with top weights and manual inspection. Alternatively, to study the level of interpatient heterogeneity of MTCs, we first merged the cancer cells from all samples, then we scaled and centered the combined normalized counts data, PCA was performed on the top 2000 highly variable genes selected by Seurat’s variance stabilizing transformation method in FindVariableFeatures. Variance explained was calculated for the first 50 PCs using R as follow: variance explained equals to square of standard deviations (SD2) divided by sum of SD2. Standard deviations for all 50 PCs were collected from the Seurat object. Similarly, for individual samples, variance was calculated using the top 50 PCs.
NMF program interrogation on primary tumors
scRNAseq data of primary tumors (Kim et al., 2020; Neftel et al., 2019; Tirosh et al., 2016a; Wu et al., 2021) were downloaded from Single Cell Portal (https://singlecell.broadinstitute.org/single_cell) or Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). Raw counts of annotated cancer cells were normalized and scaled as described in “Data Preprocessing with Seurat Package”. The expression of the top 30 genes of each NMF metaprogram described in this study was then visualized in heatmaps using R.
Cell cycle analysis
Cell cycle phases of cancer cells were inferred based on G1/S and G2/M expression signature using Seurat’s CellCycleScoring function. The Cycling Score was defined as the mean of G1/S and G2/M score. To identify transcriptome difference between cycling and non-cycling cancer cells shared by brain metastasis samples, we utilized a “leave-one-out” approach. Excluding one sample at a time, we performed differential expression analysis 15 times on cycling and non-cycling cells in the remaining samples (Seurat FindMarkers function). Genes expressed in at least 50% of the cycling and non-cycling cells were included in the analysis. We took the intersection of significant genes (LFC > 0.25, Bonferroni corrected p-val < 0.1) of the 15 tests as the final list of shared differentially expressed genes between cycling and non-cycling cells. This approach minimized the impact of specific samples with very large or small number of cycling cells. For the list of shared highly expressed genes in cycling cells, we removed genes annotated as R-HSA-1640170 Cell Cycle genes by Metascape (Zhou et al., 2019).
Correlation analysis in MET500 cohort
We downloaded the log2(fpkm) RNAseq data of MET500 from UCSC Xena (Goldman et al., 2020). We calculated the MimmScore, an aggregate measure of immune infiltration, of all samples in MET500 as described in the original publication (Robinson et al., 2017). Briefly, inverse normal transformation was applied to the 141 immune signature genes (Yoshihara et al., 2013). The MimmScore was calculated by summing the normalized expression of these genes. Pearson correlation coefficients were calculated between the inverse normal transformed expression of the mentioned signatures and the MimmScore.
We used the CIBERSORTx tool (Newman et al., 2019), an updated version of the CIBERSORT tool used by the original publication, to estimate the relative proportions of 22 types of infiltrating immune cells in MET500 samples. CIBERSORTx uses a set of reference gene expression values representing each cell type (signature matrix). We used LM22 downloaded from CIBERSORTX, a 547 gene expression matrix distinguishing 22 human hematopoietic cell phenotypes, as the signature matrix. B-mode batch correction was applied to minimize the platform difference. We ran CIBERSORTx using the default setting with 1000 permutations. Only samples with high immune composition estimation confidence (empirical p-val <0.05) were included in the correlation between the expression of mentioned signatures.
Single cell RNAseq analysis of experimental brain metastasis
The Seurat pipeline was applied to each experimental metastasis sample (Butler et al., 2018). Genes that were expressed in less than 5 cells and cells that expressing less than 500, or more than 9,000 genes (outliers), or with a percentage of mitochondrial genes higher than 10% were excluded from further analyses. For the model 4T1Br (syngeneic BrM model), the count matrix of cells that passed the quality control was then normalized using global scaling log normalization followed by PCA on the variable genes selected by mean variability plot method in FindVariableFeatures. UMAP dimensionality reduction (Becht et al., 2018) was used to visualize cells in the 2D space with the first 10 PCs as input, this step allows the identification and exclusion of contaminating residual immune or non-immune stromal cells before proceed with NMF pipeline.
As previously described in human data analysis, for both experimental models, Non-Negative Matrix Factorization (Matlab nnmf function, with number of factors set to 10) was applied to scaled and centered expression data (Seurat ScaleData function), after converting all negative values to zero. For each of the resulting 10 factors, 50 genes with the highest NMF scores were defined as a mouse program, followed by functional annotation evaluated by Metascape (Zhou et al., 2019), gene lists with no significant biological processes were excluded.
RNA velocity analysis
We used scVelo 0.2.2 (Bergen et al., 2020) to subpopulation kinetics in myeloid cells with the default setting. ScVelo is a more robust and scalable inference of the original RNA velocity (La Manno et al., 2018). Briefly, we used the loom files prepared by the Cell Ranger, filtered lowly expressed genes with less than 30 counts, and kept top 2000 highly variable genes for downstream analysis. RNA velocities were inferred and projected as field onto the pre-computed diffusion map embedding for visualization. This pipeline was implemented in Python 3.6.9.
Mass cytometry analyses
Cell staining and preparation:
Cells were resuspended in cell-staining media (CSM, PBS with 0.5% BSA and 0.02% NaN3), and human Fc block (Biolegend) was added for 5 min at room temperature on a shaker to block Fc receptors. Surface marker antibodies were then added, and cells were stained in a final colume of 400 ul of CSM for 30 min at room temperature on a shaker. After staining, cells were washed two times with cell-staining media and then permeabilized with transcription factor permeabilization solution (eBioscience, Thermo Fisher). Cells were then stained with intracellular antibodies in a final volume of 400 ul of transcription factor permeabilization solution for 30 min at room temperature on a shaker. Cells were washed once in transcription factor permeabilization solution and once in CSM and then stained with 1 ml of 1:4,000 191/193Ir DNA intercalator (Fluidigm) diluted in PBS with 1.6% PFA overnight. Cells were then washed once with cell-staining media and then two times with double-deionized (dd) water. Care was taken to assure that buffers preceding analysis were not contaminated with metals in the mass range above 100 Da. Mass cytometry samples were diluted in dd water containing bead standards (see below) to approximately 106 cells per ml and then analyzed on a CyTOF 2 mass cytometer (Fluidigm) equilibrated with dd water.
Mass cytometry bead standard data normalization:
Data normalization was performed as previously described (Finck et al., 2013). Briefly, just before analysis, the stained and intercalated cell pellet was resuspended in freshly prepared dd water containing the bead standard (Fluidigm). The mixture of beads and cells was filtered through filter cap FACS tubes (BD Biosciences) before analysis. All mass cytometry files were normalized together using the mass cytometry data normalization algorithm (same citation as above), which uses the intensity values of a sliding window of these bead standards to correct for instrument fluctuations over time and between samples.
Mass cytometry gating strategy and analysis:
After normalization and debarcoding of files, singlets were gated by Event Length and DNA. Live cells were identified as cisplatin-negative cells. All positive and negative populations and antibody-staining concentrations were determined by titration on positive and negative control cell populations. Gating was performed using CellEngine software, and downstream analyses performed in R.
Clustering:
Manually gated CD4 and CD8 T cells were downloaded as FCS files from CellEngine. The R package Premessa (https://github.com/ParkerICI/premessa/) was used to harmonize panels between experiments and flowCore (Hahne et al., 2009) was used to import FCS files into R. The FlowSOM clustering algorithm (Van Gassen et al., 2015), available through the CATALYST R/Bioconductor package (Chevrier et al., 2018), was used to generate clusters based on CD4 and CD8 T cell specific markers. Plots were produced with the R packages CATALYST and ggplot2.
Supplementary Material
Figure S1. Molecular Survey of the Human Brain Metastatic Niche, Related to Figure 1 (A) Single sample clustering visualized in UMAP plots for all samples included in the study. Cells are colored by Seurat clusters (STAR Methods). (B) IHC staining showing the expression of KRT19 (carcinoma cells) or Melan-A (melanoma cells) in BrMs (Scale bars, 60 μm). (C) IHC staining showing the expression of GFAP in selected BrMs (Scale bars, 100 μm). In carcinoma and melanoma BrMs, astrocytes represented 1.04% and 1.1% of the total stromal fraction, respectively, confirmed by immunostaining (Figure S1C) and in line with other studies (Priego et al., 2018). (D) Separated visualization of melanoma and carcinoma BrM-associated non-malignant cells. Cells are colored by sample. (E) Quantification and comparisons of non-malignant cell type composition in melanomas and carcinoma BrMs. (F) UMAP projection of BrM-associated stromal cells including immune and non-immune fractions, ambiguous clusters that were excluded from further analyses are shown. (G) Visualization of 6,325 cells from a case of parenchymal rhabdomyosarcoma BrM using UMAP embedding. See also Figure 1.
Figure S2. Cell Types That Integrate the Blood-Tumor Interface in Patients, Related to Figure 2 (A) Spatial validation of RGS5+ cells using multiplex ISH (RNAscope). One representative staining in sample Melan-2 is shown. Same patterns were observed in lung, breast and ovarian BrMs. Scale bar, 75 μm. (B) Immunohistochemical staining of the endothelial marker PECAM1 (CD31) in 6 BrM samples is shown. Scale bars, 50 μm. (C) Heatmap of mural vascular cells and mesenchymal progenitor clusters, each containing a unique set of signature genes, with red arrows pointing to selected markers. Information of the distribution across samples is colored for each cell. (D) Spatial validation of CTHRC1+ cells using multiplex ISH (RNAscope). One representative staining in sample Breast-2 is shown. Scale bar, 75 μm. (E) Violin plots show key differentially expressed markers that differentiate between mural vascular cells (RGS5+) and mesenchymal progenitor markers (CTHRC1+ and ISLR+). (F) UMAP projection of 3,292 endothelial cells colored by clusters. The arteriovenous axis is shown. (G) Bar charts show the confirmation that markers GJA5 and ACKR1 are expressed in arterial and venous endothelial cells respectively using the molecular atlas of brain vasculature (Vanlandewijck et al., 2018). (H) Heatmap reporting the expression of top 10 marker genes in endothelial clusters. (I-J) Heatmaps reporting the average expression of curated gene signatures in each endothelial cell cluster in all and individual samples. (See also Figure 2).
Figure S3. Details of Immune Annotations and Immune States, Related to Figure 3 (A) Frequencies of the indicated immune clusters in BrM samples separated by cancer type. (B) Heatmap reporting the average expression of curated gene signatures used for T cell annotation (Table S2) and selected markers in each T cell cluster. (C) Heatmap of immune clusters described in this study, each containing a unique set of signature genes. Selected top differentially expressed markers on T cells are highlighted. Information of the distribution across samples is colored for each cell. (D) UMAP projections of scaled expression of selected immune markers. (E) Heatmap reporting the average expression of the top 50 positively correlated genes with diffusion component 1 in each T cell cluster. (F) Biological processes and pathways associated with the top 50 differentially expressed genes between the two macrophages clusters, processes were annotated based on Metascape (Zhou et al., 2019). See also Figure 3 and Table S2.
Figure S5. Characterization of Transcriptional and Genetic Diversity of Metastatic Cells, Related to Figure 5 (A) Visualization of Metastatic Tumor Cells (MTCs) using UMAP embedding. Cells are colored by sample (STAR Methods). (B) Percentage of variance explained for the top 25 PCA components (STAR Methods). (C) 3D projections represent the top 3 diffusion components for malignant cells from samples Breast-1, Lung-1 and Melan-2. The trajectories were functionally annotated using the top 50 positively/negatively correlated genes. Bottom panels show the normalized expression of selected markers projected in 3D diffusion map plots. (D) Percentage of variance explained for the top 2 PCA components for the MTCs of samples analyzed in figures 5A and S5C. (E) Heatmap showing the inferred CNV profiles in metastatic cells and their comparison with non-malignant cells. This analysis shows that MTCs are predominantly clonal and that metaprograms observed can’t be explained by genetic diversity. We identified subclones in samples Melan-3, Lung-1 and Colorectal. (F) Sub-clonal populations in samples Melan-3 (gain in chromosome 8), Lung-1 (grain in chromosome 2) and Colorectal (loss in chromosome 10), revealed by inferred CNV profiles. Black arrows point to affected chromosomes. See also Figure 5.
Figure S6. Characterization of Metastatic Cell Archetypes, Related to Figure 5 (A, B) Scatter plots comparing the median score value on MTCs for metaprograms P8 or P2, and the composition of immune cell clusters in each sample. (C) Heatmaps showing the expression of the top 30 genes of each MTC metaprogram on primary tumors, using publically available single cell datasets (See STAR Methods). See also Figure 5.
Figure S7. Case Study: Interrogation of the MDA-MB-231Br and 4T1 Br Models in Recapitulating in vivo the Functional Transcriptional Programs Observed in Patients, Related to Figure 5 (A) Heatmap showing the normalized expression of mouse NMF programs identified by analyzing 3,194 MDA-MB-231Br cells at endpoint (4 weeks) (STAR Methods). (B) Annotation and selected top genes for each NMF program identified in the MDA-MB-231Br model. (C) Schematic illustration of the murine syngeneic 4T1Br brain metastasis model and scRNAseq analysis approach (STAR Methods). (D) Heatmap showing the normalized expression of mouse NMF programs identified by analyzing 1,472 4T1Br cells at endpoint (12 days). (E) Annotation and selected top genes for each NMF program identified in the 4T1Br model. (F) Immunofluorescent staining in floating mouse brain sections of the marker mCherry expressed by metastatic tumor cells and CD45. Nuclei were counterstained with DAPI. Scale bars, 100 μm. See also Figure 5 and Table S4.
Table S1: Clinical characteristics of the patients, Related to Figure 1
Figure S4. Mass Cytometry Analysis of BrM-associated CD4+ T Cells, Related to Figure 4 (A) CD4+ T cell population clustered by FlowSOM and visualized by UMAP (see STAR Methods). (B) Functional and phenotypic median expression profiles for each CD4+ T cell clusters. (C) CD4+ T cell cluster proportions by patients. See also Figure 4 and Table S3.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Full list of CyTOF antibodies | This paper | Table S3 |
| Mouse CD45-FITC | Biolegend | Cat# 103108 |
| Mouse Ter119-FITC | eBioscience | Cat# 11-5921-82 |
| Mouse CD31-FITC | eBioscience | Cat# 11-0311-85 |
| Human CD298-PE | Biolegend | Cat# 341704 |
| Human KRT19 | Sigma-Aldrich | Cat# HPA002465 |
| Human PECAM1/CD31 | Invitrogen | Cat# MA5-16337 |
| Human PEG10 | Proteintech | Cat# 14412-I-AP |
| Human CD63 | Sigma-Aldrich | Cat# HPA010088 |
| Human S100A6 | Proteintech | Cat# 10245-I-AP |
| Human CD45 | DACO Agilent | Cat# GA75161-2 |
| Human Ki67 | DACO Agilent | Cat# GA62661-2 |
| Human Melan-A | Invitrogen | Cat# MA5-14168 |
| Mouse CD45 | Thermo Fisher | Cat# BDB550539 |
| Anti-mCherry | Abcam | Cat# Ab167453 |
| Biological samples | ||
| Fresh Human Brain Metastases | This Study | Table S1 |
| Critical commercial assays | ||
| Chromiun Controller and the Single Cell Reagent kit 3’ v2 | 10X Genomics | Cat# PN-120237 |
| RNAscope Fluorescent Mutiplex Assay | ACD | Cat# 320850 |
| Deposited data | ||
| Raw and analyzed data | This paper | GSE186344 |
| MET500 Cohort Dataset | (Robinson et al., 2017) | http://met500.path.med.umich.edu |
| Glioblastoma | (Neftel et al., 2019) | GSE131928 |
| Primary Breast Cancer | (Wu et al., 2021) | GSE176078 |
| Primary Lung Cancer | (Kim et al., 2020) | GSE131907 |
| Primary Melanoma | (Tirosh et al., 2016a) | GSE72056, GSE77940 |
| Experimental models: Cell lines | ||
| MDA-MB-231BR | (Zhang et al., 2015) | NA |
| 4T1 | ATCC | CRL-2539 |
| Experimental models: Organisms/strains | ||
| Female Nude mice | Charles River | Strain Code: 088 |
| Female BALB/c mice | Charles River | Strain Code: 028 |
| Recombinant DNA | ||
| PLKO-mCherry-Luc | Addgene | Cat# 29783 |
| Software and algorithms | ||
| Seurat version: 4.0.0 | (Butler et al., 2018) | https://satijalab.org/seurat/index.html |
| R version: 4.0.3 | NA | https://cran.r-project.org/ |
| Cell Ranger version 3.0.2 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/tutorial_ov |
| CONICSmat | (Muller et al., 2018) | https://github.com/diazlab/CONICS |
| Destiny Package version 2.14.0 | (Angerer et al., 2016) | https://github.com/theislab/destiny |
| CIBERSORTx tool | (Newman et al., 2019) | https://cibersortx.stanford.edu/ |
| PRINSEQ-lite 0.20.4 | NA | https://edwards.sdsu.edu/cgi-bin/prinseq/prinseq.cgi |
| STAR 2.7.5a | NA | https://github.com/alexdobin/STAR |
| GSVA | (Hanzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| scVelo 0.2.2 | (Bergen et al., 2020) | https://pypi.org/project/scvelo/ |
| CellEngine | NA | https://cellcarta.com/cellenginesoftware/ |
Highlights.
Single-cell analysis of human BrMs, reveal conserved features across multiple cancers.
Metastasis-associated macrophages, exhibit two well defined states.
Eight metaprograms define two coexisting archetypes of metastatic tumor cells (MTCs).
An immune evasive program supports MTCs proliferation.
ACKNOWLEDGMENTS
This study is dedicated to the memory of our friend, teacher, and mentor Zena Werb, who first envisioned the potential of this study. Our deepest gratitude for her encouragement, support, brilliance, and kindness. We thank the patients and their families for their generosity in contributing to this study, surgeons and all members of the UCSF Brain Tumor SPORE Biorepository, and patient advocate Susan Samson for insightful discussions. This study was supported by grants from the National Cancer Institute (CA057621, CA180039, CA199315, CA190851 and 5U01CA199315-05 to Z.W.) the NIH Office of the Director (OD023056 to M.H.S.), and Oncology Research Fund (to Z.W. and H.G.). General infrastructure in the Roose lab has been supported by grants from the NIH/NCI (R01 - CA187318), NIH/NIAID (R01-AI104789 and P01-AI091580), and the NIH/NHLBI (R01 - HL120724) (all to JPR). This specific project was supported by a Mark Foundation for Cancer Research Endeavor Program grant (A136299) and California Breast Cancer Research grants (B26IB1494) (both to JPR). The Becas Chile Post-Doctoral Fellowship (to H.G.), the Parker Institute for Immunotherapy (to M.H.S and Z.W.); The Emerson Collective Cancer Research Fund (to H.G and Z.W); The National Institutes of Health (5P50CA097257-18) (to J.J.P); by funds from Swedish Society of Medicine, Gunnar Nilsson Cancer Foundation, Swedish Society for Medical Research and governmental funding of clinical research within the National Health Services (ALF) (to C.H). The Carlsberg Foundation (to T.L.H.O).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
REFERENCES
- Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, Peters S, Arvold ND, Harsh GR, Steeg PS, et al. (2019). Brain metastases. Nat Rev Dis Primers 5, 5. [DOI] [PubMed] [Google Scholar]
- Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. [DOI] [PubMed] [Google Scholar]
- Arvanitis CD, Ferraro GB, and Jain RK (2020). The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer 20, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308 e1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Bessho Y, Hojo M, and Kageyama R (2000). The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development 127, 2933–2943. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. [DOI] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bova GS (2017). Cancer genomics: Human metastases under scrutiny. Nature 548, 287–288. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, et al. (2016). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, and Bodenmiller B (2018). Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst 6, 612–620 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coifman RR, Lafon S, Lee AB, Maggioni M, Nadler B, Warner F, and Zucker SW (2005). Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proc Natl Acad Sci U S A 102, 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, and Bendall SC (2013). Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebel E, Kapolou K, Unger S, Nunez NG, Utz S, Rushing EJ, Regli L, Weller M, Greter M, Tugues S, et al. (2020). Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642 e1620. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Contreras F, Prado C, Elgueta D, Franz D, Bernales S, and Pacheco R (2013). Dopamine receptor D3 expressed on CD4+ T cells favors neurodegeneration of dopaminergic neurons during Parkinson’s disease. J Immunol 190, 5048–5056. [DOI] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, and Werb Z (2018a). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Robles I, and Werb Z (2018b). Innate and acquired immune surveillance in the postdissemination phase of metastasis. FEBS J 285, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gril B, Paranjape AN, Woditschka S, Hua E, Dolan EL, Hanson J, Wu X, Kloc W, Izycka-Swieszewska E, Duchnowska R, et al. (2018). Reactive astrocytic S1P3 signaling modulates the blood-tumor barrier in brain metastases. Nat Commun 9, 2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerling C, Gonzalez H, Salari K, Wang CY, Lin C, Robles I, van Gogh M, Dejmek A, Jirstrom K, and Werb Z (2019). Immune effector monocyte-neutrophil cooperation induced by the primary tumor prevents metastatic progression of breast cancer. Proc Natl Acad Sci U S A 116, 21704–21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Buettner F, and Theis FJ (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998. [DOI] [PubMed] [Google Scholar]
- Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, and Gentleman R (2009). flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 10, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall WA, Djalilian HR, Nussbaum ES, and Cho KH (2000). Long-term survival with metastatic cancer to the brain. Med Oncol 17, 279–286. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, and Krummel MF (2016). Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirashima M, and Suda T (2006). Differentiation of arterial and venous endothelial cells and vascular morphogenesis. Endothelium 13, 137–145. [DOI] [PubMed] [Google Scholar]
- Ishii S, Yamashita K, Harada H, Ushiku H, Tanaka T, Nishizawa N, Yokoi K, Washio M, Ema A, Mieno H, et al. (2017). The H19-PEG10/IGF2BP3 axis promotes gastric cancer progression in patients with high lymph node ratios. Oncotarget 8, 74567–74581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp MA, Slotty PJ, Cornelius JF, Steiger HJ, Rapp M, and Sabel M (2018). The impact of cerebral metastases growth pattern on neurosurgical treatment. Neurosurg Rev 41, 77–86. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, and Werb Z (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, Lee JI, Suh YL, Ku BM, Eum HH, et al. (2020). Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C, Tabar V, et al. (2020). Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660 e1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak T, Wang F, Deng H, Condamine T, Kumar V, Perego M, Kossenkov A, Montaner LJ, Xu X, Xu W, et al. (2020). Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep 33, 108571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169, 750–765 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xiao R, Tembo K, Hao L, Xiong M, Pan S, Yang X, Yuan W, Xiong J, and Zhang Q (2016). PEG10 promotes human breast cancer cell proliferation, migration and invasion. Int J Oncol 48, 1933–1942. [DOI] [PubMed] [Google Scholar]
- Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, Zahnow CA, Nickels ML, Liu F, Tantawy MN, et al. (2018). DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun 9, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Enomoto A, Hara A, Asai N, Kobayashi T, Horinouchi A, Maruyama S, Ishikawa Y, Nishiyama T, Kiyoi H, et al. (2016). Identification of Meflin as a Potential Marker for Mesenchymal Stromal Cells. Sci Rep 6, 22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Cho A, Liu SJ, Lim DA, and Diaz A (2018). CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones. Bioinformatics 34, 3217–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieblas-Bedolla E, Nayyar N, Singh M, Sullivan RJ, and Brastianos PK (2021). Emerging Immunotherapies in the Treatment of Brain Metastases. Oncologist 26, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IS, Dervan PA, Broderick D, Harrison M, Miller N, Delany E, O’Shea D, Costello P, McGoldrick A, Keating G, et al. (1999). Frequent loss of imprinting of PEG1/MEST in invasive breast cancer. Cancer Res 59, 5449–5451. [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, Doglio L, Martinez L, Martinez-Saez E, Ramon YCS, et al. (2018). STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 24, 1024–1035. [DOI] [PubMed] [Google Scholar]
- Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, Duyvesteyn K, Haidari S, van Hoeck A, Onstenk W, et al. (2019). Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, and Pollard JW (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JG, Hung WT, Lee IH, Nagpal S, Giunta P, Degner S, Liu G, Wassenaar ECE, Jeck WR, Taylor MS, et al. (2020). Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nat Genet 52, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. (2017). Integrative clinical genomics of metastatic cancer. Nature 548, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, Ashenberg O, Cerami E, Coffey RJ, Demir E, et al. (2020). The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, et al. (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol 22, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalova M, Lee JK, Li Y, Vander Griend DJ, Coleman IM, Nelson PS, Dehm SM, and LeBeau AM (2019). PEG10 Promoter-Driven Expression of Reporter Genes Enables Molecular Imaging of Lethal Prostate Cancer. Cancer Res 79, 5668–5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Jiang YZ, Wei Y, Ell B, Sheng X, Esposito M, Kang J, Hang X, Zheng H, Rowicki M, et al. (2019). Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell 35, 64–80 e67. [DOI] [PubMed] [Google Scholar]
- Shojania KG, Burton EC, McDonald KM, and Goldman L (2003). Changes in rates of autopsy-detected diagnostic errors over time: a systematic review. JAMA 289, 2849–2856. [DOI] [PubMed] [Google Scholar]
- Sprowls SA, Arsiwala TA, Bumgarner JR, Shah N, Lateef SS, Kielkowski BN, and Lockman PR (2019). Improving CNS Delivery to Brain Metastases by Blood-Tumor Barrier Disruption. Trends Cancer 5, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, et al. (2014). Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al. (2020). Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol 21, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal AC, Henry NM, Murphy SK, Oneko O, Nye M, Bartlett JA, Overcash F, Huang Z, Wang F, Mlay P, et al. (2014). PEG1/MEST and IGF2 DNA methylation in CIN and in cervical cancer. Clin Transl Oncol 16, 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner NB, Weide B, Gries M, Reith M, Tarnanidis K, Schuermans V, Kemper C, Kehrel C, Funder A, Lichtenberger R, et al. (2019). Tumor microenvironment-derived S100A8/A9 is a novel prognostic biomarker for advanced melanoma patients and during immunotherapy with anti-PD-1 antibodies. J Immunother Cancer 7, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, Menk AV, Rittenhouse NL, DePeaux K, Whetstone RD, et al. (2021). Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N, et al. (2021). A single-cell and spatially resolved atlas of human breast cancers. Nat Genet 53, 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Nishimura O, Misaki K, Nishita M, Minami Y, Yonemura S, Tarui H, and Sasaki H (2008). Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev Cell 15, 23–36. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galvan JA, Robinson HPC, Zlobec I, et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459 e429. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, et al. (2015). Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845 e820. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Eichten A, Parveen A, Adler C, Huang Y, Wang W, Ding Y, Adler A, Nevins T, Ni M, et al. (2018). Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res 78, 2370–2382. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunder ER, Finck R, Behbehani GK, Amir el AD, Krishnaswamy S, Gonzalez VD, Lorang CG, Bjornson Z, Spitzer MH, Bodenmiller B, et al. (2015). Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc 10, 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Molecular Survey of the Human Brain Metastatic Niche, Related to Figure 1 (A) Single sample clustering visualized in UMAP plots for all samples included in the study. Cells are colored by Seurat clusters (STAR Methods). (B) IHC staining showing the expression of KRT19 (carcinoma cells) or Melan-A (melanoma cells) in BrMs (Scale bars, 60 μm). (C) IHC staining showing the expression of GFAP in selected BrMs (Scale bars, 100 μm). In carcinoma and melanoma BrMs, astrocytes represented 1.04% and 1.1% of the total stromal fraction, respectively, confirmed by immunostaining (Figure S1C) and in line with other studies (Priego et al., 2018). (D) Separated visualization of melanoma and carcinoma BrM-associated non-malignant cells. Cells are colored by sample. (E) Quantification and comparisons of non-malignant cell type composition in melanomas and carcinoma BrMs. (F) UMAP projection of BrM-associated stromal cells including immune and non-immune fractions, ambiguous clusters that were excluded from further analyses are shown. (G) Visualization of 6,325 cells from a case of parenchymal rhabdomyosarcoma BrM using UMAP embedding. See also Figure 1.
Figure S2. Cell Types That Integrate the Blood-Tumor Interface in Patients, Related to Figure 2 (A) Spatial validation of RGS5+ cells using multiplex ISH (RNAscope). One representative staining in sample Melan-2 is shown. Same patterns were observed in lung, breast and ovarian BrMs. Scale bar, 75 μm. (B) Immunohistochemical staining of the endothelial marker PECAM1 (CD31) in 6 BrM samples is shown. Scale bars, 50 μm. (C) Heatmap of mural vascular cells and mesenchymal progenitor clusters, each containing a unique set of signature genes, with red arrows pointing to selected markers. Information of the distribution across samples is colored for each cell. (D) Spatial validation of CTHRC1+ cells using multiplex ISH (RNAscope). One representative staining in sample Breast-2 is shown. Scale bar, 75 μm. (E) Violin plots show key differentially expressed markers that differentiate between mural vascular cells (RGS5+) and mesenchymal progenitor markers (CTHRC1+ and ISLR+). (F) UMAP projection of 3,292 endothelial cells colored by clusters. The arteriovenous axis is shown. (G) Bar charts show the confirmation that markers GJA5 and ACKR1 are expressed in arterial and venous endothelial cells respectively using the molecular atlas of brain vasculature (Vanlandewijck et al., 2018). (H) Heatmap reporting the expression of top 10 marker genes in endothelial clusters. (I-J) Heatmaps reporting the average expression of curated gene signatures in each endothelial cell cluster in all and individual samples. (See also Figure 2).
Figure S3. Details of Immune Annotations and Immune States, Related to Figure 3 (A) Frequencies of the indicated immune clusters in BrM samples separated by cancer type. (B) Heatmap reporting the average expression of curated gene signatures used for T cell annotation (Table S2) and selected markers in each T cell cluster. (C) Heatmap of immune clusters described in this study, each containing a unique set of signature genes. Selected top differentially expressed markers on T cells are highlighted. Information of the distribution across samples is colored for each cell. (D) UMAP projections of scaled expression of selected immune markers. (E) Heatmap reporting the average expression of the top 50 positively correlated genes with diffusion component 1 in each T cell cluster. (F) Biological processes and pathways associated with the top 50 differentially expressed genes between the two macrophages clusters, processes were annotated based on Metascape (Zhou et al., 2019). See also Figure 3 and Table S2.
Figure S5. Characterization of Transcriptional and Genetic Diversity of Metastatic Cells, Related to Figure 5 (A) Visualization of Metastatic Tumor Cells (MTCs) using UMAP embedding. Cells are colored by sample (STAR Methods). (B) Percentage of variance explained for the top 25 PCA components (STAR Methods). (C) 3D projections represent the top 3 diffusion components for malignant cells from samples Breast-1, Lung-1 and Melan-2. The trajectories were functionally annotated using the top 50 positively/negatively correlated genes. Bottom panels show the normalized expression of selected markers projected in 3D diffusion map plots. (D) Percentage of variance explained for the top 2 PCA components for the MTCs of samples analyzed in figures 5A and S5C. (E) Heatmap showing the inferred CNV profiles in metastatic cells and their comparison with non-malignant cells. This analysis shows that MTCs are predominantly clonal and that metaprograms observed can’t be explained by genetic diversity. We identified subclones in samples Melan-3, Lung-1 and Colorectal. (F) Sub-clonal populations in samples Melan-3 (gain in chromosome 8), Lung-1 (grain in chromosome 2) and Colorectal (loss in chromosome 10), revealed by inferred CNV profiles. Black arrows point to affected chromosomes. See also Figure 5.
Figure S6. Characterization of Metastatic Cell Archetypes, Related to Figure 5 (A, B) Scatter plots comparing the median score value on MTCs for metaprograms P8 or P2, and the composition of immune cell clusters in each sample. (C) Heatmaps showing the expression of the top 30 genes of each MTC metaprogram on primary tumors, using publically available single cell datasets (See STAR Methods). See also Figure 5.
Figure S7. Case Study: Interrogation of the MDA-MB-231Br and 4T1 Br Models in Recapitulating in vivo the Functional Transcriptional Programs Observed in Patients, Related to Figure 5 (A) Heatmap showing the normalized expression of mouse NMF programs identified by analyzing 3,194 MDA-MB-231Br cells at endpoint (4 weeks) (STAR Methods). (B) Annotation and selected top genes for each NMF program identified in the MDA-MB-231Br model. (C) Schematic illustration of the murine syngeneic 4T1Br brain metastasis model and scRNAseq analysis approach (STAR Methods). (D) Heatmap showing the normalized expression of mouse NMF programs identified by analyzing 1,472 4T1Br cells at endpoint (12 days). (E) Annotation and selected top genes for each NMF program identified in the 4T1Br model. (F) Immunofluorescent staining in floating mouse brain sections of the marker mCherry expressed by metastatic tumor cells and CD45. Nuclei were counterstained with DAPI. Scale bars, 100 μm. See also Figure 5 and Table S4.
Table S1: Clinical characteristics of the patients, Related to Figure 1
Figure S4. Mass Cytometry Analysis of BrM-associated CD4+ T Cells, Related to Figure 4 (A) CD4+ T cell population clustered by FlowSOM and visualized by UMAP (see STAR Methods). (B) Functional and phenotypic median expression profiles for each CD4+ T cell clusters. (C) CD4+ T cell cluster proportions by patients. See also Figure 4 and Table S3.
Data Availability Statement
Raw mapped counts from patients, raw mapped counts and FASTQ data from experimental mouse models, and normalized expression data with cell type annotations can be found in the National Center for Biotechnology information Gene Expression Omibus (GEO) (accession number GSE186344).
We did not generate original codes in this study. Codes used are available upon request without restrictions.
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.